")
Back to Journals » Journal of Blood Medicine » Volume 6
Recent trends in the gene therapy of β-thalassemia
Authors Finotti A, Breda L, Lederer C, Bianchi N , Zuccato C, Klenathous M, Rivella S, Gambari R
Received 28 August 2014
Accepted for publication 28 October 2014
Published 19 February 2015 Volume 2015:6 Pages 69—85
DOI https://doi.org/10.2147/JBM.S46256
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin Bluth
Alessia Finotti,1–3 Laura Breda,4 Carsten W Lederer,6,7 Nicoletta Bianchi,1–3 Cristina Zuccato,1–3 Marina Kleanthous,6,7 Stefano Rivella,4,5 Roberto Gambari1–3
1Laboratory for the Development of Gene and Pharmacogenomic Therapy of Thalassaemia, Biotechnology Centre of Ferrara University, Ferrara, Italy; 2Associazione Veneta per la Lotta alla Talassemia, Rovigo, Italy; 3Department of Life Sciences and Biotechnology, Section of Biochemistry and Molecular Biology, Ferrara University, Ferrara, Italy; 4Department of Pediatrics, Division of Haematology/Oncology, Weill Cornell Medical College, New York, NY, USA; 5Department of Cell and Development Biology, Weill Cornell Medical College, New York, NY, USA; 6Department of Molecular Genetics Thalassaemia, The Cyprus Institute of Neurology and Genetics, Nicosia, Cyprus; 7Cyprus School of Molecular Medicine, Nicosia, Cyprus
Abstract: The β-thalassemias are a group of hereditary hematological diseases caused by over 300 mutations of the adult β-globin gene. Together with sickle cell anemia, thalassemia syndromes are among the most impactful diseases in developing countries, in which the lack of genetic counseling and prenatal diagnosis have contributed to the maintenance of a very high frequency of these genetic diseases in the population. Gene therapy for β-thalassemia has recently seen steadily accelerating progress and has reached a crossroads in its development. Presently, data from past and ongoing clinical trials guide the design of further clinical and preclinical studies based on gene augmentation, while fundamental insights into globin switching and new technology developments have inspired the investigation of novel gene-therapy approaches. Moreover, human erythropoietic stem cells from β-thalassemia patients have been the cellular targets of choice to date whereas future gene-therapy studies might increasingly draw on induced pluripotent stem cells. Herein, we summarize the most significant developments in β-thalassemia gene therapy over the last decade, with a strong emphasis on the most recent findings, for β-thalassemia model systems; for β-, γ-, and anti-sickling β-globin gene addition and combinatorial approaches including the latest results of clinical trials; and for novel approaches, such as transgene-mediated activation of γ-globin and genome editing using designer nucleases.
Keywords: Thalassemia, gene therapy, HbF induction, transcription factors, induced pluripotent stem cells, genome editing, TALEN, CRISPR, ZFN
Introduction
The β-thalassemias are a group of hereditary hematological diseases caused by over 300 mutations of the adult β-globin gene,1 with excellent reviews providing background information outlining genetics,2–4 pathophysiology,5,6 and therapeutics7 of β-thalassemia that is beyond the scope of this review. In brief, β-thalassemia is brought about by mutations reducing or abrogating β-globin expression, which thus lead to reduced adult hemoglobin ([HbA] an α2β2 heterotetramer) and excess α-globin content in erythroid cells, in turn resulting in ineffective erythropoiesis and apoptosis in the