Back to Journals » Drug Design, Development and Therapy » Volume 11
Redox-responsive and pH-sensitive nanoparticles enhanced stability and anticancer ability of erlotinib to treat lung cancer in vivo
Received 11 September 2017
Accepted for publication 3 November 2017
Published 8 December 2017 Volume 2017:11 Pages 3519—3529
DOI https://doi.org/10.2147/DDDT.S151422
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Sheng Tan, Guoxiang Wang
Department of Thoracic Surgery, The Affiliated Hospital of Xuzhou Medical University, Xuzhou, Jiangsu, People’s Republic of China
Purpose: Erlotinib (ETB) is a well-established therapeutic for non-small-cell lung cancer (NSCLC). To overcome drug resistance and severe toxicities in the clinical application, redox-responsive and pH-sensitive nanoparticle drug delivery systems were designed for the encapsulation of ETB.
Methods: Poly(acrylic acid)-cystamine-oleic acid (PAA-ss-OA) was synthesized. PAA-ss-OA-modified ETB-loaded lipid nanoparticles (PAA-ETB-NPs) were prepared using the emulsification and solvent evaporation method. The tumor inhibition efficacy of PAA-ETB-NPs was compared with that of ETB-loaded lipid nanoparticles (ETB-NPs) and free ETB anticancer drugs in tumor-bearing mice.
Results: PAA-ETB-NPs had a size of 170 nm, with a zeta potential of -32 mV. The encapsulation efficiency and drug loading capacity of PAA-ETB-NPs were over 85% and 2.6%, respectively. In vitro cytotoxicity of ETB-NPs were higher than that of ETB solution. The cytotoxicity of PAA-ETB-NPs was the highest. The in vivo tumor growth inhibition by PAA-ETB-NP treatment was significantly higher than that by ETB-NPs and ETB solution. No obvious weight loss was observed in any of the treatment groups, indicating that all the treatments were well tolerated.
Conclusion: PAA-ETB-NPs could enhance the stability and anti-cancer ability of ETB to treat lung cancer and are a promising drug delivery system for lung cancer treatment.
Keywords: epidermal growth factor receptor, kinase inhibitor, pH-sensitive, redox-responsive, poly(acrylic acid)
Introduction
Non-small-cell lung cancer (NSCLC), the leading cause of cancer-related death, was responsible for 1 million new cases and 900,000 deaths every year worldwide.1 Epidermal growth factor receptor (EGFR) mutation-positive NSCLC accounts for ~40%–80% of NSCLC.2,3 Unfortunately, the majority of NSCLC patients are diagnosed in the late stage, and prognosis remains poor with a median over survival of around 1 year.4 Therefore, the main treatment counts on chemotherapy or maintenance treatment.5,6
Conventional platinum-based chemotherapies include platinum alone or combination with the third-generation cytotoxic drugs such as pemetrexed, paclitaxel, docetaxel, gemcitabine, and vinorelbine.7,8 It is well known that these therapies bring adverse toxic effects due to lack of selectivity for tumor cells.9 Tyrosine kinase plays an important role in many cell signaling pathways, and the inhibition of their receptors could block the pathways and tumor activity.10 The introduction of small molecule targeted therapy like kinase inhibitors for the therapy of NSCLC has landmark significance.11 Recent clinical data revealed that EGFR tyrosine kinase inhibitors are applied as the standard first-line treatments for advanced nonsquamous NSCLC with activating EGFR mutations.5,12
Erlotinib (ETB), a kinase inhibitor, is a well-established therapeutic.12,13 Several clinical researchers have reported that ETB showed a significantly longer median progression-free survival compared with a standard chemotherapy group with European and Eastern Asians patients with EGFR mutation-positive NSCLC.12,13 The US-FDA has approved ETB for the treatment of metastatic NSCLC and pancreatic cancer. ETB is available in the market as tablets for oral administration. However, its clinical application is hindered by drug resistance and severe toxicities such as skin rash due to oral delivery, diarrhea, Stevens–Johnson syndrome, gastrointestinal perforations, and so on. Therefore, there is an unmet need to develop a novel strategy for the effective delivery of ETB to the cancer zone to reduce side effects and overcome drug resistance.
Nanoscale drug delivery systems have been investigated widely in oncotherapy due to their merits including the ability to target to the tumor sites, reduction in effective dose, and less toxic to healthy cells.14 Compared with passive targeted nanocarriers, nanoparticles with structures that respond to external stimuli (including reduction, pH, light, and enzyme activities) have attracted considerable attention in the field of cancer therapy. The extracellular environment of tumors is more acidic (in the range of pH 6.5–6.9), while endosomes and lysosomes have even lower pH values of 5.0–5.5.15 Thus, pH-sensitive nanoparticles represent an effective strategy for cancer therapies. In comparison, redox-responsive nanoparticles are mostly intended to release drugs in the cytoplasm in which the concentration of glutathione (GSH) is higher than in the plasma.16,17 In the present study, GSH- and pH-sensitive nanoscale drug delivery systems were designed and evaluated.
Poly(acrylic acid) (PAA) is a pH-sensitive and biocompatible polymer.18,19 The disulfide bonds of cystamine (ss) could stabilize the nanoparticles during systemic circulation and be broken in the cytoplasm where the GSH concentrations are usually 1,000-fold higher.20 Therefore, PAA-ss-oleic acid (PAA-ss-OA) was developed as a novel material for the surface modification of lipid carrier for the delivery of ETB. The tumor inhibition efficacy of PAA-ss-OA-modified ETB-loaded lipid nanoparticles (PAA-ETB-NPs) was compared with that of ETB-loaded lipid nanoparticles (ETB-NPs) and free ETB anticancer drugs in tumor-bearing mice.
Materials and methods
Materials
ETB, PAA (mw =1,800), ss, OA, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), N-hydroxysuccinimide (NHS), dimethyl sulfoxide (DMSO), glyceryl monostearate (GMS), fetal bovine serum (FBS), Dulbecco’s Modified Eagle’s Medium (DMEM), and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). All other chemicals and reagents were of analytical grade or high-performance liquid chromatography (HPLC) grade and used without further purification.
Cell line and culture
Adenocarcinomic human alveolar basal epithelial cells (A549 cell line human, A549 cells), NCI-H460 cell line (NCI-H460 cells), and human umbilical vein endothelial cells (HUVECs) were purchased from Sigma-Aldrich Co. and were maintained in DMEM and supplemented with 10% FBS at 37°C in the presence of 5% CO2 and 95% relative humidity.
