Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
Reduced MBD2 expression enhances airway inflammation in bronchial epithelium in COPD
Authors Zeng ZL, Li M, Chen JK, Li QH, Ning Q, Zhao JP, Xu YJ, Xie JG ![]() , Yu J
, Yu J
Received 8 August 2017
Accepted for publication 9 October 2017
Published 28 February 2018 Volume 2018:13 Pages 703—715
DOI https://doi.org/10.2147/COPD.S148595
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chunxue Bai
Zhilin Zeng,1,2 Miao Li,1 Jinkun Chen,3 Qinghai Li,1 Qin Ning,2 Jianping Zhao,1 Yongjian Xu,1 Jungang Xie,1 Jun Yu4
1Department of Respiratory and Critical Care Medicine, National Clinical Research Center of Respiratory Disease, 2Department of Infectious Disease, Institute of Infectious Disease, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China; 3Acadia Junior High School, Winnipeg, MB, Canada; 4Department of Thoracic Surgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China
Background: Chronic obstructive pulmonary disease (COPD) is a common inflammatory lung disease characterized by inflammatory cells activation and production of inflammatory mediators. Methyl-CpG-binding domain protein 2 (MBD2) plays an important role in diverse immunological disorders by regulating immune cell functions, such as differentiation and mediator secretion. However, the role of MBD2 in COPD remains unknown.
Methods: MBD2 protein expression in lung tissues of patients with COPD and cigarette smoke (CS)-exposed mice were evaluated by Western blot and immunohistochemistry. The role of MBD2 in cigarette smoke extract (CSE)-induction of inflammatory mediator expression in the human bronchial epithelial (HBE) cell line was assessed by silencing MBD2 expression in vitro. The involvement of signaling pathways in mediation of inflammation was tested with signaling inhibitors.
Results: Compared with controls, MBD2 expression was distinctly reduced in the bronchial epithelium of both patients with COPD and CS-exposed mice. Moreover, MBD2 expression was decreased in HBE after CSE stimulation in vitro. Moreover, MBD2 knockdown enhanced interleukin (IL)-6 and IL-8 expression in HBE in the presence and absence of CSE treatment by the ERK signaling pathway.
Conclusion: MBD2 protein expression was reduced in the airway epithelium of COPD. In HBE, this reduced expression was associated with increased levels of IL-6 and IL-8 mediated by the ERK pathway. These results suggest that MBD2 could contribute to chronic airway inflammation in COPD.
Keywords: chronic obstructive pulmonary disease, methyl-CpG-binding domain protein 2, airway inflammation
Introduction
Chronic obstructive pulmonary disease (COPD), characterized by irreversible and progressive expiratory airflow limitation, is a worldwide public health problem that accounts for 5% of total deaths each year and is predicted to be the third leading cause of death by 2020.1–3 Cigarette smoking is the main pathological driver in COPD development by causing aberrant inflammation, protease–anti-protease imbalance, oxidative stress, apoptosis, and accelerated lung aging.4–6 Inflammation, as one of the central features of COPD, causes activation and/or recruitment of immune cells. Human bronchial epithelial (HBE) cells, as the first defensive barrier and major effector in lung tissue, can trigger immune and inflammatory responses by secreting mediators.7 Ongoing inflammation in airways can subsequently activate and/or recruit more immune cells, leading to production of cytokines, chemokines, reactive oxygen species, and enzymes that together contribute to the development and progression of COPD.8 However, the underlying mechanisms of smoking-induced airway inflammation remain largely undefined.
Methyl-CpG-binding domain protein 2 (MBD2) is a member of the MBD protein family that responds to DNA methylation signals.9 MBD2 regulates gene transcription by acting as a methylation-dependent reader that participates in the recruitment of silencing complexes, nucleosome remodeling deacetylases, and histone deacetylases.10,11 Cigarette smoking is associated with elevated serum homocysteine – a well-established risk factor for atherosclerosis.12,13 Yideng et al found that homocysteine could reduce MBD2 protein expression in vascular smooth muscle cells, which suggests that cigarette smoking might regulate MBD2 protein expression.14 Furthermore, accumulating evidence suggests that MBD2 is associated with numerous cancers, as well as several immunological disorders, including systemic lupus erythematosus, autoimmune encephalomyelitis, sclerosis, and dermatomyositis.15–23 MBD2 overexpression was reported to increase interleukin (IL)-10 and TGF-β levels, but reduce the amount of IL-6 in MBD2−/− Treg cells.24 Additionally, MBD2 was recognized with implications in Th1 differentiation through repression of interferon gamma (IFN-γ) expression.25,26 Besides, MBD2 knockdown activated ERK1/2 and p38 MAPK pathways, which are involved in regulating cytokine production.27 Taken together, MBD2 may play an instrumental role in modulating expression of inflammatory mediators.
We hypothesize that MBD2 may contribute to airway inflammation in COPD pathogenesis. The current study was conducted to determine MBD2 expression in the airway epithelium of COPD patients and CS-exposed mice. In in vitro experiments, we analyzed the effects of cigarette smoking on MBD2 protein levels in HBE. Further, we silenced MBD2 expression in HBE to explore the function of MBD2 and the underlying mechanisms involved in modulating the secretion of inflammatory mediators.
Materials and methods
Subjects
Normal lung tissue specimens were obtained from 34 patients with operable nonmalignant and solitary pulmonary nodule, who were recruited from Tongji Hospital, Wuhan, People’s Republic of China. All participants were assigned into three groups: COPD group, non-COPD smoker group, and non-smoker according to their smoking history and pulmonary function. COPD cases were diagnosed before surgery based on a post-bronchodilator forced expiratory volume in 1 second (FEV1)/forced vital capacity ratio less than 70% according to the criteria of the Global Initiative for Chronic Obstructive Lung Disease. Smokers were patients with smoking history of more than 20 pack-years. Main exclusion criteria were malignant tumors, asthma, severe lung infections, or other obstructive lung diseases. Written informed consent was obtained from all subjects. This study was approved by the Research Ethics Committee of Huazhong University of Science and Technology.
Animals and CS exposure
Six-week-old male C57BL/6 mice were purchased from the Animal Experiment Center of Wuhan University. Mice were housed in sterilized cages with filter tops in specific pathogen-free conditions at Tongji Medical College, Huazhong University of Science and Technology. All animal experiments were approved by the Huazhong University Animal Experiment Ethics Committee and were conducted in accordance with the animal experimentation guidelines of Huazhong University.
Mice were randomly divided into two groups: CS-exposed mice and healthy controls (n=8 per group). CS-exposed mice were exposed to smoke in a PAB-S200 Animal Passive Smoking Exposure System (BioLab Technology Co. Ltd., Beijing, People’s Republic of China) using a modified method of 10 cigarettes for two separate 2.5-h periods per day, 5 days per week for continuously for 8 months.28 Simultaneously, healthy controls were exposed to room air.
Lung function measurement and histological analysis
The lung function of all mice was measured using AniRes 2005 lung function system (Bestlab, Beijing, People’s Republic of China) as previously described.29 Mice were anesthetized intraperitoneally with 90 mg/kg sodium pentobarbital (Servicebio, Wuhan, People’s Republic of China), and a tracheal cannula was inserted via tracheotomy. Then, the mouse was quickly placed supine in a sealed whole-body plethysmograph and connected to a computer-controlled ventilator via a tracheal cannula. The respiratory rate was preset at 90 breaths/min with a tidal volume of 5 mL/kg and the expiration/inspiration time ratio was set at 1.5:1. When all the parameters were stable, lung resistance (RL) was recorded. After the mouse was allowed to acclimatize for at least 30 s, dynamic lung compliance (Cdyn), forced expiratory volume in 0.1 s (FEV0.1s), and ratio of FEV0.1s to forced vital capacity (FEV0.1s/FVC) were evaluated as follows: immediately after the ending of the last expiration of volume history, which was automatically detected by the system, the lungs was inflated to 30 cm H2O tracheal pressure (TLC level) and held for a brief period (0.4 s). Following this, mice was exposed to a negative pressure reservoir (−30 cm H2O), forcing rapid exhalation as quickly as possible.
