Back to Journals » International Journal of Women's Health » Volume 6
Refractory fallopian tube carcinoma – current perspectives in pathogenesis and management
Authors Sama A, Schilder R
Received 28 September 2013
Accepted for publication 28 November 2013
Published 28 January 2014 Volume 2014:6 Pages 149—157
DOI https://doi.org/10.2147/IJWH.S40889
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Ashwin R Sama, Russell J Schilder
Department of Medical Oncology, Thomas Jefferson University, Philadelphia, PA, USA
Abstract: Fallopian tube carcinoma (FTC) is considered a rare malignancy, but recent evidence shows that its incidence may have been underestimated. Risk-reducing salpingo-oophorectomy (RRSO) in breast cancer susceptibility gene (BRCA)-positive women has provided a unique opportunity to study the pathogenesis of FTC and ovarian carcinomas. Newer data now suggest that most high-grade serous cancers of the ovary originate in the fimbrial end of the fallopian tube. Due to the presumed rarity of FTC, most current and more recent ovarian cancer clinical trials have now included patients with FTC. The treatment guidelines recommend similar overall management and that the same chemotherapy regimens be used for epithelial ovarian cancers and FTC.
Keywords: high grade serous cancers, serous tubal intraepithelial carcinomas, fallopian tube carcinoma
Introduction
Fewer than 500 patients are diagnosed with fallopian tube carcinoma (FTC) every year, representing about 0.2% of cancers diagnosed in women in the United States annually.1 The incidence rate of FTC is between 3.72 and 4.1 per one million women in the United States.1,2 The incidence rate is higher in Caucasian women than in women of African American, Hispanic, or Asian descent.1 Recent evidence shows that the incidence of FTC may have been grossly underestimated.3 In this article, we will highlight the recent literature and understanding of the pathogenesis of FTC that explains the underestimation of its incidence, and the current perspectives in the management of FTC.
Pathogenesis
The origin of epithelial ovarian cancer (EOC) was thought to be the neoplastic transformation of cells in the cortical inclusion cysts of the ovary.4–6 The incessant ovulation theory postulated that there is a stepwise accumulation of genetic mutations in the ovarian epithelial lining as a result of repeated damage and repair during ovulation, leading to EOC.6–9 However, it has never been demonstrated conclusively that there exists a precursor lesion in the ovary that progresses to high-grade serous cancer (HGSC).10,11 Another hypothesis is that HGSC originates, not from the surface of the ovary but, from the epithelial layer of the neighboring fimbrial end of the fallopian tube.3–5,8,12–27 Newer data now suggest that HGSC of ovary, primary peritoneal carcinoma (PPC), and FTC have similar pathogenesis and molecular biomarkers and that these cancers should more accurately be called pelvic serous carcinomas.14,28 The epidemiological data also support a common etiology of ovarian, fallopian, and primary peritoneal cancers. There are racial, ethnic, and geographic similarities in the incidence of these three cancers, and the higher incidence of all three cancers in non-Hispanic white women further suggests a common origin.2
In general, most epithelial cancers in the body arise as a consequence of a series of events and progress from a benign epithelium to an invasive cancer.18 A proposed stepwise sequence to the development of invasive cancers in the fallopian tube is shown in Figure 1.4,8,16,19,29
 | Figure 1 A proposed stepwise progression to invasive cancer in the fallopian tube. |
The earliest abnormality noted in the fallopian tube epithelium is called secretory cell outgrowth (SCOUT). SCOUTs are discretely localized alterations commonly containing an altered expression of multiple genes within a histologically benign tubal epithelium. Immunohistochemistry shows low expression of paired box 2 (PAX2), low Ki67 index, and, in most cases, no tumor protein p53 (TP53) gene mutations.19 Next, somatic tumor protein 53 (p53) mutations arising from deoxyribonucleic acid (DNA) damage appear in the benign tubal epithelium, with foci of strong p53 immunostaining, termed “p53 signatures”.30 The “p53 signature” is defined as 12 or more consecutive cells with strongly positive p53 nuclear staining within a benign-appearing epithelium.15,16,18 In the presence of a p53 signature, proliferative lesions appear, which are called serous tubal intraepithelial lesions (STILs)31 or transitional intraepithelial lesions of the tube (TILTs).22 These lesions are dysplastic and give rise to serous tubal intraepithelial carcinoma (STIC), also called tubal intraepithelial carcinoma (TIC), which is a precursor for invasive cancer.4,8,31 STIC is composed of secreting cells in the distal fallopian tube, with cytologic atypia, a high proliferative index, and strong nuclear staining for p53.17 The pathogenesis of HGSC from ovarian surface epithelium is unclear as a similar step-wise carcinogenic sequence has never been identified. Recent observations show that STIC may be a precursor lesion for most fallopian tube, ovarian, and peritoneal HGSCs.5,8,14,25
The conventional pathologic classification of pelvic serous cancers mostly as ovarian cancer has contributed to the underreporting of FTCs. FTC is diagnosed only when there is no mass in the ovary or endometrium.14 The presence of a dominant tubal mass and a precursor lesion in the fallopian tube is a prerequisite for classification of a tumor’s origin in the fallopian tube, while the presence of a precursor lesion is not a requirement to diagnose a tumor of ovarian origin.5 Similarly, PPC is diagnosed only when no mass is found on the ovary, fallopian tube, or endometrium. The convention has been to classify serous tumors in the pelvis as ovarian cancer when the origin is unclear.3 This has led to significant underreporting of fallopian tube cancers as many cases of FTC also had tumors on the surface of the ovary and therefore, were classified as serous ovarian cancers.3,23
Molecular pathways
Advances in molecular diagnostics have led to the discovery of different mechanisms driving the EOC histological subtypes.17,32 Type 1 tumors include clear cell, mucinous, and low-grade serous and endometrioid tumors.8,17 These tumors show mutations in the mismatch repair genes, Kirsten rat sarcoma vial oncogene homolog (KRAS), v-raf murine sarcoma viral oncogene homolog B (BRAF), catenin (cadherin-associated protein), beta 1, 88kDa (CTNNB), phosphatidylinositol-4,5-biphosphate 3-kinase, catalytic subunit alpha (PIK3CA), adenine-thymine (AT)-rich interactive domain-containing protein 1A (ARIDIA), protein phosphatase 2, regulatory subunit A, alpha (PPP2R1A), and phosphatase and tensin homolog (PTEN).12,17,20 These tumors have a slow stepwise progression from cortical inclusion cysts to invasive cancer.4,8,12,18 In contrast, type 2 ovarian tumors harbor mutations (as shown in Figure 2) in the TP53 gene and are usually HGSC or (some) high-grade endometrioid tumors.12,17 These tumors are aggressive, usually present in advanced stages, and have a worse prognosis.
