Back to Journals » International Medical Case Reports Journal » Volume 19
Recessive Dystrophic Epidermolysis Bullosa in a Resource-Limited Setting: A Case Report From Mohamed Adam Sheikh Children’s Teaching Hospital Hargeisa, Somaliland
Authors Aw Hashi MO, Ahmed SI ![]() , Derie AM
, Derie AM ![]() , Abdirahman RM, Abdullahi RR
, Abdirahman RM, Abdullahi RR
Received 31 January 2026
Accepted for publication 21 April 2026
Published 23 April 2026 Volume 2026:19 600282
DOI https://doi.org/10.2147/IMCRJ.S600282
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Thomas E Hutson
Mohamed Osman Aw Hashi,1,2 Sadam Ismail Ahmed,3 Ahmed M Derie,4 Radia Mohamed Abdirahman,5 Riham Rashid Abdullahi5
1Department of Medicine and Surgery, University of Hargeisa, Hargeisa, Somaliland; 2Department of Dermatology and Venereology, Hargeisa Group Hospital, Hargeisa, Somaliland; 3Department of Academic and Research, Hargeisa Group Hospital, Hargeisa, Somaliland; 4Department of Medicine, Gargaar Multispecialty and Teaching Hospital, Hargeisa, Somaliland; 5Department of Emergency, Hargeisa Group Hospital, Hargeisa, Somaliland
Correspondence: Sadam Ismail Ahmed, Email [email protected]
Introduction: Epidermolysis Bullosa (EB) is a rare, genetically determined mechanobullous disorder characterized by profound skin and mucosal fragility. The management of EB is complex, multidisciplinary, and palliative, posing extreme challenges in resource-limited settings where diagnostic capabilities and specialized care are scarce. This report highlights the utility of clinical acumen and the devastating impact of socioeconomic barriers on disease progression.
Case Presentation: A 4-year-old male from rural Somaliland presented to a tertiary children’s hospital with a lifelong history of trauma-induced blistering pseudohypertrophic scarring, and digital contractures. Born to consanguineous parents, who presented with a lifelong history of trauma-induced fragile blisters, erosions, and crustations affecting his limbs, abdomen, and oral mucosa. Examination revealed flaccid bullae, erosions, hemorrhagic crusts, secondary bacterial infection, and disabling finger contractures. A sibling had died in infancy with similar symptoms. A clinical diagnosis of severe, recessive dystrophic EB was made. Diagnostic confirmation via skin biopsy was declined by the caregivers. Management with oral antibiotics, topical wound care, and education was initiated, however, the patient was lost to follow-up due to extreme poverty and geographical isolation. The patient was lost to follow-up after discharge due to socioeconomic barriers and a return to a remote village, highlighting the fragility of care continuity.
Conclusion: This case epitomizes the profound challenges of managing a severe genodermatosis within a fragile health system. It underscores the critical reliance on clinical acumen in the absence of diagnostic tools and illustrates how poverty, social disruption, and geographical barriers converge to exacerbate morbidity and preclude long-term management. The report calls for context-specific strategies, including training in basic EB wound care, development of low-cost care protocols, and enhanced support systems for patients and caregivers in underserved regions.
