Back to Journals » Neuropsychiatric Disease and Treatment » Volume 13
Recombinant human brain-derived neurotrophic factor prevents neuronal apoptosis in a novel in vitro model of subarachnoid hemorrhage
Authors Li M, Wang Y, Wang W, Zou C, Wang X, Chen Q
Received 22 November 2016
Accepted for publication 25 January 2017
Published 3 April 2017 Volume 2017:13 Pages 1013—1021
DOI https://doi.org/10.2147/NDT.S128442
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Wai Kwong Tang
Mingchang Li,1 Yuefei Wang,1 Wei Wang,1 Changlin Zou,1 Xin Wang,2 Qianxue Chen1
1Department of Neurosurgery, Renmin Hospital of Wuhan University, Wuhan, People’s Republic of China; 2Department of Neurosurgery, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA
Abstract: Subarachnoid hemorrhage (SAH) is a hemorrhagic stroke with high mortality and morbidity. An animal model for SAH was established by directly injecting a hemolysate into the subarachnoid space of rats or mice. However, the in vitro applications of the hemolysate SAH model have not been reported, and the mechanisms remain unclear. In this study, we established an in vitro SAH model by treating cortical pyramidal neurons with hemolysate. Using this model, we assessed the effects of recombinant human brain-derived neurotrophic factor (rhBDNF) on hemolysate-induced cell death and related mechanisms. Cortical neurons were treated with 10 ng/mL or 100 ng/mL rhBDNF prior to application of hemolysate. Hemolysate treatment markedly increased cell loss, triggered apoptosis, and promoted the expression of caspase-8, caspase-9, and cleaved caspase-3. rhBDNF significantly inhibited hemolysate-induced cell loss, neuronal apoptosis, and expression of caspase-8, caspase-9, and cleaved caspase-3. Our data revealed a previously unrecognized protective activity of rhBDNF against hemolysate-induced cell death, potentially via regulation of caspase-9-, caspase-8-, and cleaved caspase-3-related apoptosis. This study implicates that hemolysate-induced cortical neuron death represents an important in vitro model of SAH.
Keywords: recombinant human brain-derived neurotrophic factor, subarachnoid hemorrhage, neuroprotection, neuron, apoptosis
Introduction
Subarachnoid hemorrhage (SAH) is a type of stroke characterized by spontaneous bleeding into the subarachnoid space following trauma.1 SAH has a high morbidity and mortality, and approximately 25% of patients die immediately after hemorrhage.2 Early brain injury (EBI) and cerebral vasospasm are two major complications of aneurismal SAH.3,4 Cerebral vasospasm was proposed to be the primary treatment target for SAH. However, the prognosis of vasospasm remains unsatisfactory, even with proper management.5 Therefore, eliminating EBI is becoming an alternative therapy of choice for SAH. Several mechanisms are involved in the pathogenesis of EBI after SAH, including neuronal apoptosis.6 Brain injury following SAH has been well modeled by applying hemolysate directly into the subarachnoid space. The mechanisms and signaling pathways involved have been well investigated.7,8 However, an effective in vitro model for SAH is not available.
Recombinant human brain-derived neurotrophic factor (rhBDNF) is a neurotrophin that binds to the high-affinity tropomyosin-related receptor kinase B (TrkB) receptor to regulate neurodevelopmental processes, including neuronal survival, neuronal differentiation, and synaptic plasticity.9–11 Huang et al12 showed that upregulation of brain-derived neurotrophic factor (BDNF) inhibited apoptosis of hippocampal neurons in a rat model of depression. Li et al13 reported that BDNF confers neuroprotection against ischemic injury. However, the protective effects of BDNF against neuronal apoptosis during EBI after SAH have not been investigated. In this study, we developed a novel in vitro model that mimicked the clinical scenario caused by SAH. In addition, we evaluated the protective effects of BDNF against EBI after SAH.
Materials and methods
Cell culture
The Institutional Animal Care and Use Committee of Wuhan University approved this study, and the Guide for the Care and Use of Laboratory Animals were the protocols followed. Newborn C57BL/6 mice (1–3 days old) were purchased from the Animal Center of Wuhan University.
Cerebral cortex tissue was obtained from newborn C57BL/6 mice as previously described.14 Briefly, cortical tissue was pooled, minced, and gently dissociated by mechanical trituration in Ca2+- and Mg2+-free Hank’s Balanced Salt Solution and then digested in 0.025% trypsin for 20 min at 37°C. The reaction was terminated by mixing the suspension with Dulbecco’s Modified Eagle’s Medium (Gibco BRL, Grand Island, NY, USA) supplemented with 20% fetal bovine serum. After that, cells were filtered through a nylon mesh and pelleted by centrifugation. Cells were plated on 100 mm dishes (precoated with 0.01 mg/mL poly-L-lysine) at a density of 7.5×106 cells/dish in Neurobasal medium supplemented with 2% B27, 0.5 mM glutamine, 50 units/mL penicillin, and 50 μg/mL streptomycin. Cells were cultured at 37°C in a humidified incubator containing 5% CO2. Culture medium was replaced twice a week. Cell cultures were routinely observed under a phase contrast microscope.