 | Figure 2 Proposed models for the development of type 1 and type 2 tumors of the ovary. |
Tumor suppressor p53 inactivation is detected in more than 95% of cases of HGSC.12 Studies reporting the p53 staining of the normal ovarian epithelium in patients with ovarian cancer or those at high risk of ovarian cancer (breast cancer susceptibility gene [BRCA]-positive) have been mixed, with some authors reporting the presence of p53 staining while others finding no evidence of p53 staining on the surface epithelium of the ovary.11,15,33 In BRCA-positive patients who underwent risk-reducing salpingo-oophorectomies (RRSO), a p53 signature was found in 38% of examined fallopian tubes, while none of the cortical inclusion cysts had a p53 signature, suggesting a fallopian origin to these cancers.15
Gene expression profiling of HGSC has shown a close correlation to normal fallopian tube epithelium, rather than ovarian epithelium.26,34 Molecular studies have also shown that the different histologies of ovarian cancer have distinct gene expression profiles. While HGSC most closely correlates with normal fallopian tube epithelium, mucinous carcinomas correlate with colonic epithelium; endometrioid and clear cell carcinomas correlate with the endometrium and carry different and specific marker genes.34 Amylase, alpha 2B (AMY2B) and chitinase-3-like protein 1 (CHI3L1) have been shown to be upregulated in serous ovarian cancer and normal fallopian tube epithelium. Endometrioid genes, like fibroblast growth factor homologous factor 9 (FHF9), stratifin (SFN), metallothionein 1G (MT1G), and Indian hedgehog (IHH), have been shown to be expressed in normal endometrium. Carcinoembryonic antigen-related cell adhesion molecule 5 (CEACAM5), lectin, galactoside-binding, soluble, 4 (LGALS4), and trefoil factor 2 (TFF2) were all shown to be distinctly expressed in mucinous carcinomas and to be upregulated in normal colon.34 ARID1A mutations are frequent in clear cell and endometrioid ovarian cancer but not in HGSC.35,36 The same mutation is found in the majority of uterine endometrioid carcinomas.37
Evidence of tubal origin of ovarian cancer
RRSO in BRCA-positive and other high-risk women has provided a unique opportunity to study the pathogenesis of ovarian carcinomas, PPC, and FTC.17,18,27,38 There has never been a clear precursor lesion of EOC found on the surface of the ovary in patients undergoing RRSO, in spite of data from thousands of patients at high risk of ovarian cancers (BRCA-positive) who have undergone this procedure. The Sectioning and Extensively Examining the Fimbriated End (SEE-FIM) protocol, used for a more detailed examination of the fallopian tubes in RRSO specimens, has led to an improved understanding of the pathogenesis of pelvis serous cancers.39,40
Retrospective studies
In patients with BRCA mutation who underwent RRSO, thorough examination of the fallopian tube revealed a tubal source 57%–100% of the time when a tumor was identified.8,13,18,38,40,41 In unselected women undergoing RRSO, a tubal origin was found 36%–47% of the time.14,28 Similarly, in a retrospective analysis of 51 patients from a single institution, 56% of patients who were originally classified as having a PPC were found to have STIC in the fallopian tubes.42 In another retrospective review, six out of 76 patients with BRCA1 mutation undergoing RRSO were found to have microscopic cancers. Four of these six patients had a tumor in the fallopian tube, one in the ovary, and the other was in the peritoneal washings.43 In the same study, 78% of patients with BRCA mutations who had stage I to II cancers were found to have a dominant ovarian mass. These data suggest that although HGSC initiation may occur in the fallopian tube, tumor growth and progression may be favored in the ovary, which could be due to a more favorable microenvironment in the ovary.43
In a prospective study of 360 high-risk patients who underwent RRSO, the pathology review included SEE-FIM of the fallopian tube to identify the primary site of disease. In this study, there were two tubal, two ovarian, and four noninvasive STICs, supporting the fallopian tube as a likely site of origin of many pelvic HGSCs.27 In patients who had HGSCs involving the ovary without evidence of a concurrent fallopian tube lesion, it is possible that the fallopian tube was not the only site of tumor initiation.
Treatment of FTC
Due to the presumed rarity of FTC and PPC, there are no large, prospective, randomized clinical trials exclusively evaluating treatment for FTC or PPC. The National Comprehensive Cancer Network guidelines recommend similar management, and the same regimens be used for EOC, FTC, and PPC.44 In fact, compared with the older clinical trials in ovarian cancer, most current and more recent ovarian cancer clinical trials have included patients with FTC and PPC, although they are considered distinct clinical entities.45–55 Since the current evidence points toward a common pathogenesis for ovarian cancer, FTC, and PPC, similar treatment of these cancers is likely to be the most appropriate management of all these tumors.