Keywords: epidermolysis bullosa, consanguinity, resource-limited setting, Somaliland, diagnostic challenge, wound care, global health dermatology
Introduction
Epidermolysis Bullosa (EB) encompasses a group of rare, genetically determined mechanobullous disorders caused by mutations in genes encoding structural proteins at the dermal-epidermal junction. The hallmark is extreme skin and mucosal fragility, leading to blistering and erosion formation following minor mechanical trauma.1 EB is classified into four major types—simplex (EBS), junctional (JEB), dystrophic (DEB), and Kindler EB syndrome—based on the ultrastructural level of tissue cleavage within the skin.2 Among these, the recessive dystrophic subtype (RDEB), often resulting from mutations in the COL7A1 gene encoding type VII collagen, is frequently severe. It is characterized by mutilating scarring, pseudosyndactyly, joint contractures, and a significantly elevated lifelong risk of developing aggressive squamous cell carcinoma.3,4
Diagnostic confirmation and precise subtyping traditionally require specialized techniques such as immunofluorescence antigen mapping (IFM) or transmission electron microscopy (TEM) of a skin biopsy, often supplemented by genetic sequencing.5 Management is inherently complex, multidisciplinary, and palliative, focusing on meticulous wound care, infection prevention, nutritional support, and surgical intervention for complications.6 The lifelong and debilitating nature of severe EB places an immense psychosocial and economic burden on patients and their families.7 This case is unique as it illustrates the intersection of a severe genodermatosis with extreme social disruption, including parental abandonment and total lack of specialized supplies. While diagnostic confirmation traditionally requires immunofluorescence mapping (IFM) or transmission electron microscopy (TEM), recent literature suggests that in resource-constrained settings, clinical matrix tools, such as those proposed by Yenamandra et al, can provide a structured pathway for diagnosis when advanced modalities are unavailable.
This burden is critically amplified in resource-limited settings, where access to diagnostic modalities, specialized dressings, and multidisciplinary care is severely constrained. This case report describes the presentation and profound management challenges of a child EB in Somaliland. It highlights the reliance on clinical acumen in the absence of confirmatory testing and underscores the devastating intersection of a severe genodermatosis with poverty, social disruption, and healthcare infrastructure limitations.
Case Presentation
A 4-year-old male presented to the Dermatology Department of Mohamed Adam Sheikh Children’s Teaching Hospital, referred from a private facility. He had a 7-day history of worsening fragile blisters with foul-smelling oozing, pus, and crustations, causing such pain that he could not move his hands or legs. On assessment, the child’s pain score was 8/10 on the FLACC scale, preventing him from moving his extremities. His nutritional status was poor, with signs of protein-energy malnutrition and a weight-for-age Z-score below −2. Dental examination revealed multiple carious teeth and limited mouth opening (microstomia).
The caregiver (his aunt) reported that skin lesions began at birth with small, fragile blisters on the lower legs and soles, later involving the abdomen and oral mucosa. The blisters typically progressed to bullae, ruptured easily, and healed with erosions, crusts, and post-inflammatory hypopigmentation without scarring, often precipitated by minor trauma or scratching.
Past Medical and Family History
The patient had no history of surgeries or blood transfusions and was on no regular medications. He was born to consanguineous parents, who are now divorced. Notably, a sibling died at one month of age with similar blistering symptoms. The patient had been abandoned by his parents and was under the care of his aunt in a rural area outside Burao city.
Physical Examination
On admission, the child was anxious but alert. Dermatological examination revealed multiple flaccid blisters and bullae with hemorrhagic crust over right thighs and right antecubital area (Figure 1). Large ulcerated and crusted purulent plaques were present on the dorsal hands (with finger contractures) and dorsal feet bilaterally (Figure 2). The abdomen showed areas of post-inflammatory hypopigmentation (Figure 3). Systemic examination was unremarkable.
