Back to Journals » International Medical Case Reports Journal » Volume 19
Primary High-Grade Intraperitoneal Sarcoma Mimicking a Germ Cell Tumor in an 8-Year-Old Girl in Somaliland: A Diagnostic Challenge
Authors Ali YA, H Ali AM, Ma'alin Qasim MN, Ibrahim KJ ![]()
Received 12 May 2026
Accepted for publication 7 July 2026
Published 10 July 2026 Volume 2026:19 623904
DOI https://doi.org/10.2147/IMCRJ.S623904
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Thomas E Hutson
Yahye Abdi Ali,1,2 Abdisamed Mohamoud H Ali,3,4 Mustafe Nouh Ma’alin Qasim,3,4 Khadar Jama Ibrahim3,4
1Department of Pathology, Hargeisa Group Hospital, Hargeisa, Somaliland; 2College of Medicine and Health Sciences, University of Hargeisa, Hargeisa, Somaliland; 3Department of Emergency Medicine, Hargeisa Group Hospital, Hargeisa, Somaliland; 4School of Medicine and Surgery, College of Health Sciences, Amoud University, Borama, Somaliland
Correspondence: Khadar Jama Ibrahim, Email [email protected]
Background: Primary intra-abdominal sarcomas in children are rare, aggressive malignancies that often present with non-specific clinical and radiological features. Differentiation from more common pediatric abdominal tumors, such as germ cell tumors, can be challenging, particularly in resource-limited settings with restricted access to advanced pathology and molecular diagnostics.
Case Presentation: We report an 8-year-old previously healthy girl from Somaliland presenting with a 2-week history of progressive abdominal distension, weight loss, and low-grade fever. Imaging revealed a large heterogeneous abdominopelvic mass suggestive of a germ cell tumor. Due to limited access to biopsy, empiric germ cell tumor–directed chemotherapy was initiated; however, no clinical or radiological response was observed after two cycles. Exploratory laparotomy identified a large intra-peritoneal mass with omental and nodal involvement, and cytoreductive surgery was performed. Histopathology showed a high-grade malignant neoplasm with pleomorphic round-to-spindle cells and rhabdoid features. Immunohistochemistry demonstrated diffuse desmin positivity and negative myogenin expression; INI1 testing was unavailable, resulting in a diagnosis of primary high-grade intra-peritoneal sarcoma. Despite postoperative chemotherapy, the disease progressed, and the patient died approximately 3 months after treatment.
Conclusion: Early histopathological confirmation is essential in atypical pediatric abdominal masses. Reliance on imaging alone may lead to misdiagnosis and delayed treatment. Limited diagnostic resources remain a major barrier to optimal care in low-resource settings.
Keywords: primary peritoneal sarcoma, paediatric sarcoma, rhabdoid tumour, rhabdomyosarcoma, diagnostic challenge, Somaliland
Introduction
Primary malignant intra-abdominal tumors in children are rare but account for a significant proportion of pediatric solid malignancies and frequently present late with large abdominal masses and systemic symptoms.1 Among these, soft tissue sarcomas and desmoplastic small round cell tumor (DSRCT) form a rare but highly aggressive subset with predilection for the peritoneal cavity and poor survival despite multimodal therapy.1,2
Pediatric abdominal masses share overlapping clinical and radiologic features, and common entities such as germ cell tumors, lymphoma, rhabdomyosarcoma, and neuroblastoma may be indistinguishable on imaging alone.3 This diagnostic overlap often leads to initial misclassification, particularly in resource-limited settings where histopathology and immunohistochemistry are restricted.4
Primitive intra-abdominal sarcomas fall within the “small round blue cell tumor” spectrum, requiring integrated histopathological, immunohistochemical, and molecular evaluation for accurate classification.5,6 In particular, entities such as rhabdomyosarcoma, DSRCT, malignant rhabdoid tumor, and undifferentiated sarcomas require specific markers and molecular alterations for definitive diagnosis.5,7
In low- and middle-income countries (LMICs), limited access to image-guided biopsy, extended immunohistochemistry panels, and molecular diagnostics often necessitates empiric treatment based on imaging alone, increasing the risk of misdiagnosis and inappropriate therapy.4,8
We report a diagnostically and therapeutically challenging case from Somaliland, in which an 8-year-old girl with a large intra-peritoneal sarcoma was initially managed empirically as a germ cell tumour before definitive diagnosis was established following surgical intervention.
