Back to Journals » OncoTargets and Therapy » Volume 16
Recurrent Glioblastoma: Ongoing Clinical Challenges and Future Prospects
Authors Pineda E, Domenech M, Hernández A, Comas S, Balaña C ![]()
Received 27 October 2022
Accepted for publication 19 January 2023
Published 25 January 2023 Volume 2023:16 Pages 71—86
DOI https://doi.org/10.2147/OTT.S366371
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Estela Pineda,1 Marta Domenech,2 Ainhoa Hernández,2 Silvia Comas,3 Carmen Balaña2
1Medical Oncology, Hospital Clínic de Barcelona, Barcelona, Spain; 2Medical Oncology, Institut Catala d’Oncologia (ICO) Badalona, Barcelona, Spain; 3Radiation Oncology, Institut Catala d’Oncologia (ICO) Badalona, Badalona, Spain
Correspondence: Carmen Balaña, Institut Catala d’Oncologia (ICO) Badalona, Carretera Canyet s/n, Badalona, 08916, Spain, Tel +34 497 89 25, Fax +34 497 89 50, Email [email protected]
Abstract: Virtually all glioblastomas treated in the first-line setting will recur in a short period of time, and the search for alternative effective treatments has so far been unsuccessful. Various obstacles remain unresolved, and no effective salvage therapy for recurrent glioblastoma can be envisaged in the short term. One of the main impediments to progress is the low incidence of the disease itself in comparison with other pathologies, which will be made even lower by the recent WHO classification of gliomas, which includes molecular alterations. This new classification helps refine patient prognosis but does not clarify the most appropriate treatment. Other impediments are related to clinical trials: glioblastoma patients are often excluded from trials due to their advanced age and limiting neurological symptoms; there is also the question of how best to measure treatment efficacy, which conditions the design of trials and can affect the acceptance of results by oncologists and medicine agencies. Other obstacles are related to the drugs themselves: most treatments cannot cross the blood-brain-barrier or the brain-to-tumor barrier to reach therapeutic drug levels in the tumor without producing toxicity; the drugs under study may have adverse metabolic interactions with those required for symptom control; identifying the target of the drug can be a complex issue. Additionally, the optimal method of treatment – local vs systemic therapy, the choice of chemotherapy, irradiation, targeted therapy, immunotherapy, or a combination thereof – is not yet clear in glioblastoma in comparison with other cancers. Finally, in addition to curing or stabilizing the disease, glioblastoma therapy should aim at maintaining the neurological status of the patients to enable them to return to their previous lifestyle. Here we review currently available treatments, obstacles in the search for new treatments, and novel lines of research that show promise for the future.
Keywords: glioblastoma, research, challenges, endpoints, recurrent, treatment
Introduction
The incidence of malignant tumors of the central nervous system (CNS) is 7.08 per 100,000 inhabitants in the USA. Glioblastoma (GB) accounts for 47.7% of all CNS malignant tumors in adults, and patients have a median overall survival (OS) of only eight months and a 5-year survival of 5.8% (95% CI 5.5–6.1).1 The incidence of GB is low compared to that of other tumors, but there is no localized and surgically curable form of GB as there is in other cancers, so all cases are considered grade 4.
The standard first-line treatment for GB was established in 2005 as maximal safe surgery followed by radiotherapy plus concomitant and adjuvant temozolomide.2 In the USA, tumor-treating fields can also be administered after chemoradiotherapy. This system delivers low-intensity, intermediate-frequency alternating fields via transducer arrays when applied to the scalp, exerting an antimitotic effect.3 Unfortunately, only around 44–50% of patients are able to receive the full standard treatment,4,5 only one fourth of those who start treatment are able to complete the six cycles of adjuvant temozolomide,5 and only 60% of these patients receive second-line treatments. Fewer than 43% of GB patients are fit enough to be included in clinical trials.6
The symptoms of GB and the sequelae of treatments often lead to functional dependence and cognitive impairment,7 making it impossible for patients to return to their normal pre-diagnosis activities and impacting negatively on family life. There is thus great concern about patient quality of life during the extra period of survival conferred by treatment, especially on the maintenance of neurocognition and the adjustment of palliative and supporting treatments, including rehabilitation.8,9
To date, the search for alternative effective treatments for GB has been unsuccessful. Here we review the standard of care and available therapies, obstacles in the search for new treatments, and new lines of treatment under investigation.
Currently Approved Treatments for Recurrent Glioblastoma
Several treatments have shown some efficacy in specific patients at recurrence, including second surgery (S-S), re-irradiation, and re-treatment with temozolomide, nitrosoureas or bevacizumab (Figure 1).
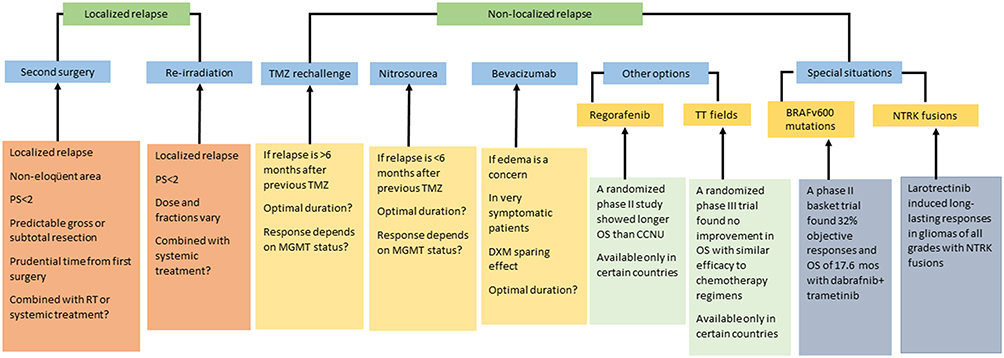 |
Figure 1 Treatment options for recurrent glioblastoma. Inclusion in a clinical trial is always the preferred option. |
Patients who benefit from S-S are those with a localized relapse in non-eloquent areas, in whom a complete or subtotal resection of the enhancing tumor can be performed. Candidates for S-S need to have a good performance status and a relatively indolent tumor history, measured as a prudential time from the first to the S-S.10–14 In general, only 20–30% of relapsed patients are eligible for S-S. Median OS after S-S is highly variable (11–17 months).12,15 In fact, the potential benefit of S-S has never been explored in a randomized study suggesting that the increased survival attributed to S-S may be due to bias in patient selection. Consensus guidelines to select candidates for S-S have been proposed by some authors,16 but there is no consensus on the administration of postoperative treatments after the procedure.
Re-irradiation could also be an option in selected patients, but again there is a lack of randomized studies. Several questions are unresolved, such as the criteria for selection of candidates, the type of recurrence that can be treated, the total dose, optimal fractionation and best volume.17 The different irradiation techniques include stereotactic radiosurgery, hypofractionated stereotactic radiotherapy, and conventionally fractionated external radiotherapy. Estimated OS and progression-free survival (PFS) from re-irradiation are 73% and 43%, respectively, at six months and 36% and 17%, respectively, at 12 months.18 Several ongoing prospective studies are testing the value of re-irradiation – either alone or in combination with systemic treatments. The main risks of re-irradiation are radionecrosis, which seems to be reduced by bevacizumab,19 and a limited tolerance to irradiation of several areas, such as the brainstem, chiasm and optic nerves. A validated scale20 has been published that can be useful when deciding on the use of re-irradiation in the clinical setting. Advances in technology have also led to new strategies, including proton therapy, intraoperative radiation therapy, and brachytherapy.17
Commonly used systemic treatments include chemotherapy with temozolomide or nitrosoureas and antiangiogenic therapy with bevacizumab. Other treatments, such as regorafenib and tumor-treating fields,3,21 have been approved in a few countries.
Temozolomide efficacy was demonstrated in early trials,22,23 where 6-month PFS was 21%, which was superior to other agents used at that time. When temozolomide became part of the standard first-line treatment, however, there was no place for its use in recurrent GB, although a rechallenge seemed to benefit some patients, especially those with the longest interval after their last cycle of standard treatment, who attained a 6-month PFS of 35.7%.24
Nitrosoureas, including lomustine, and procarbazine, were widely used before the availability of temozolomide, but their use is now limited to recurrent GB. Because of their liposolubility, they are able to cross the blood-brain barrier (BBB). Lomustine has become the standard of care at relapse in Europe and is the treatment in the control arm of most trials.25 However, its benefit is limited: objective response rate around 10%; median PFS <2 months; 6-month PFS 20%; and OS 6–9 months. Thrombocytopenia is the most frequent limiting toxicity.26 Procarbazine was found to be less effective than temozolomide in a randomized trial.22 Fotemustine, a third-generation intravenous nitrosourea with lower rates of thrombocytopenia,27,28 is used in several countries at different schedules.
Bevacizumab created a great expectation after the first reports showing a high objective response rate even when combined with an inactive drug in gliomas like irinotecan. This led to an accelerated US FDA approval for the treatment of recurrent GB.29,30 Several later randomized studies in both the first-line (standard therapy with or without temozolomide)31,32 and second-line settings (lomustine versus lomustine-plus-bevacizumab)33 found that the addition of bevacizumab did not improve OS but did increase PFS and decreased the need for corticosteroids to treat cerebral edema. The efficacy of bevacizumab has been questioned precisely because of its antiangiogenic effect, which decreases contrast uptake on magnetic resonance imaging (MRI) but does not prevent tumor infiltration of normal brain tissue. This situation has led to different decisions by national health authorities and irregular availability around the world. A randomized trial comparing lomustine alone versus bevacizumab-plus-lomustine reported objective responses of 41.5% and median PFS of 3.8 months for the bevacizumab group, compared to 13.9% and 1.5 months with lomustine alone.34
Regorafenib is a multikinase inhibitor that blocks tyrosine kinases that are active in angiogenesis (VEGFR-1, VEGFR-2, VEGFR-3 and TIE2), cancer development and growth (KIT, RET, RAF1, BRAF and BFRAFV600E), and maintenance of the tumor microenvironment (PDGFR and FGFR). As some of these genes are altered in GB, there was a rationale for exploring the activity of this drug. The REGOMA trial was a Phase II randomized study comparing regorafenib and lomustine in 124 patients. There was a benefit in OS for the patients treated with regorafenib (HR 0.50; P=0.0009) although median OS was only 7.4 months in the regorafenib arm and 5.6 months in the lomustine arm.35 Based on these findings, regorafenib was approved for treatment of recurrent GB in Italy and is included as an alternative treatment in the NCCN guidelines (https://www.nccn.org/professionals/physician_gls/pdf/cns.pdf). It is currently being tested in the phase II/III Adaptive, Global, Innovative Learning Environment (AGILE) trial (ClinicalTrials.gov NCT03970447).