First-line treatment
The first-line treatment of ovarian cancer, FTC, and PPC is a combination of a platinum agent and a taxane.7,56 The combination of carboplatin and paclitaxel was compared with the combination of cisplatin and paclitaxel in the Gynecologic Oncology Group (COG) 158 trial. There was no statistical difference in progression-free survival (PFS) or overall survival (OS) between carboplatin and paclitaxel compared with cisplatin and paclitaxel; however, the carboplatin arm was better tolerated, with fewer nonhematologic side effects.56
The combination of dose-dense weekly paclitaxel and carboplatin every 3 weeks was compared with the standard regimen of both paclitaxel and carboplatin every 3 weeks, in a study done by the Japanese Gynecologic Oncology Group. The median PFS was longer in the dose-dense arm (28.0 months versus [vs] 17.2 months) (hazard ratio [HR] 0.71) (P=0.0015). The OS at 3 years was also higher in the dose-dense regimen group (72.1%) than in the conventional treatment group (65.1%) (HR 0.75) (P=0.03) However, there were more hematologic toxicities and treatment discontinuations with the dose-dense paclitaxel, although other toxicities were similar in both arms.57 The recently reported update to this trial, with over 6 years of follow-up, showed a median overall survival greater than 100 months in the dose-dense arm.58
In a Phase III trial (GOG 172) evaluating intraperitoneal therapy, patients with optimally debulked, newly diagnosed ovarian cancer or PPC received intravenous paclitaxel over 24 hours and were randomized to receive either intravenous cisplatin or intraperitoneal cisplatin and intraperitoneal paclitaxel. Although the median PFS (23.8 vs 18.3 months) and OS (65.6 vs 49.7 months) were longer in the intraperitoneal arm when compared with the intravenous arm, there were significantly more grade 3 and 4 toxicities, including pain, fatigue, and gastrointestinal (GI), hematologic, and neurologic toxicities, with only 42% of patients receiving all six cycles of the assigned intraperitoneal therapy.46 Therefore, intraperitoneal therapy should only be considered in patients with very good performance status who are willing to accept the increased toxicity of the regimen. The recently completed GOG 252 trial is evaluating a modified GOG 172 intraperitoneal regimen (which can be given in an outpatient setting) including intraperitoneal carboplatin compared with intravenous carboplatin and paclitaxel, and the results are pending (arm 1: IV paclitaxel, IV carboplatin and IV bevacizumab, arm 2: IV paclitaxel, IP carboplatin and IV bevacizumab, arm 3: IV placlitaxel, IP cisplatin, IP paclitaxel and IV bevacizumab).
The combination of docetaxel plus carboplatin59 is another reasonable first-line option. Due to ease of administration, favorable side-effect profile, long track record, and convenience to patients, the combination of carboplatin and paclitaxel has emerged as the worldwide standard of care in the first-line treatment of advanced ovarian cancer, FTC, and PPC.7,45
The GOG 218 and International Collaboration on Ovarian Neoplasms (ICON) 7 trials evaluated the addition of bevacizumab to front-line chemotherapy with carboplatin and paclitaxel. There was improvement in PFS in both trials but no OS benefit.48,49 There were more GI perforations in the patients receiving bevacizumab; however, this was not statistically different. The improved PFS was only observed among patients receiving bevacizumab with chemotherapy and as extended-maintenance treatment and in high-risk patients (36.6 months vs 28.8 months).49
Second-line treatment
Second-line treatment is determined in part by the time since the prior regimen. The platinum-free interval is an important predictor of the response to second-line treatment.60 Patients whose cancer was controlled for greater than 6 months from the last therapy are considered to be platinum sensitive.61 These patients are retreated with a platinum doublet, depending on their comorbidities, prior toxicities, and performance status. Carboplatin, in combination with paclitaxel (ICON4/Arbeitsgemeinschaft Gynaekologische Onkologie-Ovarian [AGO-OVAR]-2.2), gemcitabine (Intergroup trial), or PEGylated liposomal doxorubicin (PLD) (Caelyx in Platinum Sensitive Ovarian [CALYPSO]) have all been shown to be reasonable options.62–65 In the CALYPSO trial, the combination of carboplatin and PLD was compared with the combination of carboplatin and paclitaxel. Although the OS was not different in the two arms, patients in the PLD arm had a statistically significant improvement in PFS, the primary endpoint of the trial.64,65 The side effect profile also favored the PLD arm.65 Although combinations, in general, have shown an improvement in response rate (RR), PFS, and OS, there is also a higher incidence of side effects. Therefore, in patients with multiple comorbidities or poor performance status, single-agent chemotherapy is appropriate.
Patients who have a recurrence within 6 months of platinum treatment are considered to be platinum resistant.60,61 Patients whose cancers progress while on treatment with platinum are considered to be platinum refractory, and their prognosis is very poor. Platinum agents are not recommended for these patients, and further treatment is based on underlying renal function, hepatic function, and other comorbidities. Patients usually receive sequential therapy with single agents rather than combination therapy. A Phase III trial of weekly paclitaxel was found to be as effective as a combination therapy of paclitaxel with carboplatin or topotecan, in resistant ovarian cancer.66 In a retrospective study, PLD was shown to prolong the platinum-free interval so that patients could be resensitized to a platinum agent.67 However, this study was small, uncontrolled, and retrospective in nature, and in the absence of prospective trials, no firm conclusions can be drawn regarding the prolongation of the platinum-free interval with PLD. Most single-agent regimens produce a RR of 10%–30%. The RRs of commonly used single-agent cytotoxic chemotherapies are reported in Table 1. Patients whose disease progresses after two or more consecutive lines of therapy should be considered refractory to platinum and are candidates for early phase clinical trials.68
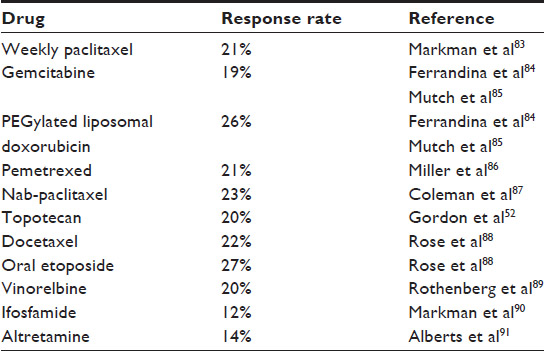 | Table 1 Response rates of single agent chemotherapies in platinum resistant and refractory setting |
Targeted agents
Like most other cancers, there has been an increased interest in using targeted therapies in ovarian cancer. The Gynecologic Oncology Group conducted multiple Phase II trials (GOG 170 series) evaluating targeted therapies in patients with refractory ovarian cancer, FTC, or PPC. Most of the targeted therapies tested had minimal activity in these unselected patients. Bevacizumab, aflibercept, and olaparib have been tested in ovarian cancer patients and had some antitumor activity. Other targeted agents examined in this disease, and, their RRs and PFS are shown in Table 2.