Bronchoalveolar lavage fluid (BALF) and histological analysis
The right lung was lavaged three times with 0.8 mL saline through the tracheal cannula. The bronchoalveolar lavage fluid (BALF) obtained was centrifuged at 1,000 g for 5 min, and supernatants of the first BALF were stored at −80°C for cytokine measurements. The left lung was not lavaged, but instead was inflated and fixed with 10% formalin at a pressure of 25 cm H2O for 24 h and then embedded in paraffin. Lung tissue sections (5 mm) were stained with hematoxylin–eosin to detect morphological alternations by measuring the mean linear intercept (MLI) under light microscopy.30 Using eight parallel lines drawn across the lung section, MLI was calculated for each sample based on eight random fields at a magnification of 100×. MLI was equal to the ratio of the lengths of cross lines over the total number of intercepts encountered in eight lines per field.
Cell culture and stimulation
Human lung bronchial epithelial cell line (HBE) were obtained from the Wuhan Boster Biotech Co., Ltd. (People’s Republic of China). Cells were maintained in RPMI 1640 medium containing 10% heat-inactivated fetal bovine serum at 37°C with 5% CO2. Cell cultures were maintained and stimulated with CSE, which was prepared as previously described.31 Where indicated, cells were pretreated for 1 h with the MAPK signal pathway inhibitors (Selleck) SB-203580 (10 μM), U-0126 (10 μM), or SP-600125 (10 μM), dissolved to the relevant concentration with dimethyl sulfoxide (DMSO).
Cell viability assay
Approximately 3,000 cells were seeded in each well of a 96-well plate (Corning, MA, USA). After treatment, 10 μL of Cell Counting Kit 8 (CCK 8) solution was added, and cells were incubated at 37°C for 3 h according to the manufacturer’s instructions (Dojindo Laboratories, Tokyo, Japan). Optical density (OD) values (measuring wavelength at 450 nm, reference wavelength at 630 nm) were obtained using an EL ×800 Universal Microplate Reader (Bio-Tek Instruments, Inc., VT, USA).
Immunohistochemical analysis
Human lung and mouse lung tissues were stained with rabbit monoclonal MBD2 antibody (1:500, Abcam, Cambridge, UK) and rabbit polyclonal MBD2 antibody (1:100, Santa Cruz Biotechnology, CA, USA), respectively. These proteins were then detected in lung sections with an IgG Streptavidin Biotin Complex kit (Boster, Wuhan, People’s Republic of China) and developed with DAB substrate according to the manufacturer’s protocol (Dako, Glostrup, Denmark). A Nikon Spot image acquisition and processing system (USA) was used for image assessment. The mean integral OD of MBD2 protein staining in the bronchiolar epithelium was measured as follows.32 We used Image-Pro Plus 4.1 professional image analysis software to open all immunohistochemical images from one sample slide. Thereafter, we activated the Magnetic Lasso tool and dragged selection areas only containing bronchial epithelium, then copy and pasted it into a new image. Then, the area of bronchial epithelium and the integral OD of MBD2-positive epithelium were measured. Finally, the mean integral OD of MBD2 protein staining in the bronchial epithelium was the ratio of the integral OD of MBD2-positive epithelium to area of bronchial epithelium. All of the images are assessed with the same parameters.
Western blot analysis
Total proteins were prepared using RIPA lysis supplemented with protease inhibitor cocktail. Equal amounts of protein (40 μg) were subjected to 10% SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Germany). Membranes were incubated with the appropriate primary antibody: anti-MBD2 (1:2,000, Santa Cruz Biotechnology, CA, USA), anti-GAPDH antibody (1:4,000, Sungene biotech, Tianjin, People’s Republic of China). After washing with Tris-buffered saline (TBS) containing 0.1% Tween-20, blots were incubated with secondary antibody conjugated to horseradish peroxidase (HRP; 1:4,000, Proteintech, Hubei, People’s Republic of China). The intensity of individual bands was quantified using ImageJ.
SiRNA transfection
MBD2 small interfering RNA (siRNA) and non-targeting negative control siRNA were synthesized by Ruibo Biotechnology, Co. (Guangzhou, People’s Republic of China). The target sequences were as follows: MBD2-siRNA sense 5′-GCGAAACGAUCCUCUCAAU dTdT-3′, MBD2-siRNA anti-sense 3′-dTdT CGCUUUGCUAGGAGAGUUA-5′. SiRNAs were transfected into HBE cells using Lipofectamine 2000 according to the manufacturer’s protocol (Invitrogen, Carlsbad, CA, USA).
RNA isolation and quantitative real-time PCR
Total RNA was extracted using TRIzol (Takara, Dalian, People’s Republic of China). RNA was reversed transcribed into cDNA using a cDNA RT–PCR kit (Takara, Dalian, People’s Republic of China). Then, cDNA was used as a template for quantitating gene expression using a SYBR Green real-time PCR kit (Takara) in an ABI PRISM Fast 7500 system (Applied Biosystems, Foster City, CA, USA). Data were analyzed using the 2−ΔΔCt method. Primers were as follows: β-actin F-5′-GCAAGCAGGACTATGACGAG-3′, β-actin R-5′-CAAATAAAGCCATGCCAATC-3′; IL-6 F-5′-CTGCTGCCTTCCCTGCC-3′, IL-6 R-5′-CCTCTTTGCTGCTTTCACACAT-3′; IL-8 F-5′-TCTGGCAACCCTAGTCTGCT-3′, IL-8 R-5′-AAACCAAGGCACAGTGGAAC-3′; MBD2 F-5′-AAGTGATCCGAAAATCTGGG C-3′, MBD2 R-5′-TGCCAACTGAGGCTTGCT TC-3′; IL-4 F-5′-CCAACTGCTTCCCCCTCTG-3′, IL-4 R-5′-TCTGTTACGGTCAACTCGGTG-3′; IL-10 F-5′-GACTTTAAGGGTTACCTGGGTTG-3′, IL-10 R-5′-TCACATGCGCCTTGATGTCTG-3′; TGF-β F-5′-GGCCAGATCCTGTCCAAGC-3′, TGF-β R-5′-GTGGGTTTCCACCATTAGCAC-3′; TNF-α F-5′-CCTCTCTCTAATCAGCCCTCTG-3′, TNF-α R-5′-GAGGACCTGGGAGTAGATGAG-3′.
Enzyme-linked immunosorbent assay
Concentrations of IL-6 and IL-8 in cell-culture supernatants were measured using IL-6 and IL-8 DuoSet ELISA kits (R&D Systems Europe, Abingdon, UK) according to the manufacturer’s instructions.33 The limits of detection for the IL-6 and IL-8 ELISA kits were 9.38 and 31.3 pg/mL, respectively. Levels of IL-6 and IL-6-inducible CXCL1 (KC) in BALF were measured using commercial mouse IL-6 and KC ELISA kits (R&D Systems Europe, Abingdon, UK). The limits of detection for the mouse IL-6 and KC ELISA kits were 1.8 and 2.0 pg/mL, respectively.
Statistical analysis
In human and mouse sample tests, normally distributed data were presented as mean ± SD and analyzed using Student’s t-test for two groups, or one-way analysis of variance (ANOVA) with Newman–Keuls post hoc test for multiple comparisons. Data which did not meet normal distribution were presented as median with range and evaluated by Mann–Whitney U-test for two groups. In the in vitro experiments, data were analyzed using Student’s t-test for two groups or one-way ANOVA with Newman–Keuls post hoc test for multiple comparisons. The relationship between MBD2 protein expression and proinflammatory cytokines was assessed by using Pearson analysis. All data were analyzed using Prism 6.0 (GraphPad Software Inc., San Diego, CA, USA). P<0.05 was considered statistically significant.
Results
Subject characteristics
The clinical characteristics of subjects are summarized in Table 1. Lung tissues were collected from 34 patients. Smoking history was similar among smokers with COPD and without COPD. Compared with non-COPD smokers or non-smokers, patients with COPD exhibited lower FEV1, FEV1% of predicted, and FEV1/FVC%. No significant differences in age were observed among the groups.