erythroid lineage.3,8,9 Most β-thalassemia patients therefore require lifelong clinical management by blood transfusion and chelation therapy,10–12 with a few having the option of curative but potentially hazardous allogeneic transplantation of hematopoietic stem and progenitor cells (HSPCs) instead.13,14 This indicates the need for alternative therapies, and the observation that high levels of the fetal β-globin-like γ-globin chain result in an ameliorated β-thalassemia phenotype15 has prompted the search for γ-globin-inducing chemical agents.16–21 Patient response to known γ-globin inducers, however, is varied,22 and the search continues for reagents with higher efficiency, consistency, and tolerability in chronic application,23 if not to cure the disease, then to reduce transfusion requirements and the significant cost of disease management. Of note, hemoglobinopathies, such as the thalassemia syndromes and sickle cell anemia (SCA; caused by the toxic β-globinE6V mutation), most severely affect low-income countries, where the lack of prevention programs and an underlying high carrier rate bring about high disease frequencies,24 although global migration has now turned hemoglobinopathies into a concern for many nonendemic countries as well.25 Globally, β-thalassemia mutations introducing gene deletions, aberrant splicing, or premature stop codons have the greatest impact in terms of global disease burden and clinical severity.26,27 Recent progress in the research of disease modifiers,28 chemical modulation of gene expression,22,29 and tools and approaches for DNA-based therapies30,31 have opened new avenues toward novel and more personalized strategies to manage or cure β-thalassemia, as we have reviewed recently.23,32 Particularly with regard to curative approaches by gene therapy, the field has come to a crossroads, with the initiation of clinical trials, the possible plateauing off of incremental improvements to gene augmentation therapy, and the increasing preclinical application of novel genome-editing tools. The objective of the present manuscript is to review the most relevant findings published in the period 2005–2014 concerning the preclinical and clinical application of gene therapy for β-thalassemia. To this end, we will describe the pertinent model systems, β-like-globin gene-addition strategies, gene addition in combination with chemical inducers of γ-globin, transgene-mediated activation of endogenous γ-globin, and the emerging use of designer nucleases for β-thalassemia gene therapy. The general flow of gene-therapy-based approaches for β-thalassemia is outlined in Figure 1.
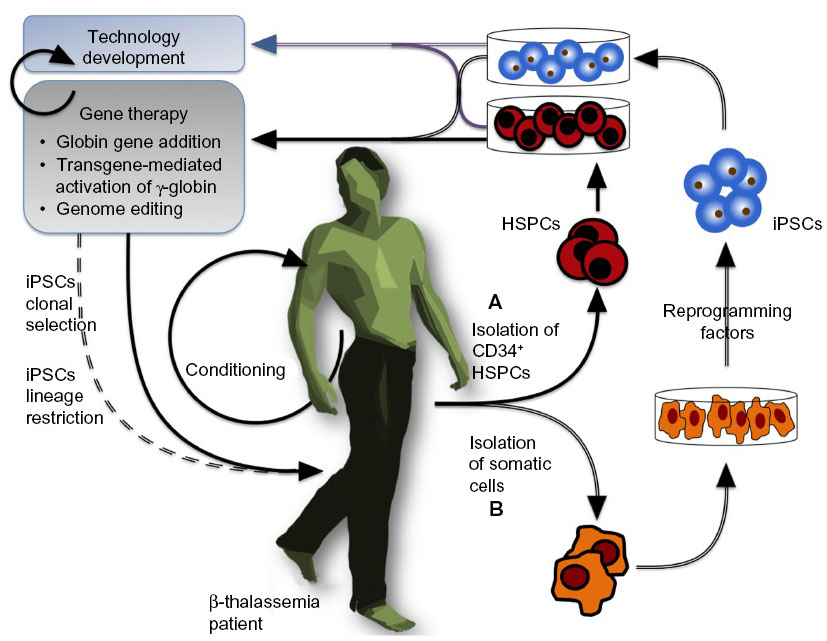 | Figure 1 General view of a gene-therapy approach for β-thalassemia. |
Experimental model systems
Several experimental systems have been developed to establish the suitability of and provide proof of principle for gene-therapy approaches to β-thalassemia. Erythroid cell lines, such as human and murine erythroleukemia cells, allow cost-effective high-throughput assessments in the erythroid lineage,33–36 and cancer-prone mouse models have been instrumental in gauging the genotoxicity of integration and genome-modification events for vector classes applied to β-thalassemia.37–39 The most informative functional studies of candidate therapies toward their clinical application, however, instead rely on thalassemic human stem cells for in vitro assessments of authentic human responses and on thalassemic murine models for long-term systemic assessments in vivo.