Animals and tumor xenografts
C57BL/6 mice (6 weeks old, male) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, People’s Republic of China) and were housed in cages with bedding and maintained under controlled conditions of 12/12 hour light/dark cycle, 20°C±2°C and 50%±15% relative humidity. The in vivo experiments complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and the Animal Care and Use Committee of Xuzhou Medical University approved all the animal protocols used (No 101017021701).
To produce the tumor xenografts, about 107 A549 cells suspended in saline (200 μL) were subcutaneously injected into the right flank of mice. Tumor volume (TV) was determined by the formula: (the largest superficial diameter)*(the smallest superficial diameter)2/2.
Synthesis and characterization of PAA-ss-OA
PAA-ss-OA was prepared by amidation of the amine groups of ss with the carboxyl groups of PAA and OA (Figure 1).19,20 Briefly, OA (1 mmol) was added to DMSO (20 mL) in the presence of EDC (1 mmol) and NHS (1 mmol) with stirring for 1 hour at room temperature. Cystamine (1 mmol) was dissolved in 10 mL of DMSO and then added dropwise into the OA mixture and stirred for 10 hours at room temperature to get ss-OA. Then PAA (1 mmol) was dissolved in 20 mL of DMSO and added dropwise into ss-OA mixtures, incubated for 12 hours at room temperature with vigorous stirring and was then dialyzed against excess ultrapure water for 3 days (MWCO, 3,500 Da). The chemical structure of PAA-ss-OA was determined by using hydrogen-1 nuclear magnetic resonance (1H-NMR) analysis at 300 MHz, after dissolving in DMSO-d6 as solvent.
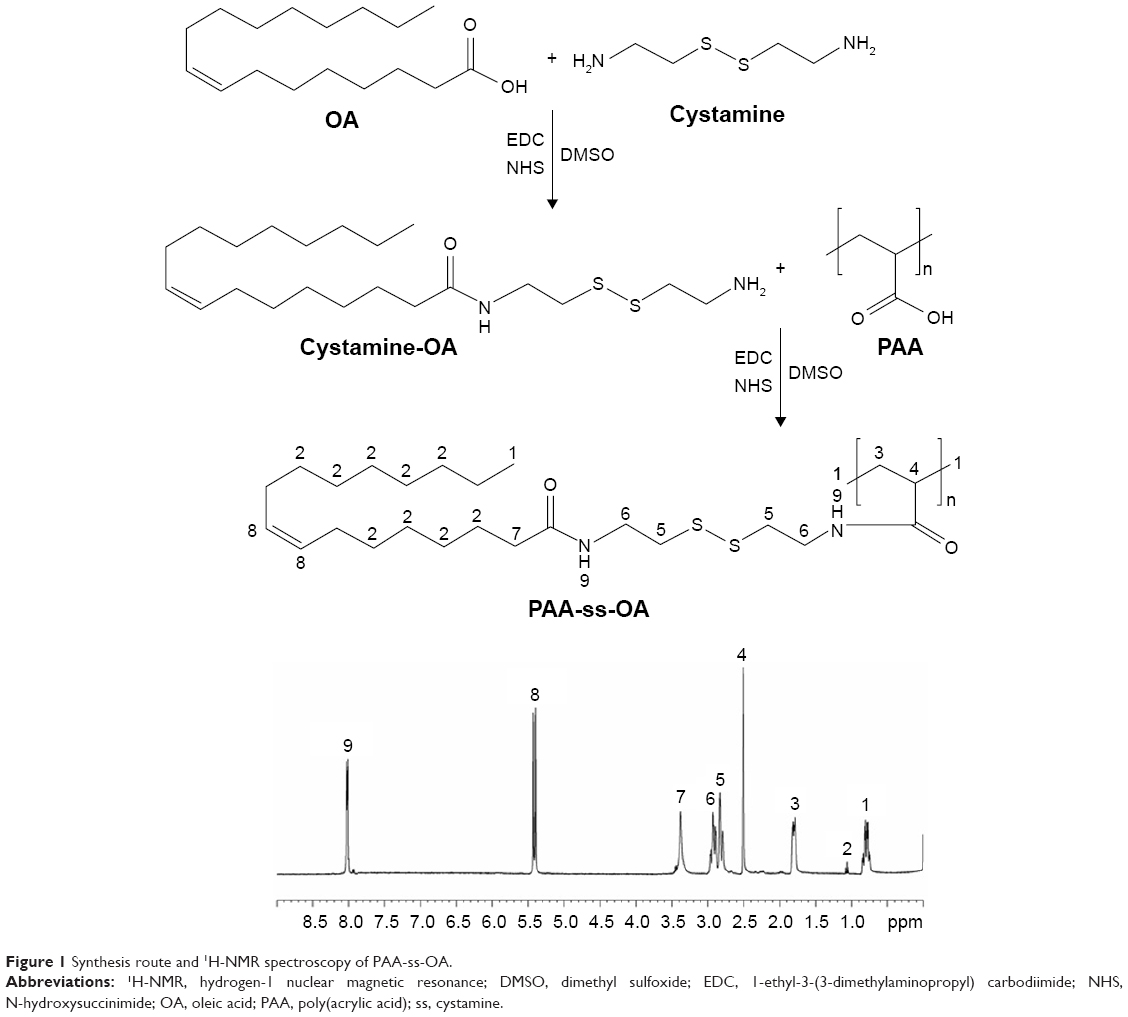 | Figure 1 Synthesis route and 1H-NMR spectroscopy of PAA-ss-OA. |
Preparation of PAA-ETB-NPs
PAA-ETB-NPs (Figure 2) were prepared by using the emulsification and solvent evaporation method.21 Briefly, ETB (50 mg), OA (50 mg), and GMS (100 mg) were dissolved in chloroform (5 mL) and added to the aqueous phase (45 mL) containing 1% Tween 80, homogenized at 15,000 rpm for 3 minutes. PAA-ss-OA (100 mg) was dissolved in chloroform (5 mL) and added to the homogenized mixture. The resulting mixture was stirred at 600 rpm for 3 hours until the complete evaporation of chloroform to obtain PAA-ETB-NPs.
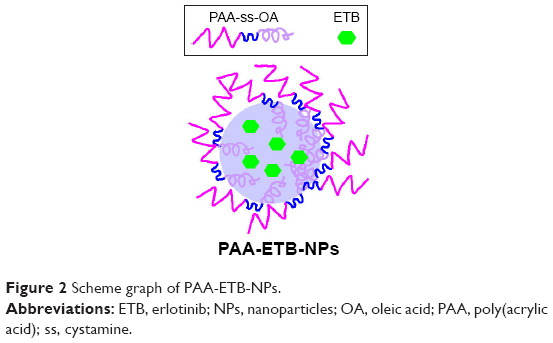 | Figure 2 Scheme graph of PAA-ETB-NPs. |
ETB-NPs were prepared by using the same method without adding PAA-ss-OA.