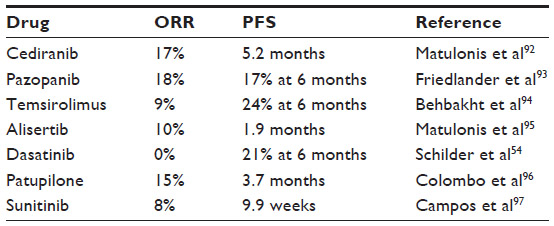 | Table 2 Response rates and progression-free survival of selected targeted agents |
Bevacizumab, a monoclonal antibody against the vascular endothelial growth factor (VEGF) receptor, in combination with chemotherapy, is useful in both the recurrent platinum-sensitive and in the platinum-resistant setting. The addition of bevacizumab to carboplatin and gemcitabine in platinum-sensitive, recurrent pelvic serous cancers has shown an improvement in RR and PFS but a slightly higher incidence of side effects, including hypertension and arterial thrombosis;50 however, no difference in the OS has been noted, which may be partly due to the subsequent therapy with bevacizumab in the patients who progressed on the placebo arm. In a Phase III trial of patients with platinum resistant disease (AURELIA), patients were randomized to receive investigator-selected chemotherapy (weekly paclitaxel, topotecan, or PLD) with or without bevacizumab.69 The primary end point was Response Evaluation Criteria in Solid Tumors (RECIST)-defined PFS. Patients in the bevacizumab plus chemotherapy arm had a PFS of 5.7 months compared with 4 months in the chemotherapy-only arm (HR 0.48, 95% confidence interval [CI]: 0.38−0.60) (P<0.001). There were also higher RRs and complete responses in the bevacizumab arm. No OS data are available yet. There were more grade 2 or higher adverse events in the bevacizumab arm, including hypertension, proteinuria, GI perforation, and fistula formation. There were also more grade 3 or higher arterial thromboembolic events.69 Bevacizumab has been tested in Phase II trials as a single agent and been found to have an RR of 21% to 26% and a median PFS of 4.4 months to 4.7 months and is a reasonable option.70,71
Aflibercept, a VEGF trap, was found to be effective in controlling malignant ascites in a double-blind, placebo-controlled Phase II trial in patients who had received a median of four prior therapies.72 The mean time to repeat paracentesis was significantly longer after aflibercept, 55 days vs 23 days. However, there were more grade 3 and 4 side effects, including dyspnea, fatigue, and dehydration. There were also three intestinal perforations with aflibercept.
Olaparib, a poly–adenosine diphosphate (ADP) ribose polymerase (PARP) inhibitor has shown activity in BRCA-mutant ovarian cancer – patients with BRCA mutations have a defect in the DNA homologous recombination repair mechanism, and PARP inhibitors prevent base excision repair.73–75 Together, these mechanisms achieve tumor cell death by preventing DNA repair via two different pathways, a process referred to as synthetic lethality. A Phase I/II trial in BRCA mutation-positive patients showed an RR of 40%, with a response duration of 28 weeks.76 Other studies have shown RRs from 13% to 33%.77,78 Even in patients with HGSC without BRCA mutation, the RR was 24%.79 Olaparib has been used as maintenance therapy in platinum-sensitive relapsed ovarian cancer and found to prolong the PFS but did not add an OS benefit.80 A Phase I study of olaparib with cediranib, a VEGF receptor tyrosine kinase inhibitor, showed an RR of 44% in recurrent ovarian cancer.81
Future directions
Radical fimbriectomy followed by surveillance has been suggested as an alternative to surveillance alone in high-risk BRCA-positive patients reluctant to undergo bilateral salpingo-oophorectomy (BSO) for risk reduction of pelvic serous carcinomas, due to concerns of prolonged menopause and other complications (like osteoporosis, vaginal atrophy, and decreased quality of life associated with menopause).21 This approach would preserve the ovaries and consequently delay menopausal symptoms until natural menopause occurs. However, a microscopic spread of the tumor, from the fallopian tube to the ovary, may occur very early; thus, salpingectomy alone may be insufficient to protect against the development of ovarian cancer and may create a false sense of protection.43 Therefore further evaluation of this procedure, with long-term follow-up, is necessary. The early detection of an STIC can help in the prevention of invasive ovarian cancers; however, no screening techniques are currently available to detect STIC. Endometrial cytological testing may detect early-stage ovarian, tubal, and peritoneal HGSCs and may be useful for ovarian cancer screening, but this study needs further validation.82 The inclusion of patients with FTC, PPC, and HGSC of the ovary in clinical trials is recommended as they have similar molecular abnormalities and likely the same cell of origin. The better understanding of FTC biology will lead to improved treatment and, ultimately, improved outcomes in FTC, PPC, and HGSC.