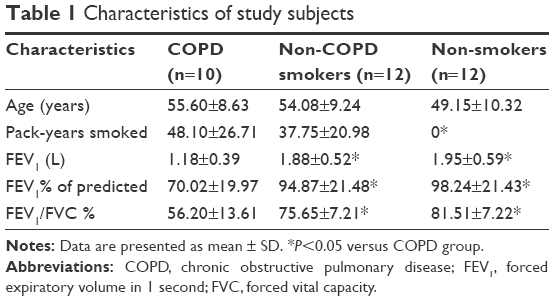 | Table 1 Characteristics of study subjects |
MBD2 protein expression was decreased in patients with COPD and CS-exposed mice
MBD2 protein expression in lung tissues was assessed by immunohistochemistry. Bronchial epithelium showed strong MBD2 protein staining in lung tissues of non-smokers on immunohistochemistry (Figure 1A). Inflammatory cells had moderate immunoreactivity, and there was low or negative MBD2 protein staining in mesenchymal cells (Figure S1). Relative to those of non-COPD smokers (Figure 1A, P<0.05) and non-smokers (Figure 1B, P<0.01), MBD2 protein levels in the epithelium were markedly decreased in patients with COPD (Figure 1C). There was no difference in MBD2 protein expression levels between non-COPD smokers and non-smokers (Figure 1E). We further analyzed MBD2 protein expression in whole-lung tissue homogenates by Western blot. Consistent with results from immunohistochemistry, the relative expression of MBD2 protein was significantly decreased in patients with COPD, as compared with the other two groups (P<0.05 for both; Figure 1F and G).
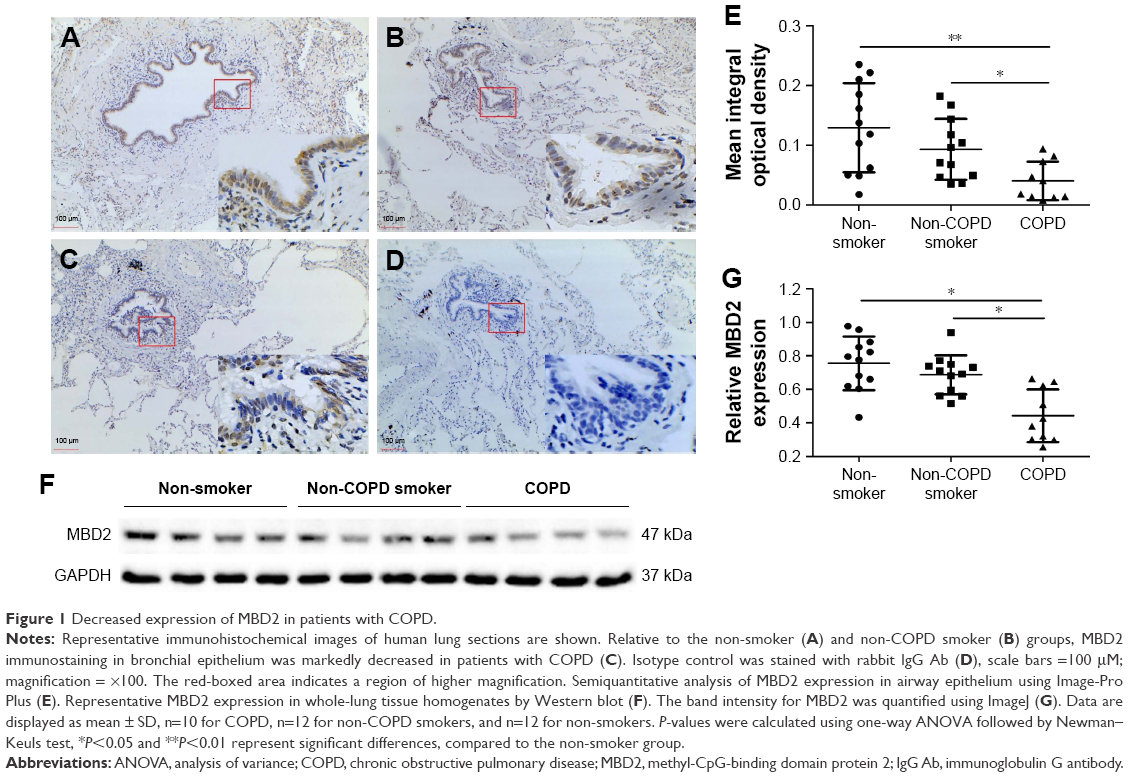 | Figure 1 Decreased expression of MBD2 in patients with COPD. |
Pulmonary function and MLI were measured to confirm the establishment of a passive smoking model. Compared with controls, forced expiratory volume in 0.1 s (FEV0.1s; P<0.01), ratio of FEV0.1s to forced vital capacity (FEV0.1s/FVC; P<0.01), and dynamic lung compliance (Cdyn; P<0.001) were significantly decreased in CS-exposed mice (Figure 2A–C), whereas lung resistance (RL) was remarkably increased (P<0.01; Figure 2D). Moreover, MLI measurement was dramatically elevated in CS-exposed mice (P<0.001; Figure 2E and F). Levels of IL-6 and KC in BALF were determined by ELISA. Compared with controls, levels of IL-6 (Figure S2A) and KC (Figure S2B) in BALF were upregulated in CS-exposed mice. These results indicate that CS-exposed mice suffer from airflow limitation, emphysema, and airway inflammation. Then, we examined MBD2 expression in lung tissues of model mice. MBD2 protein expression was significantly downregulated in CS-exposed mice compared with that of controls on evaluation by immunohistochemistry (Figure 3A–D) and by immunoblot (Figure 3E and F). MBD2 expression in the bronchial epithelium were negatively correlated with IL-6 (P=0.036, R=-0.53, Figure S3A) and KC (P=0.026, R=0.55, Figure S3B) levels in BALF of mice. However, there was no significant difference in MBD2 mRNA levels between the CS-exposed and control groups (Figure S4A).