In addition, alternating tumor-treating fields yielded similar outcomes to other treatments, which led to US FDA approval for recurrent GB patients.36
Special Patient Populations
Glioblastoma patients are often included in clinical trials of agents targeting tumor-agnostic biomarkers. Glioblastoma tumors may have alterations of BRAF (v-Raf murine sarcoma viral oncogene homolog B1) in 9% of cases. Drugs targeting BRAF v600 mutations, such as dabrafenib, vemurafenib and encorafenib, combined with MEK1/2 kinase inhibitors (as used in melanoma) have proven to be beneficial in other gliomas, including pilocytic astrocytoma and pediatric diffuse gliomas. In GBs and relapsed high-grade gliomas, the combination of dabrafenib plus trametinib attained substantially superior results to other commonly used treatments: 32% objective responses; 19% stable disease by independent radiology review; median duration of response 13.6 months; median PFS of 4.5 months; OS 17.6 months.37 Other BRAF and MET1/2 kinase inhibitors are currently being studied in gliomas with BRAF v600 mutations (CliinicalTrials.gov NCT03973918).
Some GBs may harbor NTRK alterations (NTRK1, NTRK2, and NTRK3), which encode the tyrosine receptor kinases TRK-A, TRK-B, and TRK-C, respectively. The most clinically relevant alterations are NTRK fusions, which lead to constitutively activated receptors or splice variants, mutations, copy number alterations and increased expression with oncogenic potential. Targetable fusions are rare in adult GB (1.7%).38 NTRK fusions are druggable with entrectinib or larotrectinib, both of which can cross the BBB.39 Entrectinib is a selective tyrosine kinase inhibitor of TRK-A, TRK-B and TRK-C, the proto-oncogene ROS1, and ALK. It induced 55% of objective responses in patients with brain metastases from non-CNS tumors.40 Larotrectinib, a TRK inhibitor, induced 30% of responses in gliomas of all grades harboring TRK fusions, including in adult patients (7 of 33). The 24-week disease control rate was 73% and 1-year PFS was 56%.41 Larotrectinib was approved by the US FDA based on overall response rate and duration of response while awaiting results of confirmatory trials.
FGFR alterations, such as gene amplification, overexpression, mutations (FGFR 1–4) and fusions (FGFR:TACC3) are found in 8% of high-grade gliomas.38,42 Several inhibitors targeting these alterations are being studied. An interim analysis of the basket trial RAGNAR reported 20.7% of responses in 29 heavily pre-treated patients with high-grade gliomas treated with erdafitinib.43 Infigratinib has been tested in a phase II trial and while the response rate was not outstanding, several patients had long-lasting responses.44 Although these are very initial results, it seems that this pathway of inhibition will continue to develop in the near future. Pending concerns are the identification of the alteration with the best response to therapy and of the agent most capable of crossing the BBB.45
Novel Lines of Research in Glioblastoma and Gliomas in General
Research in glioblastoma encompasses not only treatment therapy but also improvements in imaging technology, the application of artificial intelligence to radiogenomics, surgical technology advances, which in turn impact on irradiation treatment technique, and other advances such as molecular sequencing, liquid biopsy and better GB models for pre-clinical research as 3D organoid -based GB model systems among others. In addition to research into how to measure the impact of treatments on patients, their quality of life and patient reported outcomes. All of these are moving forward intertwined in an interconnected network. In the present review we will focus on the specific treatment of the disease. Figure 2 shows some of the focuses of current GB research.
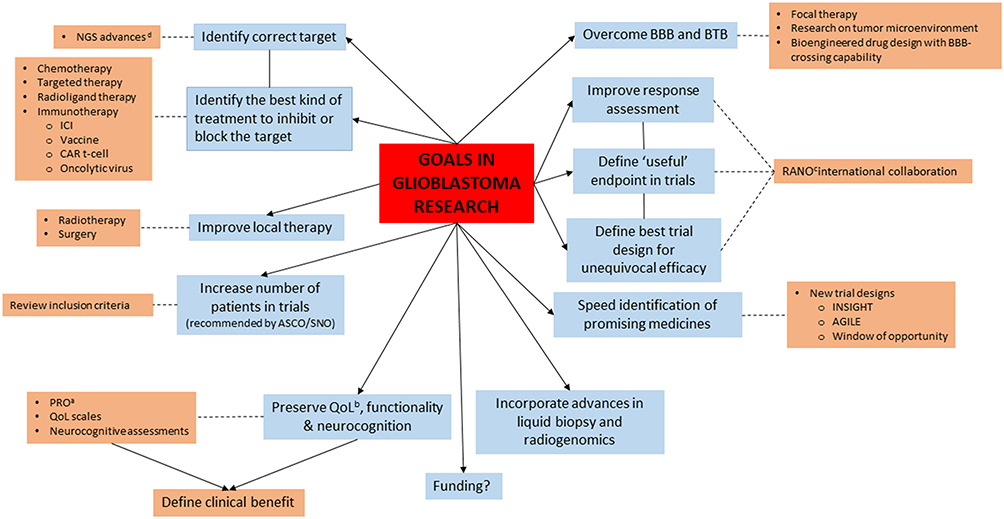 |
Figure 2 Goals of glioblastoma research and the means by which they can be achieved. aPatient reported outcomes, bQuality of life, cResponse assessment in Neuro-oncology, dNext generation sequencing. |
Overcoming the BBB
One line of research has focused on disrupting the BBB/BTB to facilitate the extravasation of drugs into the brain parenchyma. This can potentially be achieved with hyperosmolar agents like mannitol or focused ultrasounds (ClinicalTrials.gov NCT04614493), which induce vasodilation and shrinkage of endothelial cells, thus opening the tight junctions and increasing the permeability of the BBB.46–49
Other research has focused on the use of lipid carriers, prodrugs, stem cells, exosomes, gene therapy, biologics or nanoparticles.50 Nanomedicine structures of 10–500-nm particles can be built with different core materials, such as polymers, lipids, dendrimers or inorganic materials, and can be loaded with different drugs and administered locally or systemically by different routes. Over the last years, a large amount of research has explored this technology, which is leading to improved solubility, stability, biodistribution, release kinetics, biocompatibility, tumor accumulation and a good toxicity profile.51–53 Early-phase trials are ongoing.54 However, the complex and heterogeneous nature of GB requires these technologies to be tested on advanced models of the disease, such as patient-derived organoids or xenografts, before considering translation to the clinic.
Another line of research is re-engineering biologics to enable them to cross the BBB with molecular Trojan horse technology using a monoclonal antibody against an endogenous BBB receptor transporter55 or designing bioengineered drugs with a high a priori ability to cross the BBB, as is the case of the ATM kinase inhibitor AZD1390.56
Local Therapy
Local therapy could potentially avoid the BBB and systemic toxicity and attain high concentration levels near the tumor, but it would only be beneficial in localized relapses. Different materials have been studied, including wafers, hydrogel depots, microparticles and fibers, and foam formulations loaded with different drugs or biologics that do not reach therapeutic concentrations after systemic administration or even direct instillation into the tumor mass by convection enhanced delivery. Only Gliadel®, an implantable intracavitary wafer loaded with carmustine, has shown benefit in randomized studies in the recurrent setting.57 Potential complications, including cerebral edema, intracranial hypertension, CSF leakage, and seizures, have prevented the standardization of this treatment.3,21 Research continues in this context and studies in newly diagnosed patients are ongoing (ClinicalTrials.gov NCT03657576, NCT03152318, NCT04135807).58
Chemotherapy
Chemotherapy seems to have been dropped from the therapeutic arsenal for GB, probably because current research is focusing on new treatment modalities like immunotherapy and targeted therapy. Nonetheless, several agents are being evaluated, including lisavanbulin (BAL101553), a microtubule-targeted agent,59 and dianhydrogalactitol (VAL083), a bi-functional alkylating agent,60 but results are still very preliminary. Berubicin (RTA744) is a novel, synthetic, second-generation anthracycline that uses natural processes to induce DNA damage in targeted cancer cells by interfering with the action of topoisomerase II, a critical enzyme enabling cell proliferation. It is designed to circumvent P glycoprotein/MRP1-mediated efflux and is not only a more potent topoisomerase II inhibitor than doxorubicin but is also able to cross the BBB.61 In a Phase I trial in recurrent GB, 12 of the 35 patients (48%) attained longer PFS.62 In 2020, the US FDA granted Orphan Drug Designation to berubicin for the treatment of recurrent GB and granted it Fast Track Designation in 2021. An ongoing phase II trial (ClinicalTrials.gov NCT04762069) is comparing berubicin versus lomustine in recurrent GB.
Targeted Therapy
EGFR alterations are present in many GBs (50% with EGFR overexpression, 40% with EGFR amplification, 6–13% with fusions).63 The most common mutation is EGFR vIII, which promotes cell proliferation, angiogenesis, and tumor invasion.64 EGFR and EGFR vIII transduce signals via the RAS/RAF/MEK/ERK, PI3K/AKT, JAK/STAT, and PKC pathways. EGFR has been targeted with multiple therapies with different mechanisms of action, including tyrosine kinase inhibitors, antibodies, vaccines, CAR T-cells, bispecific T-cell engaging antibodies targeting EGFR vIII, and RNA-based therapies. To date, however, none of these targeted therapies has improved patient outcome.65,66 Nevertheless, the importance of EGFR alterations is clear and new agents are being tested, for example in the INSIGhT trial and in a phase I trial of RO7428731 (ClinicalTrials.gov NCT05187624).
The PI3K/AKT/mTOR pathway is also frequently altered. Different attempts have been made to target PI3K, AKT and/or mTOR with different drugs, but none has succeeded in improving outcome, meeting study endpoints, or translating promising results from cell line or xenograft models to convincing results in randomized studies, probably due to molecular compensatory mechanisms, crosstalk between signal pathways, or tumor heterogeneity.67–70 Nevertheless, due to the importance of this pathway in GB, research on new agents continues in early-phase trials,71,72 and other agents, such as CC-115, are being tested in the INSIGhT trial and in several phase I trials (ClinicalTrials.gov NCT02133183, NCT02142803).