Case Presentation
History
An 8-year-old previously healthy girl presented with a 2-week history of progressively increasing abdominal distension. This was associated with intermittent low-grade fever, anorexia, and significant unintentional weight loss. There was no history of vomiting, constipation, jaundice, urinary symptoms, or prior similar episodes. There was no reported history of malignancy, significant family history of cancer, or known congenital disorders. Immunization history was up to date, and developmental milestones were appropriate for age.
Physical Examination
On admission, the patient appeared chronically ill-looking and cachectic but was conscious and hemodynamically stable Her vital signs were as follows: respiratory rate 22 breaths per minute, heart rate 90 beats per minute, temperature 37°C, and blood pressure 115/78 mmHg.
Abdominal examination revealed marked distension with a large, firm, non-tender abdominopelvic mass occupying most of the abdomen. The mass had an irregular surface and was not clearly mobile. There was no clinically evident hepatosplenomegaly separate from the mass. Bowel sounds were present.
Examination of the lymphatic system revealed multiple discrete, firm, non-tender lymph nodes in the cervical, axillary, and inguinal regions.
There was no peripheral edema, jaundice, or cyanosis. Cardiovascular, respiratory, and neurological examinations were unremarkable.
Diagnostic Assessments
Basic laboratory investigations revealed mild anemia with hemoglobin of 9.5g/dl, white blood cell count of 11.5 × 109/L and platelet count of 420 × 109/L. Serum lactate dehydrogenase (LDH) was elevated at 680 U/L. Renal function tests showed mildly elevated serum creatinine of 1.2 mg/dL.
Abdominal ultrasonography demonstrated a large heterogeneous solid-cystic intra-abdominal mass measuring approximately 27 × 22 cm with displacement of bowel loops.
Contrast-enhanced CT imaging revealed a large heterogeneous enhancing abdominopelvic mass measuring approximately 22 × 14×17 cm, with predominantly solid components and focal non-enhancing areas suggestive of necrosis. The lesion displaced bowel loops laterally and exerted mass effect on adjacent abdominal organs, including the stomach and pancreas, and was associated with bilateral hydronephrosis, likely secondary to ureteric compression, as shown in (Figure 1).
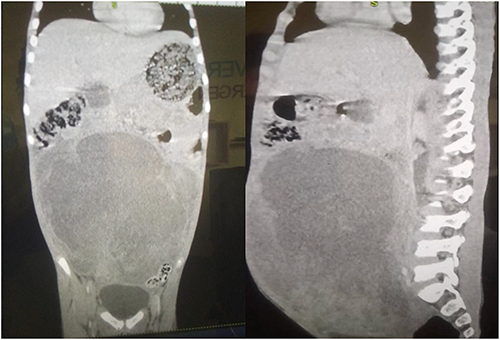 |
Figure 1 Contrast-enhanced CT of the abdomen and pelvis demonstrating a large heterogeneous enhancing abdominopelvic mass with areas of necrosis, causing displacement of bowel loops and mass effect on adjacent organs, with associated bilateral hydronephrosis. |
Differential Diagnosis
Based on clinical and radiological findings, the differential diagnoses included:
- Germ cell tumour
- Desmoplastic small round cell tumour (DSRCT)
- Rhabdomyosarcoma
- Extrarenal malignant rhabdoid tumour
- Lymphoma
Initial Management
Based primarily on the clinical presentation and radiological findings, a germ cell tumour was initially considered the leading diagnosis. Histopathological confirmation before treatment was considered; however, image-guided core needle biopsy was not available at our institution. Referral to another center for biopsy would have resulted in a substantial delay in diagnosis and treatment. Furthermore, the patient had a rapidly enlarging abdominal mass associated with cachexia and bilateral hydronephrosis secondary to ureteric compression, raising concerns that further delay could lead to clinical deterioration. Following multidisciplinary team discussion and considering the available diagnostic resources, empiric germ cell tumour-directed chemotherapy was initiated based on the most likely radiological diagnosis. Despite completing two cycles of chemotherapy, there was no meaningful clinical or radiological response, prompting exploratory laparotomy to obtain definitive tissue diagnosis and guide subsequent management.
