Back to Journals » OncoTargets and Therapy » Volume 12
Receptor-Interacting Protein Kinase 1 Promotes Cholangiocarcinoma Proliferation And Lymphangiogenesis Through The Activation Protein 1 Pathway
Authors Li CZ ![]() , Lin YX, Huang TC, Pan JY, Wang GX
, Lin YX, Huang TC, Pan JY, Wang GX
Received 10 May 2019
Accepted for publication 21 August 2019
Published 1 November 2019 Volume 2019:12 Pages 9029—9040
DOI https://doi.org/10.2147/OTT.S215276
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Carlos E Vigil
Cheng-Zong Li,1,* Yu-Xiang Lin,2,* Tian-Cong Huang,1 Jun-Yong Pan,1 Gao-Xiong Wang1
1Department of Hepatobiliary and Pancreatic Surgery, The Second Affiliated Hospital of Fujian Medical University (Donghai District), Quanzhou 36200, People’s Republic of China; 2Department of Surgery Ward 6, The Second Affiliated Hospital of Fujian Medical University (Licheng District), Quanzhou 36200, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Gao-Xiong Wang
Department of Hepatobiliary and Pancreatic Surgery, The Second Affiliated Hospital Of Fujian Medical University (Donghai District), 950 Donghai Avenue, Fengze, Quanzhou 36200, People’s Republic of China
Tel +86-133-1387-8789
Fax +86-0595-22770258
Email [email protected]
Purpose: Receptor-interacting protein kinase 1 (RIPK1) is an important upstream regulator of multiple cell signaling pathways including inflammatory signals. RIPK1 is reported to be closely associated with the prognostic implications of cancer, especially epithelial tumors. But its role in proliferation and lymphangiogenesis in cholangiocarcinoma (CCA) remains unclear and requires further investigation.
Patients and methods: Expression of RIPK1 in human CCA tissues and CCA cell lines (QBC939, HUH28 and CCPL-1) was measured using qPCR, immunoblotting and immunohistochemistry. Silencing of RIPK1 was achieved by transduction of CCA cells via lentiviral plasmids (LV3-H1/GFP&Puro) encapsulating RIPK1 shRNA (LV-shRIPK1) or negative control shRNA (LV-shNC), and puromycin was used to select stable colonies. Proliferation and lymphangiogenesis were assessed in vitro by CCK-8 and matrigel-based tube formation assays, respectively. Activity of the activation protein-1 (AP-1) was evaluated by double-luciferase reporter gene assay. Protein expression of JNK, P38MAPK, ERK1/2, AP-1, P-AP-1, E-cadherin, N-cadherin and vascular endothelial growth factor-C (VEGF-C) was measured by immunoblotting or ELISA. An orthotopic CCA model in null mice was generated by transplanting QBC939 LV-shRIPK1, LV-shNC and control cells to further evaluate the role of RIPK1 on lymphangiogenesis in vivo. Immunohistochemistry was utilized to evaluate the expression of RIPK1 and VEGF-C, and tumor lymphatic vessels in the CCA model mice.
Results: Upregulated expression of RIPK1 in CCA tissues was closely related to tumor size, lymph node metastasis and poor prognosis. RIPK1 promoted proliferation and lymphangiogenesis in CCA cells, and regulated the activation of JNK and P38MAPK-mediated AP-1/VEGF-C pathway. Finally, in vivo animal experiments in the orthotopic CCA mouse model further confirmed the function of RIPK1 in lymphangiogenesis.
Conclusion: This is the first report demonstrating the role of RIPK1 in proliferation and lymphangiogenesis through the MAPK (JNK and P38MAPK)- AP-1 pathway in CCA.
Keywords: cholangiocarcinoma, RIPK1, AP-1, VEGF-C, proliferation, lymphangiogenesis
Introduction
Cholangiocarcinoma (CCA) is the second most common malignant tumor of the biliary tract and its incidence has been increasing for 40 years in the world.1,2 Approximately 80–90% of CCA are derived from extrahepatic bile duct, and early atypical symptoms make it difficult to detect until the advanced stages.3 Only about 35% of patients can be diagnosed early and surgically treated, and thus the 5-year survival rate does not exceed 10%.4,5 Also, regional lymph node metastasis is an important factor affecting the prognosis of CCA. The 5-year survival rate of patients with negative lymph nodes is 30%, while the survival rate of patients with local lymph node metastasis is less than 15%.6 With the advent of state-of-the-art molecular biology techniques, research on exploring genes and molecules associated with CCA has made slow but steady progress.7–9 But the apparent inter- and intra-tumoral heterogeneity of CCA has stymied the discovery of effective and targeted therapeutics.3 Therefore, discovering more effective targets to CAA proliferation and lymphangiogenesis has attracted attention worldwide.
Inflammation promotes the growth and metastasis of cancer.10 Clinical studies have found that chronic cholangitis caused by primary sclerosing cholangitis, cholelithiasis and clonorchiasis infection are important risk factors of CCA.11 Several mechanistic studies have revealed that modulation of genes such as K-ras, p53, p14AFR and p16INK4a by inflammatory factors including TNF-α, IL-6, TGF-β and PDGF may lead to the occurrence and development of CCA.12–14 Taken together, these findings have confirmed that inflammatory signals are important in the pathogenesis of CCA.
Receptor-interacting protein kinase 1 (RIPK1) is an critical upstream regulator of diverse cell signaling pathways, including TNF-α, IL-6 and TLR3/4 inflammatory signals.15 And the presence of multiple domains including C-terminal death domain, intermediate domain and N-terminal Ser/Thr kinase enables RIPK1 to control inflammation and cell survival by nuclear factor-κB (NF-κB) and MAPK signaling or death by apoptosis and necroptosis.15,16 Activation of NF-κB or MAPK-mediated activation protein 1 (AP-1) – a critical mediator of inflammation – is a common event during tumorigenesis.17,18 Previous studies have reported that RIPK1 is dispensable for NF-κB activation in some cancer cells, but reports on the cooperation of RIPK1 with AP-1 signaling in cancer are scarce. Recent studies in RIPK1 deficient mice have clearly indicated its importance in the formation and development of lymphoid system in normal tissues.19 In addition, RIPK1 has been implicated in proliferation, and lymphangiogenesis in malignant melanoma,20 gallbladder cancer21 and breast cancer.22 But the function of RIPK1 and RIPK1-AP-1 inflammatory signaling in the proliferation and lymphangiogenesis of CCA is still unknown.
In this study, we found that RIPK1 upregulation in human CCA tissues was associated with poor prognosis. Both in vitro and in vivo results further demonstrated that RIPK1 regulated proliferation and lymphangiogenesis through AP-1 signaling in CCA. This is the first report demonstrating the participation of RIPK1 in CCA.