The pRB cell cycle control pathway, which controls cell cycle progression, is often disrupted due to CDK4/6 amplification or CDKN2A/B loss or mutation.73 The CDK4 and CDK6 complex with cyclin D drives the phosphorylation of the retinoblastoma protein, which allows the cell to commit to division.74,75 Palbociclib, a CDK4/6 inhibitor approved for the treatment of breast cancer76 failed to provide a benefit in a phase II trial.75 Other CDK4/6 inhibitors are being tested, such as zotiraciclib (ClinicalTrials.gov NCT03224104, NCT02942264), and abemaciclib is being examined in the INSIGhT trial77 and in a phase I trial (ClinicalTrials.gov NCT04074785).
TP53 is involved in cell cycle progression and induction of apoptosis. Since the key function of its gene product, P53, is to arrest cells in G0/1 or to trigger apoptosis in response to genotoxic stress, various approaches have tried to restore P53 function but without success. The MDM2 protein binds to P53, preventing its action and transporting it from the nucleus to the cytosol. MDM2 also covalently attaches ubiquitin to P53, thus marking P53 for degradation by the proteasome. Several ongoing trials are focusing on neutralizing MDM2 and MDM4 in GB patients who lack P53 function due to MDM2 or MDM4 gene amplification.78
Mutations of mismatch repair (MMR) genes, especially MSH6, are common in relapsed GB and may impact response to temozolomide. Loss of expression of MSH6 and other MMR proteins was found in about 25% of GB patients at relapse but rarely at diagnosis, and there was no correlation with microsatellite instability (MSI). Totally deficient MMR (dMMR) tumors had a hypermutated genome,79 suggesting that loss of MMR proteins may be a surrogate of hypermutation in high-grade gliomas. Patients with dMMR and high MSI are immunogenic as they harbor thousands of somatic mutations that trigger the upregulation of immune checkpoint proteins. Pembrolizumab is active in non-colorectal dMMR/MSI tumors but not in relapsed dMMR GB patients.80 It is currently being tested in combination with radiotherapy and with other agents (ClinicalTrials.gov NCT03661723).
Proteasomes regulate cell survival, cell cycle progression, gene transcription, antigen presentation, and DNA repair.81 Proteasome inhibitors, such as bortezomib, carfilzomib, ixazomib and marizomib, interfere with this activity and may disrupt various cellular processes, stopping proliferation and leading to cell death. Bortezomib was tested in GB without success, probably because of low penetration into the brain.82 However, marizomib, an irreversible inhibitor of the beta-lactone class that binds to the catalytic moieties of the proteasome, can cross the BBB and has shown promising activity in preclinical and xenograft models. The encouraging results of a phase I trial of marizomib in combination with bevacizumab (ClinicalTrials.gov NCT02330562) led to a phase II study (ClinicalTrials.gov NCT02903069) and then to the randomized Phase III Mirage trial in newly diagnosed GB, where it was added to the standard radiotherapy/temozolomide regimen. Unfortunately the results of this trial have been clearly disappointing regarding outcome, and marizomib-related neurological and psychiatric toxicities were quite significant.83
IDH1 and IDH2 mutations are present in about 10% of GBs diagnosed according to the WHO 2016 classification,84 but these tumors are now classified as grade 4 astrocytoma85 and have a better prognosis even in the recurrent setting.86 Nevertheless, due to the importance of this alteration in gliomas, research on targeting IDH is continuing while the new classification is adopted for pathological diagnosis.63,87 Several early-phase studies targeting IDH are ongoing; most include low-grade gliomas but not all of them limit inclusion to this grade. Ongoing or recently finished phase I studies have examined first-generation IDH1 inhibitors like ivosidenib (AG-120), BAY-1436032, DS-1001b, IDH305 and olutasidenib (FT2102), the second-generation IDH1 inhibitor LY3410738, the IDH2 inhibitor enasidenib (AG-221), and the dual IDH1/2 inhibitor vorasidenib (AG-881). In addition, several peptide vaccines, such as the IDH1(R132H) peptide vaccine PEPIDH1M, and an autologous dendritic cell vaccine are testing the safety and efficacy of targeting IDH1/2.88–90
PARP proteins are involved in the base excision repair (BER) pathway. It has been postulated that PARP inhibitors could enhance the efficacy of the standard treatment in GB since the combination of temozolomide and PARP inhibitors could increase cytotoxic effects.91 BER repairs most radiation-induced single-strand DNA breaks, suggesting that PARP inhibitors could also act as radiosensitizers.92,93 Moreover, since GB stem cells increase tumor aggressiveness, confer chemo- and radioresistance, and have a high DNA repair capacity, PARP inhibition could also be a good strategy to ameliorate these effects. Several studies have demonstrated that PARP inhibitors have the ability to reach GB margins94,95 and some early-phase clinical trials have been carried out, but with variable results (ClinicalTrials.gov NCT04614909, NCT03914742, NCT03749187, NCT05076513).96,97
HIF-1 is a transcription factor regulated by the presence or absence of oxygen. Under hypoxic conditions, HIF-1α is stabilized and dimerizes with HIF-1β, after which it translocates to the nucleus to act as a transcription factor controlling the expression of hundreds of genes.98,99 HIF-1 promotes the expression of proangiogenic factors, such as VEGF, angiopoietin, and erythropoietin,100 promotes tumor cell migration, and enhances tumor cell autophagy.101 Several approaches to inhibit the hypoxia pathway have been explored. EZN-2698, a HIF-1 α inhibitor failed to demonstrate a clinical benefit in solid tumors.102 Panobinostat and vorinostat effectively induced HIF-1 degradation,103 but they inhibit all class I histone deacetylases, making them very non-specific. Other approaches have looked at blocking the interaction of HIF-1α with its co-activators, thus antagonizing hypoxia-inducible transcription.98 However, despite these diverse strategies, only a few molecules have shown a good safety profile in early-phase studies, and efficacy is limited in refractory solid tumors (ClinicalTrials.gov NCT02974738).
Other targets are being explored in preclinical models and phase I studies. Some are not specific to GB but are commonly altered in cancer. Some of the alterations that are currently being studied are: Bruton tyrosine kinase, arginine, the XPO protein, C-Met, BET, AXL-UFO, farnesyltransferase, STAT, cerebron, SHH, DRD2 antagonist/ClpP agonist, SMO/SHH, tryptophan and its associated enzymes, and IDO1/2. However, these studies are in the initial stages and no results are yet available. At the same time, other potential targets in GB, such as TERT promoter mutations, which are involved in cell proliferation and longevity, are yet to be explored.73 At present, unfortunately, no agent is envisioned to succeed in a phase III study and become a standard of care.
Radioligand and Boron Neutron Capture Therapy (BCNT)
Radioligand therapy is an innovative approach that combines a radioligand, which locates cancer cells, with a radioisotope, which is a therapeutic radioactive particle. The radioligand binds to cancer cells anywhere in the body and emits targeted radiation, directly attacking the tumor. Radioligand therapy is well established as a treatment modality for advanced gastroenteropancreatic neuroendocrine tumors and is being studied for some types of prostate cancer and other solid tumors, including breast, lung and brain. [177Lu]Lu-NeoB binds to the gastrin-releasing peptide receptor (GRPR) through the NeoB peptide, which contains a DOTA metal chelator that allows radiolabeling with different radionuclides.104 GRPR is present in various glioma cell lines and a correlation has been observed in glioma patients between SUV and GRPR expression.105 A phase I study is being completed (ClinicalTrials.gov NCT05109728) and a randomized trial comparing [177Lu]Lu-NeoB versus carmustine in recurrent GB is planned.
Boron neutron capture therapy (BNCT) combines biological targeting and low-energy thermal neutrons. Kawabata et al demonstrated acceptable safety and prolonged survival for recurrent GB in a phase II trial involving 27 patients compared with results with bevacizumab.106 Although preliminary clinical results of BNCT are promising, further research is needed in order to find more selective boron compounds.107
Immunotherapy
Unfortunately, initial results with immunotherapy have been disappointing. Several factors have been postulated as the cause of this resistance to immunotherapy: low tumor burden; immunosuppressive microenvironment (macrophages, myeloid-derived suppressor cells, microglia, T cell exhaustion); tumor heterogeneity (antigen escape); the BBB; and systemic immunosuppression (corticosteroids, lymphopenia). A better understanding of resistance mechanisms to immunotherapy would provide insight into effective strategies.108–111 Four different types have been studied in GB: immune checkpoint inhibitors (ICIs), vaccines, chimeric antigen receptor (CAR) T-cell therapy and oncolytic viruses.
Immune Checkpoint Inhibitors
Three phase III randomized trials have explored the benefit of ICIs in GB – one in the recurrent setting112 and two in newly diagnosed GBs.113,114 In a trial comparing nivolumab with bevacizumab in the recurrent setting, no differences in survival were found although more durable responses were observed in the nivolumab arm (10 vs 5 months). However, fewer than 10% of patients in the nivolumab arm attained a radiographic response compared to 23% in the bevacizumab arm.112 Neither of the two trials in newly diagnosed GB met their primary endpoint of differences in OS.113,114 Several phase II trials have also examined ICIs in GB. A study comparing pembrolizumab alone versus pembrolizumab plus bevacizumab in recurrent GB found that pembrolizumab was ineffective both as monotherapy and in combination with bevacizumab.115 Neoadjuvant trials of ICIs have reported intriguing results, showing clonal expansion of T cells and overexpression of interferon-gamma and T cell related genes, suggesting a potential benefit in this setting.116
Vaccine Therapy
Several vaccine therapies have been explored in GB, including peptide/multipeptide vaccines and dendritic cells. The aim of vaccine therapy is to induce an immune response against tumor antigens in order to eliminate GB cells. There are two types of antigens: tumor-associated antigens (TAA), which are expressed in both normal and tumor cells, and tumor-specific antigens, which are expressed only in tumor cells.117,118 Some TAAs have been explored in clinical trials with the aim of activating GB immunogenicity, which depends on the affinity of the antigen for major histocompatibility complex and the sequence length. A vaccine against EGFR vIII, rindopepimut®, showed promising results in early clinical trials but failed to demonstrate any benefit in a phase III trial.65 A vaccine against IDH1(R132H) that was tested in a phase I trial in newly diagnosed GB induced T cell and B cell immune responses, and vaccine-induced immune responses were observed in 93.3% of patients.119 A randomized trial is warranted to confirm these results. Multipeptide vaccines may be more active. A phase I study of IMA950, a GB-specific vaccine containing 11 tumor-associated peptides, met its primary immunogenicity endpoint.120 Several other trials of multipeptide vaccines are ongoing but in initial stages.