Acknowledgment
The authors are grateful to Mahmoud Gaballa, MD, Resident in Internal Medicine at Thomas Jefferson University, for the excellent artwork he provided for this article.
Disclosure
The authors report no conflicts of interest in this work.
References
Stewart SL, Wike JM, Foster SL, Michaud F. The incidence of primary fallopian tube cancer in the United States. Gynecol Oncol. 2007;107(3):392−397. | |
Goodman MT, Shvetsov YB. Incidence of ovarian, peritoneal, and fallopian tube carcinomas in the United States, 1995–2004. Cancer Epidemiol Biomarkers Prev. 2009;18(1):132–139. | |
Berek JS, Crum C, Friedlander M. Cancer of the ovary, fallopian tube, and peritoneum. Int J Gynaecol Obstet. 2012;119 Suppl 2: S118–S129. | |
Crum CP, Drapkin R, Kindelberger D, Medeiros F, Miron A, Lee Y. Lessons from BRCA: the tubal fimbria emerges as an origin for pelvic serous cancer. Clin Med Res. 2007;5(1):35–44. | |
Crum CP, Drapkin R, Miron A, et al. The distal fallopian tube: a new model for pelvic serous carcinogenesis. Curr Opin Obstet Gynecol. 2007;19(1):3–9. | |
Katabuchi H, Okamura H. Cell biology of human ovarian surface epithelial cells and ovarian carcinogenesis. Med Electron Microsc. 2003;36(2):74–86. | |
Hennessy BT, Coleman RL, Markman M. Ovarian cancer. Lancet. 2009;374(9698):1371−1382. | |
Chene G, Dauplat J, Radosevic-Robin N, Cayre A, Penault-Llorca F. Tu-be or not tu-be: that is the question … about serous ovarian carcinogenesis. Crit Rev Oncol Hematol. 2013;88(1):134–143. | |
Fathalla MF. Incessant ovulation – a factor in ovarian neoplasia? Lancet. 1971;2(7716):163. | |
Casey MJ, Bewtra C, Hoehne LL, Tatpati AD, Lynch HT, Watson P. Histology of prophylactically removed ovaries from BRCA1 and BRCA2 mutation carriers compared with noncarriers in hereditary breast ovarian cancer syndrome kindreds. Gynecol Oncol. 2000;78(3 Pt 1):278–287. | |
Barakat RR, Federici MG, Saigo PE, Robson ME, Offit K, Boyd J. Absence of premalignant histologic, molecular, or cell biologic alterations in prophylactic oophorectomy specimens from BRCA1 heterozygotes. Cancer. 2000;89(2):383–390. | |
Kessler M, Fotopoulou C, Meyer T. The molecular fingerprint of high grade serous ovarian cancer reflects its fallopian tube origin. Int J Mol Sci. 2013;14(4):6571–6596. | |
Callahan MJ, Crum CP, Medeiros F, et al. Primary fallopian tube malignancies in BRCA-positive women undergoing surgery for ovarian cancer risk reduction. J Clin Oncol. 2007;25(25):3985–3990. | |
Kindelberger DW, Lee Y, Miron A, et al. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: Evidence for a causal relationship. Am J Surg Pathol. 2007;31(2):161–169. | |
Folkins AK, Jarboe EA, Saleemuddin A, et al. A candidate precursor to pelvic serous cancer (p53 signature) and its prevalence in ovaries and fallopian tubes from women with BRCA mutations. Gynecol Oncol. 2008;109(2):168–173. | |
Jarboe EA, Folkins AK, Drapkin R, Ince TA, Agoston ES, Crum CP. Tubal and ovarian pathways to pelvic epithelial cancer: a pathological perspective. Histopathology. 2008;53(2):127–138. | |
Crum CP. Intercepting pelvic cancer in the distal fallopian tube: theories and realities. Mol Oncol. 2009;3(2):165–170. | |
Folkins AK, Jarboe EA, Roh MH, Crum CP. Precursors to pelvic serous carcinoma and their clinical implications. Gynecol Oncol. 2009;113(3):391–396. | |
Chen EY, Mehra K, Mehrad M, et al. Secretory cell outgrowth, PAX2 and serous carcinogenesis in the Fallopian tube. J Pathol. 2010;222(1):110–116. | |
Kurman RJ, Shih IM. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer – shifting the paradigm. Hum Pathol. 2011;42(7):918–931. | |
Leblanc E, Narducci F, Farre I, et al. Radical fimbriectomy: a reasonable temporary risk-reducing surgery for selected women with a germ line mutation of BRCA 1 or 2 genes? Rationale and preliminary development. Gynecol Oncol. 2011;121(3):472–476. | |
Leonhardt K, Einenkel J, Sohr S, Engeland K, Horn LC. p53 signature and serous tubal in-situ carcinoma in cases of primary tubal and peritoneal carcinomas and serous borderline tumors of the ovary. Int J Gynecol Pathol. 2011;30(5):417–424. | |
Crum CP, McKeon FD, Xian W. The oviduct and ovarian cancer: causality, clinical implications, and “targeted prevention”. Clin Obstet Gynecol. 2012;55(1):24–35. | |
Mingels MJ, Roelofsen T, van der Laak JA, et al. Tubal epithelial lesions in salpingo-oophorectomy specimens of BRCA-mutation carriers and controls. Gynecol Oncol. 2012;127(1):88–93. | |
Gao FF, Bhargava R, Yang H, Li Z, Zhao C. Clinicopathologic study of serous tubal intraepithelial carcinoma with invasive carcinoma: is serous tubal intraepithelial carcinoma a reliable feature for determining the organ of origin? Hum Pathol. 2013;44(8):1534–1543. | |
O’Shannessy DJ, Jackson SM, Twine NC, et al. Gene expression analyses support fallopian tube epithelium as the cell of origin of epithelial ovarian cancer. Int J Mol Sci. 2013;14(7):13687–13703. | |