Definitive Intervention
Following treatment failure, exploratory laparotomy was performed. Intraoperative findings, as shown in (Figure 2), included:
- A large 30 × 30 cm intra-peritoneal mass
- Omental encasement
- Sigmoid colon involvement
- Approximately 10 cc ascitic fluid
- Multiple pelvic nodules
- Pelvic lymphadenopathy
- Grossly uninvolved uterus and adnexa
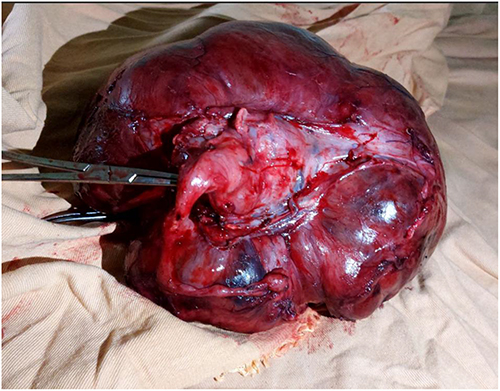 |
Figure 2 Intraoperative photograph showing a large intra-peritoneal tumor with extensive omental involvement and multiple peritoneal nodules. |
Procedures included:
- Tumor debulking/cytoreductive surgery
- Omentectomy
- Pelvic lymph node biopsy
Pathological Findings
Gross Pathology
The principal tumor specimen measured 25×25 cm after fixation and demonstrated a nodular external surface with hemorrhagic, necrotic and cystic cut surfaces. Additional cul-de-sac nodules and lymph node specimens were also submitted (Figure 3).
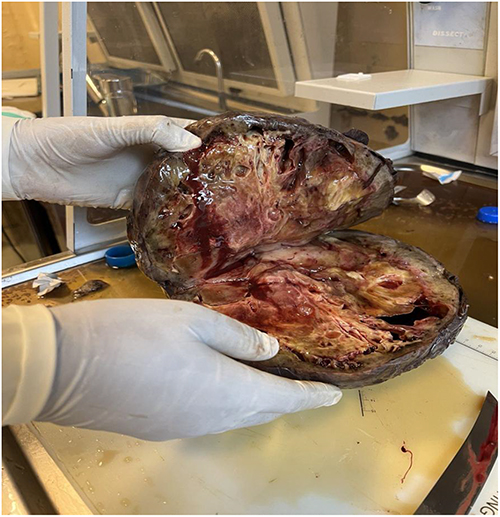 |
Figure 3 Gross pathological specimen of the resected tumor demonstrating a large nodular mass with hemorrhagic, necrotic and cystic cut surfaces. |
Histopathology
Microscopy demonstrated sheets and dispersed pleomorphic round-to-spindle cells with rhabdoid morphology, eccentric eosinophilic cytoplasm, necrosis, microcystic change and mitotic activity. As shown in (Figure 4) Similar tumor infiltration was identified in pelvic nodules and lymph nodes.
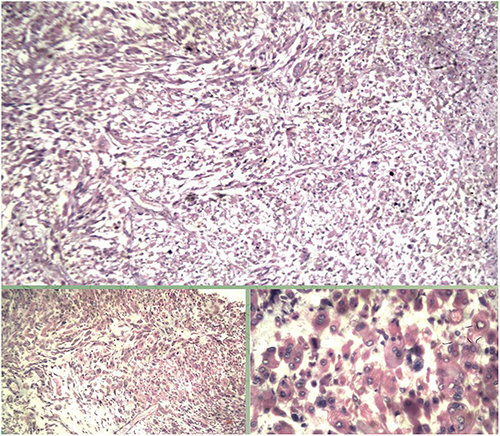 |
Figure 4 Histopathological image (H&E stain) showing pleomorphic round-to-spindle tumor cells with rhabdoid features, including eccentric nuclei and abundant eosinophilic cytoplasm. |
Immunohistochemistry
- Desmin: Diffusely positive
- Myogenin: Negative
- INI1: Not available
Pathological Differential Diagnosis
- Poorly differentiated rhabdomyosarcoma
- Extrarenal malignant rhabdoid tumour
The absence of myogenin expression reduced support for conventional rhabdomyosarcoma but did not completely exclude poorly differentiated rhabdomyosarcoma. Similarly, the lack of INI1 (SMARCB1) immunohistochemistry prevented definitive exclusion of malignant rhabdoid tumour, while the absence of WT1 immunohistochemistry and EWSR1–WT1 fusion testing precluded confirmation or exclusion of desmoplastic small round cell tumour (DSRCT). Based on the available histomorphological and immunohistochemical findings, the lesion was therefore classified as a primary high-grade intra-peritoneal sarcoma with myogenic differentiation. Definitive subclassification was not possible because advanced immunohistochemical and molecular diagnostic testing was unavailable.