Drug-free (non-ETB-loaded) lipid nanoparticles (free NPs) were prepared by using the same method without adding PAA-ss-OA and ETB.
Characterization of PAA-ETB-NPs
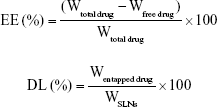
The size, polydispersity index (PDI), and zeta potential of PAA-ETB-NPs, ETB-NPs, and free NPs were measured by a Malvern Zetasizer Nano ZS90 (Malvern Instruments, Malvern, UK).22 The drug encapsulation efficiency (EE) and drug loading capacity (DL) of ETB were measured by HPLC with mobile phase of acetonitrile/KH2PO4 buffer (6:4, v:v), flow 1 mL/min, detection 345 nm, injected volume 20 μL and C18 column.23 The EE and DL were calculated according to the following formulas:
|
Plasma stability of PAA-ETB-NPs
The colloidal stability of PAA-ETB-NPs, ETB-NPs, and free NPs in plasma was evaluated.24 NPs were suspended in the serum of the tumor-bearing mice and then dispersed in phosphate-buffered solution (PBS) with a final nanoparticle concentration of 1 mg/mL. The NPs were washed in PBS by using centrifuge filter molecular weight cutoff 30 kDa and concentrated to 2 mg/mL. An equal volume of mouse plasma was then added. Samples were incubated at 37°C under mild shaking. At predetermined time points (0, 0.5, 1, 3, 6, 12, 24, 48, and 72hours), an aliquot of NPs was collected to measure the mean particle size, PDI, and EE by the methods described in the “Characterization of PAA-ETB-NPs” section.
In vitro drug release
For the in vitro release study, 3 mL of PAA-ETB-NPs, ETB-NPs, or ETB solution (1 mg/mL) was placed into a dialysis bag with a 2 kDa cutoff.25 It was then immersed into 47 mL of PBS (pH 7.4) at 37°C to confirm the temperature-sensitive release behavior of the synthesized micelles in physiological condition. Aliquots (2 mL) were taken out from the solution periodically, and total volume of the solution was kept constant by adding 2 mL of PBS after each sampling. Reduction responsive release media was produced by adding GSH to PBS buffer (pH =7.4) at final concentrations of 20 mM. The amount of ETB released from NPs was analyzed by using the methods described in the “Characterization of PAA-ETB-NPs” section.
In vitro cytotoxicity
The in vitro cytotoxicity of PAA-ETB-NPs, ETB-NPs, Free NPs, ETB solution, and 0.9% saline control samples on A549 cells, NCI-H460 cells, and HUVEC were assessed by MTT assay.26 A549 cells were seeded in 96-well plates at 7,000 cells per well in 100 mL of DMEM and incubated at 37°C in a 5% CO2 atmosphere for 24 hours, followed by removing culture medium and then adding samples at different concentrations. The cells were subjected to MTT assay after being incubated for another 72 hours. The absorbency of the solution was measured with a Bio-Rad 680 microplate reader at 492 nm. The relative cell viability was calculated by the formula: (the absorbance of the sample well)/(the absorbance of the control well)*100.
In vivo tissue distribution study
Tumor xenografts were given intravenous injections of PAA-ETB-NPs, ETB-NPs, or ETB solution by tail vein and killed at predetermined times.27 Various tissues (tumor, heart, liver, lung, and kidney) were immediately harvested, kept in saline solution to remove the blood and contents, blotted on filter paper, and then weighed for wet weight. After homogenization by using a 3-fold volume of 0.1 M phosphate buffer (pH 7.4), tissue samples were stored at 20°C until assay. The amount of ETB distributed in tissues was analyzed by using the methods described in the “Characterization of PAA-ETB-NPs” section.
In vivo tumor inhibition study
Tumor xenografts were randomly divided into five groups (eight mice per group). Each mouse was treated with PAA-ETB-NPs, ETB-NPs, free NPs, ETB solution, and 0.9% saline control intravenously by tail vein every 3 days.28 The tumor size was measured every 3 days across its two perpendicular diameters, and TV was estimated by using the following formula: (the largest superficial diameter)*(the smallest superficial diameter)2/2. The tumor inhibition rate (%) was then calculated by using the formula: (tumor weight of the control group – tumor weight of drug treated groups)/ (tumor weight of the control group)*100. Body weight lost of each group was summarized to evaluate the systematic toxicity of the systems.
Statistical analysis
The data of the studies presented in this article were expressed as mean ± standard deviation for at least three replicates; multiple groups were compared by using a one-way analysis of variance and between two groups by Student’s t-test analysis. The mean differences were considered significant in all experiments valued at *p<0.05, **p<0.01, and ***p<0.001.
Results
Synthesis and characterization of PAA-ss-OA
The chemical structure of PAA-ss-OA determined by 1H-NMR spectroscopy was presented in Figure 1, and the chemical structure shifts were marked by numbers: (1) 0.79 (−CH3 of PAA), (2) 1.05 (−CH2− of OA), (3) 1.78 (−CH2− of PAA), (4) 2.49 (−CH− of PAA), (5) 2.84 (−CH2−S−S−CH2− of ss), (6) 2.93 (−C(=O)−N−CH2− of amido linkage), (7) 3.37 (−N−C(=O)−CH2− of amido linkage), (8) 5.38 (−CH=CH− of OA), (9) 8.03 (−NH−C(=O)−). The chemical shifts of the amido linkages, PAA, ss, and OA could determine the formation of PAA-ss-OA.
Characterization of NPs
Table 1 summarizes the size, PDI, zeta potential, EE, and DL of NPs. The size of free NPs and ETB-NPs was about 110 nm, with a zeta potential of around −20 mV. The size of PAA-ETB-NPs was 170 nm, and the size of NPs was enlarged after PAA modification. The zeta potential of PAA-ETB-NPs was −32 mV, which were more negatively charged after PAA coating. The PDI of NPs varies from 0.10 to 0.18. The EE of ETB-NPs was over 80%. The DL of PAA-ETB-NPs and ETB-NPs was 2.6% and 4.3%, respectively.