In vitro experimental systems: erythroid precursor cells from β-thalassemia patients
HSPCs are the substrate for clinical gene-therapy application, so that in vitro assessment of HSPC-derived erythroid precursor cells (ErPCs) is highly informative for toxicity and efficacy of any therapeutic intervention (Figure 1A). ErPCs from peripheral blood are widely used, while access to bone marrow and mobilized blood,40,41 which, incidentally, contain the cells preferentially used in clinical applications,42,43 is more restricted. Using peripheral-blood-derived ErPCs, it is possible to obtain large cultures of relatively pure and synchronized erythroid cell populations in which compounds can be added at specific stages of maturation. In the procedure developed by Fibach et al,44,45 the culture is divided into two phases: first, an erythropoietin (EPO)-independent proliferation phase, in which peripheral blood cells are first cultured in the presence of a combination of growth factors, but in the absence of EPO; and, second, a differentiation phase, when the culture, supplemented with EPO, generates orthochromatic normoblasts and enucleated erythrocytes, with cells decreasing in size and accumulating hemoglobin (Hb) and large cellular clusters assuming a reddish color and giving brown-red pellets upon centrifugation.45,46 This system recapitulates many aspects of in vivo erythropoiesis, including globin RNA metabolism, cell cycle kinetics, expression of cell surface antigens, iron and ferritin metabolism, and recruitment of transcription factors,45 and allows analysis of Hb content by a variety of techniques, such as alkaline denaturation, benzidine staining, capillary electrophoresis, cation-exchange high-performance liquid chromatography for hemoglobins, and reversed-phase high-performance liquid chromatography for globin chains.45,46
In vitro experimental systems: human embryonic stem cells and induced pluripotent stem cells
Human embryonic stem cells (hESCs) have been used extensively to study the early phases of hematopoietic and erythroid development.47 In this approach, after 5 to 7 days of in vitro cell culture, a blastocyst is generated, showing a clearly visible and easily accessible inner cell mass, from which pluripotent stem cells can be isolated, giving rise to in vitro hESC lines. From these cell lines, embryoid bodies can be developed and used for further tissue-specific differentiation. hESCs themselves have only a minor role in the preclinical study of therapies for hemoglobinopathies,48,49 and their clinical application would suffer due to the ethical repugnance of their origin and from the same incompatibilities seen for allogeneic HSPC transplantations. However, the underlying hESC methodology is being reemployed in the culture of induced pluripotent stem cells (iPSCs),50–53 which closely mimic hESCs and represent a potential cornucopia for cell-based therapies in general. The creation of iPSCs from somatic cells with the use of reprogramming factors (originally Oct3/4, Sox2, c-Myc, and Klf450) represented a paradigm shift in our understanding of developmental biology and in the conception of novel therapeutic approaches, not least because their use avoids the ethical concerns associated with hESCs and creates a patient-specific, histocompatible substrate for cell therapy. Human iPSCs retain embryonic and fetal characteristics of gene expression even upon erythroid differentiation in vitro, so that the hope arose that patient-derived iPSCs for β-thalassemia or SCA might be therapeutic in their own right via the maintenance of high levels of γ-globin expression (Figure 1B).54,55 However, according to recent in vivo findings after transplantation into immunodeficient mice, in which a gradual switch to the adult β-globin gene was observed,56,57 this hope appears to be unfounded. Notwithstanding this apparent setback, iPSCs are a promising substrate for gene therapy, as they can be amplified in vitro indefinitely (where they are, alas, still subject to the same mutation rates and potentially undesirable changes as any other cell type) and thus allow the clonal selection of rare events of therapeutic interest. Since its inception, iPSC technology has been used extensively in innovative studies on β-thalassemia and other hemoglobinopathies, as will be detailed for specific gene-addition and genome-editing approaches.