Immunofluorescence
Cortical neurons were grown on 12 mm glass coverslips and washed with Dulbecco’s phosphate-buffered saline (DPBS, pH 7.4) before fixation with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) for 15 min. Cells were permeablized in 0.1% Triton X-100 for 5 min and washed three times with DPBS for 5 min. Nonspecific binding sites were blocked with 10% normal goat serum for 1 h at room temperature. Cortical cells were incubated in a NeuN antibody (1:100; Millipore, Temecula, CA, USA) overnight at 4°C followed by incubation with an appropriate fluorophore-conjugated secondary antibody. The nucleus was stained with 4′,6-diamidino-2-phenylindole.
Preparation of hemolysate
Hemolysate was prepared from C57BL/6 mouse arterial blood as previously described.15 Briefly, the erythrocytic fraction was removed from whole blood and lysed by freezing on dry ice for 10 min followed by complete thawing twice. Hemolysate was kept at −80°C until use.
Cell density assay
Cell viability was determined by counting the number of adherent cells as described previously.16 The cells were seeded at a density of 5×105 cells/well in 24-well plates. The cells were treated with hemolysate diluted in Neurobasal medium (1:10, 1:100, 1:200, 1:500, and 1:1,000) for 24 h. Then, cells were incubated with hemolysate (1:100) and BDNF (10 ng/mL and 100 ng/mL) for 24 h. Nonadherent cells were removed by two washes with PBS. Adherent cells were harvested by trypsinization. Cells were counted using a hemocytometer. Cell viability in experimental groups was normalized to controls.
Hoechst 33342 staining
After treatment, cells were stained by Hoechst 33342. The stained cells were washed twice with PBS and imaged using a confocal laser microscope (Leica TCS SP5, Leica Biosystems, Wetzlar, Germany) with the following parameters: excitation: 340 nm and emission: 510 nm.
Flow cytometry analysis
Apoptosis was detected by flow cytometry with fluorescin isothiocyanate-Annexin V and propidium iodide (PI) staining. Four quadrants in Annexin V/PI dot plots represented live cells (Annexin V-/PI-), early/primary apoptotic cells (Annexin V+/PI-), late/secondary apoptotic cells (Annexin V+/PI+), and necrotic cells (Annexin V-/PI+). After treatment, cells were trypsinized and centrifuged for 5 min, then incubated with Annexin V and PI for 5 min in the dark at room temperature. Fluorescent distribution was analyzed by flow cytometry (BD Immunocytometry Systems, San Jose, CA, USA).
Western blotting
After treatment, cells were lysed in lysis buffer (50 mM Tris–HCl, pH 8.0, 100 mM NaCl, 1 mM ethylenediaminetetraacetic acid, 1 mM dithiothreitol, 1% Triton X-100, 0.1% sodium dodecyl sulfate, 50 mM sodium fluoride, and 1 mM sodium vanadate). The lysate was incubated on ice for 30 min and then centrifuged at 12,000× g for 10 min at 4°C. The supernatant was collected, and the protein concentration was determined using a BCA kit (Beyotime, Ningbo, China). Equal amounts of protein were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. After blocking with 5% nonfat milk, membranes were incubated in the following primary antibodies overnight at 4°C: anti-GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anticaspase-9, anticaspase-8, and anticleaved caspase-3 (Cell Signaling Technology, Danvers, MA, USA). After three washes with PBS, membranes were labeled with specific horseradish peroxidase (HRP)-coupled secondary antibodies (antimouse IgG HRP or antirabbit IgG HRP). Protein bands were visualized by staining with a chemiluminescent substrate detection reagent. Grayscale analysis of target bands was performed using ImageJ software.
Statistical analyses
Data were analyzed by SPSS v. 13.0 (SPSS Inc., IBM, Armonk, NY, USA). The data were presented as mean ± SD for at least three independent experiments. Statistical significance was analyzed by one-way analysis of variance, and a P-value of <0.05 was considered to be statistically significant.
Results
rhBDNF promotes neuronal viability after hemolysate treatment
In this study, we established a novel in vitro model that mimics the clinical scenario caused by SAH. Cortical neuron growth is presented in Figure 1, and cortical neurons were identified by positive NeuN staining (Figure 1D). Hemolysate treatment caused obvious cell loss in a dose-dependent manner, but not until 24 h after incubation, according to the cell viability assay. After treatment with different hemolysate concentrations (1:10, 1:100, 1:200, 1:500, and 1:1,000) for 24 h, cell numbers decreased to 50.33%, 57.67%, 80.67%, 83.33%, and 86.67%, respectively. Based on these findings, we selected a hemolysate concentration of 1:100 for subsequent experiments.