Outcome and Follow-Up
The postoperative course was initially uneventful, and the patient was referred for pediatric oncology evaluation. Following surgical debulking and histopathological diagnosis, she received three cycles of sarcoma-directed chemotherapy. Unfortunately, the exact chemotherapeutic agents, dosages, treatment intervals, treatment-related toxicities, and any dose modifications could not be retrospectively retrieved because treatment was administered at another oncology center and the available medical records were incomplete. This limitation restricted detailed evaluation of the therapeutic regimen and its tolerability. Despite combined surgical and systemic treatment, the disease progressed, and the patient died approximately 3 months after completion of postoperative chemotherapy. Further molecular characterization remained unavailable because of local resource limitations. The chronological clinical course is summarized in (Table 1).
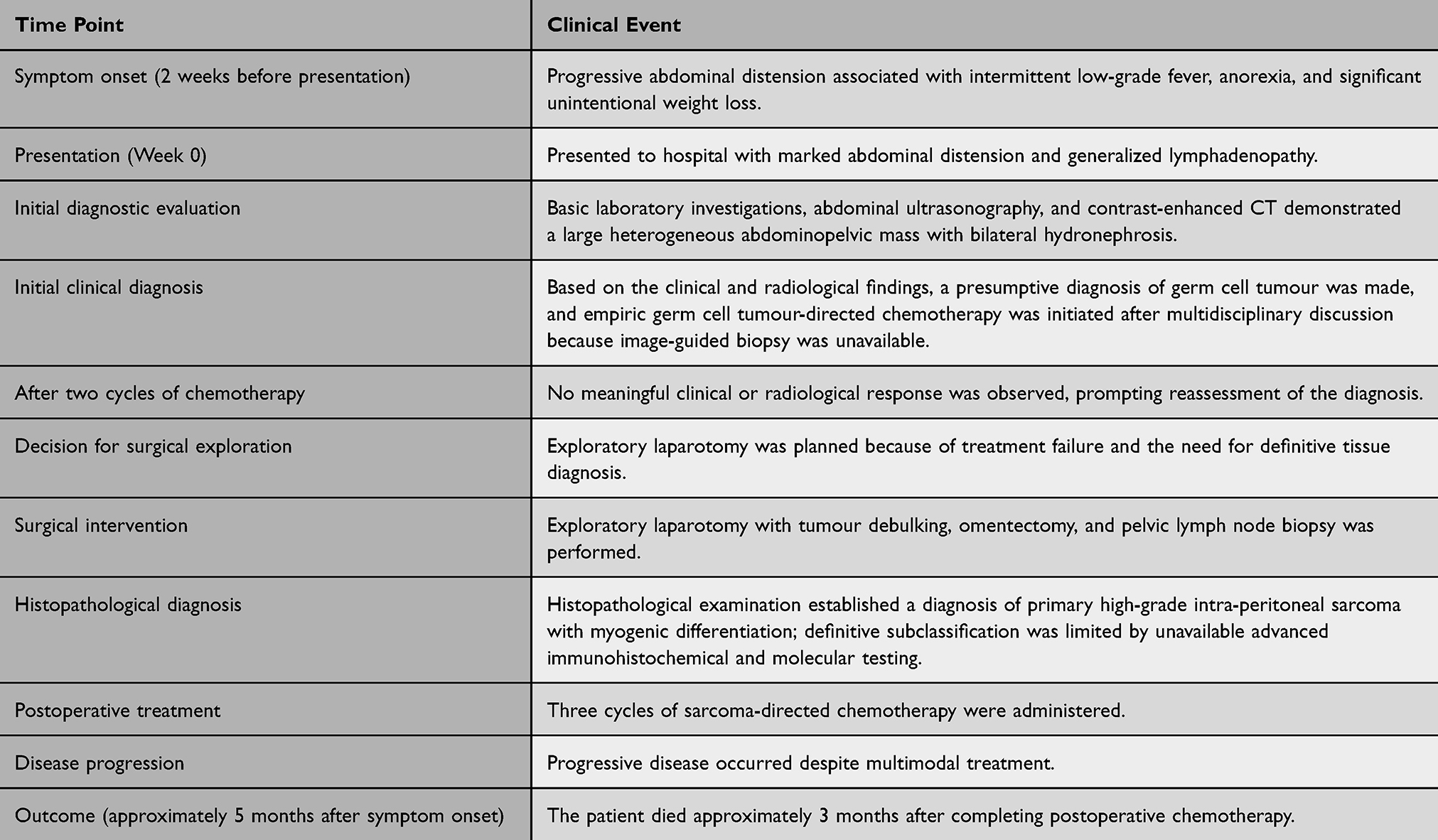 |
Table 1 Chronological Summary of the Patient’s Clinical Presentation, Diagnostic Evaluation, Treatment, and Outcome |
Discussion
High-grade intra-abdominal sarcomas in children represent a heterogeneous group of aggressive malignancies, including rhabdomyosarcoma, desmoplastic small round cell tumor (DSRCT), malignant rhabdoid tumor, and undifferentiated sarcomas.1,2 These tumors often share a “small round blue cell” morphology and exhibit overlapping clinical and radiologic features, making accurate diagnosis challenging without tissue confirmation.5
The patient’s presentation with progressive abdominal distension, weight loss, and a large heterogeneous intra-abdominal mass with necrosis is consistent with reported patterns of pediatric abdominal sarcomas and DSRCT.1 Imaging findings such as a large solid-cystic lesion with mass effect and peritoneal involvement are non-specific and overlap with germ cell tumors and lymphoma.3,9 However, diffuse peritoneal involvement and omental disease at surgery are more characteristic of aggressive sarcomatous processes than germ cell tumors.2