In vivo experimental systems: mouse models
Thalassemic mouse models provide the most economical option for gauging the putative and systemic effects of gene-therapy approaches in thalassemic patients. Of note, the regulation of β-like globin chains in humans comprises a switch in utero from the embryonic (ε) to the fetal (γ) chain, followed by an HbA switch perinatally up to 6 months after birth,58,59 which also allows the birth and early postnatal development of homozygous β0 patients without disease management. In contrast, the murine β-globin locus encodes four functional β-like globin genes: βh1 and εy (transcribed only during the embryonic phase of development up to E14–E15 of a total gestation period of approximately 21 days), and the b1 (βmajor) and b2 (βminor) genes, which are transcriptionally activated in utero around 11 days after conception.59 Accordingly, mice homozygous for (β0) mutations that prevent expression of the adult β-globin genes die perinatally, owing to a complete lack of expression of any Hb.59 The most widely used, non-humanized adult murine models of β-thalassemia therefore need to retain some β-globin expression and thus show features similar to those observed for β-thalassemia intermedia patients, who carry moderate to mild (β+) mutations,60 although a β0 surgical model of murine β-thalassemia major has also been developed.60,61
In order to test the activity of novel mutation-specific approaches in vivo, humanized mouse models needed to be developed,58 with those combining absence of murine β-like globin genes with the presence of a human β-globin gene cluster and mutated β-globin gene being of the greatest utility. For instance, Vadolas et al62 reported generation of a humanized mouse model carrying the common β+ IVSI-110 splicing mutation on a bacterial artificial chromosome carrying the human β-globin locus. Comparison of heterozygous β-globin knockout mice carrying either the IVSI-110 or the normal human β-globin locus showed a 90% decrease in human β-globin chain synthesis in the IVSI-110 mouse model. The model, moreover, accurately recapitulates the splicing defect found in β-thalassemia patients and is thus a suitable platform on which to test approaches for the restoration of normal splicing. Similarly, a humanized mouse model carrying the common G26A (HbE) mutation, frequently co-inherited with β-thalassemia in Southeast Asia, has been developed, which allows in vivo analysis in mouse of therapies for HbE/β-thalassemia.63 Mouse models (whether of a wild-type or thalassemic background) carrying all or parts of the human β-globin locus have also proven an essential resource for the analysis of globin switching and therapeutic approaches for β-thalassemia.64–66 Finally, a keen interest in the study of developmental gene regulation, γ-globin induction, and therapies for β-thalassemia major has prompted the development of further humanized transgenic mice as models for β-thalassemia major.67 These mice carry a mutated human β-globin gene and are born viable due to the prolonged expression of human fetal hemoglobin (HbF), but require chronic transfusion for survival and are not yet widely available in the community.67–69
Globin gene addition
Over the last 2 decades, major efforts have been made to achieve therapeutic levels of exogenous β-like globin chains in β-thalassemia and SCA. These finally came to fruition when a switch from γ-retroviral vectors to lentiviral vectors allowed the efficient transduction of nondividing cells with a sufficiently large expression cassette,70 encouraging numerous research groups to work toward vectors expressing β-globin, anti-sickling variants of β-globin and γ-globin.
Lentiviral expression of exogenous β-globin