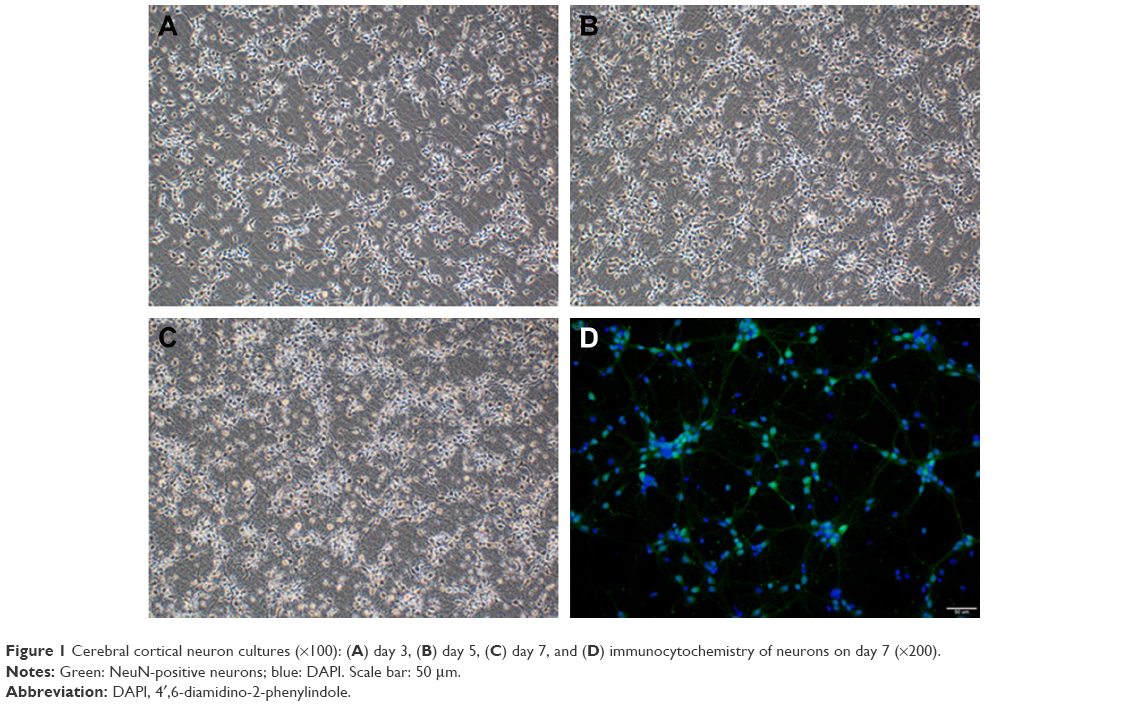 | Figure 1 Cerebral cortical neuron cultures (×100): (A) day 3, (B) day 5, (C) day 7, and (D) immunocytochemistry of neurons on day 7 (×200). |
As shown in Figure 2, 10 ng/mL rhBDNF mitigated hemolysate (1:100)-induced cell loss, but this was not significant (P>0.05). A high concentration of rhBDNF (100 ng/mL) significantly eliminated hemolysate-induced cell loss (Figure 2).
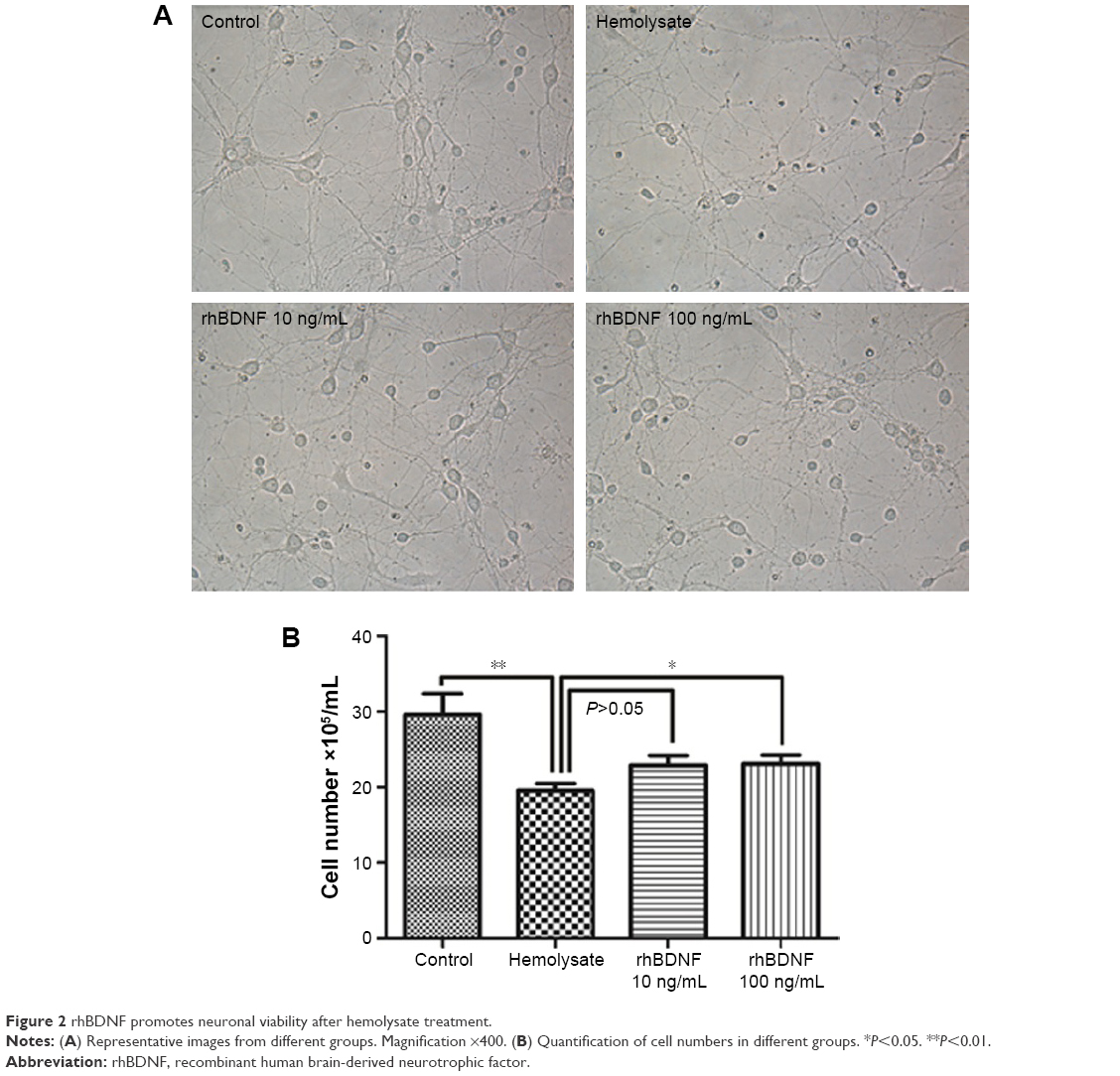 | Figure 2 rhBDNF promotes neuronal viability after hemolysate treatment. |
rhBDNF inhibits hemolysate-induced neuronal apoptosis
The effects of rhBDNF on primary cortical neuronal apoptosis induced by hemolysate were evaluated by Hoechst staining. As shown in Figure 3, cell nuclei had regular contours and were round or oval in shape in control cells. In contrast, most hemolysate-exposed cells had condensed chromatin, nuclear shrinkage, and contained apoptotic bodies. Interestingly, 10 ng/mL or 100 ng/mL rhBDNF significantly improved these hemolysate-mediated effects.