Diagnostic Challenge
Initial empiric treatment for a presumed germ cell tumor reflects a common diagnostic pitfall in pediatric oncology: anchoring bias based on imaging and age profile.3,9 Current guidelines emphasize that solid pediatric abdominal masses without definitive radiologic origin should undergo tissue biopsy prior to chemotherapy whenever feasible.6,10 Lack of response after two cycles of chemotherapy in this case appropriately triggered diagnostic reassessment and surgical exploration.
Pathological Interpretation
Histopathology showed pleomorphic round-to-spindle cells with rhabdoid morphology and necrosis. Immunohistochemistry revealed diffuse desmin positivity and negative myogenin expression.
Desmin positivity supports myogenic or mesenchymal differentiation but is not specific, as it may be seen in rhabdomyosarcoma and DSRCT.5 However, the absence of myogenin/MyoD1 expression argues strongly against conventional embryonal or alveolar rhabdomyosarcoma, where nuclear myogenin positivity is a key diagnostic feature.7
Importantly, INI1 (SMARCB1) testing was not available. Loss of INI1 is a defining feature of malignant rhabdoid tumor and some undifferentiated sarcomas; therefore, its absence limits definitive exclusion of these entities.8,11
This diagnostic uncertainty places the tumor within the category of high-grade unclassified intra-abdominal sarcoma, which reflects modern WHO classification when molecular confirmation is unavailable.7
Molecular Diagnostic Limitations
Modern classification of primitive intra-abdominal sarcomas relies on integrated morphology, immunohistochemistry, and molecular profiling, including:
- EWSR1–WT1 fusion in DSRCT12
- SMARCB1/INI1 loss in rhabdoid tumor8
- MYOD1 mutations in aggressive rhabdomyosarcoma7
- CIC or BCOR rearrangements in undifferentiated sarcomas6
The absence of these investigations in this case reflects a major diagnostic gap in resource-limited settings and directly impacts therapeutic precision.