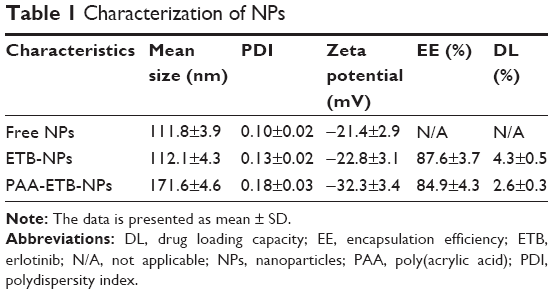 | Table 1 Characterization of NPs |
Serum stability
The serum stability of NPs in tumor-bearing mouse plasma was tested at 37°C for 72 hours. Free NPs and ETB-NPs were stable in plasma retaining their physicochemical characteristics for 24 hours (Figure 3). After 24 hours, the mean size and PDI of NPs gradually increased from about 110 to about 290 nm until 72 hours of incubation. For PAA-ETB-NPs, the mean size and PDI remains stable until 48 hours of incubation. The EE of ETB-NPs remains over 80% in the first 48 hours, while the EE of PAA-ETB-NPs was steady until the end of 72 hours without obvious reduction.
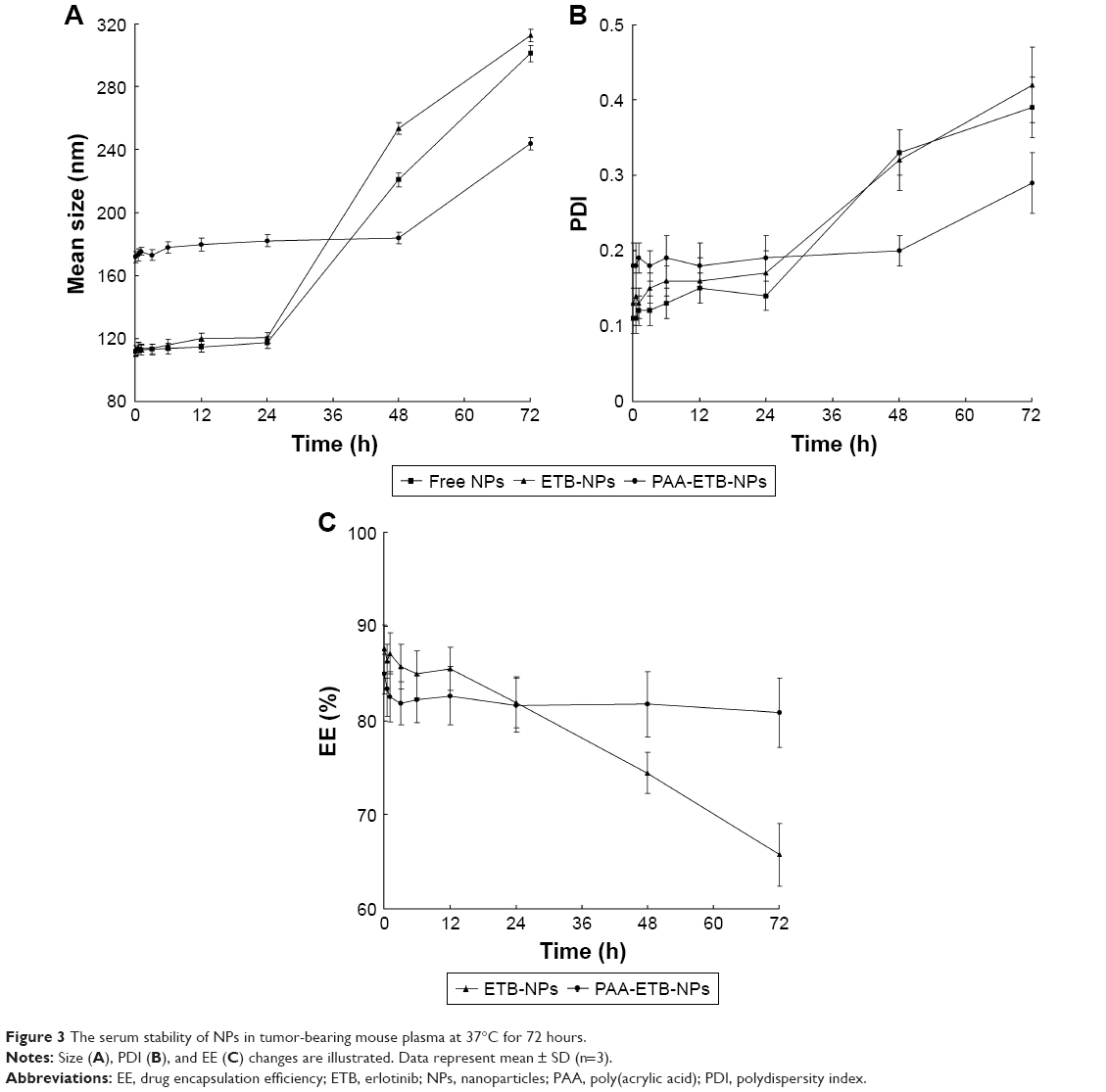 | Figure 3 The serum stability of NPs in tumor-bearing mouse plasma at 37°C for 72 hours. |
In vitro drug release
The in vitro release of ETB was calculated, and the cumulative drug release from the NPs and solution was plotted against time (Figure 4). There was no burst release from NPs, and cumulative drug release occurred with a sustained behavior. The release of ETB from PAA-ETB-NPs over a period of 72 hours was significantly slower than that of PAA-ETB-NPs (p<0.05). However, in reduction-responsive release media (containing 20 mM GSH), the release of PAA-ETB-NPs was faster than that of ETB-NPs. In contrast, ETB solution had a rapid drug release which reached over 80% of cumulative ETB release within the first 4 hours.