The efforts of the groups working in this field have been dedicated to achieving highly efficient and stable transduction of HSPCs, to optimizing transgene expression (erythroid- and stage-specific, elevated, position-independent, and sustained over time), and to correcting the β-thalassemia phenotype in preclinical models with minimal genotoxicity.35,36,40,42,71–75 While the field has reached a high level of optimization, incremental improvements to procedures and vectors continue to be made. These include the use of rapamycin to enhance LV transduction76 and the recent inclusion of chromatin opening elements77–79 or an ankyrin insulator72 for improved vector-derived expression, with an ongoing search for and evaluation of alternative insulators80 to prevent transgene silencing and minimize host gene perturbation while avoiding the reduction of vector titer during production that is associated with the most widely used chicken HS4 insulator.81 It has also been demonstrated that, in order to avoid insertional mutagenesis, it is possible to select suitable clones with insertions in inert (“safe harbor”) genome sites, in combination with iPSC technology.82 Several recent reviews on gene therapy of thalassemia and related hemoglobinopathies point out the state of the art with respect to the structure of β-globin-carrying lentiviral vectors,70,83,84 and Figure 2A depicts a number of therapeutic lentiviral vectors that produce high levels of β-globin in human or murine β-thalassemic erythroid cells.35,36,40,42,71–75 Figure 2B and C illustrates how one such vector, T9W, generates high-level HbA production in ex vivo culture of HSPC-derived cells isolated from a β0-thalassemia patient upon differentiation.46
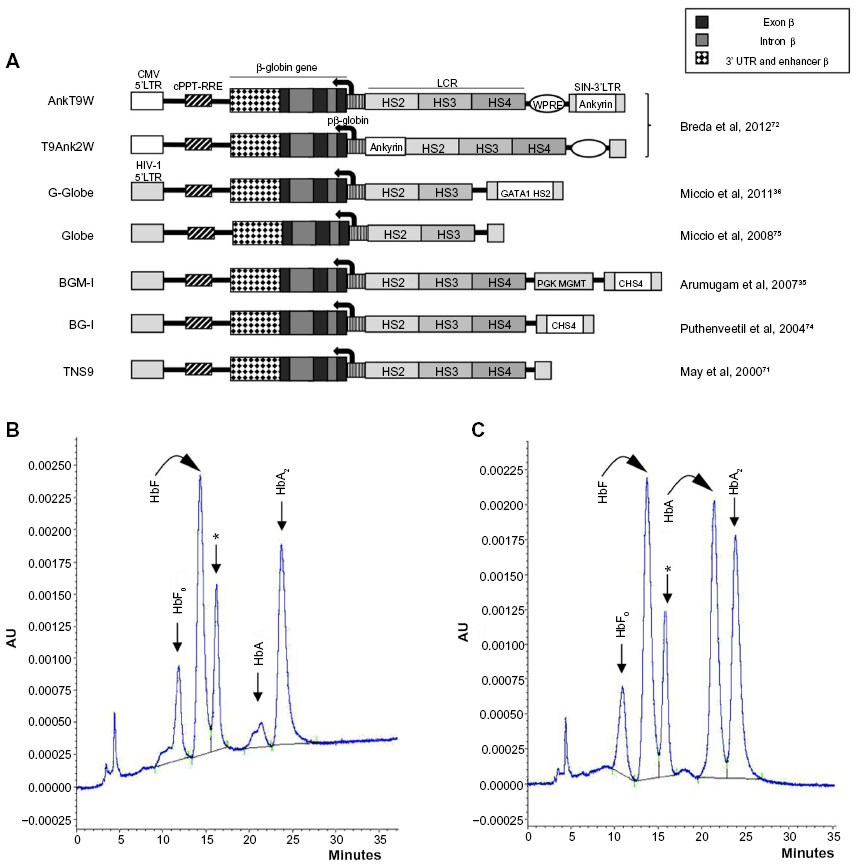 | Figure 2 Lentiviral vectors expressing exogenous β-globin. |
Lentiviral expression of anti-sickling β-globin and exogenous γ-globin
With a view to applying the same vector for β-thalassemia and SCA, β-globin vectors have also been modified to approach or even exceed the anti-sickling activity shown by γ- and δ-globin. Of note are the HPV569 and BB305 LentiGlobin® vectors,85,86 which feature in βE/β0 clinical trials86–89 (see Clinical trials below) and carry β-globinT87Q, and which are expected to provide some anti-sickling activity90 and thus be suitable for SCA therapy. Particularly important in this context, however, are the anti-sickling β(AS3) β-globin designed by Townes et al91,92 and pertaining lentiviral vectors.93 The combination of three amino acid changes (creating the artificial β-globinG16D, E22A, T87Q variant) confers anti-sickling activity exceeding that of γ-globin to β(AS3) and therefore renders the mutant transgene particularly suitable for the therapy of SCA and β-thalassemia/β-globinE6V compound heterozygotes. Independently, and primarily with clinical application for SCA in mind, numerous groups have also developed retroviral vectors encoding γ-globin instead of β-globin (Figure 3).41,94–101 Of note, Wilber et al41 used lentiviral vectors encoding the human γ-globin gene with or without an insulator, which were tested on erythroid progeny of normal CD34+ cells and resulted in high levels of HbF production, suggesting that lentiviral-mediated treatments have the potential to provide therapeutic HbF levels to patients. These findings are corroborated by several independent research groups that work on γ-globin-based lentiviral (and γ-retroviral) therapy of SCA and β-thalassemia.41,94–101 Figure 3A shows the structure of some of the corresponding vectors.41,96–99 All vectors intended for gene augmentation described here, be it for the expression of β-globin, anti-sickling β-globin, or γ-globin, have overlapping fields of application. Further preclinical and clinical studies will show which vector may be most suitable for specific disease conditions, with the vector itself as a key factor, but with all components of the treatment protocol, including conditioning, HSPC source and isolation, transduction protocol, and general culture conditions, playing a critical role in the outcome and in the comparison of vector performance.