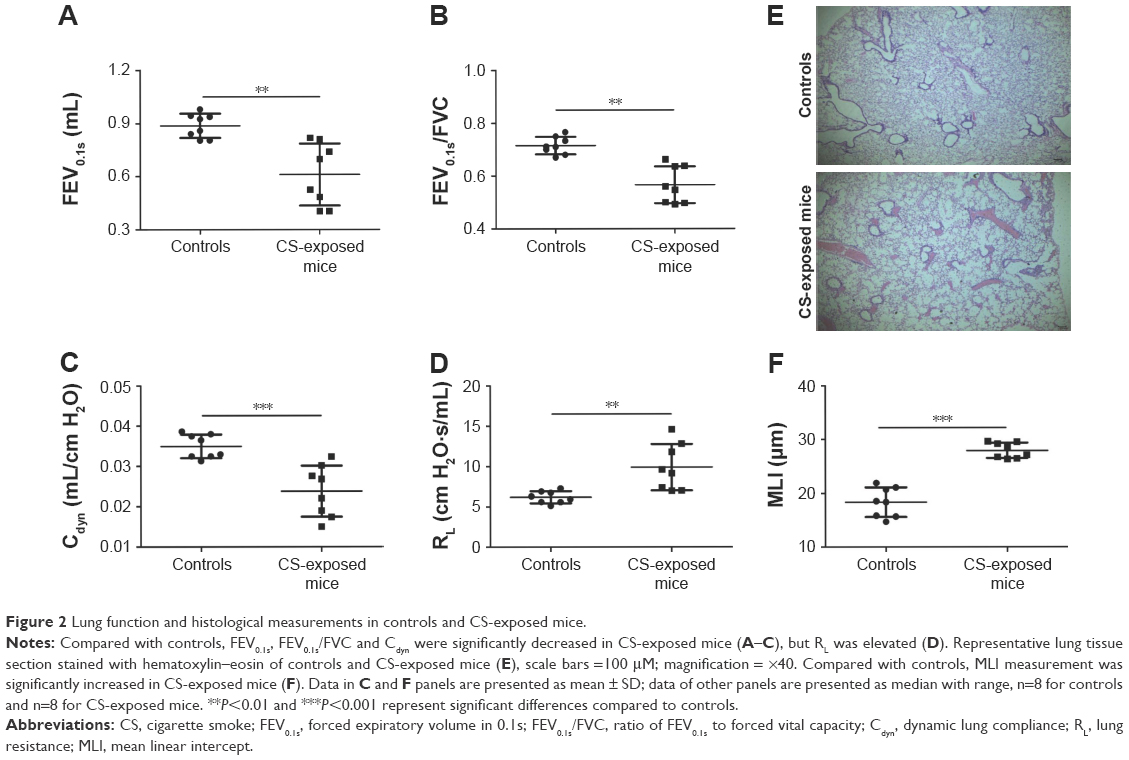 | Figure 2 Lung function and histological measurements in controls and CS-exposed mice. |
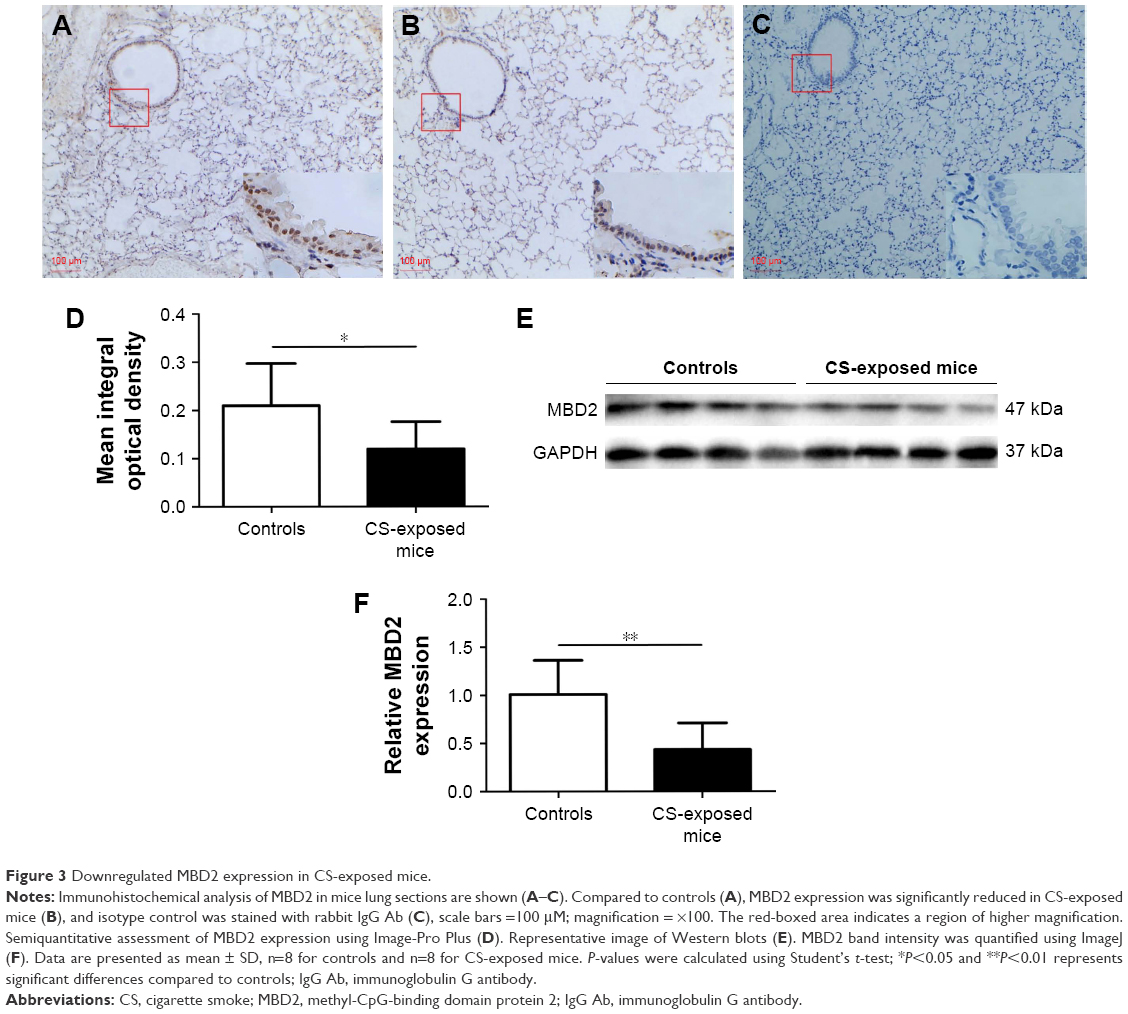 | Figure 3 Downregulated MBD2 expression in CS-exposed mice. |
CSE attenuates MBD2 protein expression in HBE
CCK8 cell-activity estimation showed that CSE at concentrations ranging from 1.25% to 10% did not affect cell viability after incubation for 24–72 h (Figure 4A).
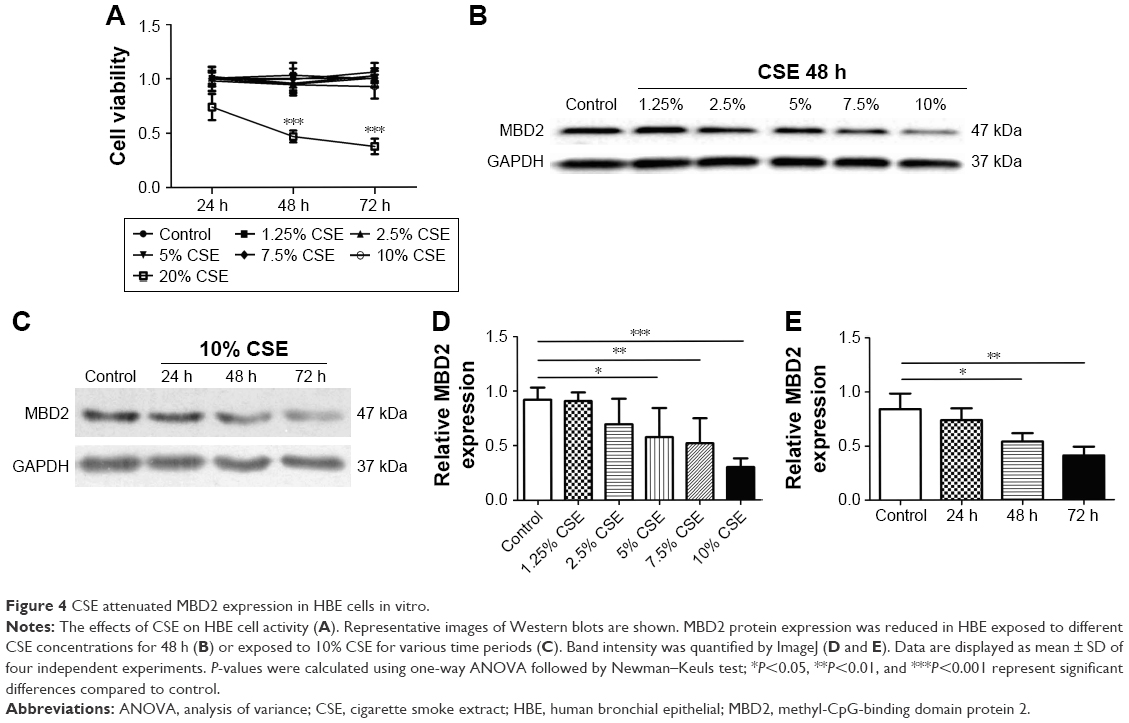 | Figure 4 CSE attenuated MBD2 expression in HBE cells in vitro. |
To determine the potential influences of CSE on MBD2 protein expression in vitro, HBE were treated with various concentrations of CSE for 48 h or with 10% CSE for 24, 48, and 72 h. MBD2 protein expression began to significantly decline when cells were treated with 5% CSE (P<0.05) for 48 h, and reached the lowest level at 10% CSE (P<0.001; Figure 4B and D). Meanwhile, a time-dependent decrease in MBD2 protein expression was found for HBE treated with 10% CSE (Figure 4C and E). However, there were no changes in MBD2 mRNA expression in HBE treated with different concentrations of CSE for different time periods (Figure S4B).