Reitsma W, de Bock GH, Oosterwijk JC, Bart J, Hollema H, Mourits MJ. Support of the ‘fallopian tube hypothesis’ in a prospective series of risk-reducing salpingo-oophorectomy specimens. Eur J Cancer. 2013;49(1):132–141. | |
Roh MH, Kindelberger D, Crum CP. Serous tubal intraepithelial carcinoma and the dominant ovarian mass: clues to serous tumor origin? Am J Surg Pathol. 2009;33(3):376–383. | |
Mehra K, Mehrad M, Ning G, et al. STICS, SCOUTs and p53 signatures; a new language for pelvic serous carcinogenesis. Front Biosci (Elite Ed). 2011;3:625–634. | |
Lee Y, Miron A, Drapkin R, et al. A candidate precursor to serous carcinoma that originates in the distal fallopian tube. J Pathol. 2007;211(1):26–35. | |
Gross AL, Kurman RJ, Vang R, Shih IeM, Visvanathan K. Precursor lesions of high-grade serous ovarian carcinoma: morphological and molecular characteristics. J Oncol. 2010;2010:126295. | |
Shen H, Fridley BL, Song H, et al; PRACTICAL Consortium; Australian Ovarian Cancer Study Group; Australian Cancer Study. Epigenetic analysis leads to identification of HNF1B as a subtype-specific susceptibility gene for ovarian cancer. Nat Commun. 2013;4:1628. | |
Hutson R, Ramsdale J, Wells M. p53 protein expression in putative precursor lesions of epithelial ovarian cancer. Histopathology. 1995;27(4):367–371. | |
Marquez RT, Baggerly KA, Patterson AP, et al. Patterns of gene expression in different histotypes of epithelial ovarian cancer correlate with those in normal fallopian tube, endometrium, and colon. Clin Cancer Res. 2005;11(17):6116–6126. | |
Wiegand KC, Shah SP, Al-Agha OM, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363(16):1532–1543. | |
Lowery WJ, Schildkraut JM, Akushevich L, et al. Loss of ARID1A-associated protein expression is a frequent event in clear cell and endometrioid ovarian cancers. Int J Gynecol Cancer. 2012;22(1):9–14. | |
Ayhan A, Mao TL, Seckin T, et al. Loss of ARID1A expression is an early molecular event in tumor progression from ovarian endometriotic cyst to clear cell and endometrioid carcinoma. Int J Gynecol Cancer. 2012;22(8):1310–1315. | |
Leeper K, Garcia R, Swisher E, Goff B, Greer B, Paley P. Pathologic findings in prophylactic oophorectomy specimens in high-risk women. Gynecol Oncol. 2002;87(1):52–56. | |
Lee Y, Medeiros F, Kindelberger D, Callahan MJ, Muto MG, Crum CP. Advances in the recognition of tubal intraepithelial carcinoma: applications to cancer screening and the pathogenesis of ovarian cancer. Adv Anat Pathol. 2006;13(1):1–7. | |
Powell CB, Kenley E, Chen LM, et al. Risk-reducing salpingo-oophorectomy in BRCA mutation carriers: role of serial sectioning in the detection of occult malignancy. J Clin Oncol. 2005;23(1):127–132. | |
Finch A, Shaw P, Rosen B, Murphy J, Narod SA, Colgan TJ. Clinical and pathologic findings of prophylactic salpingo-oophorectomies in 159 BRCA1 and BRCA2 carriers. Gynecol Oncol. 2006;100(1):58–64. | |
Seidman JD, Zhao P, Yemelyanova A. “Primary peritoneal” high-grade serous carcinoma is very likely metastatic from serous tubal intraepithelial carcinoma: assessing the new paradigm of ovarian and pelvic serous carcinogenesis and its implications for screening for ovarian cancer. Gynecol Oncol. 2011;120(3):470–473. | |
Yates MS, Meyer LA, Deavers MT, et al. Microscopic and early-stage ovarian cancers in BRCA1/2 mutation carriers: building a model for early BRCA-associated tumorigenesis. Cancer Prev Res (Phila). 2011;4(3):463–470. | |
Morgan RJ, Alvarez RD, Armstrong DK, et al; National Comprehensive Cancer Network. Ovarian cancer, version 3.2012. J Natl Compr Canc Netw. 2012;10(11):1339–1349. | |
Ozols RF, Bundy BN, Greer BE, et al; Gynecologic Oncology Group. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol. 2003;21(17):3194–3200. | |
Armstrong DK, Bundy B, Wenzel L, et al; Gynecologic Oncology Group. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med. 2006;354(1):34−43. | |
Markman M, Moon J, Wilczynski S, et al. Single agent carboplatin versus carboplatin plus pegylated liposomal doxorubicin in recurrent ovarian cancer: final survival results of a SWOG (S0200) phase 3 randomized trial. Gynecol Oncol. 2010;116(3):323–325. | |
Burger RA, Brady MF, Bookman MA, et al; Gynecologic Oncology Group. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365(26):2473–2483. | |
Perren TJ, Swart AM, Pfisterer J, et al; ICON7 Investigators. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365(26):2484−2496. | |
Aghajanian C, Blank SV, Goff BA, et al. OCEANS: a randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol. 2012;30(17):2039–2045. | |
Alberts DS, Liu PY, Hannigan EV, et al. Intraperitoneal cisplatin plus intravenous cyclophosphamide versus intravenous cisplatin plus intravenous cyclophosphamide for stage III ovarian cancer. N Engl J Med. 1996;335(26):1950–1955. | |
Gordon AN, Tonda M, Sun S, Rackoff W; Doxil Study 30-49 Investigators. Long-term survival advantage for women treated with pegylated liposomal doxorubicin compared with topotecan in a phase 3 randomized study of recurrent and refractory epithelial ovarian cancer. Gynecol Oncol. 2004;95(1):1–8. | |