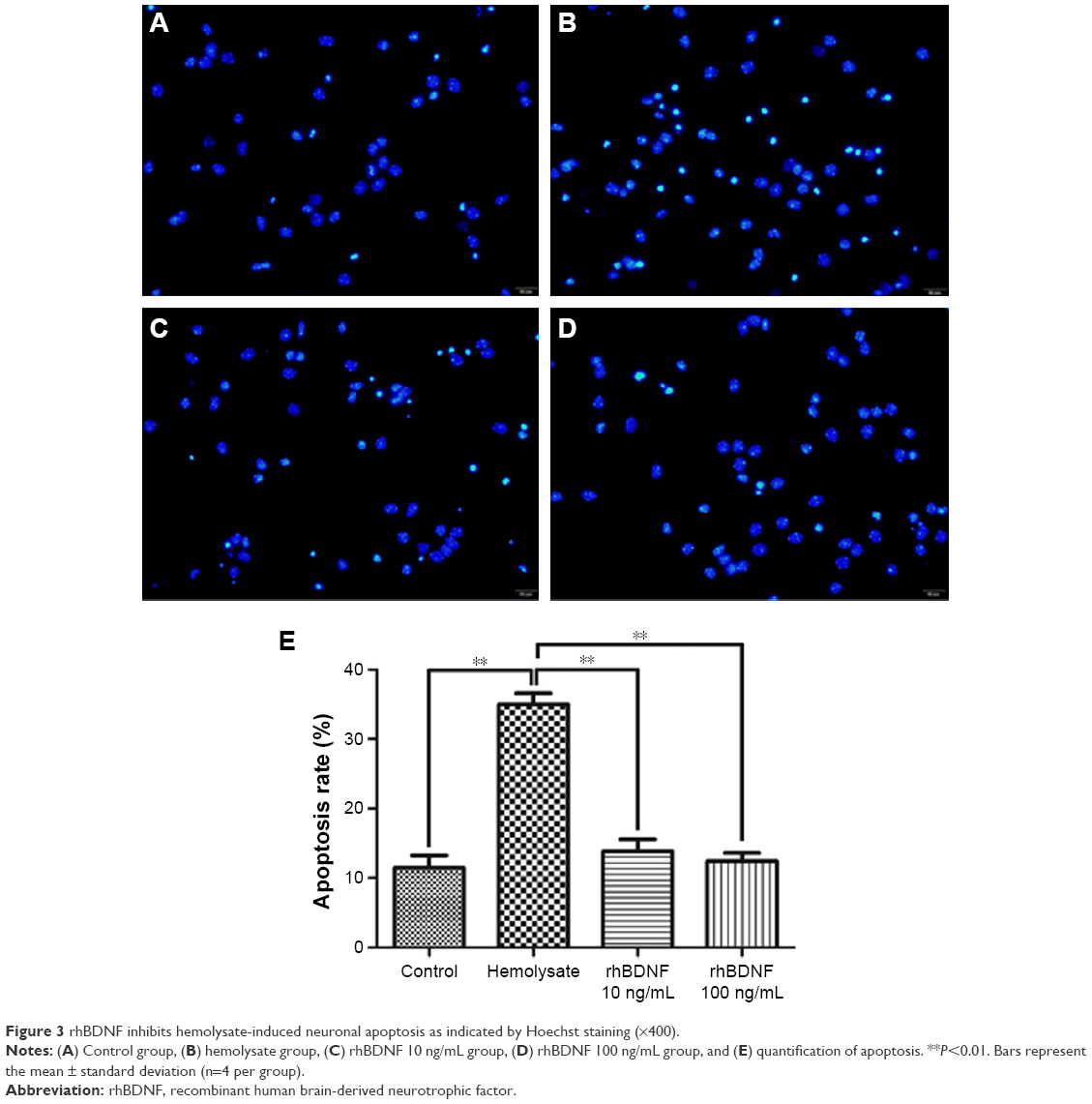 | Figure 3 rhBDNF inhibits hemolysate-induced neuronal apoptosis as indicated by Hoechst staining (×400). |
To further confirm the effects of rhBDNF on hemolysate-induced neuronal apoptosis, we performed flow cytometry. Compared with controls, exposure to hemolysates for 48 h significantly triggered apoptosis in cortical neurons (Figure 4). However, hemolysate-induced neuronal apoptosis was dramatically decreased by treatment with 10 ng/mL or 100 ng/mL rhBDNF.