Materials And Methods
Cell Lines And Human Tissue
Human CCA cell lines (HUH28, CCPL-1 and QBC939) and human extrahepatic biliary epithelial cells (HEBEpics) were purchased from Sciencell (San Diego, California, USA). All CCA cell lines and HEBEpics were cultured in Dulbecco minimum essential medium (Gibco, Grand Island, USA) with 10% fetal bovine serum (Gibco, Grand Island, USA). Human dermal lymphatic endothelial cells (HDLECs) obtained from Sciencell (San Diego, California, USA) were maintained in endothelial cell growth medium (Sciencell, San Diego, California, USA). Forty-two CCA samples and matched non-cancerous bile duct specimens were obtained from the Department of Hepatobiliary Surgery, the Second Affiliated Hospital of Fujian Medical University. Written informed consent was agreed by patients before the surgical removal. All studies were approved by the Ethics Committee of the Medical Faculty of the Second Affiliated Hospital of Fujian Medical University, in accordance with the Declaration of Helsinki.
Quantitative PCR (qPCR)
Total RNA was extracted from samples by using TRIzol reagent (Life Technologies, USA). The purity and concentration of total RNA was measured on a UV3000 ultraviolet spectrophotometer. Reverse transcription of RNA was performed using Revert Aid TM First Strand cDNA Synthesis Kit (Life Technologies, USA). qPCR reactions were performed using SYBR Green qPCR Kit (Life Technologies, USA), and the fluorescence was observed using RT-PCR System (Applied Biosystems). PCR primer sequences used for RIPK1 were as follows: forward, 5ʹ-GTCCTGGTTTGCTCCTTCCC-3ʹ, reverse, and 5ʹ-GTCTCCTTTCCTCCTCTCTGTT G-3ʹ. Each condition was performed in triplicate. Relative expression was calculated by the -∆∆Ct and 2—∆Ct methods, and GAPDH was used as an internal control.
Immunohistochemistry
Five micrometer paraffin-embedded sections were stained for RIPK1 (Abcam, Cambridge, UK, 1:500), vascular endothelial growth factor-C (VEGF-C) (Santa Cruz, New York, USA, 1:350) and LYVE-1 (R&D Systems, Minneapolis, USA, 1:250) using MaxVisionTM HRP-Polymer IHC Kit according to the manufacturer’s instructions (Maixin, Fuzhou, People’s Republic of China). Five high-power fields (400×) were randomly selected from each slice. RIPK1 and VEGF-C protein expression in each slice was semiquantitatively analyzed by calculating the mean optical density (MOD) using the Image-Pro plus 6.0 software (Media Cybernetics, Maryland, USA). Expression intensity of RIPK1 was categorized as high or low based on the MOD medians.
Immunoblotting
After washing with ice-cold PBS, cells were lysed on ice in 100μL RIPA buffer containing 100mM PMSF. Cell lysates were collected and centrifuged at 14,000 rpm for 10 mins at 4°C. The protein lysates were then mixed with 5×loading buffer and denatured by boiling for 5 mins at 100°C, separated on 10% polyacrylamide gels (Invitrogen), and transferred to PVDF (polyvinylidene fluoride) membranes. Membranes were blocked with 5% skim milk in PBS containing 0.1% Tween 20 (PBS-T) for 2.5 hrs. Thereafter, membranes were incubated overnight in antibodies targeting RIPK1 (Abcam, Cambridge, UK, 1:1000), AP-1 (Abcam, Cambridge, UK, 1:1000), P-AP-1 (Abcam, Cambridge, UK, 1:1000), JNK (Abcam, Cambridge, UK, 1:1000), P38MAPK (Abcam, Cambridge, UK, 1:1000), ERK1/2 (Abcam, Cambridge, UK, 1:1000), E-cadherin (Abcam, Cambridge, UK, 1:500) N-Cadherin (Abcam, Cambridge, UK, 1:1000) and VEGF-C (Santa Cruz, New York, USA, 1:1000) at 4°C. After conjugating with respective HRP-coupled secondary antibodies, the protein–antibody complexes were detected using chemiluminescence (Life Technologies, USA) and recorded on Hyperfine-ECI detection film. Target protein expression was semi-quantified relative to GAPDH (loading control) expression.
siRNA, shRNA And Transfection
RIPK1-siRNA (sequence: 5′-GCCTGAGAATATCCTCGTT-3′ and 5′-GCACAAATACGAACTTCAA-3′), RIPK1-shRNA and lentiviral plasmids (LV3-H1/GFP&Puro) were purchased from Suzhou GenePharma (People’s Republic of China). Sequences extracted from the GenBank (GenBank accession no NM_003804) was used to construct two RIPK1 and one negative control siRNA plasmids. Transfection of siRNA plasmids was carried out using OptiMEM medium (Invitrogen, USA) and Lp2000 (Lipofectamine 2000, Invitrogen, USA). The same siRNA sequences were used to generate lentiviral shRNA plasmids. Lentiviral plasmids (LV3-H1/GFP&Puro) encapsulating RIPK1 shRNA (LV-shRIPK1) or negative control shRNA (LV-shNC) were used to transduce CCA cells, and puromycin was used to select stable colonies. Additionally, CCA cells were transfected with VEGF-C-siRNA (Santa Cruz biotechnology, New York, USA) using Lp2000 (Invitrogen, USA).
Cell Proliferation Assay
The cell proliferation assay was performed using CCK-8 Kit (Cell Signaling Technology, New York, USA) according to the manufacturer’s protocols. 5×103 cells were seeded in each well of a 96-well plate and 200 μL fresh medium containing 10μl CCK-8 reagent was added into each well 24, 48, 36 and 72-hr post-transfection and incubated for 4 hrs at 37°C. OD was measured at 450 nm on a plate reader (Bio-Rad, Hercules, CA, USA). All experiments were performed in triplicate.
Matrigel-Based Tube Formation Assay
For lymphangiogenesis of CCA, QBC939 cell lines were co-incubated with HDLECs in a 3D co-culture system. HDLECs were first labeled with DiI (Beyotime, Shanghai, People’s Republic of China). And then, 7.5×103/well CCA cells and 7.5× 103/well HDLECs were added together into Matrigel-coated microwell plate (ibidi, Martinsried, Germany) and incubated in a 37°C incubator for 30 mins. Tubes were imaged after 1, 3, 6, 10 and 24 hrs under a fluorescence microscope and analyzed using AxioVision Rel 4.1 software (Carl Zeiss AG, Jena, Germany).
Luciferase Reporter Assay
Transduced QBC939 cell groups (control, siNC, siRIPK1-1, siRIPK1-2) were cultured in 24-well plates (1×105/well). And these cell groups were transduced with pRL-TK (10ng) and AP-1-luc (50ng) plasmids (Yeasen Biotechnology, Shanghai, People’s Republic of China) using Lipofectamine2000 (Lp2000, Invitrogen, USA). Luciferase activity was measured by Dual-Luciferase Reporter Assay System (Promega, Madison, USA) using a Microplate Luminometer (Promega, Madison, USA) following the manufacturer’s protocols. Each condition was performed in triplicate.
ELISA
Concentration of VEGF-C in the culture supernatant was detected by using an ELISA Kit (R&D Systems, Minneapolis, MN, USA) following the manufacturer’s protocols. Each condition was performed in triplicate.