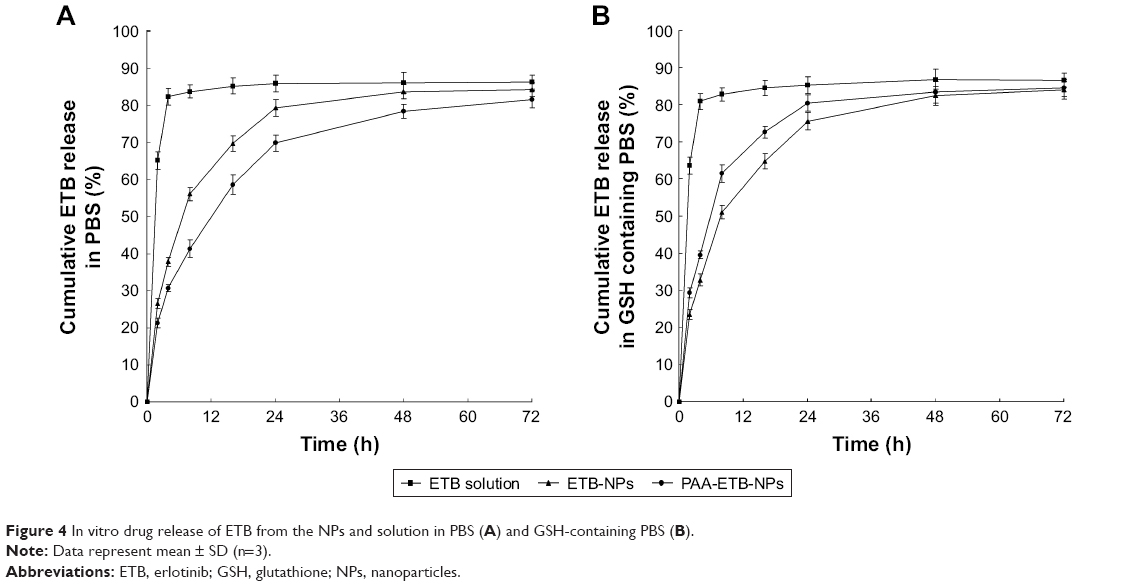 | Figure 4 In vitro drug release of ETB from the NPs and solution in PBS (A) and GSH-containing PBS (B). |
In vitro cytotoxicity
The in vitro cytotoxicity of the NPs was evaluated by using MTT assay (Figure 5). The cell viabilities of A549 and NCI-H460 cells treated with free NPs after 72 hours of incubation were all above 80%, indicating that the NPs had good safety and biocompatibility. At all the studied drug concentrations, the cytotoxicity of ETB-NPs were higher than that of ETB solution (p<0.05). The cytotoxicity of PAA-ETB-NPs was the highest. The cytotoxicity of PAA-ETB-NPs and ETB-NPs on HUVEC showed no obvious higher than ETB solution. The IC50 values of PAA-ETB-NPs, ETB-NPs, and ETB solution on different cells are summarized in Table 2.
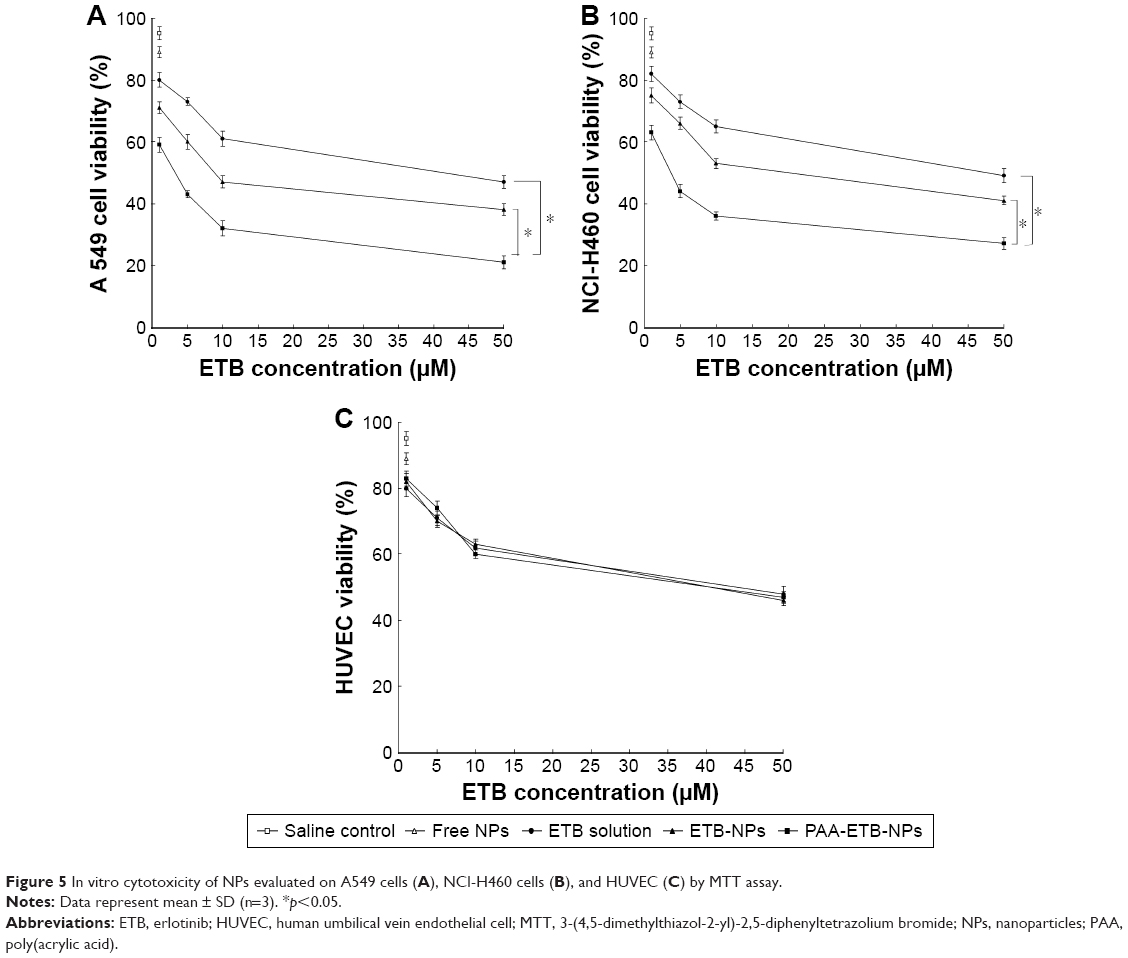 | Figure 5 In vitro cytotoxicity of NPs evaluated on A549 cells (A), NCI-H460 cells (B), and HUVEC (C) by MTT assay. |
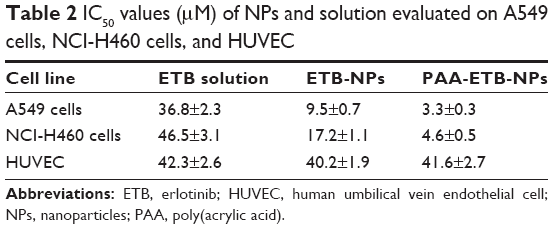 | Table 2 IC50 values (μM) of NPs and solution evaluated on A549 cells, NCI-H460 cells, and HUVEC |
In vivo tissue distribution study
The in vivo tissue distribution of the ETB-NPs and solutions are illustrated in Figure 6. The distribution of ETB in the tumor following the injection of PAA-ETB-NPs and ETB-NPs was higher than that following the injection of ETB solution. On the contrary, the distribution in heart and kidney after injection of ETB-NPs was lower than that of ETB solution. To be noticed, the drug distribution of PAA-ETB-NPs group remained high in the tumor during the 72 hours of study, while the drug distribution of ETB-NPs group reduced after 48 hours of administration.