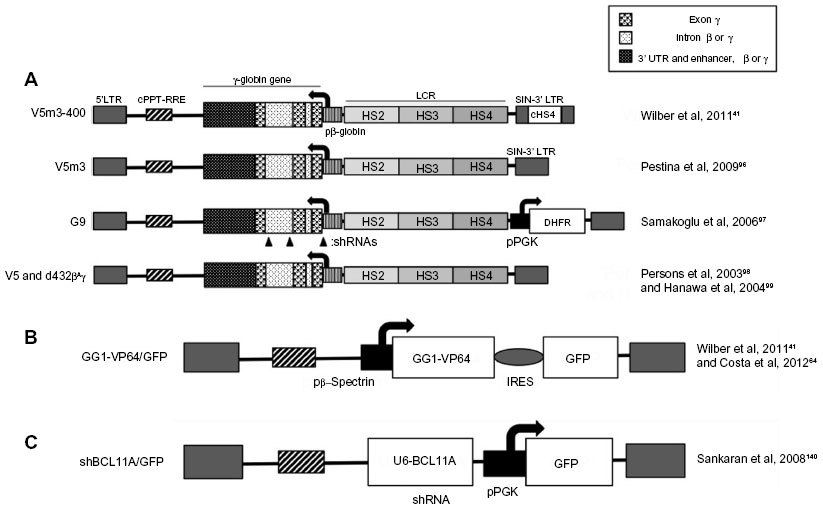 | Figure 3 Lentiviral vectors expressing exogenous γ-globin or inducing endogenous γ-globin. |
Combination therapy of gene addition with HbF inducers
Induction of endogenous HbF is one of the most widely applied therapeutic strategies for β-thalassemia and SCA, as indicated by several recent studies and reviews.102–110 Lending additional significance to preclinical studies, it has been shown that the level of γ-globin mRNA and in vitro induction of HbF in primary ErPCs isolated from β-thalassemia patients is predictive of the hydroxyurea response in vivo.111,112 While most of the recent studies in the field still focus on low-molecular-weight HbF inducers,102–110,113 the innovative strategy of combining them with vector-derived β-globin has lately been investigated and reviewed.30,114 The combined treatment induces an increase of both HbA (by gene addition) and HbF (by chemical HbF induction) with important therapeutic implications, given that β-like globin transfer in some β-thalassemia major ErPCs has been unable to reach physiological levels of Hb in vitro and might thus only lead to partial phenotypic correction in vivo as well. Since increased production of HbF in β-thalassemia is undoubtedly beneficial, the one-off application of gene therapy combined with chronic application of HbF inducers appears to be a pertinent strategy to achieve clinical benefits not achievable with either strategy alone. Representative results for this approach are depicted in Figure 4 on ErPCs from a β0-thalassemia patient carrying the codon-39 stop codon mutation (β039) (Figure 4A) and a β039/β-IVSI-110-thalassemia patient (Figure 4B). The results demonstrate that this combination strategy achieves high levels of functional Hb in β-thalassemic cells and a concomitant sharp decrease of excess α-globin, with significant scope for further improvements for what is as yet a nascent field of research.
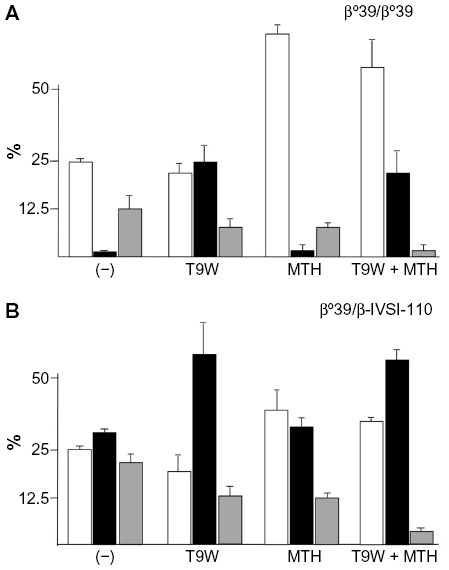 | Figure 4 Combination therapy using lentiviral-vector-derived β-globin expression and chemical γ-globin induction. |
Clinical trials
To date, there are a total of seven patients who have been treated successfully or for whom longer follow-up is pending in three clinical trials for β-thalassemia, all of which have used β-globin-expressing lentiviral vectors. The first successful gene therapy trial for β-thalassemia85 was reported in the manuscript published by Cavazzana-Calvo et al in 201087 and commented on by Kaiser.115 The pertaining β-globinT87Q vector (LentiGlobin® HPV569) holds a tandem copy of the 250-bp cHS4 insulator in its 3′ long terminal repeat (LTR) as a safety feature and bears a T87Q amino acid, which, besides its conferring anti-sickling activity,90 makes it distinguishable from transfusion-derived β-globin and thus allows the quantification of vector-derived β-globin during follow-up. Three patients with severe βE/β0-thalassemia have been treated to date. In the first patient, engraftment of treated bone marrow failed after full myeloablation, requiring reinfusion of backup bone marrow. For the second patient, however, transfusion independence was achieved at 12 months after treatment and continues to date. At 36-month follow-up, of 24 detectable clones in peripheral blood, one clone with cross-lineage dominance held a proviral integration in the high mobility group AT-hook 2 (HMGA2) gene, whose expression is associated with tumor metastasis and proliferation,116,117 in a position that removed posttranscriptional control elements and thus increased HMGA2 mRNA stability. This clone, moreover, showed a recombination event that had removed one of the cHS4 copies and possibly exacerbated transcriptional enhancement of HMGA2 