Orthotopic Xenograft Mouse Model
All animal experiments were conducted in accordance with the Ethics Committee of Fujian Medical University, the Animal Welfare Act of Health guidelines, and all protocols were approved by the Institutional Animal Care and Use Committee. Male nude mice of 5–6 weeks old were purchased from the Shanghai SLAC laboratory Animals Co. Ltd (Shanghai, People’s Republic of China) and raised in the specific pathogen-free laboratory animal room. The orthotopic xenograft mouse model was constructed based on the method as previously described.23 Briefly, nude mice were transplanted orthotopically with QBC939, or QBC939 cells transduced with LV-shRIPK1 or LV-shNC. After 4 weeks, tumors specimens were collected, fixed in formalin and processed for immunohistochemical examinations.
Statistical Analyses
Results were presented as means±SD. Student’s t-test (two groups), one-way analysis of variance and Fisher’s exact tests (quantitative data) were used to analyze the data. A p-value <0.05 was considered significant.
Results
RIPK1 Is Upregulated In CCA Tissues
Transcript abundance of RIPK1 in human CCA tissues was significantly higher than the matched non-neoplastic tissues (Figure 1A and B). Of the 42 CCA tissues, two samples showed strong upregulation in RIPK1 mRNA as well as protein expression (Figure 1C). Similarly, immunohistochemistry revealed cytoplasmic localization of RIPK1 in CCA tissues (Figure 1D–G). These results indicated enrichment of RIPK1 in CCA tissues.
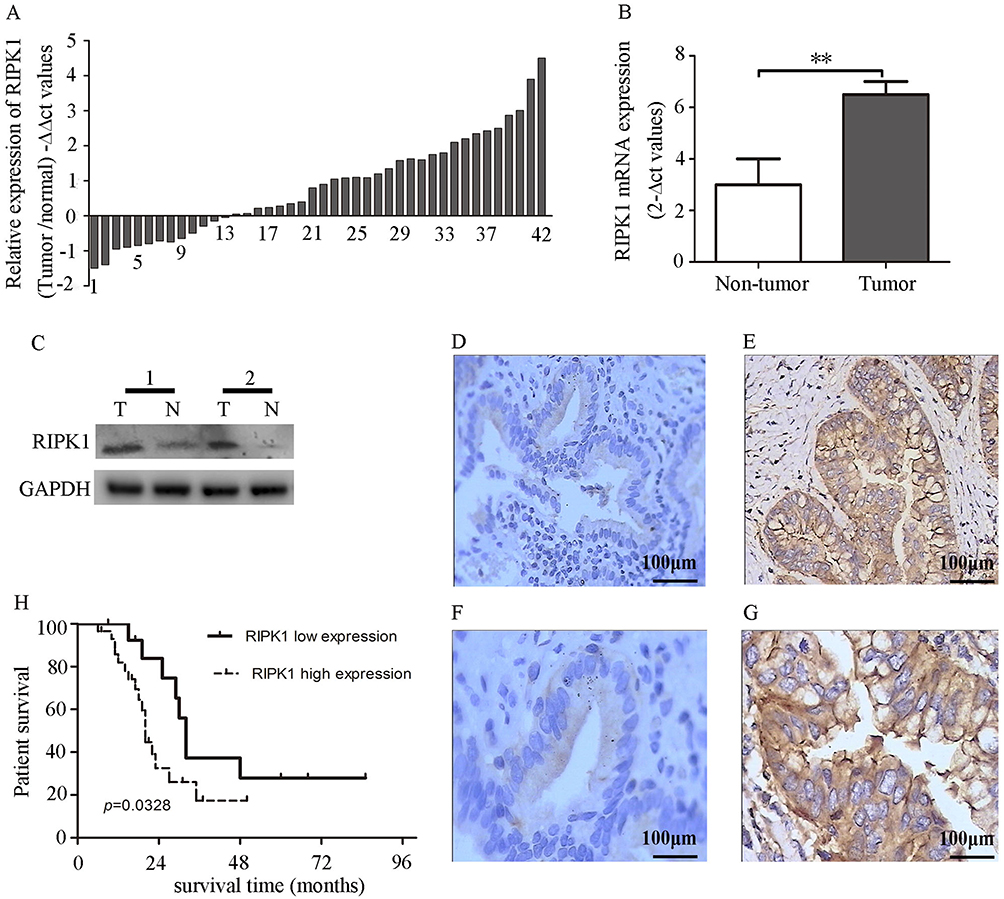 |
Figure 1 Upregulation of RIPK1 in CCA is associated with poor prognosis. (A–B) -∆∆Ct values of RIPK1 mRNA levels in CCA and matched nonneoplastic tissues. (C) Western blots showing RIPK1 protein expression in 2 paired bile duct tissues. (D–G) Representative photomicrographs showing higher expression of RIPK1 in the cytoplasm of CCA tissues (e, 200× and g, 400×) than non-neoplastic tissues (d, 200× and f, 400×). (H) Correlation between RIPK1 protein expression and overall survival rate in CCA patients. **P<0.01. |
Correlation Between RIPK1 Expression And Clinicopathological Features And Survival
The relationship between the MOD of RIPK1 protein expression as assessed by immunohistochemistry and clinicopathological features of CCA is shown in Table 1. We observed that tumor size and lymph node metastasis were the only indicators closely associated with overexpression of RIPK1. A Kaplan–Meier curve analysis revealed that higher expression of RIPK1 significantly lowered the overall survival rate of patients with CCA (Figure 1H).