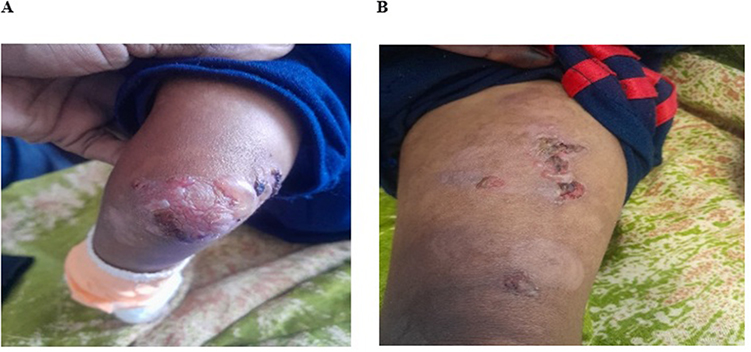 |
Figure 1 Day1, Bullae with crust on the right knee and left antecubital area. (A) shows large bullae with hemorrhagic crust over left knee. (B) shows several erosions with crust and few blisters over left thigh. |
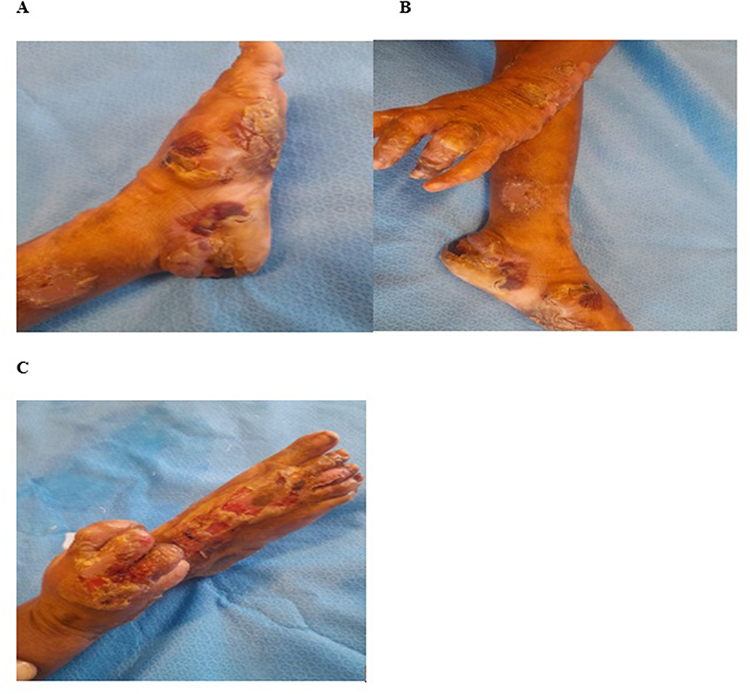 |
Figure 2 Day 1. Large crusted plaques and ulcers on the bilateral dorsal feet and dorsal hands. (A) shows large crusted plaque with bullae over left sole and dorsal feet with scarred area over left inner lower leg. (B) shows ruptured bullae and crustations over left dorsal fingers with contractures and crusted plaque over left sole and scarred area over left lower leg. (C) shows large ulcers with yellowish purulent crust and contractures over right dorsal hands, fingers and foot. |
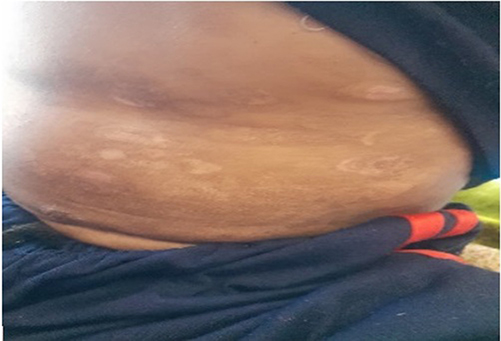 |
Figure 3 Post-inflammatory hypopigmentation on the abdomen. |
Investigations and Diagnosis
Based on the classic history of trauma-induced blistering since birth, consanguineous parentage, similar fatal history in a sibling, and clinical findings of contractures mutilating scarring and contractures, a clinical diagnosis of severe, recessive dystrophic epidermolysis bullosa was made. Recommended investigations—complete blood count, renal function tests, C-reactive protein were normal and a confirmatory skin biopsy—were declined by the caregiver due to fear and financial constraints.
Management and Outcome
Management focused on treating secondary bacterial infection and wound care. The patient was started on oral antibiotics (amoxicillin-clavulanate) Pain was managed with scheduled paracetamol and ibuprofen, as opioids were unavailable and daily local wound care with topical antibiotic ointment (fusidic acid), non-adherent Vaseline-impregnated gauze, and mometasone furoate 0.1% cream for inflammatory borders. Nutritional supplementation with high-protein local foods was initiated. On day 3 follow up, Additionally new blisters and bullae with erosions and hemorrhagic crustations were noted on the bilateral lower legs, forearms, palms and soles (Figure 4) and there is local wound care dressing on both upper and lower extremities (Figure 5). He showed initial improvement and was discharged on the third day with detailed wound care instructions, a supply of dressings, and an appointment for regular follow-up.