Piccart MJ, Bertelsen K, James K, et al. Randomized intergroup trial of cisplatin-paclitaxel versus cisplatin-cyclophosphamide in women with advanced epithelial ovarian cancer: three-year results. J Natl Cancer Inst. 2000;92(9):699–708. | |
Schilder RJ, Brady WE, Lankes HA, et al. Phase II evaluation of dasatinib in the treatment of recurrent or persistent epithelial ovarian or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol. 2012;127(1):70–74. | |
Seamon LG, Richardson DL, Copeland LJ. Evolution of the Gynecologic Oncology Group protocols in the treatment of epithelial ovarian cancer. Clin Obstet Gynecol. 2012;55(1):131–155. | |
Bookman MA, Greer BE, Ozols RF. Optimal therapy of advanced ovarian cancer: carboplatin and paclitaxel vs cisplatin and paclitaxel (GOG 158) and an update on GOG0 182-ICON5. Int J Gynecol Cancer. 2003;13(6):735–740. | |
Katsumata N, Yasuda M, Takahashi F, et al; Japanese Gynecologic Oncology Group. Dose-dense paclitaxel once a week in combination with carboplatin every 3 weeks for advanced ovarian cancer: a phase 3, open-label, randomised controlled trial. Lancet. 2009;374(9698):1331–1338. | |
Katsumata N, Yasuda M, Isonishi S, et al; Japanese Gynecologic Oncology Group. Long-term results of dose-dense paclitaxel and carboplatin versus conventional paclitaxel and carboplatin for treatment of advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer (JGOG 3016): a randomised, controlled, open-label trial. Lancet Oncol. 2013;14(10):1020–1026. | |
Vasey PA, Jayson GC, Gordon A, et al; Scottish Gynaecological Cancer Trials Group. Phase III randomized trial of docetaxel-carboplatin versus paclitaxel-carboplatin as first-line chemotherapy for ovarian carcinoma. J Natl Cancer Inst. 2004;96(22):1682–1691. | |
Markman M, Rothman R, Hakes T, et al. Second-line platinum therapy in patients with ovarian cancer previously treated with cisplatin. J Clin Oncol. 1991;9(3):389−393. | |
Rose PG, Fusco N, Fluellen L, Rodriguez M. Second-line therapy with paclitaxel and carboplatin for recurrent disease following first-line therapy with paclitaxel and platinum in ovarian or peritoneal carcinoma. J Clin Oncol. 1998;16(4):1494–1497. | |
Parmar MK, Ledermann JA, Colombo N, et al; ICON and AGO Collaborators. Paclitaxel plus platinum-based chemotherapy versus conventional platinum-based chemotherapy in women with relapsed ovarian cancer: the ICON4/AGO-OVAR-2.2 trial. Lancet. 2003;361(9375):2099–2106. | |
Pfisterer J, Plante M, Vergote I, et al; AGO-OVAR; NCIC CTG; EORTC GCG. Gemcitabine plus carboplatin compared with carboplatin in patients with platinum-sensitive recurrent ovarian cancer: an intergroup trial of the AGO-OVAR, the NCIC CTG, and the EORTC GCG. J Clin Oncol. 2006;24(29):4699–4707. | |
Wagner U, Marth C, Largillier R, et al. Final overall survival results of phase III GCIG CALYPSO trial of pegylated liposomal doxorubicin and carboplatin vs paclitaxel and carboplatin in platinum-sensitive ovarian cancer patients. Br J Cancer. 2012;107(4):588–591. | |
Gladieff L, Ferrero A, De Rauglaudre G, et al. Carboplatin and pegylated liposomal doxorubicin versus carboplatin and paclitaxel in partially platinum-sensitive ovarian cancer patients: results from a subset analysis of the CALYPSO phase III trial. Ann Oncol. 2012;23(5):1185–1189. | |
Lortholary A, Largillier R, Weber B, et al; GINECO group France. Weekly paclitaxel as a single agent or in combination with carboplatin or weekly topotecan in patients with resistant ovarian cancer: the CARTAXHY randomized phase II trial from Groupe d’Investigateurs Nationaux pour l’Etude des Cancers Ovariens (GINECO). Ann Oncol. 2012;23(2):346–352. | |
Tanguay JS, Ansari J, Buckley L, Fernando I. Epithelial ovarian cancer: role of pegylated liposomal Doxorubicin in prolonging the platinum-free interval and cancer antigen 125 trends during treatment. Int J Gynecol Cancer. 2009;19(3):361–366. | |
Griffiths RW, Zee YK, Evans S, et al. Outcomes after multiple lines of chemotherapy for platinum-resistant epithelial cancers of the ovary, peritoneum, and fallopian tube. Int J Gynecol Cancer. 2011;21(1):58–65. | |
Pujade-Lauraine, Hilpert F, Weber B, et al; AURELIA Investigators. AURELIA: A randomized phase III trial evaluating bevacizumab (BEV) plus chemotherapy (CT) for platinum (PT)-resistant recurrent ovarian cancer (OC). J Clin Oncol. 2012;30(Suppl 18):LBA5002. Abstract. | |
Cannistra SA, Matulonis UA, Penson RT, et al. Phase II study of bevacizumab in patients with platinum-resistant ovarian cancer or peritoneal serous cancer. J Clin Oncol. 2007;25(33):5180–5186. | |
Burger RA, Sill MW, Monk BJ, Greer BE, Sorosky JI. Phase II trial of bevacizumab in persistent or recurrent epithelial ovarian cancer or primary peritoneal cancer: a Gynecologic Oncology Group Study. J Clin Oncol. 2007;25(33):5165–5171. | |
Gotlieb WH, Amant F, Advani S, et al. Intravenous aflibercept for treatment of recurrent symptomatic malignant ascites in patients with advanced ovarian cancer: a phase 2, randomised, double-blind, placebo-controlled study. Lancet Oncol. 2012;13(2):154–162. | |