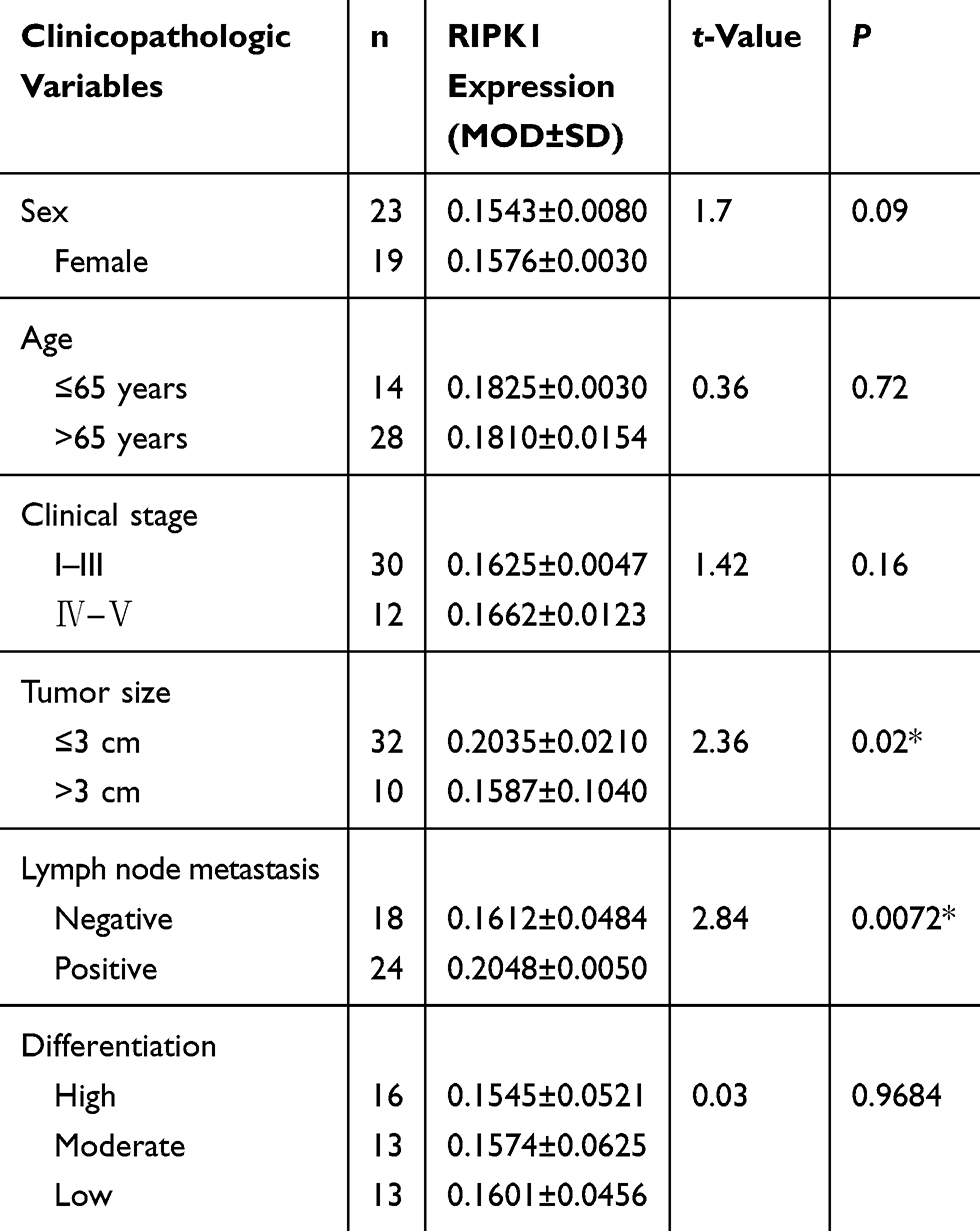 |
Table 1 Correlation Between RIPK1 Protein Expression And Clinicopathological Features |
Transcript And Protein Expression Of RIPK1 In CCA Cell Lines
Results showed that RIPK1 transcript abundance and protein expression were significantly upregulated in CCA lines than HEBEpics. Meanwhile, we found that RIPK1 expression was higher in QBC939 cells compared with HUH28 or CCPL-1 cells (Figure 2A1-3). Henceforth, QBC939 cells were selected for further experimentation.
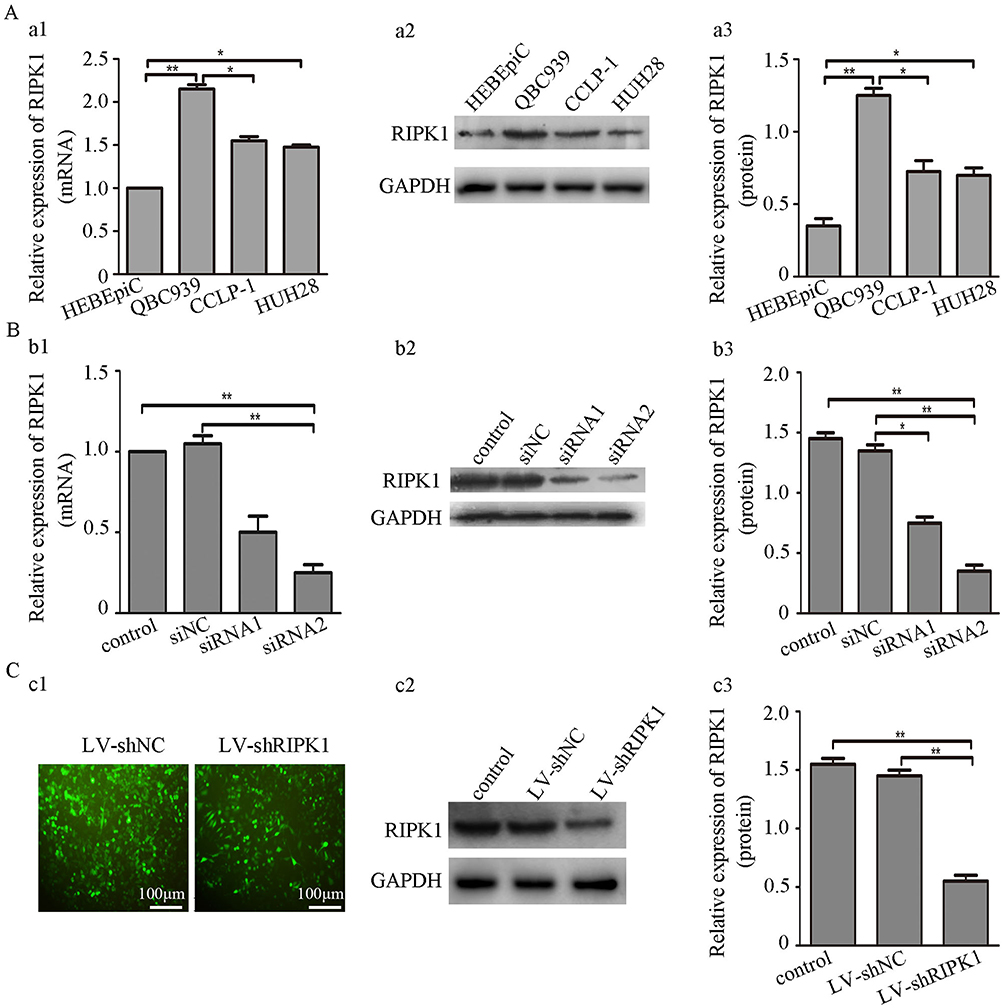 |
Figure 2 (A) RIPK1 expression in CCA cell lines and HEBEpics. (a1-3) RIPK1 mRNA and protein levels were significantly higher in QBC939 than HUH28, CCPL-1 and HEBEpics. (B) siRNA mediated silencing of RIPK1 in QBC939 cells. (b1-3) siRIPK1-1 or siRIPK1-2 mediated silencing of RIPK1 mRNA and protein expression in QBC939 cells. (C) Establishment of stable siRIPK1-2 QBC939 cell line. (c1) Representative images showing transduction efficiency of LV-shRIPK1 and LV-siNC in QBC939 cells. (c2-3) Western blots showing RIPK1 expression in QBC939 cells stably infected with LV-shRIPK1 or LV-shNC. *P<0.05, **P<0.01. |
Stable Expression Of siRIPK1 In QBC939 Cells
QBC939 cells were transfected with siRIPK1-1, siRIPK1-2 and siNC (negative control) following sequence verification in NCBI BLAST. Both siRIPK1-1 and siRIPK1-2 significantly silenced mRNA and protein expression of RIPK1 as compared with the siNC and non-transfected control plasmids (Figure 2B1-3). As siRIPK1-2 was more efficient than siRIPK1-1, siRIPK1-2 was used for generating lentiviral shRIPK1 (LV-shRIPK1). LV-shRIPK1 and shNC (LV-shNC) plasmid containing the non-targeting siRNA sequence were used to generate stable QBC939 cell lines through puromycin screening (Figure 2C1). Validation by immunoblotting confirmed that RIPK1 was significantly silenced in LV-shRIPK1-2 transfected cells (Figure 2C2-3). Further experimentation was carried out on the stably transfected cell line.