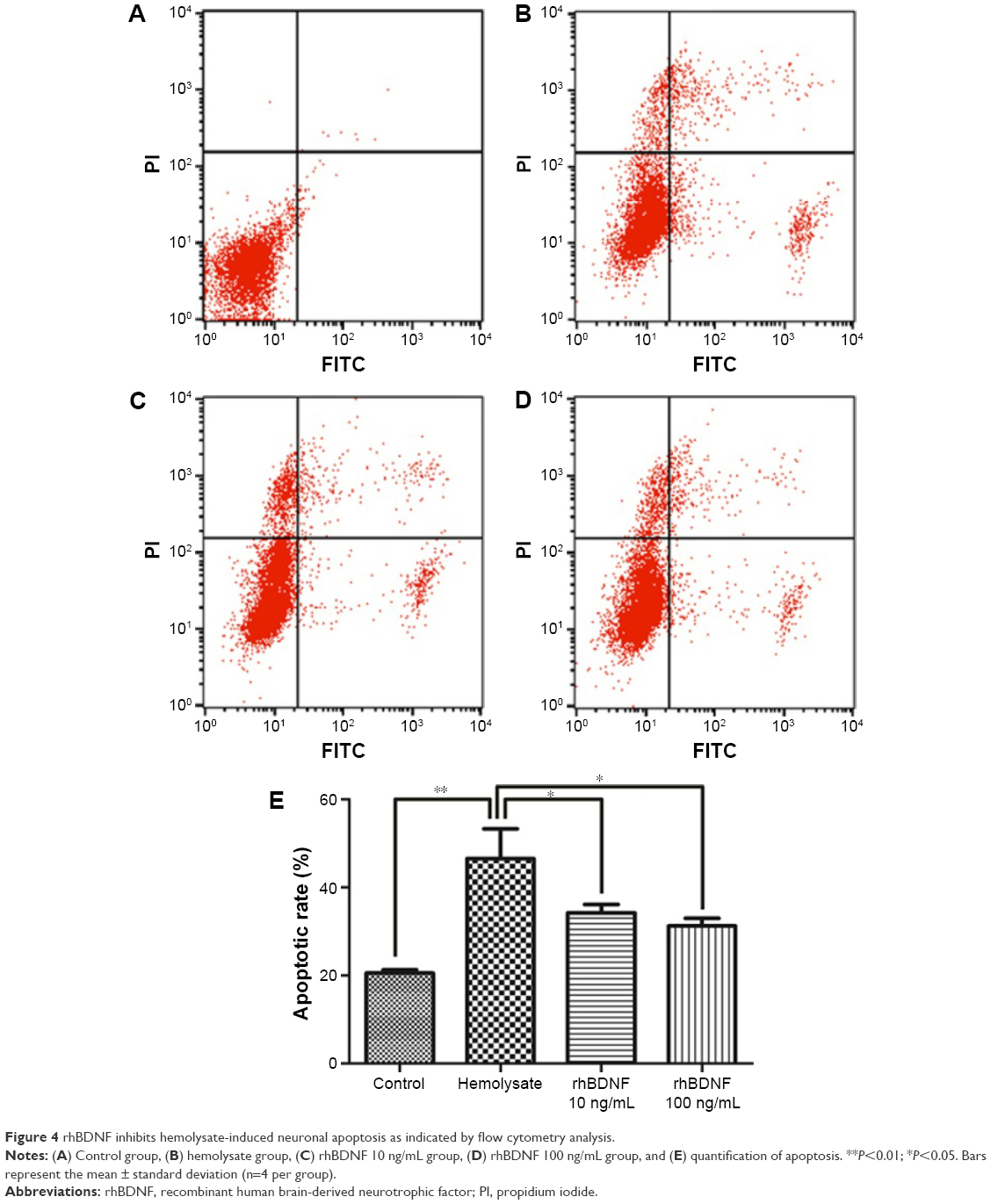 | Figure 4 rhBDNF inhibits hemolysate-induced neuronal apoptosis as indicated by flow cytometry analysis. |
rhBDNF suppresses expression of caspase-9, caspase-8, and cleaved caspase-3
Caspase signaling pathways participate in cell apoptosis and are involved in SAH.17 We measured the expression of caspase-9, caspase-8, and cleaved caspase-3 in primary cortical neurons. As shown in Figure 5, caspase-9, caspase-8, and cleaved caspase-3 were significantly upregulated after hemolysate treatment. In contrast, 10 ng/mL and 100 ng/mL rhBDNF significantly decreased the expression of caspase-8, caspase-9, and cleaved caspase-3 in hemolysate-treated cortical neurons (vs hemolysate alone, P<0.05).