MBD2 knockdown increases cytokine gene expression in HBE
To investigate whether MBD2 participates in modulating cytokines expression in airway epithelium, we silenced the MBD2 gene in HBE using siRNA, and then tested cytokine expression. As expected, after transfecting HBE with MBD2 siRNA (MBD2-siRNA), there was a significant decrease in MBD2 mRNA and protein expression, compared to cells transfected with negative control siRNA (Con-siRNA; Figure 5A and B).
 | Figure 5 MBD2 knockdown increased IL-6 and IL-8 expression in HBE. |
We, next, examined levels of the inflammatory cytokines IL-4, IL-6, IL-8, IL-10, TGF-β, and TNF-α in MBD2-silenced HBE, both at 24 and 48 h after transfection. Compared with Con-siRNA-transfected HBE, mRNA levels of IL-6 (P<0.01 for both) and IL-8 (P<0.05 for 24 h and P<0.01 for 48 h) were significantly increased in MBD2-siRNA-transfected HBE (Figure 5C and D). However, there were no differences in TGF-β, and TNF-α mRNA between the two groups (data not shown). IL-4 and IL-10 mRNA expression was near or below the detection limit of real-time PCR. Consistent with the results for mRNA levels, IL-6 (P<0.05) and IL-8 (P<0.01) protein expression was significantly elevated in MBD2-knockdown HBE (Figure 5E and F).
Decreased MBD2 expression enhanced IL-6 and IL-8 secretion promoted by the ERK1/2 pathway in HBE
After MBD2-siRNA transfection for 24 h, HBE were treated with or without 10% CSE for 24 h. The mRNA expression levels of IL-6 (P<0.001) and IL-8 (P<0.05) were markedly increased after 10% CSE simulation (Figure 6A and B). Similar results were detected in IL-6 (P<0.05) and IL-8 (P<0.05) protein levels (Figure 6C and D). These data further verified that MBD2 knockdown amplified IL-6 and IL-8 secretion by HBE in a CSE inflammatory milieu.
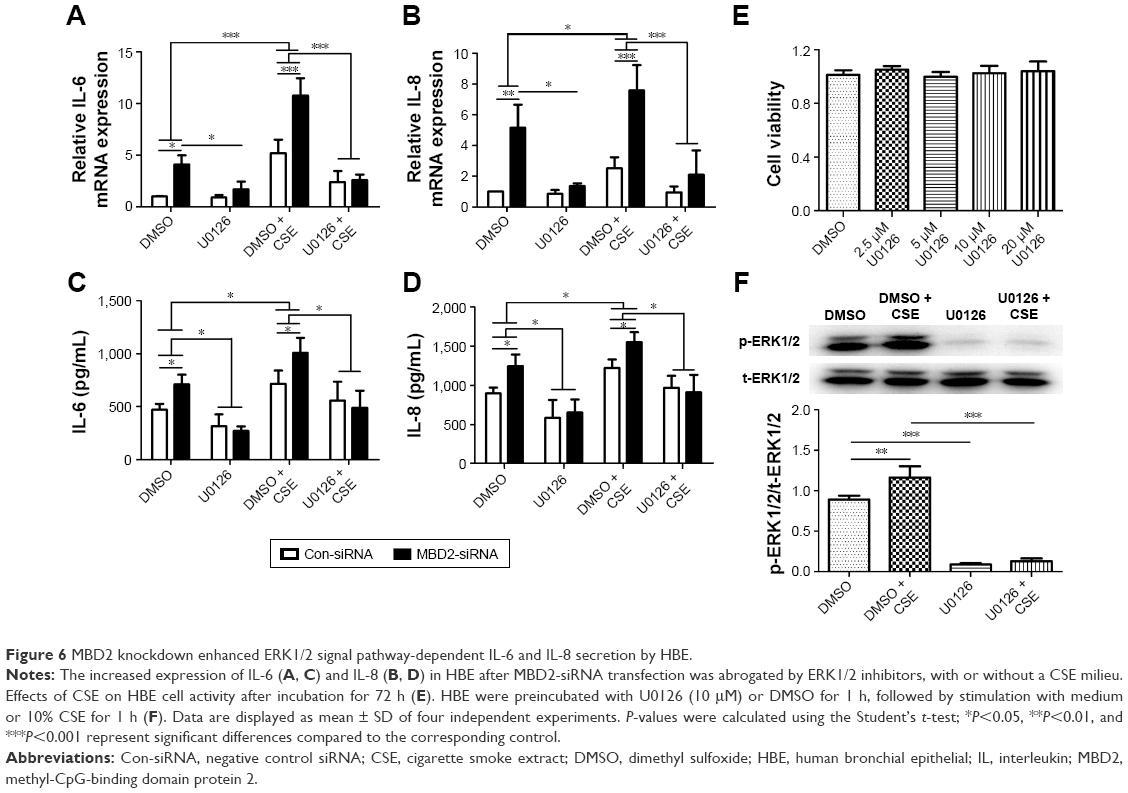 | Figure 6 MBD2 knockdown enhanced ERK1/2 signal pathway-dependent IL-6 and IL-8 secretion by HBE. |
To evaluate the underlying mechanisms by which MBD2 affects cytokine production, we next explored whether MAPK signaling was activated in HBE after MBD2 knockdown by using the inhibitors U-0126, SP600125, and SB239063 to specifically block the ERK1/2, JNK, and p38 MAPK pathways, respectively. CCK8 cell-activity estimation showed that U0126 at concentrations ranging from 2.5 to 20 μM did not affect cell viability after incubation for 72 h (Figure 6E). HBE were preincubated with 10 μM U0126 or vehicle control (DMSO) for 1 h, followed by treatment with or without 10% CSE for 1 h (Figure 6F). As demonstrated in Figure 6B, 10% CSE increased the phosphorylation of ERK1/2 (p-ERK1/2), and U0126 could diminish the CSE-induced activation of ERK1/2 in HBE. Only U-0126, but not SP600125 or SB239063 (data not shown), could significantly inhibit IL-6 and IL-8 expression in MBD2-knockdown HBE, with or without CSE treatment (Figure 6A–D). Those results indicated that decreased MBD2 levels could enhance secretion of IL-6 and IL-8 via the ERK1/2 pathway.
Discussion
In the present study, we demonstrated for the first time that MBD2 protein expression in airway epithelium from patients with COPD was decreased relative to non-COPD smokers and non-smokers. MBD2 expression was, likewise, distinctly reduced in CS-exposed mice and CSE-stimulated HBE in vitro. As an additional functional verification, we found that MBD2 knockdown promoted HBE to release more IL-6 and IL-8 via ERK signaling. These results indicate that MBD2 might play an instrumental role in modulating airway inflammation associated with COPD.