Dendritic cells can be used to activate innate and acquired immunity. Monocytes need to be isolated from a patient’s peripheral blood, after which GM-CSF and IL-4 are used to induce immature dendritic cells. Then tumor antigens or patient-derived tumor lysates are loaded into the cells.121 A randomized phase III trial in newly diagnosed GB patients found that (DCVax®-L) conferred longer OS in all patients, especially in those with MGMT methylation. However, crossover had led to 90% of the patients receiving the vaccine, making it difficult to draw definite conclusions.122 In a recent report the 64 patients with recurrent WBC who crossed over to DCVAX treatment had an mOS of 13.2 (95% CI, 9.7–16.8) months from relapse vs 7.8 (95% CI, 7.2–8.2) months among control patients (HR, 0.58; 98% CI, 0.00–0.76; P < 0.001).123 Methodological problems in the study design obscure these results.124 ICT-107 is an autologous dendritic cell vaccine pulsed with six synthetic peptide epitopes targeting GB tumor/stem cell-associated antigens (MAGE-1, HER-2, AIM-2, TRP-2, gp100, and IL13Ra2). A randomized placebo-controlled phase II trial of ICT-107 in newly diagnosed GB patients showed a two-month benefit in PFS but no impact on OS.125 Two recent meta-analyses of dendritic cell vaccines in GB have reported conflicting conclusions as to the benefit of these vaccines.126,127 In summary, vaccine therapy may prove to be a beneficial strategy in GB but results to date are not promising. Moreover, the high cost of vaccines and difficulties associated with their manufacture could hinder their use in clinical practice.
Chimeric Antigen Receptor (CAR) T-Cell Therapy
CAR T-cell therapy relies on genetically modified T cells that express CARs engineered to recognize cancer-associated cell surface antigens. CAR T-cell therapy has revolutionized the treatment of lymphoma and acute leukemia and is now being explored in other hematological and solid tumors. Several ongoing early-phase trials evaluating the safety and feasibility of CAR T-cell therapy in GB have found an acceptable safety profile when administered intravenously or intracranially but efficacy data are limited.128 Different antigens have been used in CAR T-cell therapy, including IL13Ralfa2, EGFRvIII, HER2 and B7-H3. Clinical benefit has been generally short-term, probably because of antigen loss relapses. Glioblastoma tumor heterogeneity is a challenge in this type of therapy and new strategies are under investigation, such as multiple antigen targeted CAR T cells with co-stimulatory molecules and immunostimulatory cytokines.129,130 Several ongoing clinical trials are exploring the combination of CAR T-cell therapy with ICI or with treatments targeting tumor-associated macrophages like anti-CSF-R1. New CAR-engineered natural killer cells are also being studied in GB.128
Oncolytic Viruses
Oncolytic viruses are genetically modified to be able to infect and replicate into the tumor mass, thus exerting a selective tumor lysis and inducing an antitumor immune response. The promotion of a “hot” tumor microenvironment could be crucial in increasing tumor responses to oncolytic viruses, either as monotherapy or in combination with other immunotherapies.131,132 Oncolytic viruses are generally administered locally during surgery to overcome the BBB although preclinical studies have demonstrated that they can be loaded on stem cell carriers and delivered to malignant glioma tumors when injected systemically.132 Several candidate viruses have been engineered to treat GB, including retrovirus, adenovirus, herpes simplex, reovirus, parvovirus, vaccinia virus, Newcastle disease and poliovirus.
Oncolytic viruses have shown promise in phase I/II trials. ONYX- 015 induced immune responses, and both the oncolytic adenovirus DNX-2401 (tasadenoturev)133 and the non-pathogenic polio-rhinovirus chimera (PVSPIRO) induced immune and clinical responses.134 These viruses had a good safety profile and promising efficacy in recurrent GB: 20% of patients survived more than 3 years.134,135 In contrast, after promising results with Toca 511 in an early phase I trial, with 20% of durable (>24 weeks) complete responses, no survival benefit was observed in a phase III trial of 403 patients.135
Recently, interesting data have been reported from phase I/II studies of two oncolytic herpes simplex virus-1 (HSV-1): a phase I trial of the HSV-1 G207 for pediatric high-grade gliomas136 and a phase II trial of the HSV-1 G47∆ for residual or recurrent GB in Japanese patients.137 The Japan Ministry of Health granted conditional approval to teserpaturev® (G47∆; Delytact), a triple-mutated, third-generation oncolytic HSV-1, for intratumoral administration in malignant glioma. G47Δ is the first oncolytic virus to receive approval for this use. However, the trial included only 19 patients, the increase in OS was only compared to historical controls, and six of the 19 patients were IDH1-mutated. Therefore, according to internationally accepted rules, a randomized phase III trial should be launched to validate these results.
Two phase II clinical trials have been carried out in the replication-deficient adenovirus mutant thymidine kinase (ADV-TK). ADV-TK administered by intra-arterial cerebral infusion in combination with ganciclovir conferred longer PFS and OS in a series of 53 recurrent GB patients.138 In a subsequent multicenter trial in the first-line setting, ADV-TK was combined with valacyclovir and administered at surgery, but only a non-significant benefit was observed, especially for patients with gross total resection.139 Several studies of different oncolytic viruses and different treatment schemes are now including patients (ClinicalTrials.gov NCT03714334, NCT02457845, NCT03896568).
Discussion and Future Prospects
Challenges in the Search for New Treatments
Identifying effective treatments in GB has long been a difficult task due to various impediments. In addition to the low incidence of the disease itself and the characteristics and symptoms of the patients that preclude their participation in clinical trials, there are other obstacles to clinical research. In fact, the prognosis of GB is so poor that – contrary to common practice in other cancers – new treatments are often tested first in newly diagnosed patients rather than in recurrent disease.
One of the main handicaps is the difficulty in delivering reasonable therapeutic doses to the tumor because of the BBB and the brain-to-tumor barrier (BTB), which are protective biological mechanisms of the brain that block potentially toxic agents from reaching the brain.140,141 The BBB is a tight network of endothelial cells, astrocytes and pericytes that regulates the delivery of nutrients, metabolites and xenobiotics from the blood to the CNS. It restricts the passage of 100% of macromolecules and almost 98% of smaller molecules into the CNS. The modifications that GB exerts on this barrier constitute the BTB, which is highly variable in structure and leads to a disorganized formation of new vessels and to a decrease in the tight junctions between the cells that form the barrier. As a result, the permeability of the BBB increases heterogeneously, as can be seen in MRI gadolinium sequences, which hinders the uniform distribution of drugs.140 In addition to the BBB and BTB, the local microenvironment can prevent the diffusion of the treatment within the brain. The microenvironment is formed by an extracellular matrix consisting of a mixture of collagen, elastin, proteoglycans and hyaluronic acid, together with low oxygen and pH levels that promote invasiveness and chemoresistance of the GB cells, thus diminishing the effect of treatment.142,143 Importantly, drug distribution into cerebrospinal fluid (CSF) is not a measure of BBB transport since drugs injected into CSF are not distributed to the inner parenchyma of the brain but to the blood, and drug concentrations in the brain are much lower than in CSF or blood.144
Given these hindrances, there will always be doubts as to whether a drug will reach the tumor in sufficient concentrations to be effective. Research on different delivery methods, including local therapies and methods to modify the permeability of the BBB, has been ongoing for years.145,146 Window of opportunity studies could play an important role in alleviating this situation because they measure drug levels in the tumor by administering the drug after biopsy and prior to surgical resection. However, the aggressiveness of two surgical procedures in the same patient together with the potential complications inherent in any cerebral surgical maneuver will hamper the widespread use of this type of trial.147
Another source of difficulty is the potential metabolic interaction between the drugs under study and the drugs that patients require to control their symptoms, such as anticonvulsants, antidepressants and corticosteroids, which can act either as inducers of metabolism, thus decreasing the effective doses of the antitumor drugs, or as inhibitors of metabolism, thus increasing the drug-related toxicity.148
In addition to problems associated with drug delivery and interaction, there is a need first to identify the molecular target whose inhibition will stop tumor growth among all the potential molecules that have been identified in GB.149,150 and then to determine the optimal treatment to inhibit or block the target.
A further challenge lies in measuring treatment response and determining clinical benefit, which can affect the choice of the primary endpoint of a clinical trial, which in turn can determine the acceptance of the trial results by the oncology community and medicine agencies. A clear example of the importance of the primary trial endpoint and the consequent difficulties in bringing a drug to the therapeutic arena is the case of bevacizumab. Despite having achieved an increase in PFS in all the studies, an objective that has been accepted in many tumors, it was not approved by the EMA because the main objective of the trials was to increase OS, not PFS. As a result, in Europe, bevacizumab is used only for compassionate use.151
Measuring response to treatment has always been difficult in GB because there are several confounding factors. On the one hand, at recurrence, all patients have already received local irradiation. The irradiated lesions are usually considered not measurable in the RECIST guidelines used for other tumors152 unless a clear progression is demonstrated. Brain irradiation produces changes on the MRI that make it difficult to differentiate between tumor progression, pseudo-progression and radionecrosis.153 In addition, neurological damage caused by the tumor does not usually subside once a response is obtained, and the size of the tumor is not related to the patient’s clinical condition, which depends more on the location of the tumor in eloquent or non-eloquent areas.154 To overcome these drawbacks, the international neuro-oncology community has developed the standardized RANO response criteria, which are based not only on MRI evaluation, including standardization of sequences, but also on the patient’s neurological status and the doses of dexamethasone needed, whose irreversible increase is usually related to tumor progression.155–157 The neuro-oncology community is continuously making an effort to determine the best design for clinical trials regarding both the selection of the primary endpoint (therapeutic response, PFS at 6 or 12 months, OS) and the magnitude of benefit that is acceptable to change standard of care.158–160
In fact, even though GB treatment guidelines recommend inclusion in clinical trials,3 only a minority of patients are fit enough to be included.6 Efforts are now directed towards identifying the barriers to inclusion and increasing participation.158,160
Another difficulty that we will encounter in the near future stems from the reclassification of GB (as defined by the 2016 WHO criteria)84 into different entities by the recent 2021 WHO classification.85 Glioblastomas with IDH1/2 mutations are now classified as grade 4 astrocytomas, with a considerable better prognosis; those located in midline structures and harboring H3p.K28M (K27M) mutations are now diffuse midline gliomas H3K27; and hemispheric GBs with H3 G34 mutations are now classified as diffuse hemispheric gliomas H3G34. Additionally, GBs with wild-type H3 and IDH1/2 are now classified as diffuse pediatric-type high-grade glioma H3 and IDH wild-type. These are mainly found in young adults and have different outcomes but may exhibit necrosis and vascular proliferation, which suggests that they could have been included in trials of GB in the past. In addition, some IDH wild-type histologically low-grade gliomas with TERT promoter mutations or EGFR gene amplification or a +7/−10 genotype are now reclassified as GBs even in the absence of vascular proliferation or necrosis. It is still not clear if these newly reclassified GBs will be included in trials of GB IDH wild-type because their prognosis is not exactly the same.63,161 The predicted treatment response and outcome of these new tumor types have been drawn from previous trials in which tumors were classified primarily by histology. However, it may well be that these subtypes will not be included in future trials, which will further reduce the number of patients who are candidates for inclusion and will make it impossible to determine if the new therapies are effective in these entities. As a result, it will be even more difficult to conduct phase III trials requiring large numbers of patients.