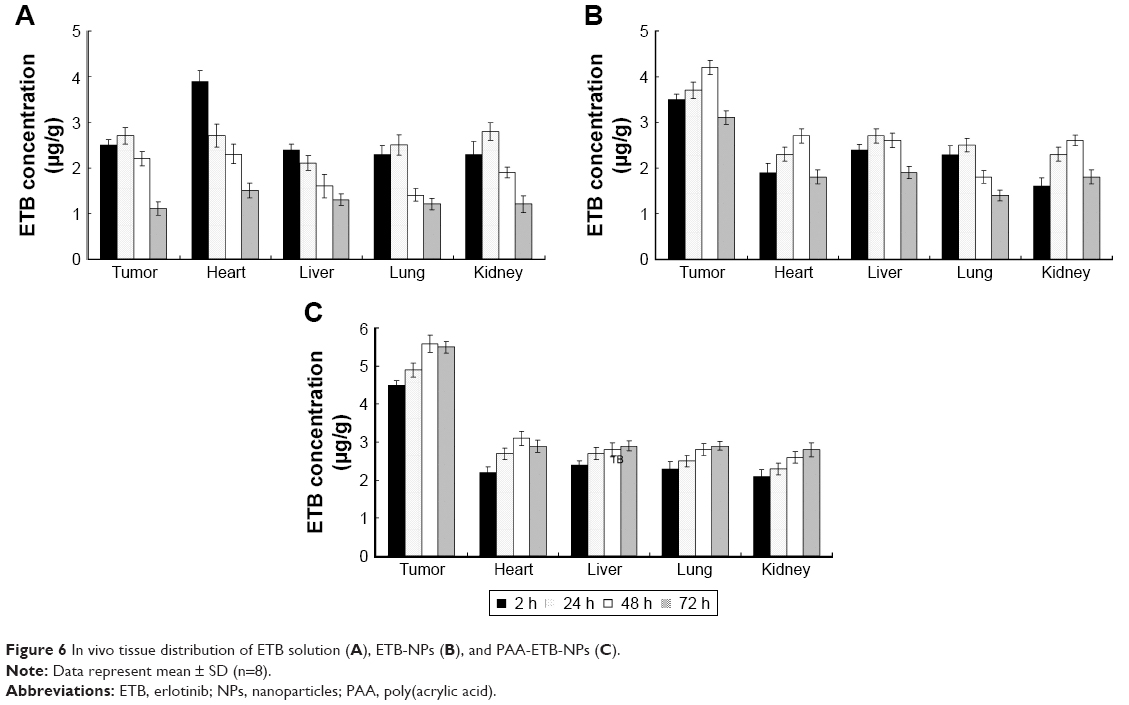 | Figure 6 In vivo tissue distribution of ETB solution (A), ETB-NPs (B), and PAA-ETB-NPs (C). |
In vivo tumor inhibition study
In vivo tumor inhibition effect of ETB-NPs were evaluated by using a xenograft nude mouse model with human lung cancer cells. TV increased rapidly when the mice were treated with saline or free NPs, with little difference between these two groups (p<0.05) (Figure 7A). After 21 days, the tumor growth inhibition by PAA-ETB-NP treatment was significantly higher than that by the ETB-NPs (p<0.05), ETB solution (p<0.01), and saline control (p<0.001). No obvious weight loss was observed in any of the treatment groups, indicating that all the treatments were well tolerated (Figure 7B). The tumor inhibition rate of PAA-ETB-NPs, ETB-NPs, and ETB solution was 84.5%, 68.7%, and 38.1%, respectively (Figure 7C).
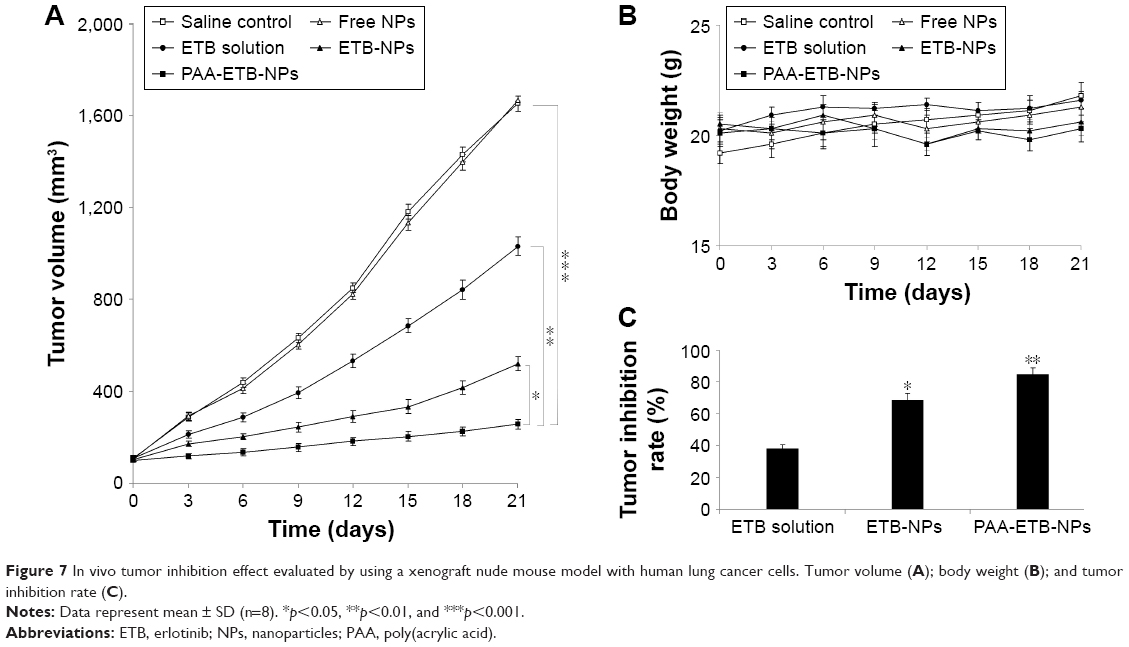 | Figure 7 In vivo tumor inhibition effect evaluated by using a xenograft nude mouse model with human lung cancer cells. Tumor volume (A); body weight (B); and tumor inhibition rate (C). |
Discussion
In the recent years, delivery of ETB encapsulated in polymeric nanoparticles has gained much attention.29 This is mainly due to the potential of nanoparticles to protect the entrapped drug molecules from the harsh conditions in vivo. In the present study, PAA-ss-OA was developed as a novel material for the surface modification of lipid carrier for the delivery of ETB. At the beginning of this research, GSH- and pH-sensitive PAA-ss-OA conjugate was designed, synthesized, and characterized. First, ss-OA was synthesized by chemical conjugation of ss to OA by using EDC chemistry. Subsequently, PAA was introduced to ss-OA conjugate in the presence of EDC and NHS, resulting in the formation of PAA-ss-OA conjugates. The chemical structure of PAA-ss-OA determined by 1H-NMR spectroscopy and the chemical structure shifts were marked by numbers (Figure 1). The chemical shifts of the amido linkages, PAA, ss, and OA could determine the formation of PAA-ss-OA.