Silencing RIPK1 Inhibited The Proliferation Of QBC939 Cells
We detected the proliferation rate of the four QBC939 cell groups (control, siNC, siRIP1-1, siRIP1-2) using CCK-8 assay. And the results showed that siRNA mediated silencing of RIPK1 significantly attenuated the proliferation of QBC939 cells compared with the siNC and non-transfected control groups (Figure 3).
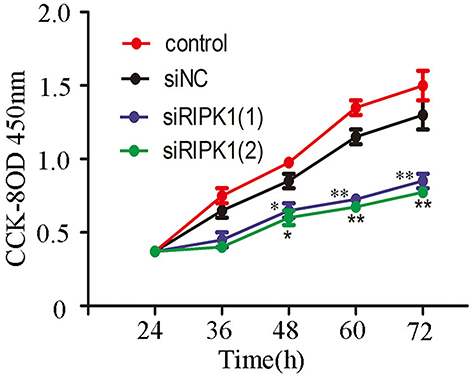 |
Figure 3 Knockdown of RIPK1 expression inhibits proliferation of QBC939 cells. Knockdown of RIPK1 significantly suppressed the proliferation of QBC939 cells compared with siNC and non-transfected control groups as measured by CCK-8 assay. *P<0.05, **P<0.01. |
Silencing RIPK1in QBC939 Inhibited Tube Formation In HDLECs In Vitro
To evaluate the role of RIPK1 in lymphangiogenesis, RIPK1 silenced QBC939 cell groups were co-cultured with HDLECs in a three-dimensional co-culture system. Tube formation, which was highest after 6 hrs of co-culturing QBC939 with HDLECs, was significantly attenuated in the siRIPK1 groups (Figure 4A and B).
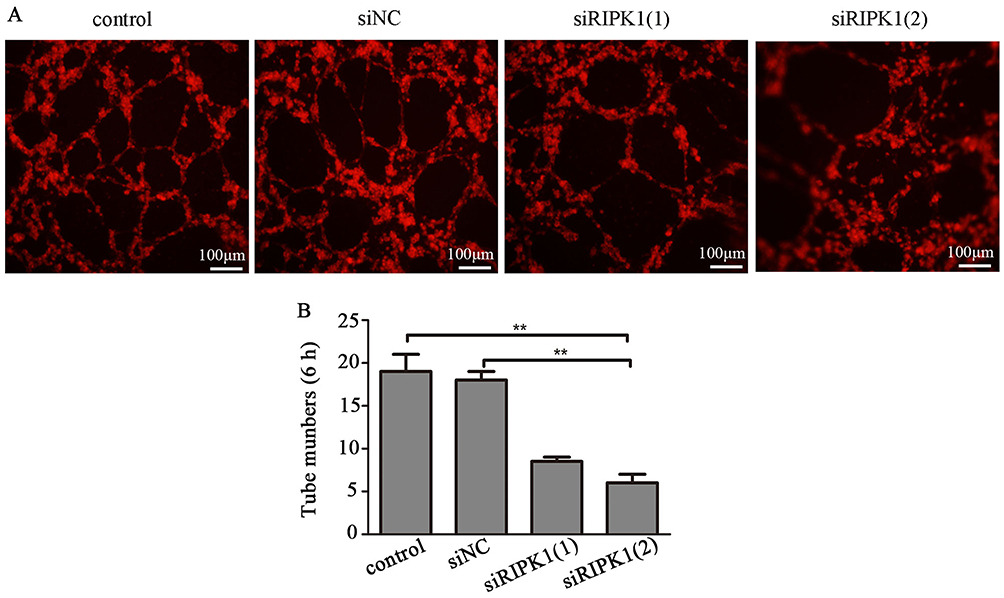 |
Figure 4 Knockdown of RIPK1 expression inhibits tube formation. (A) Dil-labeled HDLECs were co-cultured with siRIPK1-1, siRIPK1-2, siNC or non-transfected QBC939 cells for 6 hrs. (B) Tube formation was analysed under a fluorescence microscope. **P<0.01. |
Abrogation Of RIPK1 Reduced AP-1 Activation By JNK And P38MAPK Signaling
The involvement of RIPK1 in activation of AP-1 in CCA was assessed by luciferase reporter assay. Results showed that AP-1 activity was lower in the siRIPK1-1 and siRIPK1-2 groups than the siNC and non-transfected control groups (Figure 5A). Western blotting showed that the silencing of RIPK1 decreased JNK, P38MAPK, AP-1 and P-AP-1 expression, but did not affect ERK1/2 expression (Figure 5B–H). Furthermore, silencing of RIPK1 decreased N-cadherin and increased E-cadherin expression – both known markers of epithelial–mesenchymal transition (EMT). Moreover, we observed that AP-1 activity was significantly attenuated with JNK and P38MAPK inhibitors (50μM PD98059 and 10μM SP600125) treatment in CCA cells (Figure 5I). These data clearly illustrated that silencing RIPK1 regulated AP-1 expression through JNK and P38MAPK pathways, and also negatively regulated EMT in CCA.