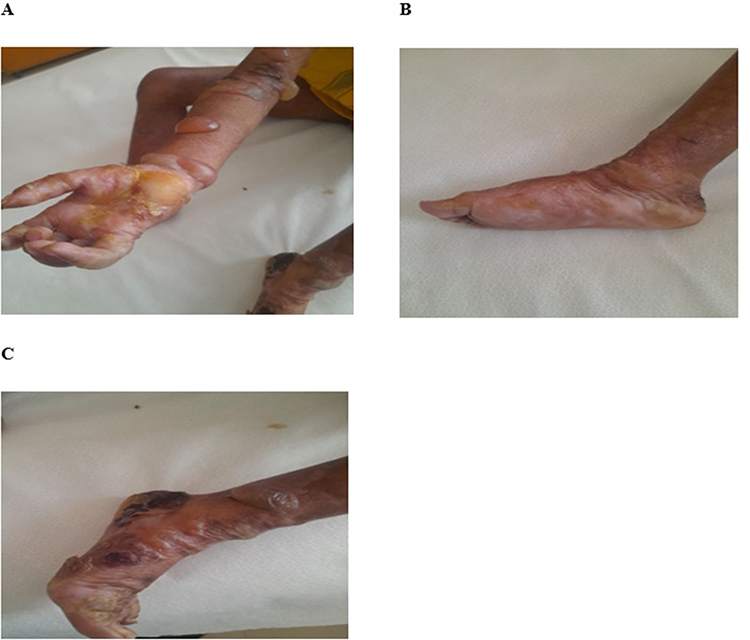 |
Figure 4 On Day 3, Bullae on forearms and lower legs with hemorrhagic and yellowish crustations on palms and soles. (A) shows ruptured bullae with yellowish crust over right palm and intact bullae over right forearm. (B) shows intact bullae over left inner and lateral sole. (C) shows large hemorrhagic crust with several blisters and contractures over left foot and lower leg. |
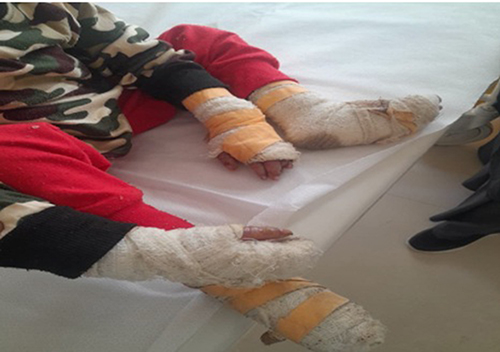 |
Figure 5 On Day 3, there is a local wound dressing around upper and lower extremities. |
However, the patient was lost to follow-up as the family returned to their remote village. A subsequent phone call to the aunt revealed that while some lesions were improving, new blisters continued to appear on knees and elbows. She was unable to provide photographic follow-up due to technological limitations.
Discussion
Epidermolysis Bullosa (EB) is a rare, heterogeneous group of inherited mechano-bullous disorders characterized by skin and mucosal fragility, leading to blistering and erosions following minor trauma. The management of EB is complex, multidisciplinary, and lifelong, posing a significant challenge even in well-resourced healthcare systems. This literature review contextualizes a case report of a 4-year-old male with suspected Epidermolysis Bullosa Congenita in Hargeisa, Somaliland, by examining key themes in the global literature: diagnostic challenges, the impact of consanguinity, multidisciplinary care deficits, and the profound socioeconomic and psychosocial burdens exacerbated in resource-limited settings.