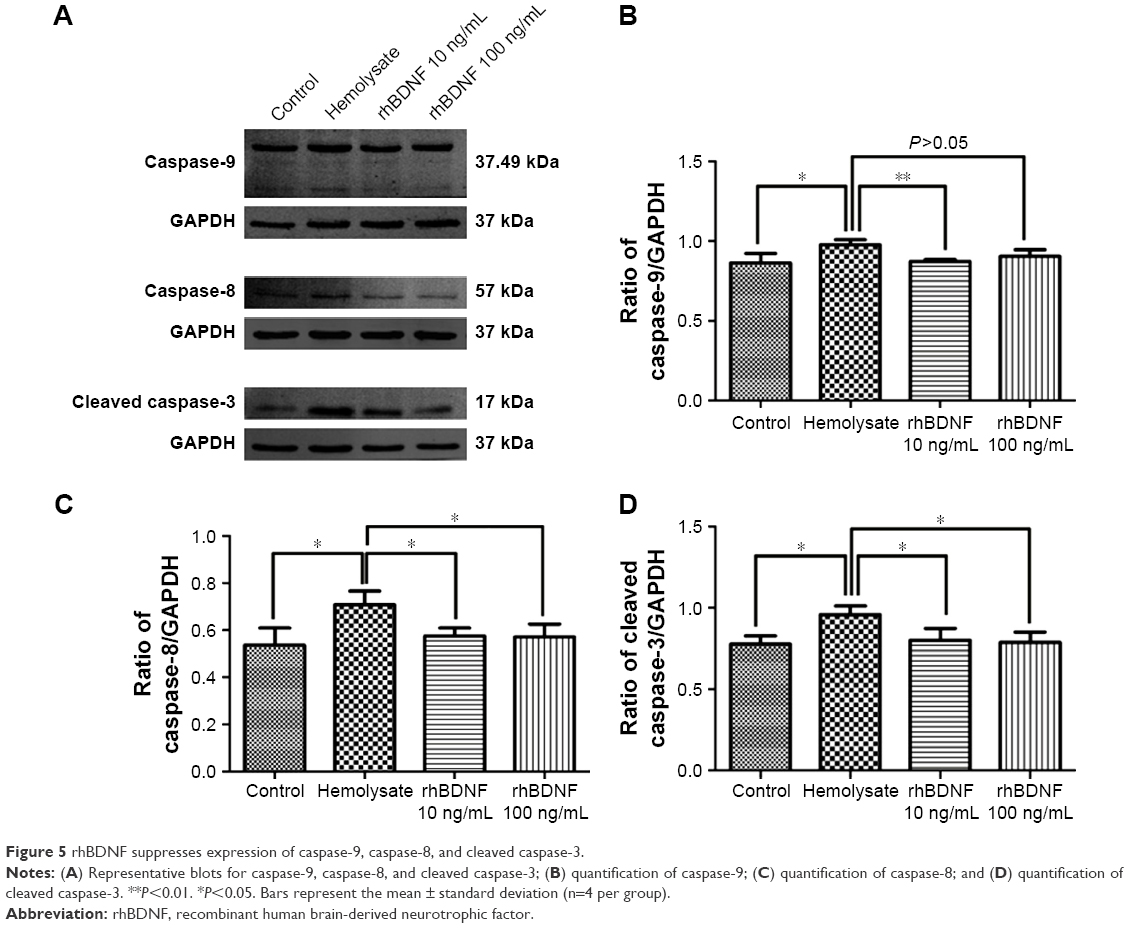 | Figure 5 rhBDNF suppresses expression of caspase-9, caspase-8, and cleaved caspase-3. |
Discussion
Hemolysates have been widely used to generate animal models of SAH.18,19 In the present study, we demonstrated that hemolysate can also be used for in vitro cortical neuron models of SAH. Moreover, we revealed that rhBDNF prevents hemolysate-induced apoptosis in cortical neurons through caspase pathways.
Three in vivo models have been used for SAH research: endovascular puncture, blood injection into the cisterna magna, and cross forebay.20 However, in vitro SAH models, such as stimulating glial cells with hemoglobin (Hb)6,21 or mimicking SAH with cytochrome oxidase,22 are still under investigation. In this present study, we established an in vitro model of SAH by application of hemolysate, which sufficiently simulated the clinical pathophysiological processes of SAH observed in vivo.18,19 Hemolysate contains multiple components that affect cell survival. For example, Hb and ATP influence vascular cells and tissues in vitro.23 A combination of Hb and a low-molecular-weight (<2 kDa) hemolysate fraction caused contraction of cerebral arteries. However, these components were also associated with neuronal apoptosis during EBI after SAH.24 Nevertheless, more factors that elicit apoptosis should be identified in the future. Our findings demonstrated that hemolysate treatment causes cortical neuron detachment in a dose-dependent manner. We also found that different concentrations of rhBDNF prevent hemolysate-induced cortical cell loss. Using this in vitro model, we confirmed the neuroprotective effects of BDNF. Protective effects of rhBDNF against SAH have not been reported, but BDNF is involved in the onset of SAH. BDNF polymorphisms predict a poor outcome in aneurysmal SAH patients.25 Moreover, BDNF was activated after cysteamine or N-acetyl serotonin derivative treatment in models of SAH.25,26 These data implicate the validity of our in vitro model, as well as the potential therapeutic effect of BDNF in SAH.
To identify the underlying mechanisms of rhBDNF-mediated protection against hemolysate-induced cortical neuron loss, we examined apoptosis. We showed that hemolysate treatment elicited apoptosis in cortical neurons. Takeda et al27 reported that BDNF protects human umbilical vein endothelial cells from tumor necrosis factor-α-induced apoptosis, and Xia et al28 showed that BDNF inhibits apoptosis in rats following spinal cord hemisection. In agreement with these findings, we found that rhBDNF mitigated hemolysate-induced apoptosis in cortical neurons. rhBDNF reduced apoptosis at lower concentrations and prevented cell detachment at higher concentrations. Taken together, these results revealed that rhBDNF protects against neuronal apoptosis in a concentration-independent manner.