Finally, immediate and delayed drug-related toxicity, especially that affecting neurocognitive function, must be taken into account when deciding on therapy. If a drug manages to break through the BBB and BTB but causes neurological damage to healthy cells, survival benefit will increase but quality of life will not be maintained and there will be no clinical benefit. Therefore, it is essential to consider quality of life, patient reported outcome, and toxicity before implementing a new treatment in clinical practice.160,162
New trial designs are aimed at a rapid identification of agents that are suitable for study in phase III trials in order to accelerate research in new clinical therapies for GB. For example, the phase II randomized INdividualized Screening Trial of Innovative Glioblastoma Therapy (INSIGhT, ClincalTrials.gov NCT02977780) is comparing a control arm of standard first-line treatment with different experimental arms, while the international, seamless phase II/III AGILE trial is an adaptive response Bayesian randomization platform designed to evaluate multiple therapies in newly diagnosed and recurrent GB.
In conclusion, although GB prognosis remains gloomy and no new treatments have proven effective, there are many ongoing lines of research. As our knowledge of genomic data grows, we will have a better understanding of resistance mechanisms, which should lead to advances in therapy. However, there is clearly still a great deal of work to be done in GB.
Disclosure
Dr Ainhoa Hernández reports speaker bureau and travel/education fees from Roche and Sanofi, speaker bureau and educational fees from BMS, grants from Janssen, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2013–2017. Neuro Oncol. 2020;22(Supplement_1):iv1–iv96. doi:10.1093/neuonc/noaa200
2. Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. doi:10.1056/NEJMoa043330
3. Mohile NA, Messersmith H, Gatson NT, et al. Therapy for diffuse astrocytic and oligodendroglial tumors in adults: ASCO-SNO guideline. J Clin Oncol. 2022;40(4):403–426. doi:10.1200/jco.21.02036
4. Hansen S, Rasmussen BK, Laursen RJ, et al. Treatment and survival of glioblastoma patients in Denmark: the Danish Neuro-Oncology Registry 2009–2014. J Neurooncol. 2018;139(2):479–489. doi:10.1007/s11060-018-2892-7
5. Fabbro-Peray P, Zouaoui S, Darlix A, et al. Association of patterns of care, prognostic factors, and use of radiotherapy–temozolomide therapy with survival in patients with newly diagnosed glioblastoma: a French national population-based study. J Neurooncol. 2019;142(1):91–101. doi:10.1007/s11060-018-03065-z
6. Skaga E, Skretteberg MA, Johannesen TB, et al. Real-world validity of randomized controlled phase III trials in newly diagnosed glioblastoma: to whom do the results of the trials apply? Neurooncol Adv. 2021;3(1):vdab008. doi:10.1093/noajnl/vdab008
7. Minaya P, Baumstarck K, Berbis J, et al. The CareGiver Oncology Quality of Life questionnaire (CarGOQoL): development and validation of an instrument to measure the quality of life of the caregivers of patients with cancer. Eur J Cancer. 2012;48(6):904–911. doi:10.1016/j.ejca.2011.09.010
8. Walbert T. Integration of palliative care into the neuro-oncology practice: patterns in the United States. Neurooncol Pract. 2014;1:3–7. doi:10.1093/nop/npt004
9. Pace A, Dirven L, Koekkoek JAF, et al. European Association for Neuro-Oncology (EANO) guidelines for palliative care in adults with glioma. Lancet Oncol. 2017;18:e330–e40. doi:10.1016/S1470-2045(17)30345-5
10. Wann A, Tully PA, Barnes EH, et al. Outcomes after second surgery for recurrent glioblastoma: a retrospective case–control study. J Neurooncol. 2018;137(2):409–415. doi:10.1007/s11060-017-2731-2
11. Lu VM, Jue TR, McDonald KL, Rovin RA. The survival effect of repeat surgery at glioblastoma recurrence and its trend: a systematic review and meta-analysis. World Neurosurg. 2018;115:453–9 e3. doi:10.1016/j.wneu.2018.04.016
12. Montemurro N, Perrini P, Blanco MO, Vannozzi R. Second surgery for recurrent glioblastoma: a concise overview of the current literature. Clin Neurol Neurosurg. 2016;142:60–64. doi:10.1016/j.clineuro.2016.01.010
13. Scoccianti S, Perna M, Olmetto E, et al. Local treatment for relapsing glioblastoma: a decision-making tree for choosing between reirradiation and second surgery. Crit Rev Oncol Hematol. 2021;157:103184. doi:10.1016/j.critrevonc.2020.103184
14. Suchorska B, Weller M, Tabatabai G, et al. Complete resection of contrast-enhancing tumor volume is associated with improved survival in recurrent glioblastoma—results from the DIRECTOR trial. Neuro-Oncology. 2016;18(4):549–556. doi:10.1093/neuonc/nov326
15. Ryken TC, Kalkanis SN, Buatti JM, Olson JJ. The role of cytoreductive surgery in the management of progressive glioblastoma: a systematic review and evidence-based clinical practice guideline. J Neurooncol. 2014;118(3):479–488. doi:10.1007/s11060-013-1336-7
16. Brennan PM, Borchert R, Coulter C, et al. Second surgery for progressive glioblastoma: a multi-centre questionnaire and cohort-based review of clinical decision-making and patient outcomes in current practice. J Neurooncol. 2021;153(1):99–107. doi:10.1007/s11060-021-03748-0
17. Garcia-Cabezas S, Rivin Del Campo E, Solivera-Vela J, Palacios-Eito A. Re-irradiation for high-grade gliomas: has anything changed? World J Clin Oncol. 2021;12(9):767–786. doi:10.5306/wjco.v12.i9.767
18. Kazmi F, Soon YY, Leong YH, Koh WY, Vellayappan B. Re-irradiation for recurrent glioblastoma (GBM): a systematic review and meta-analysis. J Neurooncol. 2019;142(1):79–90. doi:10.1007/s11060-018-03064-0
19. Fleischmann DF, Jenn J, Corradini S, et al. Bevacizumab reduces toxicity of reirradiation in recurrent high-grade glioma. Radiother Oncol. 2019;138:99–105. doi:10.1016/j.radonc.2019.06.009
20. Chapman CH, Hara JH, Molinaro AM, et al. Reirradiation of recurrent high-grade glioma and development of prognostic scores for progression and survival. Neuro-Oncology Practice. 2019;6(5):364–374. doi:10.1093/nop/npz017
21. Weller M, van den Bent M, Preusser M, et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat Rev Clin Oncol. 2021;18(3):170–186. doi:10.1038/s41571-020-00447-z
22. Yung WK, Albright RE, Olson J, et al. A phase II study of temozolomide vs. procarbazine in patients with glioblastoma multiforme at first relapse. Br J Cancer. 2000;83(5):588–593. doi:10.1054/bjoc.2000.1316
23. Brada M, Hoang-Xuan K, Rampling R, et al. Multicenter phase II trial of temozolomide in patients with glioblastoma multiforme at first relapse. Ann Oncol. 2001;12(2):259–266. doi:10.1023/A:1008382516636
24. Perry JR, Belanger K, Mason WP, et al. Phase II trial of continuous dose-intense temozolomide in recurrent malignant glioma: RESCUE study. J Clin Oncol. 2010;28(12):2051–2057. doi:10.1200/JCO.2009.26.5520
25. Alexander BM, Ba S, Berger MS, et al. Adaptive global innovative learning environment for glioblastoma: GBM AGILE. Clin Cancer Res. 2018;24(4):737–743. doi:10.1158/1078-0432.CCR-17-0764
26. Weller M, Le Rhun E. How did lomustine become standard of care in recurrent glioblastoma? Cancer Treat Rev. 2020;87:102029. doi:10.1016/j.ctrv.2020.102029
27. Perez-Segura P, Manneh R, Ceballos I, et al. GEINOFOTE: efficacy and safety of fotemustine in patients with high-grade recurrent gliomas and poor performance status. Clin Transl Oncol. 2016;18(8):805–812. doi:10.1007/s12094-015-1444-2
28. Addeo R, Lamberti G, Simonetti G, et al. Biweekly fotemustine schedule for recurrent glioblastoma in the elderly: activity and toxicity assessment of a multicenter study. CNS Oncology. 2019;8(2):CNS32. doi:10.2217/cns-2019-0004
29. Vredenburgh JJ, Desjardins A, Herndon JE, et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol. 2007;25(30):4722–4729. doi:10.1200/JCO.2007.12.2440
30. Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27(28):4733–4740. doi:10.1200/JCO.2008.19.8721
31. Chinot OL, Wick W, Mason W, et al. Bevacizumab plus Radiotherapy–temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370(8):709–722. doi:10.1056/NEJMoa1308345
32. Gilbert MR, Dignam JJ, Armstrong TS, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370(8):699–708. doi:10.1056/NEJMoa1308573
33. Taal W, Oosterkamp HM, Walenkamp AME, et al. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): a randomised controlled Phase 2 trial. Lancet Oncol. 2014;15(9):943–953. doi:10.1016/S1470-2045(14)70314-6
34. Wick W, Gorlia T, Bendszus M, et al. Lomustine and bevacizumab in progressive glioblastoma. N Engl J Med. 2017;377(20):1954–1963. doi:10.1056/NEJMoa1707358
35. Lombardi G, De Salvo GL, Brandes AA, et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): a multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2019;20(1):110–119. doi:10.1016/S1470-2045(18)30675-2
36. Stupp R, Wong ET, Kanner AA, et al. NovoTTF-100A versus physician’s choice chemotherapy in recurrent glioblastoma: a randomised phase III trial of a novel treatment modality. Eur J Cancer. 2012;48(14):2192–2202. doi:10.1016/j.ejca.2012.04.011
37. Wen PY, Stein A, van den Bent M, et al. Dabrafenib plus trametinib in patients with BRAFV600E-mutant low-grade and high-grade glioma (ROAR): a multicentre, open-label, single-arm, phase 2, basket trial. Lancet Oncol. 2022;23(1):53–64. doi:10.1016/s1470-2045(21)00578-7
38. Ferguson SD, Zhou S, Huse JT, et al. Targetable gene fusions associate with the IDH wild-type astrocytic lineage in adult gliomas. J Neuropathol Exp Neurol. 2018;77(6):437–442. doi:10.1093/jnen/nly022