from the proviral β-globin locus control region (LCR), with transcriptional and posttranscriptional effects combined resulting in 10,000-fold HMGA2 expression. Clonal dominance of this clone (peaking at 22% of nucleated cells after 48 months) dropped to 6.8% 7 years after treatment. Notably, at 36 months, only one-third of the total Hb was vector derived, with endogenous HbE and unexpectedly high HbF constituting the other two-thirds, so that the patient might have failed to become transfusion independent in the absence of endogenous HbE and elevated γ-globin expression and if mild conditioning instead of full myeloablation had been applied. Finally, engraftment with HPV569-treated cells of the third adult patient for this trial was also successful. However, the patient remains transfusion dependent, with a low vector copy number (VCN) in the originally engrafted cell material (VCN 0.3) and a low VCN in nucleated cells (VCN in neutrophils 0.016), and with vector-derived Hb accounting for only approximately 5% of total Hb more than 2 years after engraftment.88 Engraftment failure for the first patient, a low VCN for the third patient, and oligoclonal reconstitution, vector recombination, and low vector-derived gene expression for the second patient provide important pointers for necessary improvements in future trials and vectors and, moreover, call for ex vivo preclinical assessment in cells from prospective trial participants, as we argue elsewhere.72
A second clinical study (HGB-205) and follow-up to the trial described above has been initiated by bluebird bio Inc. in France and utilizes the third-generation lentiviral LentiGlobin® BB305 vector.86 Compared to HPV569, BB305 holds a cytomegalovirus (CMV) promoter instead of the U3 promoter/enhancer in its 5′ LTR and no longer bears cHS4 insulator elements in its 3′ LTR. Preliminary results obtained for two βE/β0-thalassemia patients, who had both been transfusion-dependent for most of their lives, were encouraging, with a VCN of 1.5 and 2.1, respectively, in the engrafted material and with a reported transfusion independence at 3.5 and 6.5 months, respectively, after treatment.88 This success has most recently also prompted the application of BB305 for gene therapy of SCA.89
Finally, an independent trial for globin gene transfer in adult patients with β-thalassemia major has been initiated (NCT01639690) by the Sadelain group and associates118,119 to study safety and efficacy, representing the first US trial for β-thalassemia. The β-globin vector used for the trial, TNS9.3.55, holds the cHS4 insulator and minor unpublished modifications compared to TNS9.60,71 A preclinical study testing TNS9.3.55 in patient HSPCs in vitro, by BFU-E assays, and, in vivo, using NOD-scid IL2rγnull mice, indicated high vector-derived expression (73% to 100% of normal hemizygous levels) and long-term repopulation potential (69% retention after 7 months) for vector-positive cells.42 In the ongoing clinical trial, five patients have been enrolled and three treated to date, using G-CSF-mobilized CD34+ cells and mild conditioning (8 mg/kg busulfan). Possibly owing to the latter, which reduces the risk for patients but also the level of donor chimerism and thus the overall efficiency of the approach, transfusion independence had not been reached 12 months after treatment in the first three patients, albeit with an ongoing rise of the average VCN in peripheral blood mononuclear cells (from, initially, 1% to 7%–9%) and without the emergence of clonal dominance. As of this writing, treatment of additional patients has been postponed until fuller evaluation of the first three patients can indicate whether dose escalation of the conditioning treatment might be required.