Caspases are closely related to the apoptosis of eukaryotic cells.29 A total of 14 caspase family members have been found and can be divided into two categories. One category activates other caspases, and this category includes caspase-1, -2, -4, -5, -8, -9, and -10. The second category executes apoptosis and includes caspase-3, -6, -7, and -14.30 The apoptosis inducers stimulate apoptosis through membrane death receptors and mitochondrial pathways (Figure 6).31 Caspase-8 expression is activated by death receptors when apoptosis signaling is induced. Caspase-9 facilitates mitochondria-related apoptosis after release of cytochrome C. Caspase-8 and caspase-9 activate caspase-3, which executes apoptosis. These caspases are representative components of apoptosis, so we examined their expression in our study. We observed upregulated expression of caspase-8, caspase-9, and cleaved caspase-3 in our in vitro model of SAH. These findings suggest that both death receptor and mitochondria-related pathways are involved in hemolysate-induced apoptosis of cortical neurons. As previously discussed, hemolysate contains multiple components, which elicit vascular contraction, or activate cell death receptors and mitochondria-related apoptosis pathways. In addition, we showed that BDNF prevented neuronal apoptosis by inhibiting caspase-3.32,33
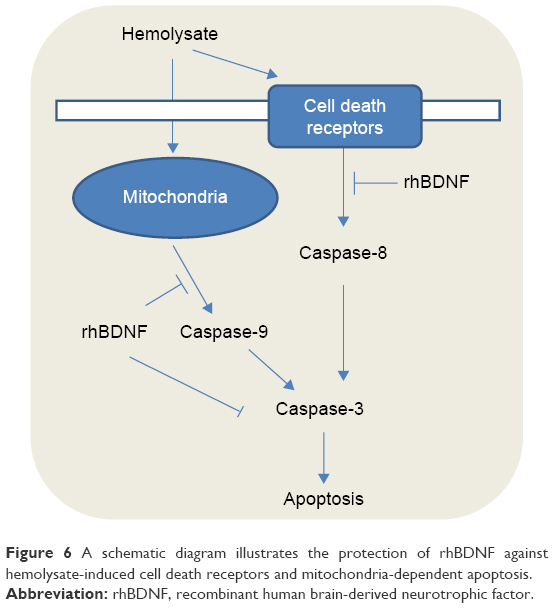 | Figure 6 A schematic diagram illustrates the protection of rhBDNF against hemolysate-induced cell death receptors and mitochondria-dependent apoptosis. |
Conclusion
We established a novel in vitro model for SAH and showed that rhBDNF inhibits neuronal apoptosis after SAH, which may represent an underlying mechanism for its neuroprotective effect against SAH. BDNF-mediated neuroprotection may be related to the activation of caspase-9, caspase-8, and cleaved caspase-3.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (81171112 and 81371272 to ML and 81372683 to QC).
Disclosure
The authors report no conflicts of interest in this work.
References
Chowdhury T, Dash HH, Cappellani RB, Daya J. Early brain injury and subarachnoid hemorrhage: where are we at present? Saudi J Anaesth. 2013;7(2):187–190. | ||
Macdonald RL, Higashida RT, Keller E, et al. Preventing vasospasm improves outcome after aneurysmal subarachnoid hemorrhage: rationale and design of CONSCIOUS-2 and CONSCIOUS-3 trials. Neurocrit Care. 2010;13(3):416–424. | ||
Hong Y, Shao A, Wang J, et al. Neuroprotective effect of hydrogen-rich saline against neurologic damage and apoptosis in early brain injury following subarachnoid hemorrhage: possible role of the Akt/GSK3beta signaling pathway. PLoS One. 2014;9(4):e96212. | ||
Zhao D, Liu Q, Ji Y, et al. Correlation between nitric oxide and early brain injury after subarachnoid hemorrhage. Int J Neurosci. 2015; 125(7):531–539. | ||
Macdonald RL, Kassell NF, Mayer S, et al; CONSCIOUS-1 Investigators. Clazosentan to overcome neurological ischemia and infarction occurring after subarachnoid hemorrhage (CONSCIOUS-1): randomized, double-blind, placebo-controlled phase 2 dose-finding trial. Stroke. 2008;39(11):3015–3021. | ||
Sun Q, Wu W, Hu YC, et al. Early release of high-mobility group box 1 (HMGB1) from neurons in experimental subarachnoid hemorrhage in vivo and in vitro. J Neuroinflammation. 2014;11:106. | ||
Zhou C, Yamaguchi M, Colohan AR, Zhang JH. Role of p53 and apoptosis in cerebral vasospasm after experimental subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2005;25(5):572–582. | ||
Zhang ZY, Sun BL, Yang MF, Li DW, Fang J, Zhang S. Carnosine attenuates early brain injury through its antioxidative and anti-apoptotic effects in a rat experimental subarachnoid hemorrhage model. Cell Mol Neurobiol. 2015;35(2):147–157. | ||
Helgager J, Huang YZ, McNamara JO. Brain-derived neurotrophic factor but not vesicular zinc promotes TrkB activation within mossy fibers of mouse hippocampus in vivo. J Comp Neurol. 2014;522(17):3885–3899. | ||
Zhu G, Li J, He L, Wang X, Hong X. MPTP-induced changes in hippocampal synaptic plasticity and memory are prevented by memantine through the BDNF-TrkB pathway. Br J Pharmacol. 2015;172(9):2354–2368. | ||
Zhu G, Liu Y, Wang Y, Bi X, Baudry M. Different patterns of electrical activity lead to long-term potentiation by activating different intracellular pathways. J Neurosci. 2015;35(2):621–633. | ||