39. Fischer H, Ullah M, de la Cruz CC, et al. Entrectinib, a TRK/ROS1 inhibitor with anti-CNS tumor activity: differentiation from other inhibitors in its class due to weak interaction with P-glycoprotein. Neuro Oncol. 2020;22(6):819–829. doi:10.1093/neuonc/noaa052
40. Doebele RC, Drilon A, Paz-Ares L, et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three Phase 1–2 trials. Lancet Oncol. 2020;21(2):271–282. doi:10.1016/s1470-2045(19)30691-6
41. Doz F, van Tilburg CM, Geoerger B, et al. Efficacy and safety of larotrectinib in TRK fusion-positive primary central nervous system tumors. Neuro Oncol. 2022;24(6):997–1007. doi:10.1093/neuonc/noab274
42. Helsten T, Elkin S, Arthur E, Tomson BN, Carter J, Kurzrock R. The FGFR landscape in cancer: analysis of 4853 tumors by next-generation sequencing. Clin Cancer Res. 2016;22(1):259–267. doi:10.1158/1078-0432.CCR-14-3212
43. Loriot Y, Schuler MH, Iyer G, et al. Tumor agnostic efficacy and safety of erdafitinib in patients (pts) with advanced solid tumors with prespecified fibroblast growth factor receptor alterations (FGFRalt) in RAGNAR: interim analysis (IA) results. J Clin Oncol. 2022;40(16_suppl):3007. doi:10.1200/JCO.2022.40.16_suppl.3007
44. Lassman AB, Sepulveda-Sanchez JM, Cloughesy TF, et al. Infigratinib in patients with recurrent gliomas and FGFR alterations: a multicenter phase II study. Clin Cancer Res. 2022;28(11):2270–2277. doi:10.1158/1078-0432.CCR-21-2664
45. Jimenez-Pascual A, Siebzehnrubl FA. Fibroblast growth factor receptor functions in glioblastoma. Cells. 2019;8(7):715. doi:10.3390/cells8070715
46. Towner RA, Saunders D, Lerner M, et al. Temporary opening of the blood-brain barrier with the nitrone compound OKN-007. Am J Nucl Med Mol Imaging. 2021;11(5):363–373.
47. Burks SR, Kersch CN, Witko JA, et al. Blood-brain barrier opening by intracarotid artery hyperosmolar mannitol induces sterile inflammatory and innate immune responses. Proc Natl Acad Sci U S A. 2021;118. doi:10.1073/pnas.2021915118
48. Sachdeva S, Persaud S, Patel M, Popard P, Colverson A, Dore S. Effects of sound interventions on the permeability of the blood–brain barrier and meningeal lymphatic clearance. Brain Sci. 2022;12(6):742. doi:10.3390/brainsci12060742
49. Roberts JW, Powlovich L, Sheybani N, LeBlang S. Focused ultrasound for the treatment of glioblastoma. J Neurooncol. 2022;157(2):237–247. doi:10.1007/s11060-022-03974-0
50. Pardridge WM. A historical review of brain drug delivery. Pharmaceutics. 2022;14. doi:10.3390/pharmaceutics14061283
51. Zhang S, Zhang S, Luo S, et al. Ultrasound-assisted brain delivery of nanomedicines for brain tumor therapy: advance and prospect. J Nanobiotechnology. 2022;20(1):287. doi:10.1186/s12951-022-01464-z
52. Khan I, Baig MH, Mahfooz S, et al. Nanomedicine for glioblastoma: progress and future prospects. Semin Cancer Biol. 2022;86:172–186. doi:10.1016/j.semcancer.2022.06.007
53. Mitchell MJ, Billingsley MM, Haley RM, Wechsler ME, Peppas NA, Langer R. Engineering precision nanoparticles for drug delivery. Nat Rev Drug Discov. 2021;20(2):101–124. doi:10.1038/s41573-020-0090-8
54. Amaral M, Cruz N, Rosa A, et al. An update of advanced nanoplatforms for glioblastoma multiforme management. Excli J. 2021;20:1544–1570. doi:10.17179/excli2021-4393
55. Pardridge WM. Delivery of biologics across the blood–brain barrier with molecular trojan horse technology. BioDrugs. 2017;31(6):503–519. doi:10.1007/s40259-017-0248-z
56. Durant ST, Zheng L, Wang Y, et al. The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci Adv. 2018;4(6):eaat1719. doi:10.1126/sciadv.aat1719
57. Brem H, Piantadosi S, Burger PC, et al. Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas. The Polymer-brain Tumor Treatment Group. Lancet. 1995;345(8956):1008–1012. doi:10.1016/S0140-6736(95)90755-6
58. van Solinge TS, Nieland L, Chiocca EA, Broekman MLD. Advances in local therapy for glioblastoma — taking the fight to the tumour. Nat Rev Neurol. 2022;18(4):221–236. doi:10.1038/s41582-022-00621-0
59. Joerger M, Stathis A, Metaxas Y, et al. A Phase 1 study of BAL101553, a novel tumor checkpoint controller targeting microtubules, administered as 48-h infusion in adult patients with advanced solid tumors. Invest New Drugs. 2020;38(4):1067–1076. doi:10.1007/s10637-019-00850-z
60. O’Brien B, Penas-Prado M, Kamiya-Matsuoka C, et al. CTNI-26. Phase 2 study of dianhydrogalactitol (VAL-083) in patients with mgmt-unmethylated, bevacizumab-naïve glioblastoma in the recurrent and adjuvant setting. Neuro-Oncol. 2021;23(Supplement_6):vi65–vi. doi:10.1093/neuonc/noab196.251
61. Kazerooni RB, Conrad CA, Johansen M, et al. Phase I clinical pharmacokinetics of RTA 744 (berubicin(B)), a blood-brain barrier penetrating anthracycline active against high grade glioma, and evaluation of its 13-hydroxy metabolite, berubicinol (B-ol). Mol Cancer Ther. 2007;6:157.
62. Silberman S, Hsu S, Muczyczenko Z, et al. RTID-03. Design and initiation of pivotal studies for berubicin, a novel, potent topoisomerase ii poison for the treatment of recurrent glioblastoma multiforme (GBM). Neuro-Oncol. 2021;23(Supplement_6):vi193–vi. doi:10.1093/neuonc/noab196.765
63. WHO Classification of Tumours Editorial Board. Central nervous system tumours.
64. An Z, Aksoy O, Zheng T, Fan Q-W, Weiss WA. Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies. Oncogene. 2018;37(12):1561–1575. doi:10.1038/s41388-017-0045-7
65. Weller M, Butowski N, Tran DD, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international Phase 3 trial. Lancet Oncol. 2017;18:1373–1385. doi:10.1016/S1470-2045(17)30517-X
66. Van Den Bent M, Eoli M, Sepulveda JM, et al. INTELLANCE 2/EORTC 1410 randomized phase II study of Depatux-M alone and with temozolomide vs temozolomide or lomustine in recurrent EGFR amplified glioblastoma. Neuro Oncol. 2020;22(5):684–693. doi:10.1093/neuonc/noz222
67. Li X, Wu C, Chen N, et al. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget. 2016;7(22):33440–33450. doi:10.18632/oncotarget.7961
68. Wen PY, Touat M, Alexander BM, et al. Buparlisib in patients with recurrent glioblastoma harboring phosphatidylinositol 3-kinase pathway activation: an open-label, multicenter, multi-arm, phase II trial. J Clin Oncol. 2019;37(9):741–750. doi:10.1200/JCO.18.01207
69. Ma DJ, Galanis E, Anderson SK, et al. A phase II trial of everolimus, temozolomide, and radiotherapy in patients with newly diagnosed glioblastoma: NCCTG N057K. Neuro Oncol. 2015;17(9):1261–1269. doi:10.1093/neuonc/nou328
70. Chang SM, Wen P, Cloughesy T, et al. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Invest New Drugs. 2005;23(4):357–361. doi:10.1007/s10637-005-1444-0
71. Omeljaniuk WJ, Kretowski R, Ratajczak-Wrona W, Jablonska E, Cechowska-Pasko M. Novel dual PI3K/mTOR inhibitor, apitolisib (GDC-0980), inhibits growth and induces apoptosis in human glioblastoma cells. Int J Mol Sci. 2021;22(21):11511. doi:10.3390/ijms222111511
72. Lapointe S, Mason W, MacNeil M, et al. A phase I study of vistusertib (dual mTORC1/2 inhibitor) in patients with previously treated glioblastoma multiforme: a CCTG study. Invest New Drugs. 2020;38(4):1137–1144. doi:10.1007/s10637-019-00875-4
73. Brennan CW, Verhaak RW, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–477. doi:10.1016/j.cell.2013.09.034
74. Xu H, Yu S, Liu Q, et al. Recent advances of highly selective CDK4/6 inhibitors in breast cancer. J Hematol Oncol. 2017;10(1):97. doi:10.1186/s13045-017-0467-2
75. Taylor JW, Parikh M, Phillips JJ, et al. Phase-2 trial of palbociclib in adult patients with recurrent RB1-positive glioblastoma. J Neurooncol. 2018;140(2):477–483. doi:10.1007/s11060-018-2977-3
76. Finn RS, Martin M, Rugo HS, et al. Palbociclib and letrozole in advanced breast cancer. N Engl J Med. 2016;375(20):1925–1936. doi:10.1056/NEJMoa1607303
77. Lee EQ, Trippa L, Fell G, et al. Preliminary results of the abemaciclib arm in the Individualized Screening Trial of Innovative Glioblastoma Therapy (INSIGhT): a phase II platform trial using Bayesian adaptive randomization. J Clin Oncol. 2021;39(15_suppl):2014. doi:10.1200/JCO.2021.39.15_suppl.2014
78. Wick W, Dettmer S, Berberich A, et al. N2M2 (NOA-20) phase I/II trial of molecularly matched targeted therapies plus radiotherapy in patients with newly diagnosed non-MGMT hypermethylated glioblastoma. Neuro Oncol. 2019;21(1):95–105. doi:10.1093/neuonc/noy161
79. Indraccolo S, Lombardi G, Fassan M, et al. Genetic, epigenetic, and immunologic profiling of MMR-deficient relapsed glioblastoma. Clin Cancer Res. 2019;25:1828–1837. doi:10.1158/1078-0432.CCR-18-1892
80. Lombardi G, Barresi V, Indraccolo S, et al. Pembrolizumab activity in recurrent high-grade gliomas with partial or complete loss of mismatch repair protein expression: a monocentric, observational and prospective pilot study. Cancers. 2020;12. doi:10.3390/cancers12082283
81. Teicher BA, Tomaszewski JE. Proteasome inhibitors. Biochem Pharmacol. 2015;96:1–9. doi:10.1016/j.bcp.2015.04.008
82. Kong XT, Nguyen NT, Choi YJ, et al. Phase 2 study of bortezomib combined with temozolomide and regional radiation therapy for upfront treatment of patients with newly diagnosed glioblastoma multiforme: safety and efficacy assessment. Int J Radiat Oncol Biol Phys. 2018;100:1195–1203. doi:10.1016/j.ijrobp.2018.01.001
83. Roth P, Gorlia T, Reijneveld JC, et al. EORTC 1709/CCTG CE.8: a phase III trial of marizomib in combination with temozolomide-based radiochemotherapy versus temozolomide-based radiochemotherapy alone in patients with newly diagnosed glioblastoma. J Clin Oncol. 2021;39:2004. doi:10.1200/JCO.2021.39.15_suppl.2004
84. Louis D. WHO Classification of Tumours of the Central Nervous System. Lyon: International Agency for Research on Cancer; 2016.