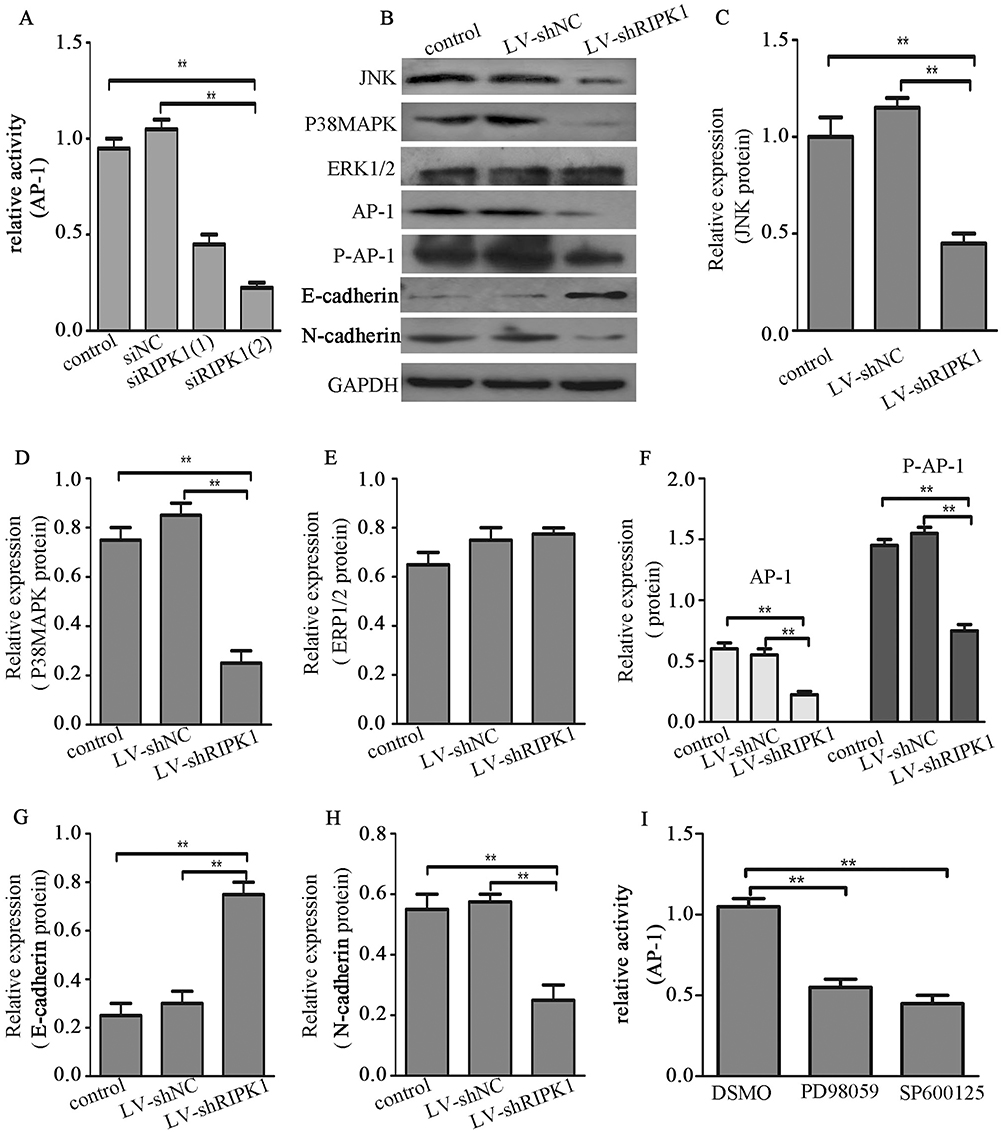 |
Figure 5 RIPK1 induces AP-1 activation via JNK and P38MAPK signaling. (A) Luciferase reporter assay for AP-1 activity in siRIPK1-1, siRIPK1-2, siNC transfected and non-transfected QBC939 cells. (B-H) Western blots showing JNK, ERK1/2, P38MAPK, AP-1, P-AP-1, E-cadherin and N-cadherin protein expression in LV-shRIPK1, LV-shNC transfected and non-transfected QBC939 cells. (I) Luciferase reporter assay for AP-1 activity in QBC939 cells treatment with 50μM PD98059 and 10μM SP600125. **P<0.01. |
Silencing Of RIPK1 Reduced VEGF-C Via AP-1 Signaling
Studies have demonstrated that VEGF-C is intimately associated with lymphangiogenesis and lymph node metastasis in CCA.24,25 We found that silencing RIPK1 decreased the protein expression of VEGF-C in CCA cells (Figure 6A–C). Furthermore, T-5224, an AP-1 inhibitor, significantly inhibited the expression of VEGF-C (Figure 6A–C). Finally, both silencing VEGF-C and inhibiting AP-1 by T-5224 significantly decreased tube formation in HDLECs (Figure 6D and E).
 |
Figure 6 RIPK1 promotes lymphangiogenesis by AP-1/VEGF-C signaling. (A–C) Expression of VEGF-C in RIPK1 silenced QBC939 cell (LV-shRIPK1, LV-shNC and control) and T-5224 treated QBC939 cells was measured by Western blotting or ELISA. (D, E) siVEGF-C transfected or T-5224 treated QBC939 cells were co-cultured with HDLECs, and tube formation in HDLECs was observed by fluorescence labeling and microscopy, and tubes number was counted. **P<0.01. |
Silencing RIPK1 Inhibited Lymphangiogenesis In Vivo
To further evaluate the role of RIPK1 on lymphangiogenesis in vivo, we generated a orthotopic CCA model in null mice by transplanting QBC939 LV-shRIPK1, LV-shNC and control cells (Figure 7A and B). Visual confirmation of lymph node metastasis was further verified by H&E staining (Figure 7C). As shown in Figure 7D and E, the number of tumor lymphatic vessels (LVN), detected by staining for LYVE-1, was remarkably reduced in the LV-shRIPK1 group compared with the LV-shNC or the control group.
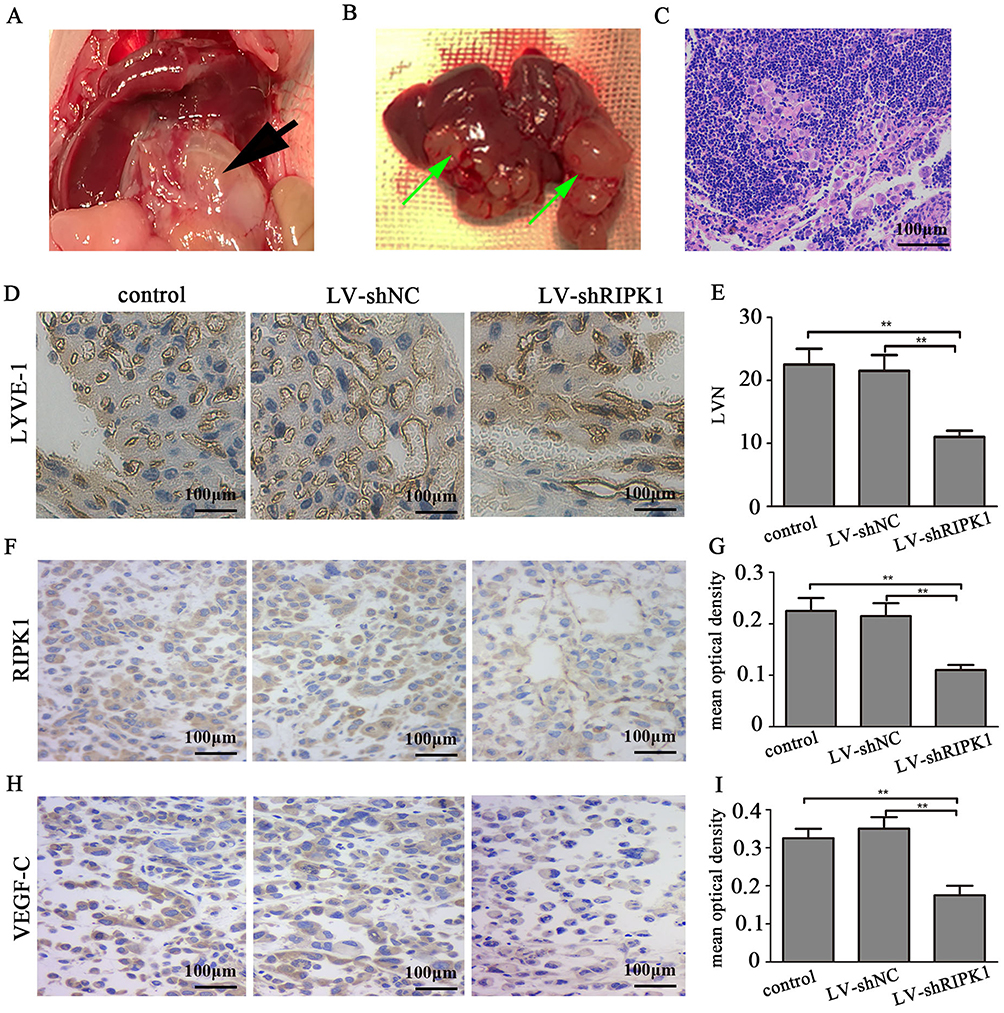 |
Figure 7 Silencing RIPK1 attenuates lymphangiogenesis in vivo. (A, B) Orthotopic transplantation model of CCA in null mice, primary lesions (black arrow) and hepatic metastases (green arrow). (C) Representative images of hematoxylin and eosin stained sections showing lymph node metastasis (100X). (D) Representative images of IHC sections showing lymphatic vessels (200×). (E) Quantification of lymphatic vessels. (F, H). Representative images RIPK1 and VEGF-C in tumors (200×). (G, I) Quantification of mean optical density of RIPK1 and VEGF-C. **P<0.01. |
Also, protein expression of RIPK1 and VEGF-C in the orthotopic tumors was remarkably reduced in the LV-shRIPK1 group compared with the LV-shNC or the control group (Figure 7F–I).