Huang X, Mao YS, Li C, Wang H, Ji JL. Venlafaxine inhibits apoptosis of hippocampal neurons by up-regulating brain-derived neurotrophic factor in a rat depression model. Int J Clin Exp Pathol. 2014;7(8):4577–4586. | ||
Li ST, Pan J, Hua XM, et al. Endothelial nitric oxide synthase protects neurons against ischemic injury through regulation of brain-derived neurotrophic factor expression. CNS Neurosci Ther. 2014;20(2):154–164. | ||
Yang L, Yang ZM, Zhang N, Tian Z, Liu SB, Zhao MG. Neuroprotective effects of vitexin by inhibition of NMDA receptors in primary cultures of mouse cerebral cortical neurons. Mol Cell Biochem. 2014;386(1–2):251–258. | ||
Matz PG, Fujimura M, Chan PH. Subarachnoid hemolysate produces DNA fragmentation in a pattern similar to apoptosis in mouse brain. Brain Res. 2000;858(2):312–319. | ||
Abbasi N, Soudi S, Hayati-Roodbari N, Dodel M, Soleimani M. The effects of plasma treated electrospun nanofibrous poly (epsilon-caprolactone) scaffolds with different orientations on mouse embryonic stem cell proliferation. Cell J. 2014;16(3):245–254. | ||
Friedrich V, Flores R, Sehba FA. Cell death starts early after subarachnoid hemorrhage. Neurosci Lett. 2012;512(1):6–11. | ||
Echigo R, Shimohata N, Karatsu K, et al. Trehalose treatment suppresses inflammation, oxidative stress, and vasospasm induced by experimental subarachnoid hemorrhage. J Transl Med. 2012;10:80. | ||
Zhou ML, Shi JX, Hang CH, et al. Potential contribution of nuclear factor-kappaB to cerebral vasospasm after experimental subarachnoid hemorrhage in rabbits. J Cereb Blood Flow Metab. 2007;27(9):1583–1592. | ||
Duris K, Manaenko A, Suzuki H, Rolland W, Tang J, Zhang JH. Sampling of CSF via the Cisterna Magna and Blood Collection via the Heart Affects Brain Water Content in a Rat SAH Model. Transl Stroke Res. 2011;2(2):232–237. | ||
You WC, Li W, Zhuang Z, et al. Biphasic activation of nuclear factor-kappa B in experimental models of subarachnoid hemorrhage in vivo and in vitro. Mediators Inflamm. 2012;2012:786242. | ||
Loftspring MC, Wurster WL, Pyne-Geithman GJ, Clark JF. An in vitro model of aneurysmal subarachnoid hemorrhage: oxidation of unconjugated bilirubin by cytochrome oxidase. J Neurochem. 2007;102(6):1990–1995. | ||
Zhang H, Weir B, Marton LS, et al. Mechanisms of hemolysate-induced [Ca2+]i elevation in cerebral smooth muscle cells. Am J Physiol. 1995;269(6 pt 2):H1874–H1890. | ||
Hasegawa Y, Suzuki H, Sozen T, Altay O, Zhang JH. Apoptotic mechanisms for neuronal cells in early brain injury after subarachnoid hemorrhage. Acta Neurochir Suppl. 2011;110(pt 1):43–48. | ||
Siironen J, Juvela S, Kanarek K, Vilkki J, Hernesniemi J, Lappalainen J. The Met allele of the BDNF Val66Met polymorphism predicts poor outcome among survivors of aneurysmal subarachnoid hemorrhage. Stroke. 2007;38(10):2858–2860. | ||
Li T, Liu H, Xue H, et al. Neuroprotective effects of hydrogen sulfide against early brain injury and secondary cognitive deficits following subarachnoid hemorrhage. Brain Pathol. 2017;27(1):51–63. | ||
Takeda K, Kermani P, Anastasia A, Obinata Y, Hempstead BL, Kurihara H. BDNF protects human vascular endothelial cells from TNFalpha-induced apoptosis. Biochem Cell Biol. 2013;91(5):341–349. | ||
Xia GN, Zou Y, Wang YC, et al. Neural stem cells grafts decrease neural apoptosis associated with caspase-7 downregulation and BDNF upregulation in rats following spinal cord hemisection. Cell Mol Neurobiol. 2013;33(7):1013–1022. | ||
Nakagawa A, Sullivan KD, Xue D. Caspase-activated phosphoinositide binding by CNT-1 promotes apoptosis by inhibiting the AKT pathway. Nat Struct Mol Biol. 2014;21(12):1082–1090. | ||
Cheng B, Zhang Y, Wang A, Dong Y, Xie Z. Vitamin C attenuates isoflurane-induced caspase-3 activation and cognitive impairment. Mol Neurobiol. 2015;52(3):1580–1589. | ||
Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. | ||
Kim DH, Zhao X. BDNF protects neurons following injury by modulation of caspase activity. Neurocrit Care. 2005;3(1):71–76. | ||
Han BH, D’Costa A, Back SA, et al. BDNF blocks caspase-3 activation in neonatal hypoxia-ischemia. Neurobiol Dis. 2000;7(1):38–53. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.