NPs can be prepared by using a variety of techniques, the most important of which are hot/cold high pressure homogenization, micro emulsion, and solvent evaporation techniques.30 In this study, emulsification combined with solvent evaporation method was used for the preparation of NPs (Figure 2). The drug is dissolved in the lipid containing organic solvent, and then added to the aqueous phase and homogenized. PAA-ss-OA was added to the homogenized mixture and stirred until the solvent was completely evaporated. Size is an important feature of NPs that play key role in their internalization by cells. NPs are known to exploit the enhanced permeability and retention (EPR) effect for targeting to tumors, thereby increasing antitumor efficiency while minimizing systemic toxicity.31 The chemotherapeutic agents delivered by nanotechnology could have advantages in selective delivering due to the size. Free NPs and ETB-NPs had a size of about 110 nm, while PAA-ETB-NPs had a size of 170 nm. The results suggested that the size of NPs was enlarged after PAA modification. The zeta potential of PAA-ETB-NPs was −32 mV, which was more negatively charged than ETB-NPs after PAA coating. This may due to the negative charge of the PAA on the surface of the PAA-ETB-NPs. The PDI of NPs were <0.2, showing the narrow distributions. The EE of ETB-NPs was over 80%; this can be the proof that the drugs were fully encapsulated in the NPs. To evaluate the stability of NPs in serum, tumor-bearing mouse plasma was used. Free NPs and ETB-NPs were stable in serum for 24 hours (Figure 3); in contrast, PAA-ETB-NPs remained stable until 48 h of incubation. The EE of PAA-ETB-NPs was steady until the end of 72 hours without obvious reduction, which was also longer than that of ETB-NPs. This phenomenon may be explained by the presence of PAA coating that enhanced the serum stability of the NPs.
In vitro drug release of the drug-loaded NPs may be controlled by the erosion, corrosion, and diffusion processes.32 Drug depot effects could be achieved by the carriers, which could lead to the sustained release of hydrophobic drugs. The more sustained-release behavior of PAA-ETB-NPs than ETB-NPs might be explained by the modification of polymeric PAA ligands that decelerated the release of ETB (Figure 4). As the ETB molecules are restrained within the NPs, the cytotoxic impacts of drug seem to be due to the internalization of the ETB-NPs, most likely through fluid-phase endocytosis/pinocytosis. Once within the endosomal compartments, the ETB molecules can be released from the NPs and induce their toxic impacts.30
In vitro cytotoxicity of the NPs was evaluated by using MTT assay. Significant improvements in cytotoxicity of drug-loaded NPs in comparison with drug solutions illustrated that the NP systems could improve the delivery of drugs thus gain better efficiency than free drug solution (Figure 5). The IC50 values of PAA-ETB-NPs, ETB-NPs, and ETB solution were 3.3, 9.5, and 36.8 μM, respectively. The cytotoxicity of PAA-ETB-NPs and ETB-NPs on HUVEC showed no obvious higher than ETB solution, this may be the proof that the NPs showed no obvious toxicity on normal cells. Higher cytotoxicity of PAA-ETB-NPs than ETB-NPs suggested that the use of PAA modification could promote the delivery of the drugs encapsulated within NPs. The cell viability during 10–50 μM was from 47% to 38% for EBT-NPs and from 32% to 21% for PAA-EBT-NPs. The rates were slightly different for these two vectors. The results may give rise the thought that “the cells get drug resistance at this dose” if so there should be increase in GSH levels in the cells, and this leads to induce more EBT release from PAA-EBT-NPs compared to EBT-NPs.
In vivo tissue distributions of PAA-ETB-NPs, ETB-NPs, and ETB solution were investigated in lung cancer-bearing mice. For ETB-NPs groups, the drug concentrations in the tumors were higher than that for the ETB solution group, while the drug concentrations in the heart and kidney for NPs groups were much lower (Figure 6). This may be because the EPR effect of tumors could let the nanoparticles passively targeted to the tumor, which resulted in the efficient drug accumulation in tumor tissue. Less drug distributions in heart and kidney may reduce the systemic toxicity, which could decrease the side effects and lead to better anti-tumor therapeutic efficiency.33
In vivo tumor inhibition efficacy of PAA-ETB-NPs, ETB-NPs, free NPs, ETB solution, and 0.9% saline control were evaluated in lung cancer-bearing mice (Figure 7). PAA-ETB-NPs, ETB-NPs, and ETB solution showed significant tumor inhibition effects, with a reduction of TV. Mice in free NPs and 0.9% saline control groups shared similar tumor growth pattern, suggesting that blank NPs were not capable of inhibiting tumor growth. After 21 days, the tumor growth inhibition by PAA-ETB-NPs treatment was significantly higher than that by ETB-NPs (p<0.05), ETB solution (p<0.01), and saline control (p<0.001). The tumor inhibition rate of PAA-ETB-NPs, ETB-NPs, and ETB solution was 84.5%, 68.7%, and 38.1%, respectively. No obvious weight loss was observed in any of the treatment groups, indicating that all the treatments were well tolerated. These results could be explained as the pH-sensitive and redox-responsive PAA ligands could promote the NPs to deliver drug into the tumor cells. The structure of NPs may delay the drug release and bring about the long-lasting drug delivery effect in tumor tissues.
Conclusion
Redox-responsive and pH-sensitive PAA ligands were synthesized and used for the surface modification of NPs for the delivery of ETB. PAA-ETB-NPs showed the highest in vitro and in vivo tumor inhibition efficacy when compared with ETB-NPs and ETB solution. The redox-responsive and pH-responsive nanoparticles could enhance the stability and anti-cancer ability of ETB to treat lung cancer and is a promising drug delivery system for lung cancer treatment.
Disclosure
The authors report no conflicts of interest in this work.