CS is a complex mixture of more than 4,700 chemical compounds, and represents a major pathogenic factor that has been implicated in numerous diseases, including cancer, cardiovascular diseases, and COPD.34 Cigarette smoking is associated with elevated serum homocysteine, and homocysteine was found to reduce MBD2 protein expression in vascular smooth muscle cells, suggesting that cigarette smoking might influence MBD2 protein expression.12–14 Our results showed that MBD2 protein expression was reduced in bronchial epithelium from patients with COPD and in CSE-treated cells in vitro. It was reported that exposure to CSE resulted in no change of MBD2 protein expression in other cells.36 The impact of CS on MBD2 protein expression appear to be cell-type specific. Given that cigarette smoking can affect gene and protein expression in both humans and mice, we further confirmed that MBD2 protein expression was decreased in bronchial epithelium from CS-exposed mice.35 These observations indicated that the decrease in MBD2 protein expression seen in the COPD bronchial epithelium is at least partially due to long-term CS exposure. Interestingly, MBD2 is not the only one of the MBD protein family altered by CSE. Mukhopadhyay showed that MBD3 and MeCP-2 protein were markedly reduced following exposure to CSE in branchial–arch-derived cells, which contribute to the formation of the lip and palate.36
However, we could not detect a distinct difference in MBD2 mRNA expression in lung tissues from CS-exposed mice and in CSE-treated HBE in vitro. Further, Tommasi et al reported no significant alteration of MBD2 mRNA between control mice and those exposed to secondhand smoke.37 In contrast, MBD2 mRNA expression was decreased in subjects who had chronic occupational exposure to benzene, toluene, and xylene, but MBD2 levels were elevated in CD4+ T cells from patients with several autoimmune diseases, including systemic erythematosus lupus, dermatomyositis, and multiple sclerosis.20–22 In addition, Tan et al confirmed a post-transcriptional role for valproate (Sigma-Aldrich, St. Louis, MO, USA) – a drug used to treat seizures – in downregulating MBD2 expression.38 Therefore, CS-induced changes in post-transcriptional regulation might explain the observed alterations in MBD2 protein expression. Post-transcriptional regulation, such as the processing, export, localization, turnover, and translation of mRNAs is an integral part of gene expression and adds substantial complexity to transcriptional control.39 Several miRNAs, including miR-520b, miR-221, and miR-290/371, were reported to regulate MBD2 expression.40–42 However, MBD2 protein-specific changes, such as cleavage, processing, and degradation, were still unknown. As such, further studies are needed to elucidate the underlying mechanisms of MBD2 reduction induced by CS.
MBD2 plays multiple and complex roles in regulating cytokine secretion. Wang et al reported that MBD2 knockdown by siRNA markedly reduced expression of IL-10, CTLA-4, and TGF-β, which are essential for normal Treg function. In a reciprocal study, retroviral transduction of MBD2−/− Treg cells with MBD2 enhanced expression of IL-10, CTLA-4, and TGF-β, but reduced IL-6 expression.24 Cheng et al showed that mRNA levels for TNF-α and MCP-1 were significantly lower in visceral adipose tissues that originated from MBD2−/− mice fed a high-fat diet as compared with that of controls.43 Moreover, researchers demonstrated that MBD2−/− CD4+ T cells manifested a significant increase in IFN-γ and IL-4 secretion, whereas dendritic cells derived from MBD2−/− mice showed a similar capacity to secrete IL-6 and TNF-α.23 So far, there were no reports on the impact of MBD2 on smoking-induced inflammatory responses. Therefore, we used siRNA to knockdown MBD2 in HBE with or without CSE treatment, and found that reduced MBD2, together with a CSE milieu, could synergistically enhance IL-6 and IL-8 secretion. Negative correlation between MBD2 protein expression in the bronchial epithelium of mice and IL-6 as well as KC level in BALF were confirmed. However, there was no difference in the expression of IL-4, IL-10, TGF-β, and TNF-α (data not shown) between MBD2-siRNA HBE and Con-siRNA controls. Impacts of MBD2 on cytokine secretion appear to be cell-type specific. Both IL-6 and IL-8 exert a role in orchestrating inflammation associated with COPD.44 Levels of these cytokines in sputum are related to lung function, disease severity, and clinical outcome of COPD.45–47 Our data implied that the smoking-induced reduction in MBD2 levels could increase secretion of some inflammatory mediators by HBE. Interestingly, this is not the first example of the MBD protein family being involved in regulating the secretion of inflammatory cytokines. Specifically, a deficiency in methyl CpG-binding protein 2 enhances the expression of IL-6 by promoting NF-κB signaling in myeloid-derived cells.48
MAPK signaling possesses critical functions in regulating COPD-related inflammation by activating transcription factors and/or stabilization of post-translational mRNA.49,50 As noted by Rao et al, p38 MAPK and ERK1/2 signals were activated in both MBD2 siRNA-transfected endothelial cells and MBD2−/− mice.27 Consistent with the Rao study, we found that ERK1/2 inhibitors could block increases in cytokine expression caused by knockdown of MBD2 in HBE with or without CSE treatment. These data suggested that MBD2 modulated HBE inflammation probably via ERK1/2 signaling. Several articles demonstrated that the Wnt/β-catenin pathway was downregulated in patients with COPD, CS-exposed mice, and in cellular models.51–53 Guo et al showed that the reduced activity of WNT/β-catenin signaling induced by CSE may promote cytokine production in airway epithelium by p38 MAPK but not by the ERK pathway.52 Although Phesse found that deficiency of MBD2 suppressed the Wnt pathway, we speculated that increased levels of IL-6 and IL-8 expression is independent of Wnt/β-catenin pathway in MBD2-siRNA transfected HBE.54
This study has some limitations. Ideally, primary bronchial epithelial cells from COPD patients, non-COPD smokers, and non-smokers should be used. Instead, we used the HBE cell line for the in vitro studies, and it is unknown whether findings obtained with these cells can be extrapolated to humans. MBD2 expression showed a tendency to decrease in non-COPD smokers compared to non-smokers; however, it failed to reach statistical significance. This might partly be due to the relatively small sample size and the many uncontrolled factors influencing MBD2 protein expression in humans. Moreover, in the future, we should identify genes that MBD2 regulates through direct binding to promoters.
In conclusion, our study demonstrated that MBD2 expression decreased in the lung bronchial epithelium of patients with COPD, and this decrease was associated with increased levels of IL-6 and IL-8 in HBE. MBD2 enhanced the secretion of those inflammatory cytokines via an ERK1/2 signal pathway-dependent mechanism. Therefore, MBD2 might contribute to COPD pathogenesis.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (grant nos 81570033, 81570047, 81470227, 81370145, and 81370156), National Key Basic Research and Development Program (973 Program, grant no 20l5CB553403), Chinese Medical Association Research Project (grant no 2013BAI09B00), and Changjiang Scholars and Innovative Research Team in University (grant no IRT_14R20).
Disclosure
The authors report no conflicts of interest in this work.