85. Louis DN, Perry A, Wesseling P, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021. doi:10.1093/neuonc/noab106
86. Miller JJ, Loebel F, Juratli TA, et al. Accelerated progression of IDH mutant glioma after first recurrence. Neuro Oncol. 2019;21:669–677. doi:10.1093/neuonc/noz016
87. Cheng W, Ren X, Zhang C, Cai J, Han S, Wu A. Gene expression profiling stratifies IDH1-mutant glioma with distinct prognoses. Mol Neurobiol. 2017;54:5996–6005. doi:10.1007/s12035-016-0150-6
88. Natsume A, Arakawa Y, Narita Y, et al. The first-in-human phase I study of a brain penetrant mutant IDH1 inhibitor DS-1001 in patients with recurrent or progressive IDH1-mutant gliomas. Neuro Oncol. 2022. doi:10.1093/neuonc/noac155
89. Mellinghoff IK, Penas-Prado M, Peters KB, et al. Vorasidenib, a dual inhibitor of mutant IDH1/2, in recurrent or progressive glioma; results of a first-in-human phase I trial. Clin Cancer Res. 2021;27:4491–4499. doi:10.1158/1078-0432.Ccr-21-0611
90. Mellinghoff IK, Ellingson BM, Touat M, et al. Ivosidenib in isocitrate dehydrogenase 1-mutated advanced glioma. J Clin Oncol. 2020;38:3398–3406. doi:10.1200/jco.19.03327
91. Gupta SK, Smith EJ, Mladek AC, et al. PARP inhibitors for sensitization of alkylation chemotherapy in glioblastoma: impact of blood-brain barrier and molecular heterogeneity. Front Oncol. 2018;8:670. doi:10.3389/fonc.2018.00670
92. Dungey FA, Loser DA, Chalmers AJ. Replication-dependent radiosensitization of human glioma cells by inhibition of poly(ADP-Ribose) polymerase: mechanisms and therapeutic potential. Int J Radiat Oncol Biol Phys. 2008;72:1188–1197. doi:10.1016/j.ijrobp.2008.07.031
93. Liu SK, Coackley C, Krause M, Jalali F, Chan N, Bristow RG. A novel poly(ADP-ribose) polymerase inhibitor, ABT-888, radiosensitizes malignant human cell lines under hypoxia. Radiother Oncol. 2008;88:258–268. doi:10.1016/j.radonc.2008.04.005
94. Gupta SK, Kizilbash SH, Carlson BL, et al. Delineation of MGMT hypermethylation as a biomarker for veliparib-mediated temozolomide-sensitizing therapy of glioblastoma. J Natl Cancer Inst. 2016;108. doi:10.1093/jnci/djv369
95. Chalmers AJ, Short S, Watts C, et al. Phase I clinical trials evaluating olaparib in combination with radiotherapy (RT) and/or temozolomide (TMZ) in glioblastoma patients: results of OPARATIC and PARADIGM phase I and early results of PARADIGM-2. J Clin Oncol. 2018;36:2018. doi:10.1200/JCO.2018.36.15_suppl.2018
96. Robins HI, Zhang P, Gilbert MR, et al. A randomized phase I/II study of ABT-888 in combination with temozolomide in recurrent temozolomide resistant glioblastoma: an NRG oncology RTOG group study. J Neurooncol. 2016;126:309–316. doi:10.1007/s11060-015-1966-z
97. Sim HW, McDonald KL, Lwin Z, et al. A randomized phase II trial of veliparib, radiotherapy, and temozolomide in patients with unmethylated MGMT glioblastoma: the VERTU study. Neuro Oncol. 2021;23:1736–1749. doi:10.1093/neuonc/noab111
98. Yin S, Kaluz S, Devi NS, et al. Arylsulfonamide KCN1 inhibits in vivo glioma growth and interferes with HIF signaling by disrupting HIF-1alpha interaction with cofactors p300/CBP. Clin Cancer Res. 2012;18:6623–6633. doi:10.1158/1078-0432.CCR-12-0861
99. Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997;11(1):72–82. doi:10.1101/gad.11.1.72
100. Zimna A, Kurpisz M. Hypoxia-inducible factor-1 in physiological and pathophysiological angiogenesis: applications and therapies. Biomed Res Int. 2015;2015:549412. doi:10.1155/2015/549412
101. Jawhari S, Ratinaud M-H, Verdier M. Glioblastoma, hypoxia and autophagy: a survival-prone ‘ménage-à-trois’. Cell Death Dis. 2016;7(10):e2434. doi:10.1038/cddis.2016.318
102. Jeong W, Rapisarda A, Park SR, et al. Pilot trial of EZN-2968, an antisense oligonucleotide inhibitor of hypoxia-inducible factor-1 alpha (HIF-1α), in patients with refractory solid tumors. Cancer Chemother Pharmacol. 2014;73(2):343–348. doi:10.1007/s00280-013-2362-z
103. Barliya T, Mandel M, Livnat T, Weinberger D, Lavie G, Tyagi AK. Degradation of HIF-1alpha under hypoxia combined with induction of Hsp90 polyubiquitination in cancer cells by hypericin: a unique cancer therapy. PLoS One. 2011;6(9):e22849. doi:10.1371/journal.pone.0022849
104. Ruigrok EAM, Verhoeven M, Konijnenberg MW, et al. Safety of [177Lu]Lu-NeoB treatment: a preclinical study characterizing absorbed dose and acute, early, and late organ toxicity. Eur J Nucl Med Mol Imaging. 2022;49(13):4440–4451. doi:10.1007/s00259-022-05926-2
105. Menegotto PR, da Costa Lopez PL, Souza BK, et al. Gastrin-releasing peptide receptor knockdown induces senescence in glioblastoma cells. Mol Neurobiol. 2017;54(2):888–894. doi:10.1007/s12035-016-9696-6
106. Kawabata S, Suzuki M, Hirose K, et al. Accelerator-based BNCT for patients with recurrent glioblastoma: a multicenter phase II study. Neurooncol Adv. 2021;3(1):vdab067. doi:10.1093/noajnl/vdab067
107. Cheng X, Li F, Liang L. Boron neutron capture therapy: clinical application and research progress. Curr Oncol. 2022;29(10):7868–7886. doi:10.3390/curroncol29100622
108. Medikonda R, Dunn G, Rahman M, Fecci P, Lim M. A review of glioblastoma immunotherapy. J Neurooncol. 2021;151(1):41–53. doi:10.1007/s11060-020-03448-1
109. Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. 2018;15(7):422–442. doi:10.1038/s41571-018-0003-5
110. Jackson CM, Choi J, Lim M. Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat Immunol. 2019;20(9):1100–1109. doi:10.1038/s41590-019-0433-y
111. Zhang M, Choi J, Lim M. Advances in immunotherapies for gliomas. Curr Neurol Neurosci Rep. 2022;22(1):1–10. doi:10.1007/s11910-022-01176-9
112. Reardon DA, Brandes AA, Omuro A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the checkmate 143 phase 3 randomized clinical trial. JAMA Oncol. 2020;6(7):1003–1010. doi:10.1001/jamaoncol.2020.1024
113. Lim M, Weller M, Idbaih A, et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol. 2022;24(11):1935–1949. doi:10.1093/neuonc/noac116
114. Omuro A, Reardon DA, Sampson JH, et al. Nivolumab plus radiotherapy with or without temozolomide in newly diagnosed glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2022;4(1). doi:10.1093/noajnl/vdac025
115. Nayak L, Molinaro AM, Peters K, et al. Randomized phase II and biomarker study of pembrolizumab plus bevacizumab versus pembrolizumab alone for patients with recurrent glioblastoma. Clin Cancer Res. 2021;27(4):1048–1057. doi:10.1158/1078-0432.CCR-20-2500
116. Cloughesy TF, Mochizuki AY, Orpilla JR, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25(3):477–486. doi:10.1038/s41591-018-0337-7
117. Daubon T, Hemadou A, Romero Garmendia I, Saleh M. Glioblastoma immune landscape and the potential of new immunotherapies. Front Immunol. 2020;11:585616. doi:10.3389/fimmu.2020.585616
118. Zhao T, Li C, Ge H, Lin Y, Kang D. Glioblastoma vaccine tumor therapy research progress. Chin Neurosurg J. 2022;8(1):2. doi:10.1186/s41016-021-00269-7
119. Platten M, Bunse L, Wick A, et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature. 2021;592(7854):463–468. doi:10.1038/s41586-021-03363-z
120. Rampling R, Peoples S, Mulholland PJ, et al. A cancer research UK first time in human phase I trial of IMA950 (novel multipeptide therapeutic vaccine) in patients with newly diagnosed glioblastoma. Clin Cancer Res. 2016;22(19):4776–4785. doi:10.1158/1078-0432.CCR-16-0506
121. Datsi A, Sorg RV. Dendritic cell vaccination of glioblastoma: road to success or dead end. Front Immunol. 2021;12:770390. doi:10.3389/fimmu.2021.770390
122. Liau LM, Ashkan K, Tran DD, et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med. 2018;16(1):142. doi:10.1186/s12967-018-1507-6
123. Liau LM, Ashkan K, Brem S, et al. Association of autologous tumor lysate-loaded dendritic cell vaccination with extension of survival among patients with newly diagnosed and recurrent glioblastoma: a phase 3 prospective externally controlled cohort trial. JAMA Oncol. 2022. doi:10.1001/jamaoncol.2022.5370