Discussion
RIPK1 is a critical regulator of multiple cell processes, including cell survival, apoptosis and necrosis in the inflammatory signaling pathway.15,26 Emerging evidence suggests that RIPK1 may be a key factor in the development and survival of the immune lymphatic system, implying its dominant physiological role in cell survival.27 However, the role of RIPK1 in tumorigenesis is controversial. Some studies have found that RIPK1 plays a dominant role in promoting liver cancer cell death and prevents liver cirrhosis from progressing to liver cancer,28 while other studies have implied that it is an initiator of several types of breast cancers,22 malignant melanoma20 and gallbladder.21 However, its role in CCA is currently unclear.
In the present study, we found that RIPK1 was highly expressed in human CCA and associated with poor prognosis, which was similar to its role in melanoma20 and gallbladder carcinoma.21 Silencing of RIPK1 significantly reduced the proliferative and lymphangiogenicity of CCA cells through AP-1 pathway. Taken together, these findings indicated that RIPK1 was an important factor in the occurrence of CCA.
Lymph node metastasis, an early event, is an important characteristic of epithelial tumors, and lymphangiogenesis is a significant mechanism of lymphatic metastasis.29,30 It was known that CCA is a malignant tumor originating from the bile duct epithelium. We observed that higher expression of RIPK1 in CCA tissue specimens tended to mean broader lymph node metastasis, and silencing RIPK1 inhibited the formation of lymphatic vessels in CCA cells in vitro, which was consistent with its role in gallbladder carcinoma31 and breast cancer.22 These results suggested that RIPK1 played an important role in lymphatic metastasis of epithelial tumors including CCA, and may provide an effective interventional target.
AP-1 activation can directly promote tumor proliferation and metastasis.18 Early studies have supported the role of RIPK1 in the activation of AP-1 signaling in mouse fibroblast cells.32 However, whether RIPK1 could promote AP-1 activation in CCA was unknown. In this study, firstly, RIPK1 silencing decreased AP-1 expression and activity in CCA cells. Secondly, JNK and P38MAPK, but not the ERK1/2 was involved in the RIPK1-AP-1 signaling in CAA cells. This was in accordance with a previous report that the MAPK family (JNK, P38MAPK and ERK1/2) is the predominant upstream regulator of AP-1 activation.33 In summary, these results demonstrated that RIPK1 can enhance AP-1 activation in CCA by activating the JNK and P38MAPK pathway.
Related studies have shown that EMT is one of the important mechanisms of local invasion, and distant metastasis of epithelial-derived tumor cells, including CCA.34,35 Present findings showed that knockdown of RIPK1 reduced the occurrence of EMT. Results also indicated that RIPK1-JNK/P38MAPK-AP-1 pathway may be an important regulator of metastasis in CCA. However, further research is required to fully understand the relationship between RIPK1-AP-1 pathway and EMT in CCA.
Elevated RIPK1 was closely related to lymph node metastasis in CCA specimens in our study, and overexpression of VEGF-C was also confirmed to be associated with lymphatic metastasis in CCA in a previous study.25,36 Therefore, we speculated that RIPK1 may affect lymphangiogenesis by regulating VEGF-C. In support of this hypothesis, we found that silencing of RIPK1 significantly decreased VEGF-C expression in vitro. Based on the relationship between RIPK1 and AP-1, the expected reduction in the expression of VEGF-C protein in response to an AP-1 inhibition further strengthened our hypothesis that AP-1 may also participate in RIPK1-mediated VEGF-C activation. Subsequently, we confirmed that the inhibition of VEGF-C and AP-1 in CCA cells decreased tube formation of HDLECs. We concluded that RIPK1 plays an important role in lymphangiogenesis and lymph node metastasis in CCA through AP-1/VEGF-C signaling.
The results of the in vitro studies were verified in vivo in an orthotopic transplanted tumor model of CCA in nude mice. Immunohistochemical results confirmed that tube formation and the protein expression of RIPK1 and VEGF-C were decreased in mice transplanted with LV-shRIPK1.
In conclusion, RIPK1 is overexpressed in human CCA and associated with unfavourable prognosis. In vivo and vitro studies revealed that RIPK1 can boost proliferation and lymphangiogenesis via the AP-1 signaling. Finally, these results suggested that RIPK1 may be a potential therapeutic target for preventing proliferation and lymphatic metastasis of CCA.
Acknowledgment
Our study was supported by the Quanzhou Science and Technology Planning Project (number Z[2014]0118).
Disclosure
The authors report no conflicts of interest in this study.