Diagnostic Challenges and Classification
The diagnosis of EB is historically clinicopathological, relying on detailed history, clinical examination, and confirmatory tests like skin biopsy for immunofluorescence mapping (IFM) or transmission electron microscopy (TEM). Genetic testing is the gold standard for precise subtyping. The presented case highlights a classic history of trauma-induced blistering since birth, with milia, scarring, and contractures, strongly suggesting dystrophic EB. However, the inability to perform biopsy or genetic testing due to patient refusal and likely system limitations is a critical barrier documented extensively in low-resource settings. Literature emphasizes that in such contexts, diagnosis often remains clinical, based on phenotypic classification (Fine et al, 2014). This can lead to prognostic uncertainty and generic management plans. The presence of consanguinity and a similarly affected deceased sibling strongly supports an autosomal recessive inheritance pattern, common in communities with high rates of consanguineous marriage.
Consanguinity and Genetic Burden
Parental consanguinity is a major risk factor for autosomal recessive forms of EB, particularly dystrophic EB (DEB) and junctional EB (JEB). Studies from regions with high rates of consanguineous marriage, such as the Middle East, North Africa, and parts of South Asia, report a higher prevalence of severe, recessive EB subtypes (Has et al, 2020). The case report’s detail of consanguineous parents and a prior neonatal death from similar symptoms is a tragic but classic epidemiological narrative in medical genetics. This underscores the urgent need for community genetic education and accessible genetic counseling services in these regions to inform family planning decisions—a need that remains largely unmet in most resource-limited settings.
Multidisciplinary Care: The Ideal vs The Reality
International guidelines stress that EB management requires a dedicated multidisciplinary team (MDT) including dermatologists, wound care specialists, nutritionists, physiotherapists, occupational therapists, and psychologists (El Hachem et al, 2018). Core principles involve meticulous wound care with non-adherent dressings, infection prevention, nutritional support (due to high catabolic demand and oral involvement), and proactive management of complications like contractures and squamous cell carcinoma.The Hargeisa case illustrates the vast gap between this ideal and reality. Management was necessarily simplified to oral antibiotics, topical agents, and basic wound care with Vaseline gauze—a pragmatic but suboptimal approach. The absence of specialized dressings (eg, silicone-based), pain management plans, nutritional assessment, and physiotherapy for contractures reflects standard constraints. The failure to follow up, common in such settings, breaks the continuity of care, leading to unchecked disease progression, chronic wounds, and severe disability.
Wound Care, Infection, and Pain: A Triad of Suffering
Chronic wounds are the hallmark of severe EB, serving as portals for infection, sources of chronic pain, and causes of scarring. Infection (as seen with pus and odor in the case) is a leading cause of morbidity and mortality. Staphylococcus aureus and Pseudomonas aeruginosa are common colonizers. In settings with limited access to culture-directed antibiotics, empirical treatment is the norm but can drive resistance.
Pain in EB is severe and under-treated globally, but critically so in low-resource contexts. The child’s inability to move due to pain is a devastating but common consequence. Literature shows pain in EB is both nociceptive (from wounds) and neuropathic, requiring a multi-modal approach rarely available in settings like Somaliland (Goldschneider & Good, 2014).
The Profound Psychosocial and Socioeconomic Impact
EB places an enormous burden on patients and families. The reported family dissolution, child abandonment to an aunt, and rural residence are extreme but not isolated outcomes. Caregiver burnout, social stigma, marital strain, and financial ruin are widely reported (Dures et al, 2011). In rural, resource-poor areas, the cost of dressings, travel to tertiary centers, and loss of income can be catastrophic. The child’s eventual loss to follow-up is a direct result of these compounded socioeconomic pressures. Furthermore, the lack of patient advocacy groups or social support systems in most limited-resource regions leaves families isolated and unsupported.