124. Preusser M, van den Bent MJ. Autologous tumor lysate-loaded dendritic cell vaccination (DCVax-L) in glioblastoma: breakthrough or fata morgana? Neuro Oncol. 2022. doi:10.1093/neuonc/noac281
125. Wen PY, Reardon DA, Armstrong TS, et al. A randomized double-blind placebo-controlled phase II trial of dendritic cell vaccine ICT-107 in newly diagnosed patients with glioblastoma. Clin Cancer Res. 2019;25(19):5799–5807. doi:10.1158/1078-0432.CCR-19-0261
126. Lv L, Huang J, Xi H, Zhou X. Efficacy and safety of dendritic cell vaccines for patients with glioblastoma: a meta-analysis of randomized controlled trials. Int Immunopharmacol. 2020;83:106336. doi:10.1016/j.intimp.2020.106336
127. Tan L, Peng J, Liu P, Wu Q. The efficacy of dendritic cell vaccine for newly diagnosed glioblastoma: a meta-analysis of randomized controlled studies. Clin Neuropharmacol. 2021;44(6):216–221. doi:10.1097/WNF.0000000000000452
128. Burger MC, Zhang C, Harter PN, et al. CAR-engineered NK cells for the treatment of glioblastoma: turning innate effectors into precision tools for cancer immunotherapy. Front Immunol. 2019;10:2683. doi:10.3389/fimmu.2019.02683
129. Brown CE, Alizadeh D, Starr R, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375(26):2561–2569. doi:10.1056/NEJMoa1610497
130. O’Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399). doi:10.1126/scitranslmed.aaa0984
131. Zeng J, Li X, Sander M, et al. Oncolytic Viro-Immunotherapy: An Emerging Option in the Treatment of Gliomas. Front Immunol. 2021;12:721830. doi:10.3389/fimmu.2021.721830
132. Zhang Q, Xiang W, Yi DY, et al. Current status and potential challenges of mesenchymal stem cell-based therapy for malignant gliomas. Stem Cell Res Ther. 2018;9:228. doi:10.1186/s13287-018-0977-z
133. Lang FF, Conrad C, Gomez-Manzano C, et al. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: replication and immunotherapeutic effects in recurrent malignant glioma. J Clin Oncol. 2018;36(14):1419–1427. doi:10.1200/jco.2017.75.8219
134. Desjardins A, Gromeier M, Herndon JE, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med. 2018;379(2):150–161. doi:10.1056/NEJMoa1716435
135. Cloughesy TF, Petrecca K, Walbert T, et al. Effect of vocimagene amiretrorepvec in combination with flucytosine vs standard of care on survival following tumor resection in patients with recurrent high-grade glioma: a randomized clinical trial. JAMA Oncol. 2020;6(12):1939–1946. doi:10.1001/jamaoncol.2020.3161
136. Friedman GK, Johnston JM, Bag AK, et al. Oncolytic HSV-1 G207 immunovirotherapy for pediatric high-grade gliomas. N Engl J Med. 2021;384(17):1613–1622. doi:10.1056/NEJMoa2024947
137. Todo T, Ito H, Ino Y, et al. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: a phase 2 trial. Nat Med. 2022;28(8):1630–1639. doi:10.1038/s41591-022-01897-x
138. Ji N, Weng D, Liu C, et al. Adenovirus-mediated delivery of herpes simplex virus thymidine kinase administration improves outcome of recurrent high-grade glioma. Oncotarget. 2016;7(4):4369–4378. doi:10.18632/oncotarget.6737
139. Wheeler LA, Manzanera AG, Bell SD, et al. Phase II multicenter study of gene-mediated cytotoxic immunotherapy as adjuvant to surgical resection for newly diagnosed malignant glioma. Neuro Oncol. 2016;18(8):1137–1145. doi:10.1093/neuonc/now002
140. Arvanitis CD, Ferraro GB, Jain RK. The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nat Rev Cancer. 2020;20(1):26–41. doi:10.1038/s41568-019-0205-x
141. Drean A, Goldwirt L, Verreault M, et al. Blood-brain barrier, cytotoxic chemotherapies and glioblastoma. Expert Rev Neurother. 2016;16(11):1285–1300. doi:10.1080/14737175.2016.1202761
142. Hatoum A, Mohammed R, Zakieh O. The unique invasiveness of glioblastoma and possible drug targets on extracellular matrix. Cancer Manag Res. 2019;11:1843–1855. doi:10.2147/CMAR.S186142
143. Domenech M, Hernandez A, Plaja A, Martinez-Balibrea E, Balana C. Hypoxia: the cornerstone of glioblastoma. Int J Mol Sci. 2021;22(22):12608. doi:10.3390/ijms222212608
144. Pardridge WM. CSF, blood-brain barrier, and brain drug delivery. Expert Opin Drug Deliv. 2016;13(7):963–975. doi:10.1517/17425247.2016.1171315
145. Bidros DS, Vogelbaum MA. Novel drug delivery strategies in neuro-oncology. Neurotherapeutics. 2009;6(3):539–546. doi:10.1016/j.nurt.2009.04.004
146. Lee D, Minko T. Nanotherapeutics for nose-to-brain drug delivery: an approach to bypass the blood brain barrier. Pharmaceutics. 2021;13(12):2049. doi:10.3390/pharmaceutics13122049
147. Vogelbaum MA, Krivosheya D, Borghei-Razavi H, et al. Phase 0 and window of opportunity clinical trial design in neuro-oncology: a RANO review. Neuro Oncol. 2020;22(11):1568–1579. doi:10.1093/neuonc/noaa149
148. Gritsch D, Gonzalez Castro LN. Relevant pharmacologic interactions in the concurrent management of brain tumor-related epilepsy and venous thromboembolism: a systematic review. J Neurooncol. 2022;157(2):285–296. doi:10.1007/s11060-022-03984-y
149. Verhaak RGW, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi:10.1016/j.ccr.2009.12.020
150. Qin A, Musket A, Musich PR, Schweitzer JB, Xie Q. Receptor tyrosine kinases as druggable targets in glioblastoma: do signaling pathways matter? Neurooncol Adv. 2021;3(1):vdab133. doi:10.1093/noajnl/vdab133
151. Balana C, Etxaniz O, Buges C, Martinez A. Approval denied by the European Medicines Agency (EMA) for bevacizumab in the treatment of high-grade glioma recurrence: a good idea or a grave error? Clin Transl Oncol. 2011;13(3):209–210. doi:10.1007/s12094-011-0642-9
152. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–247. doi:10.1016/j.ejca.2008.10.026
153. Ellingson BM, Chung C, Pope WB, Boxerman JL, Kaufmann TJ. Pseudoprogression, radionecrosis, inflammation or true tumor progression? Challenges associated with glioblastoma response assessment in an evolving therapeutic landscape. J Neurooncol. 2017;134(3):495–504. doi:10.1007/s11060-017-2375-2
154. Alentorn A, Hoang-Xuan K, Mikkelsen T. Presenting signs and symptoms in brain tumors. Handb Clin Neurol. 2016;134:19–26. doi:10.1016/b978-0-12-802997-8.00002-5
155. Ellingson BM, Bendszus M, Boxerman J, et al. Consensus recommendations for a standardized Brain Tumor Imaging Protocol in clinical trials. Neuro Oncol. 2015;17(9):1188–1198. doi:10.1093/neuonc/nov095
156. Okada H, Weller M, Huang R, et al. Immunotherapy response assessment in neuro-oncology: a report of the RANO working group. Lancet Oncol. 2015;16(15):e534–e42. doi:10.1016/S1470-2045(15)00088-1
157. Wen PY, Chang SM, Van den Bent MJ, Vogelbaum MA, Macdonald DR, Lee EQ. Response assessment in neuro-oncology clinical trials. J Clin Oncol. 2017;35(21):2439–2449. doi:10.1200/JCO.2017.72.7511
158. Bagley SJ, Kothari S, Rahman R, et al. Glioblastoma clinical trials: current landscape and opportunities for improvement. Clin Cancer Res. 2022;28(4):594–602. doi:10.1158/1078-0432.CCR-21-2750
159. Galanis E, Wu W, Cloughesy T, et al. Phase 2 trial design in neuro-oncology revisited: a report from the RANO group. Lancet Oncol. 2012;13(5):e196–204. doi:10.1016/S1470-2045(11)70406-5
160. Lee EQ, Weller M, Sul J, et al. Optimizing eligibility criteria and clinical trial conduct to enhance clinical trial participation for primary brain tumor patients. Neuro Oncol. 2020;22(5):601–612. doi:10.1093/neuonc/noaa015
161. Berzero G, Di stefano AL, Ronchi S, et al. IDH -wildtype lower-grade diffuse gliomas: the importance of histological grade and molecular assessment for prognostic stratification. Neuro Oncol. 2021;23(6):955–966. doi:10.1093/neuonc/noaa258
162. Garnier L, Charton E, Falcoz A, et al. Quality of patient-reported outcome reporting according to the CONSORT statement in randomized controlled trials with glioblastoma patients. Neuro-Oncol Pract. 2021;8(2):148–159. doi:10.1093/nop/npaa074
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.