Underhill C, Toulmonde M, Bonnefoi H. A review of PARP inhibitors: from bench to bedside. Ann Oncol. 2011;22(2):268–279. | |
McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66(16):8109–8115. | |
Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. | |
Fong PC, Yap TA, Boss DS, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28(15):2512–2519. | |
Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376(9737):245–251. | |
Kaye SB, Lubinski J, Matulonis U, et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J Clin Oncol. 2012;30(4):372–379. | |
Gelmon KA, Tischkowitz M, Mackay H, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12(9):852–861. | |
Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366(15):1382–1392. | |
Liu JF, Tolaney SM, Birrer M, et al. A Phase 1 trial of the poly(ADP-ribose) polymerase inhibitor olaparib (AZD2281) in combination with the anti-angiogenic cediranib (AZD2171) in recurrent epithelial ovarian or triple-negative breast cancer. Eur J Cancer. 2013;49(14):2972–2978. | |
Otsuka I, Kameda S, Hoshi K. Early detection of ovarian and fallopian tube cancer by examination of cytological samples from the endometrial cavity. Br J Cancer. 2013;109(3):603–609. | |
Markman M, Blessing J, Rubin SC, Connor J, Hanjani P, Waggoner S; Gynecologic Oncology Group. Phase II trial of weekly paclitaxel (80 mg/m2) in platinum and paclitaxel-resistant ovarian and primary peritoneal cancers: a Gynecologic Oncology Group study. Gynecol Oncol. 2006;101(3):436–440. | |
Ferrandina G, Ludovisi M, Lorusso D, et al. Phase III trial of gemcitabine compared with pegylated liposomal doxorubicin in progressive or recurrent ovarian cancer. J Clin Oncol. 2008;26(6):890–896. | |
Mutch DG, Orlando M, Goss T, et al. Randomized phase III trial of gemcitabine compared with pegylated liposomal doxorubicin in patients with platinum-resistant ovarian cancer. J Clin Oncol. 2007;25(19):2811–2818. | |
Miller DS, Blessing JA, Krasner CN, et al. Phase II evaluation of pemetrexed in the treatment of recurrent or persistent platinum-resistant ovarian or primary peritoneal carcinoma: a study of the Gynecologic Oncology Group. J Clin Oncol. 2009;27(16):2686–2691. | |
Coleman RL, Brady WE, McMeekin DS, et al. A phase II evaluation of nanoparticle, albumin-bound (nab) paclitaxel in the treatment of recurrent or persistent platinum-resistant ovarian, fallopian tube, or primary peritoneal cancer: a Gynecologic Oncology Group study. Gynecol Oncol. 2011;122(1):111–115. | |
Rose PG, Blessing JA, Ball HG, et al. A phase II study of docetaxel in paclitaxel-resistant ovarian and peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol. 2003;88(2):130–135. | |
Rothenberg ML, Liu PY, Wilczynski S, et al. Phase II trial of vinorelbine for relapsed ovarian cancer: a Southwest Oncology Group study. Gynecol Oncol. 2004;95(3):506–512. | |
Markman M, Hakes T, Reichman B, et al. Ifosfamide and mesna in previously treated advanced epithelial ovarian cancer: activity in platinum-resistant disease. J Clin Oncol. 1992;10(2):243–248. | |
Alberts DS, Jiang C, Liu PY, Wilczynski S, Markman M, Rothenberg ML. Long-term follow-up of a phase II trial of oral altretamine for consolidation of clinical complete remission in women with stage III epithelial ovarian cancer in the Southwest Oncology Group. Int J Gynecol Cancer. 2004;14(2):224–228. | |
Matulonis UA, Berlin S, Ivy P, et al. Cediranib, an oral inhibitor of vascular endothelial growth factor receptor kinases, is an active drug in recurrent epithelial ovarian, fallopian tube, and peritoneal cancer. J Clin Oncol. 2009;27(33):5601–5606. | |
Friedlander M, Hancock KC, Rischin D, et al. A Phase II, open-label study evaluating pazopanib in patients with recurrent ovarian cancer. Gynecol Oncol. 2010;119(1):32–37. | |
Behbakht K, Sill MW, Darcy KM, et al. Phase II trial of the mTOR inhibitor, temsirolimus and evaluation of circulating tumor cells and tumor biomarkers in persistent and recurrent epithelial ovarian and primary peritoneal malignancies: a Gynecologic Oncology Group study. Gynecol Oncol. 2011;123(1):19–26. | |
Matulonis UA, Sharma S, Ghamande S, et al. Phase II study of MLN8237 (alisertib), an investigational Aurora A kinase inhibitor, in patients with platinum-resistant or -refractory epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. Gynecol Oncol. 2012;127(1):63–69. | |
Colombo N, Kutarska E, Dimopoulos M, et al. Randomized, open-label, phase III study comparing patupilone (EPO906) with pegylated liposomal doxorubicin in platinum-refractory or -resistant patients with recurrent epithelial ovarian, primary fallopian tube, or primary peritoneal cancer. J Clin Oncol. 2012;30(31):3841–3847. | |
Campos SM, Penson RT, Matulonis U, et al. A phase II trial of Sunitinib malate in recurrent and refractory ovarian, fallopian tube and peritoneal carcinoma. Gynecol Oncol. 2013;128(2):215–220. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.