References
Barnes PJ. Chronic obstructive pulmonary disease. N Engl J Med. 2000;343(4):269–280. | ||
Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study. Lancet. 1997;349(9064):1498–1504. | ||
Pauwels RA, Rabe KF. Burden and clinical features of chronic obstructive pulmonary disease (COPD). Lancet. 2004;364(9434):613–620. | ||
Fischer BM, Pavlisko E, Voynow JA. Pathogenic triad in COPD: oxidative stress, protease-antiprotease imbalance, and inflammation. Int J Chron Obstruct Pulmon Dis. 2011;6:413–421. | ||
Ito K, Barnes PJ. COPD as a disease of accelerated lung aging. Chest. 2009;135(1):173–180. | ||
Plataki M, Tzortzaki E, Rytila P, Demosthenes M, Koutsopoulos A, Siafakas NM. Apoptotic mechanisms in the pathogenesis of COPD. Int J Chron Obstruct Pulmon Dis. 2006;1(2):161–171. | ||
Gao W, Li L, Wang Y, et al. Bronchial epithelial cells: the key effector cells in the pathogenesis of chronic obstructive pulmonary disease? Respirology. 2015;20(5):722–729. | ||
Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J. 2003;22(4):672–688. | ||
Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31(2):89–97. | ||
Bird AP, Wolffe AP. Methylation-induced repression – belts, braces, and chromatin. Cell. 1999;99(5):451–454. | ||
Ng HH, Zhang Y, Hendrich B, et al. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat Genet. 1999;23(1):58–61. | ||
Davis JA, Brown AT, Chen H, et al. Cigarette smoke increases intimal hyperplasia and homocysteine in a rat carotid endarterectomy. J Surg Res. 2004;121(1):69–75. | ||
Venn A, Britton J. Exposure to secondhand smoke and biomarkers of cardiovascular disease risk in never-smoking adults. Circulation. 2007;115(8):990–995. | ||
Yideng J, Jianzhong Z, Ying H, et al. Homocysteine-mediated expression of SAHH, DNMTs, MBD2, and DNA hypomethylation potential pathogenic mechanism in VSMCs. DNA Cell Biol. 2007;26(8):603–611. | ||
Cheishvili D, Chik F, Li CC, et al. Synergistic effects of combined DNA methyltransferase inhibition and MBD2 depletion on breast cancer cells; MBD2 depletion blocks 5-aza-2′-deoxycytidine-triggered invasiveness. Carcinogenesis. 2014;35(11):2436–2446. | ||
Pontes TB, Chen ES, Gigek CO, et al. Reduced mRNA expression levels of MBD2 and MBD3 in gastric carcinogenesis. Tumour Biol. 2014;35(4):3447–3453. | ||
Pulukuri SM, Rao JS. CpG island promoter methylation and silencing of 14-3-3sigma gene expression in LNCaP and Tramp-C1 prostate cancer cell lines is associated with methyl-CpG-binding protein MBD2. Oncogene. 2006;25(33):4559–4572. | ||
Sansom OJ, Berger J, Bishop SM, Hendrich B, Bird A, Clarke AR. Deficiency of Mbd2 suppresses intestinal tumorigenesis. Nat Genet. 2003;34(2):145–147. | ||
Sapkota Y, Robson P, Lai R, Cass CE, Mackey JR, Damaraju S. A two-stage association study identifies methyl-CpG-binding domain protein 2 gene polymorphisms as candidates for breast cancer susceptibility. Eur J Hum Genet. 2012;20(6):682–689. | ||
Balada E, Ordi-Ros J, Serrano-Acedo S, Martinez-Lostao L, Vilardell-Tarrés M. Transcript overexpression of the MBD2 and MBD4 genes in CD4+ T cells from systemic lupus erythematosus patients. J Leukoc Biol. 2007;81(6):1609–1616. | ||
Fagone P, Mangano K, Di Marco R, et al. Expression of DNA methylation genes in secondary progressive multiple sclerosis. J Neuroimmunol. 2016;290:66–69. | ||
Lei W, Luo Y, Lei W, et al. Abnormal DNA methylation in CD4+ T cells from patients with systemic lupus erythematosus, systemic sclerosis, and dermatomyositis. Scand J Rheumatol. 2009;38(5):369–374. | ||
Zhong J, Yu Q, Yang P, et al. MBD2 regulates TH17 differentiation and experimental autoimmune encephalomyelitis by controlling the homeostasis of T-bet/Hlx axis. J Autoimmun. 2014;53:95–104. | ||
Wang L, Liu Y, Han R, et al. Mbd2 promotes foxp3 demethylation and T-regulatory-cell function. Mol Cell Biol. 2013;33(20):4106–4115. | ||
Hutchins AS, Artis D, Hendrich BD, Bird AP, Scott P, Reiner SL. Cutting edge: a critical role for gene silencing in preventing excessive type 1 immunity. J Immunol. 2005;175(9):5606–5610. | ||
Hutchins AS, Mullen AC, Lee HW, et al. Gene silencing quantitatively controls the function of a developmental trans-activator. Mol Cell. 2002;10(1):81–91. | ||
Rao X, Zhong J, Zhang S, et al. Loss of methyl-CpG-binding domain protein 2 enhances endothelial angiogenesis and protects mice against hind-limb ischemic injury. Circulation. 2011;123(25):2964–2974. | ||
Seimetz M, Parajuli N, Pichl A, et al. Inducible NOS inhibition reverses tobacco-smoke-induced emphysema and pulmonary hypertension in mice. Cell. 2011;147(2):293–305. | ||
Wu H, Yang S, Wu X, et al. Interleukin-33/ST2 signaling promotes production of interleukin-6 and interleukin-8 in systemic inflammation in cigarette smoke-induced chronic obstructive pulmonary disease mice. Biochem Biophys Res Commun. 2014;450(1):110–116. | ||