Management Considerations
Ideally, pediatric solid abdominal tumors should undergo early image-guided core biopsy prior to systemic therapy initiation.10 Ultrasound or contrast-enhanced biopsy approaches have high diagnostic accuracy and safety even in large necrotic masses.13
However, in this case, cytoreductive surgery served both diagnostic and therapeutic purposes after failure of empiric chemotherapy. While complete oncologic staging and molecular characterization were not possible, surgical debulking allowed definitive tissue diagnosis and reduction of tumor burden. Despite surgical debulking and subsequent sarcoma-directed chemotherapy, the patient’s death within months of treatment completion underscores the aggressive biology and poor prognosis associated with advanced pediatric intra-peritoneal sarcomas, particularly when diagnosis and definitive subtype classification are delayed.
Similar diagnostic challenges have been described in pediatric intra-abdominal sarcomas where delayed histological classification leads to inappropriate initial therapy and poor outcomes.2,5 Even with aggressive multimodal therapy, prognosis remains poor in advanced peritoneal sarcomas, particularly when diagnosis is delayed or incomplete molecular classification is not possible.1,2
This case highlights the importance of considering early biopsy or surgical exploration in children with atypical, rapidly enlarging abdominal masses that do not respond to standard empiric regimens.
This report also contributes valuable data from Somaliland, where published pediatric oncology literature remains limited. Documenting such rare presentations is essential to improving regional awareness, encouraging earlier referral and advocating for improved pathology infrastructure, including expanded immunohistochemistry and molecular diagnostics. Greater collaboration with international pathology centers may also help bridge diagnostic gaps for rare pediatric malignancies.
This case illustrates several challenges commonly encountered in low- and middle-income countries, where limited access to pediatric oncology services, image-guided biopsy, comprehensive immunohistochemistry, and molecular diagnostics can delay definitive diagnosis and appropriate treatment. Similar diagnostic difficulties have been reported in other resource-constrained settings, where reliance on clinical and radiological findings alone may lead to inappropriate initial management and poorer outcomes. Strengthening referral pathways, expanding access to diagnostic pathology services, developing regional pathology networks, and establishing telepathology collaborations with specialized centers may substantially improve diagnostic accuracy and facilitate earlier multidisciplinary management of rare pediatric malignancies. For Somaliland, continued investment in pathology infrastructure and international collaborative partnerships represents an important step toward improving outcomes for children with complex oncological diseases.
Conclusion
Primary high-grade intra-peritoneal sarcoma in children is an exceptionally rare and highly aggressive malignancy that can closely mimic more common pediatric abdominal tumors on imaging. This case illustrates the limitations of imaging-based diagnosis alone and reinforces the necessity of early histopathological confirmation before initiating definitive therapy whenever possible. In resource-limited settings, restricted access to advanced immunohistochemistry may complicate accurate subclassification, potentially affecting treatment decisions. Early multidisciplinary evaluation, timely tissue diagnosis and consideration of aggressive cytoreductive strategies in selected patients are essential to optimizing outcomes. The fatal outcome in this case further highlights the aggressive nature of these malignancies and the urgent need for earlier diagnostic precision. Increased reporting of such cases from underrepresented regions is critical to expanding global understanding of pediatric peritoneal sarcomas and improving future care pathways.
Learning Points
- Primary pediatric intra-peritoneal sarcomas are rare and diagnostically challenging.
- Imaging alone may be insufficient for definitive diagnosis in large pediatric abdominal masses.
- Histopathological confirmation should be prioritized before treatment initiation whenever feasible.
- Limited immunohistochemical resources may significantly hinder accurate sarcoma subclassification.
- Early multidisciplinary evaluation may improve management of rare pediatric malignancies.
Patient’s Perspective
The family initially believed the abdominal swelling was due to a common gastrointestinal illness, but rapid progression, incomplete diagnostic certainty and lack of sustained response to treatment created significant emotional distress. Definitive surgery provided greater diagnostic clarity, although the disease ultimately progressed aggressively.