The Somaliland Context and Implications for Global Health
This case is a stark example of the plight of patients with rare, chronic diseases in fragile health systems. Somaliland, recovering from conflict, faces severe healthcare workforce and infrastructure shortages. Specialized services for rare diseases are virtually non-existent. The case report thus transcends a single diagnosis; it highlights systemic issues: the lack of diagnostic facilities, the absence of an MDT, medication and dressing shortages, and the insurmountable barriers to follow-up for rural populations. Global health literature on EB in Africa is sparse, indicating a large hidden burden. This case argues for context-specific interventions: training frontline health workers in basic EB wound care and diagnosis, creating low-cost dressing protocols, developing telemedicine links for remote consultation (though limited by digital access, as seen when the aunt could not send photos), and integrating palliative care principles to address pain and quality of life.
Conclusion
The case of this 4-year-old boy in Hargeisa epitomizes the immense challenges of managing severe Epidermolysis Bullosa Congenita in a resource-limited setting.
The management of this 4-year-old boy highlights several critical takeaways for global health dermatology:
- Clinical Acumen: In the absence of genetic testing, phenotypic markers like scarring and contractures are sufficient for a working diagnosis of RDEB.
- Multidisciplinary Necessity: Pain control and nutritional support are as vital as wound care but are often neglected in resource-poor settings.
- Actionable Strategy: There is an urgent need to train frontline workers in Somaliland on “EB-friendly” handling and low-cost dressing techniques.
- Socioeconomic Support: Medical care is futile if geographical and financial barriers prevent follow-up; telemedicine or community-based nursing could bridge this gap.
It mirrors global literature on the clinical course of EB while exposing the acute vulnerabilities created by diagnostic limitations, consanguinity, poverty, and fragile health systems. Effective care requires moving beyond a purely biomedical model to address the genetic, socioeconomic, and psychosocial dimensions of the disease. This case serves as a compelling call for focused research, context-adapted guideline development, and international collaboration to build capacity for rare skin diseases in the world’s most underserved regions.
Ethical Approval
In our institute, the ethical approval is not required for publication of case reports, so our hospital is waived for case reports.
Consent for Publication
Written informed consent was obtained from the patient’s Parent (Primary care giver) for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
Informed Consent
Informed consent was obtained from the patient’s Parent (Primary cgiver) for publication of this case report.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
No funding for this case.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Fine JD, Bruckner-Tuderman L, Eady RAJ, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol. 2014;70(6):1103–8. doi:10.1016/j.jaad.2014.01.903
2. Has C, Bauer JW, Bodemer C, et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183(4):614–627. doi:10.1111/bjd.18921
3. Bruckner-Tuderman L, McGrath JA, Robinson EC, Uitto J. Progress in epidermolysis bullosa research: toward treatment and cure. J Invest Dermatol. 2010;130(7):1778–1784. doi:10.1038/jid.2010.90
4. Yenamandra VK, Sreenivas V, Moss C, et al. Development and validation of a clinical diagnostic matrix for epidermolysis bullosa in resource-limited settings. Orphanet J Rare Dis. 2017;12(1):190.
5. Pope E, Lara-Corrales I, Mellerio J, et al. A consensus approach to wound care in epidermolysis bullosa. J Am Acad Dermatol. 2012;67(5):904–917. doi:10.1016/j.jaad.2012.01.016
6. Koshhal S, Al-Qahtani A, Al-Otaibi S. The burden of epidermolysis bullosa in the Middle East: a systematic review. Int J Dermatol. 2022;61(4):410–418.
7. Goldschneider KR, Good J. Pain management in epidermolysis bullosa. Dermatol Clin. 2014;32(2):293–301.
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Severe Mitral Stenosis in Term Pregnancy Presenting During Labor in a Resource-Limited Setting: A Case Report from Borama, Awdal Region of Somalia

Jama AM, Osman AA, Deria HM, Mousa MM, Jama AM, Ali AO
International Medical Case Reports Journal 2026, 19:564292
Published Date: 26 February 2026
Primary High-Grade Intraperitoneal Sarcoma Mimicking a Germ Cell Tumor in an 8-Year-Old Girl in Somaliland: A Diagnostic Challenge
Ali YA, H Ali AM, Ma’alin Qasim MN, Ibrahim KJ
International Medical Case Reports Journal 2026, 19:623904
Published Date: 10 July 2026