References
1. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. Ca A Cancer J Clin. 2016;66(2):115–132. doi:10.3322/caac.21338
2. Saha SK, Zhu AX, Fuchs CS, Brooks GA. Forty-year trends in cholangiocarcinoma incidence in the U.S.: intrahepatic disease on the rise. Oncologist. 2016;21(5):594–599. doi:10.1634/theoncologist.2015-0446
3. Rizvi S, Khan SA, Hallemeier CL, Kelley RK, Gores GJ. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol. 2018;15(2):95–111. doi:10.1038/nrclinonc.2017.157
4. Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet. 2014;383(9935):2168–2179. doi:10.1016/S0140-6736(13)61903-0
5. Everhart JE, Ruhl CE. Burden of digestive diseases in the United States part III: liver, biliary tract, and pancreas. Gastroenterology. 2009;136(4):1134–1144. doi:10.1053/j.gastro.2009.02.038
6. Kitagawa Y, Nagino M, Kamiya J, et al. Lymph node metastasis from hilar cholangiocarcinoma: audit of 110 patients who underwent regional and paraaortic node dissection. Ann Surg. 2001;233(3):385–392. doi:10.1097/00000658-200103000-00013
7. Yoon JH, Higuchi H, Werneburg NW, Kaufmann SH, Gores GJ. Bile acids induce cyclooxygenase-2 expression via the epidermal growth factor receptor in a human cholangiocarcinoma cell line. Gastroenterology. 2002;122(4):985–993. doi:10.1053/gast.2002.32410
8. Chan-On W, Nairismägi ML, Ong CK, et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat Genet. 2013;45(12):1100–1474. doi:10.1038/ng.2806
9. Voss JS, Holtegaard LM, Kerr SE, et al. Molecular profiling of cholangiocarcinoma shows potential for targeted therapy treatment decisions. Hum Pathol. 2013;44(7):1216–1222. doi:10.1016/j.humpath.2012.11.006
10. Dalgleish AG, Ken O. Byrne. Inflammation and Cancer. Nature. 2002;420(6917):860–867. doi:10.1038/nature01322
11. Chapman MH, Webster GJ, Bannoo S, Johnson G, Wittmann J, Pereira SP. Cholangiocarcinoma and dominant strictures in patients with primary sclerosing cholangitis; a 25 year single centre experience. Eur J Gastroenterol Hepatol. 2012;24(9):1051–1058. doi:10.1097/MEG.0b013e3283554bbf
12. Chen TC, Jan YY, Yeh TS. K-ras mutation is strongly associated with perineural invasion and represents an independent prognostic factor of intrahepatic cholangiocarcinoma after hepatectomy. Ann Surg Oncol. 2012;19(3):675–681. doi:10.1245/s10434-012-2224-7
13. Sasaki M, Nakanuma Y. Cellular senescence in biliary pathology. Special emphasis on expression of a polycomb group protein EZH2 and a senescent marker p16INK4a in bile ductular tumors and lesions. Histol Histopathol. 2014;30(3):267.
14. Al-Bahrani R, Abuetabh Y, Zeitouni N, Sergi C. Cholangiocarcinoma: risk factors, environmental influences and oncogenesis. Ann Clin Lab Sci. 2013;43(2):195–210.
15. Christofferson DE, Li Y, Yuan J. Control of life-or-death decisions by RIP1 kinase. Annu Rev Physiol. 2014;76:129–150. doi:10.1146/annurev-physiol-021113-170259
16. Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol. 2013;14(11):727–736. doi:10.1038/nrm3683
17. Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2(4):301. doi:10.1038/nrc780
18. Matthews CP, Colburn NH, Young MR. AP-1 a target for cancer prevention. Curr Cancer Drug Targets. 2007;7(4). doi:10.2174/156800907780809723
19. Haibing Z, Xiaohui Z, Thomas MQ, Jinghe L, Francis Ka-Ming C, Jianke Z. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature. 2011;471(7338):373–376. doi:10.1038/nature09878
20. Liu XY, Lai F, Yan XG, et al. RIP1 kinase is an oncogenic driver in melanoma. Cancer Res. 2015;75(8):1736–1748. doi:10.1158/0008-5472.CAN-14-2199
21. Zhu G, Chen X, Wang X, et al. Expression of the RIP-1 gene and its role in growth and invasion of human gallbladder carcinoma. Cell Physiol Biochem. 2014;34(4):1152–1165. doi:10.1159/000366328
22. Bist P, Leow SC, Phua QH, et al. Annexin-1 interacts with NEMO and RIP1 to constitutively activate IKK complex and NF-kappaB: implication in breast cancer metastasis. Oncogene. 2011;30(28):3174–3185. doi:10.1038/onc.2011.28
23. LI Cheng-Gang LC-H, Ai-Qun ZHANG, Jia-Hong DONG. Establishment of human hilar cholangiocarcinoma model in nude mice. J Chinese PLA Postgrad Med Sch. 2011;32(5):511–512.
24. Xu LB, Liu C, Gao GQ, Yu XH, Zhang R, Wang J. Nerve growth factor-beta expression is associated with lymph node metastasis and nerve infiltration in human hilar cholangiocarcinoma. World J Surg. 2010;34(34):1039–1045. doi:10.1007/s00268-010-0417-4
25. Wang WB, Li YH, Liu B, Wang HS, Cui AR, Zhnag XH. Correlation between PPARgamma and VEGF-C expression in extrahepatic cholangioadenocarcinoma (EHCAC) and their prognostic significance. Zhonghua Zhong Liu Za Zhi. 2009;31(10):773–777.
26. Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol. 2015;15(6):362–374. doi:10.1038/nri3834
27. Zhang J, Zhang H, Li J, et al. RIP1-mediated regulation of lymphocyte survival and death responses. Immunol Res. 2011;51(2–3):227–236. doi:10.1007/s12026-011-8249-3
28. Schneider AT, Gautheron J, Feoktistova M, et al. RIPK1 suppresses a TRAF2-dependent pathway to liver cancer. Cancer Cell. 2017;31(1):94–109. doi:10.1016/j.ccell.2016.11.009
29. Ran S, Montgomery KE. Macrophage-mediated lymphangiogenesis: the emerging role of macrophages as lymphatic endothelial progenitors. Cancers. 2012;4(3):618–657. doi:10.3390/cancers4030618
30. Peng LI, Shen Y. Lymphangiogenesis and lymphatic vessel remodelling in cancer. J Med Postgrad. 2015;14(3):159–172.
31. Li CZ, Jiang XJ, Lin B, et al. RIP1 regulates TNF-α-mediated lymphangiogenesis and lymphatic metastasis in gallbladder cancer by modulating the NF-κB-VEGF-C pathway. Oncotargets Ther. 2018;11:2875–2890. doi:10.2147/OTT.S159026
32. Devin A, Lin Y, Liu ZG. The role of the death-domain kinase RIP in tumour-necrosis-factor-induced activation of mitogen-activated protein kinases. EMBO Rep. 2003;4(6):623–627. doi:10.1038/sj.embor.embor854
33. Robinson MJ, Stippec SA, Goldsmith E, White MA, Cobb MH. A constitutively active and nuclear form of the MAP kinase ERK2 is sufficient for neurite outgrowth and cell transformation. Curr Biol. 1998;8(21):1141–1150. doi:10.1016/s0960-9822(07)00485-x
34. Ombrato L, Malanchi I. The EMT universe: space between cancer cell dissemination and metastasis initiation. Crit Rev Oncog. 2014;19(5):349. doi:10.1615/CritRevOncog.2014011802
35. Suk RH, Jin-Haeng C, Kyoungbun L, et al. Overexpression of epithelial-mesenchymal transition-related markers according to cell dedifferentiation: clinical implications as an independent predictor of poor prognosis in cholangiocarcinoma. Hum Pathol. 2012;43(12):2360–2370. doi:10.1016/j.humpath.2012.07.004
36. Aishima S, Nishihara Y, Iguchi T, et al. Lymphatic spread is related to VEGF-C expression and D2-40-positive myofibroblasts in intrahepatic cholangiocarcinoma. Mod Pathol. 2008;21(3):256. doi:10.1038/modpathol.3800985
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.