Ethical Approval
Ethical approval was obtained from the Hargeisa Group Hospital Research Center (Reference No. RC-HGHMAY2026-010) before publication of this case report.
Informed Consent
Written informed consent was obtained from the patient’s legal guardian for publication of this case report and any accompanying images.
Author Contributions
All authors made substantial contributions to the conception and design of the study, acquisition of data, or analysis and interpretation of data; participated in drafting the article or revising it critically for important intellectual content; approved the final version of the manuscript to be published; and agree to be accountable for all aspects of the work.
Funding
No funding was received for this case report.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Srinivasan A, Parikh A, Pace E, et al. Imaging of pediatric abdominal soft tissue tumors. Pediatr Blood Cancer. 2023;70(5):e30245. doi:10.1002/pbc.30341
2. Wang L, Ji Z, Gao Y, et al. Cytoreductive surgery and hyperthermic intraperitoneal chemotherapy in desmoplastic small round cell tumor. World J Surg Oncol. 2021;19(1):146. doi:10.1186/s12957-021-02261-y
3. Kim HHR, Hull NC, Lee EY, Phillips GS. Imaging of pediatric abdominal masses. Radiol Clin North Am. 2022;60(4):679–9. doi:10.1016/j.rcl.2021.08.008
4. Joseph N, Rai S, Singhal K, et al. Clinico-histopathological profile of primary pediatric intra-abdominal tumors in a resource-limited setting. Indian J Surg Oncol. 2021;12(2):345–352. doi:10.1007/s13193-021-01365-x
5. Magro G, Longo F, Angelico G, et al. Immunohistochemistry as a diagnostic pitfall in solid tumors of children and adolescents. Acta Histochem. 2015;117(7):589–598. doi:10.1016/j.acthis.2015.03.011
6. Miele E, de Vito R, Ciolfi A, et al. DNA methylation profiling for diagnosing undifferentiated sarcoma with CIC alterations. Int J Mol Sci. 2020;21(5):1818. doi:10.3390/ijms21051818
7. WHO Classification of Tumours Editorial Board. Soft Tissue and Bone Tumours.
8. Biegel JA, Kalpana G, Knudsen ES, et al. The role of SMARCB1/INI1 in the pathogenesis of malignant rhabdoid tumors. Nat Rev Cancer. 2014;14(12):789–801.
9. Ní Cheallaigh L, Liu JF, Ball-Gamble A, et al. Spotting childhood abdominal tumours: clinical presentation and diagnostic clues. Arch Dis Child. 2025;110(2):150–156.
10. Kim A, Brown CN, Kang L, et al. Image-guided biopsy in pediatric solid tumors: indications, safety, and diagnostic yield. J Pediatr Surg. 2022;57(6):1087–1093. doi:10.1016/j.jpedsurg.2022.01.029
11. Hasselblatt M, Isken S, Linge A, et al. High-resolution genomic analysis of malignant rhabdoid tumors reveals characteristic alterations and INI1 loss. Brain Pathol. 2011;21(4):417–425.
12. Gerald WL, Ladanyi M, de Alava E, et al. Clinical, pathologic, and molecular spectrum of tumors associated with t(11;22)(p13;q12): desmoplastic small round cell tumor and related lesions. Proc Natl Acad Sci U S A. 1995;92(4):1028–1032. doi:10.1073/pnas.92.4.1028
13. Liu M, Liu Y, Zhou W, et al. Contrast-enhanced ultrasound–guided versus conventional ultrasound-guided biopsy for abdominopelvic neoplasms in pediatric patients. Eur Radiol. 2024;34(2):1123–1132. doi:10.1007/s00330-023-10148-7
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Recessive Dystrophic Epidermolysis Bullosa in a Resource-Limited Setting: A Case Report From Mohamed Adam Sheikh Children’s Teaching Hospital Hargeisa, Somaliland
Aw Hashi MO, Ahmed SI, Derie AM, Abdirahman RM, Abdullahi RR
International Medical Case Reports Journal 2026, 19:600282
Published Date: 23 April 2026