References
Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60(5):277–300. | ||
Kanthala S, Pallerla S, Jois S. Current and future targeted therapies for non-small-cell lung cancers with aberrant EGF receptors. Future Oncol. 2015;11(5):865–878. | ||
Sridhar SS, Seymour L, Shepherd FA. Inhibitors of epidermal-growth-factor receptors: a review of clinical research with a focus on non-small-cell lung cancer. Lancet Oncol. 2003;4(7):397–406. | ||
Pallis AG, Syrigos K. Targeted (and chemotherapeutic) agents as maintenance treatment in patients with metastatic non-small-cell lung cancer: current status and future challenges. Cancer Treat Rev. 2012;38(7):861–867. | ||
Greenhalgh J, Dwan K, Boland A, et al. First-line treatment of advanced epidermal growth factor receptor (EGFR) mutation positive non-squamous non-small cell lung cancer. Cochrane Database Syst Rev. 2016;(5):CD010383. | ||
Wu YL, Lee JS, Thongprasert S, et al. Intercalated combination of chemotherapy and erlotinib for patients with advanced stage non-small-cell lung cancer (FASTACT-2): a randomised, double-blind trial. Lancet Oncol. 2013;14(8):777–786. | ||
Abernethy AP, Arunachalam A, Burke T, et al. Real-world first-line treatment and overall survival in non-small cell lung cancer without known EGFR mutations or ALK rearrangements in US community oncology setting. PLoS One. 2017;12(6):e0178420. | ||
Heist RS. First-line systemic therapy for non-small cell lung cancer. Hematol Oncol Clin North Am. 2017;31(1):59–70. | ||
Wang S, Peng L, Li J, et al. A trial-based cost-effectiveness analysis of erlotinib alone versus platinum-based doublet chemotherapy as first-line therapy for Eastern Asian nonsquamous non-small-cell lung cancer. PLoS One. 2013;8(3):e55917. | ||
Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–137. | ||
Liu TC, Jin X, Wang Y, Wang K. Role of epidermal growth factor receptor in lung cancer and targeted therapies. Am J Cancer Res. 2017;7(2):187–202. eCollection 2017. | ||
Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–246. | ||
Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12(8):735–742. | ||
Madni A, Batool A, Noreen S, et al. Novel nanoparticulate systems for lung cancer therapy: an updated review. J Drug Target. 2017;25(6):499–512. | ||
Wojtkowiak JW, Verduzco D, Schramm KJ, Gillies RJ. Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Mol Pharm. 2011;8(6):2032–2038. | ||
Cheng R, Feng F, Meng F, Deng C, Feijen J, Zhong Z. Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. J Control Release. 2011;152(1):2–12. | ||
Cai Z, Zhang H, Wei Y, Wei Y, Xie Y, Cong F. Reduction- and pH-sensitive hyaluronan nanoparticles for delivery of iridium(III) anticancer drugs. Biomacromolecules. 2017;18(7):2102–2117. | ||
Wang Y, Wang J, Yuan Z, et al. Chitosan cross-linked poly(acrylic acid) hydrogels: drug release control and mechanism. Colloids Surf B Biointerfaces. 2017;152:252–259. | ||
Zhang X, Wang Y, Zhao Y, Sun L. pH-responsive drug release and real-time fluorescence detection of porous silica nanoparticles. Mater Sci Eng C Mater Biol Appl. 2017;77:19–26. | ||
Kommareddy S, Amiji M. Poly(ethylene glycol)-modified thiolated gelatin nanoparticles for glutathione-responsive intracellular DNA delivery. Nanomedicine. 2007;3(1):32–42. | ||
Liu B, Han L, Liu J, Han S, Chen Z, Jiang L. Co-delivery of paclitaxel and TOS-cisplatin via TAT-targeted solid lipid nanoparticles with synergistic antitumor activity against cervical cancer. Int J Nanomedicine. 2017;12:955–968. | ||
Li C, Li H, Wang Q, et al. pH-sensitive polymeric micelles for targeted delivery to inflamed joints. J Control Release. 2017;246:133–141. | ||
He Y, Su Z, Xue L, Xu H, Zhang C. Co-delivery of erlotinib and doxorubicin by pH-sensitive charge conversion nanocarrier for synergistic therapy. J Control Release. 2016;229:80–92. | ||
Mandal B, Mittal NK, Balabathula P, Thoma LA, Wood GC. Development and in vitro evaluation of core-shell type lipid-polymer hybrid nanoparticles for the delivery of erlotinib in non-small cell lung cancer. Eur J Pharm Sci. 2016;81:162–171. | ||
Fathi M, Zangabad PS, Aghanejad A, Barar J, Erfan-Niya H, Omidi Y. Folate-conjugated thermosensitive O-maleoyl modified chitosan micellar nanoparticles for targeted delivery of erlotinib. Carbohydr Polym. 2017;172:130–141. | ||
Lv S, Tang Z, Li M, et al. Co-delivery of doxorubicin and paclitaxel by PEG-polypeptide nanovehicle for the treatment of non-small cell lung cancer. Biomaterials. 2014;35(23):6118–6129. | ||
Lin X, Wang ZJ, Wang S, et al. Comparison of tissue distribution of a PEGylated radix ophiopogonis polysaccharide in mice with normal and ischemic myocardium. Eur J Pharm Biopharm. 2011;79(3):621–626. | ||
Kim CE, Lim SK, Kim JS. In vivo antitumor effect of cromolyn in PEGylated liposomes for pancreatic cancer. J Control Release. 2012;157(2):190–195. | ||
Marslin G, Sheeba CJ, Kalaichelvan VK, Manavalan R, Reddy PN, Franklin G. Poly(D,L-lactic-co-glycolic acid) nanoencapsulation reduces Erlotinib-induced subacute toxicity in rat. J Biomed Nanotechnol. 2009;5(5):464–471. | ||
Bakhtiary Z, Barar J, Aghanejad A, et al. Microparticles containing erlotinib-loaded solid lipid nanoparticles for treatment of non-small cell lung cancer. Drug Dev Ind Pharm. 2017;43(8):1244–1253. | ||
Yan J, Wang Y, Zhang X, Liu S, Tian C, Wang H. Targeted nanomedicine for prostate cancer therapy: docetaxel and curcumin co-encapsulated lipid-polymer hybrid nanoparticles for the enhanced anti-tumor activity in vitro and in vivo. Drug Deliv. 2016;23(5):1757–1762. | ||
Lu Z, Su J, Li Z, Zhan Y, Ye D. Hyaluronic acid-coated, prodrug-based nanostructured lipid carriers for enhanced pancreatic cancer therapy. Drug Dev Ind Pharm. 2017;43(1):160–170. | ||
Ni S, Qiu L, Zhang G, Zhou H, Han Y. Lymph cancer chemotherapy: delivery of doxorubicin-gemcitabine prodrug and vincristine by nanostructured lipid carriers. Int J Nanomedicine. 2017;12:1565–1576. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.