Thurlbeck WM. Measurement of pulmonary emphysema. Am Rev Respir Dis. 1967;95(5):752–764. | ||
Xu Y, Li H, Bajrami B, et al. Cigarette smoke (CS) and nicotine delay neutrophil spontaneous death via suppressing production of diphosphoinositol pentakisphosphate. Proc Natl Acad Sci U S A. 2013;110(19):7726–7731. | ||
Lappas M, Odumetse TL, Riley C, et al. Pre-labour fetal membranes overlying the cervix display alterations in inflammation and NF-kappaB signalling pathways. Placenta. 2008;29(12):995–1002. | ||
Wang M, Gao P, Wu X, et al. Impaired anti-inflammatory action of glucocorticoid in neutrophil from patients with steroid-resistant asthma. Respir Res. 2016;17(1):153. | ||
Stedman RL. The chemical composition of tobacco and tobacco smoke. Chem Rev. 1968;68(2):153–207. | ||
Morissette MC, Lamontagne M, Bérubé JC, et al. Impact of cigarette smoke on the human and mouse lungs: a gene-expression comparison study. PLoS One. 2014;9(3):e92498. | ||
Mukhopadhyay P, Greene RM, Pisano MM. Cigarette smoke induces proteasomal-mediated degradation of DNA methyltransferases and methyl CpG-/CpG domain-binding proteins in embryonic orofacial cells. Reprod Toxicol. 2015;58:140–148. | ||
Tommasi S, Zheng A, Besaratinia A. Expression of epigenetic modifiers is not significantly altered by exposure to secondhand smoke. Lung Cancer. 2015;90(3):598–603. | ||
Tan NN, Tang HL, Lin GW, et al. Epigenetic downregulation of Scn3a expression by valproate: a possible role in its anticonvulsant activity. Mol Neurobiol. 2016;54(4):2831–2842. | ||
Mata J, Marguerat S, Bähler J. Post-transcriptional control of gene expression: a genome-wide perspective. Trends Biochem Sci. 2005;30(9):506–514. | ||
Cui S, Liu L, Wan T, Jiang L, Shi Y, Luo L. MiR-520b inhibits the development of glioma by directly targeting MBD2. Am J Cancer Res. 2017;7(7):1528–1539. eCollection 2017. | ||
He S, Lai R, Chen D, et al. Downregulation of miR-221 inhibits cell migration and invasion through targeting methyl-CpG binding domain protein 2 in human oral squamous cell carcinoma cells. Biomed Res Int. 2015;2015:751672. | ||
Cao Y, Guo WT, Tian S, et al. miR-290/371-Mbd2-Myc circuit regulates glycolytic metabolism to promote pluripotency. EMBO J. 2015;34(5):609–623. | ||
Cheng J, Song J, He X, et al. Loss of Mbd2 protects mice against high-fat diet-induced obesity and insulin resistance by regulating the homeostasis of energy storage and expenditure. Diabetes. 2016;65(11):3384–3395. | ||
Decramer M, Rennard S, Troosters T, et al. COPD as a lung disease with systemic consequences – clinical impact, mechanisms, and potential for early intervention. COPD. 2008;5(4):235–256. | ||
Agustí A, Edwards LD, Rennard SI, et al; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PLoS One. 2012;7(5):e37483. | ||
Franciosi LG, Page CP, Celli BR, et al. Markers of disease severity in chronic obstructive pulmonary disease. Pulm Pharmacol Ther. 2006;19(3):189–199. | ||
Gao P, Zhang J, He X, Hao Y, Wang K, Gibson PG. Sputum inflammatory cell-based classification of patients with acute exacerbation of chronic obstructive pulmonary disease. PLoS One. 2013;8(5):e57678. | ||
O’Driscoll CM, Lima MP, Kaufmann WE, Bressler JP. Methyl CpG binding protein 2 deficiency enhances expression of inflammatory cytokines by sustaining NF-kB signaling in myeloid derived cells. J Neuroimmunol. 2015;283:23–29. | ||
Kracht M, Saklatvala J. Transcriptional and post-transcriptional control of gene expression in inflammation. Cytokine. 2002;20(3):91–106. | ||
Rahman I. Oxidative stress in pathogenesis of chronic obstructive pulmonary disease: cellular and molecular mechanisms. Cell Biochem Biophys. 2005;43(1):167–188. | ||
Wang R, Ahmed J, Wang G, et al. Down-regulation of the canonical Wnt β-catenin pathway in the airway epithelium of healthy smokers and smokers with COPD. PLoS One. 2011;6(4):e14793. | ||
Guo L, Wang T, Wu Y, et al. WNT/β-catenin signaling regulates cigarette smoke-induced airway inflammation via the PPARδ/p38 pathway. Lab Invest. 2016;96(2):218–229. | ||
Kneidinger N, Yildirim AÖ, Callegari J, et al. Activation of the WNT/β-catenin pathway attenuates experimental emphysema. Am J Respir Crit Care Med. 2011;183(6):723–733. | ||
Phesse TJ, Parry L, Reed KR, et al. Deficiency of Mbd2 attenuates Wnt signaling. Mol Cell Biol. 2008;28(19):6094–6103. |
Supplementary materials
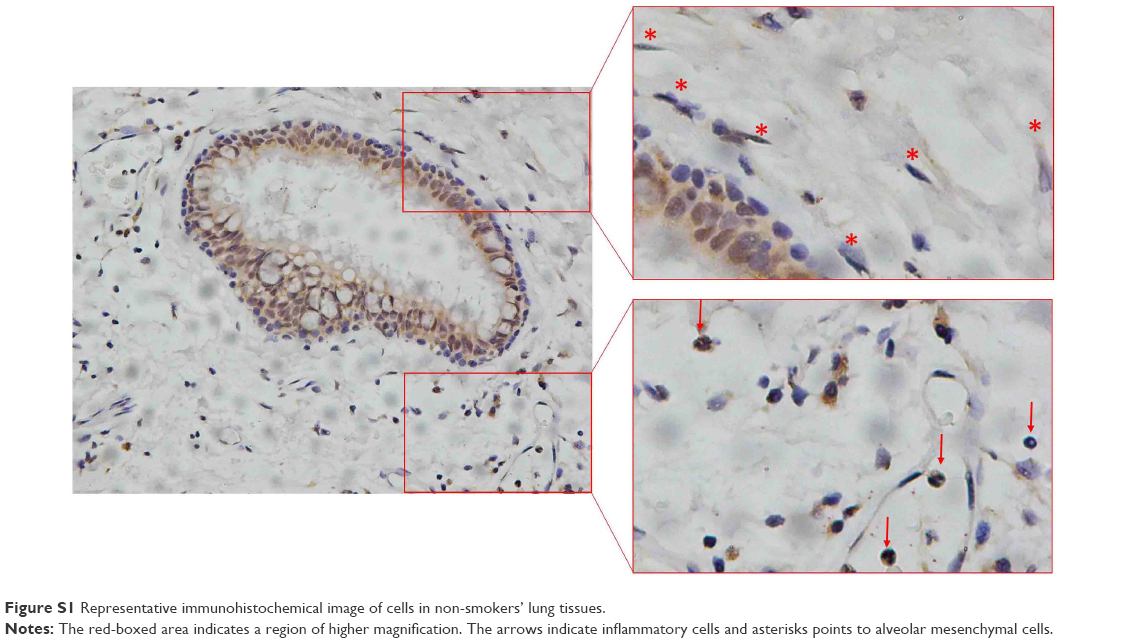 | Figure S1 Representative immunohistochemical image of cells in non-smokers’ lung tissues. |
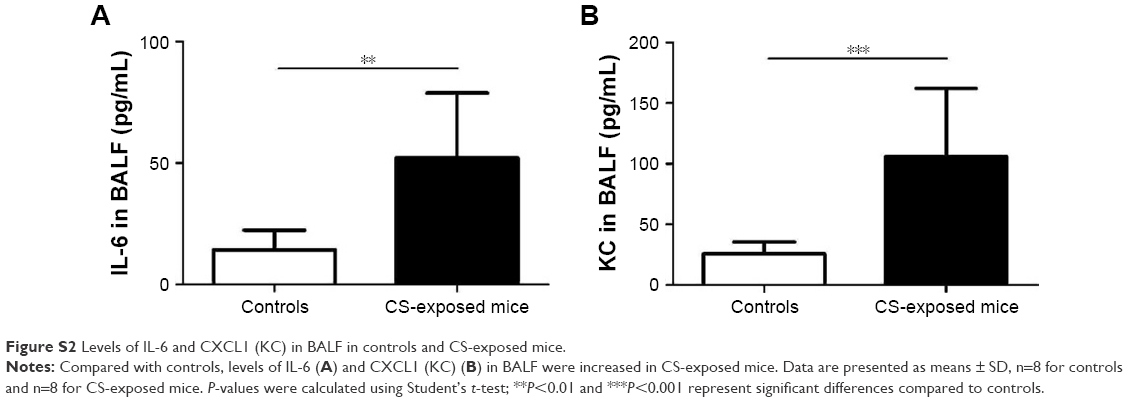 | Figure S2 Levels of IL-6 and CXCL1 (KC) in BALF in controls and CS-exposed mice. |
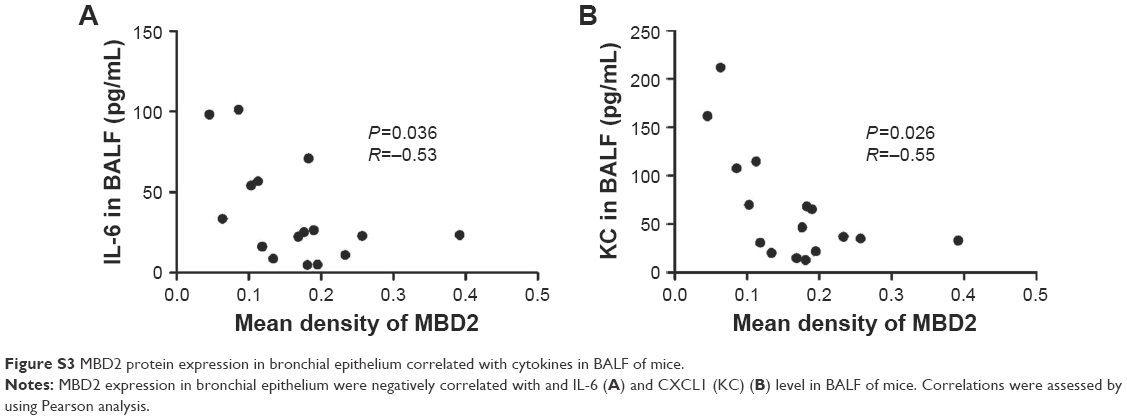 | Figure S3 MBD2 protein expression in bronchial epithelium correlated with cytokines in BALF of mice. |
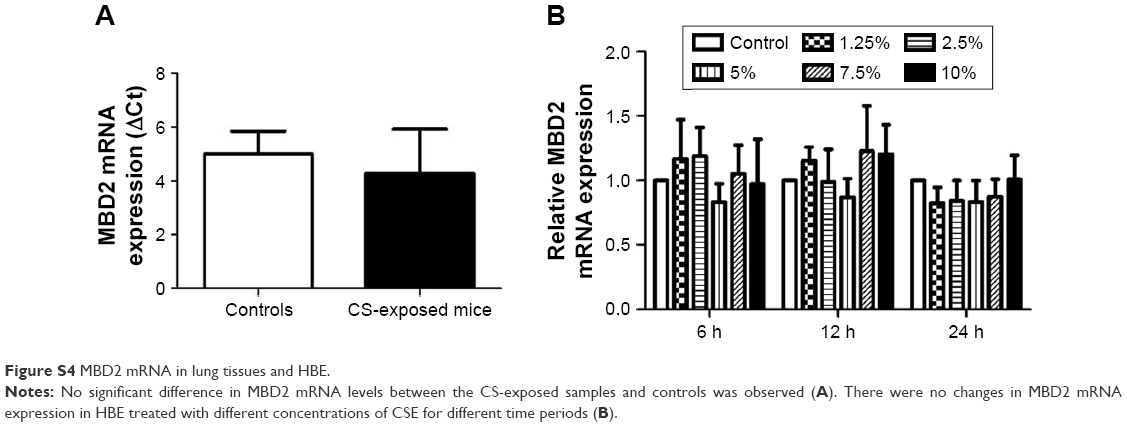 | Figure S4 MBD2 mRNA in lung tissues and HBE. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.