Transgene-mediated activation of endogenous γ-globin genes
Inspired by chemical induction of HbF as a therapeutic approach,102–109 and enabled by the burgeoning fields of engineered transcription factors and RNA interference,23,32 a relatively novel approach to the therapy of β-thalassemia is the transgenic activation of γ-globin, either by the overexpression of γ-globin-activating transcription factors or by the stable knockdown of γ-globin repressors.
Overexpression of γ-globin-activating transcription factors
The β-type globin genes are activated through dynamic interactions with a distal upstream enhancer, the LCR. The LCR physically contacts the developmental stage-appropriate globin gene via chromatin looping, a process partially dependent on the protein Ldb1. Deng et al showed that tethering Ldb1 to the murine β-globin promoter with a custom-designed zinc finger protein (ZF-Ldb1) can induce loop formation and β-globin transcription in an erythroid cell line.120 Further work using a similar approach showed that forced chromatin looping can be exploited to potently reactivate fetal globin gene expression in adult human erythroid cells.121 For this work, a fusion protein that brings together a zinc finger protein, which recognizes a specific sequence at the γ-globin promoter, and Ldb1 was created.120,122 Insertion of a lentiviral vector carrying this fusion protein into adult primary human erythroid cells strongly activated the γ-globin gene, whose transcription accounted for nearly 90% of total β-like globins and led to concomitant reduction of β-globin. This approach would therefore be particularly suitable for the therapy of SCA, by increasing anti-sickling γ-globin, while at the same time reducing βs expression (see also97).
Alternatively, engineered zinc-finger-based transcription factors can be used to reactivate developmentally silenced γ-globin genes in adult cells. Figure 3B shows the structure of a lentiviral vector expressing the artificial zinc finger protein GG1-VP64, which was designed to interact with the −117 region of the Aγ-globin gene proximal promoter and led to a significant increase in γ-globin gene expression in K562 cells.123 Moreover, Wilber et al124 and Costa et al64 reported increased γ-globin gene expression following transfection with GG1-VP64 constructs, with significantly increased HbF levels in CD34+ erythroid progenitor cells from normal human donors and β-thalassemia patients. These results provide new insights into the mechanism of γ-globin silencing and may translate into mechanism-based, improved therapies for β-thalassemia and related SCA.
Transgene-mediated silencing of β-thalassemia modifiers
With the advent of the concept of RNA interference, efforts began to utilize short interfering RNAs (siRNAs) and short hairpin RNAs (shRNAs) in the therapy of β-thalassemia. Early efforts, mindful of the amelioration of β-thalassemia pathology by a reduction in α-globin excess, knocked down α-globin mRNA to achieve a moderate but significant reduction in disease parameters,34,125–127 an approach superior to the alternative strategy of sequestering excess α-globin protein by overexpression of its private chaperone, AHSP.128 Recently, regulation by RNA interference has also become an option for the activation of endogenous γ-globin expression. This was made possible through a growing understanding of the regulation of globin switching, also by regulatory microRNAs (miRNAs), and of transcriptional repressors of γ-globin as therapeutic targets (see Figure 5).129–144 Among the candidate target genes for knockdown is the zinc finger transcription factor Krüppel-like factor 1 (KLF1, also known as the erythroid Krüppel-like factor, EKLF), which acts as an erythroid-specific master switch of globin gene expression145 and whose autonomy in directing globin gene expression is underlined by the observation that the mere insertion of a KLF1 binding site into the human δ-globin promoter confers developmental inducibility and a reduction of the thalassemia phenotype in mice.146 Besides KLF1, Oct-1,138 MYB,139 and BCL11A129,133,140 have been identified as repressors of γ-globin gene transcription. For instance, the zinc finger transcription factor BCL11A has recently been shown to function as a repressor of HbF expression, with transgenic deactivation of BCL11A reactivating HbF and correcting a humanized sickle Hb mouse model147 and with BCL11A knockdown leading to significant HbF induction in human cells,129,133,140,144 similar to knockdown of its positive regulator KLF1.132 Moreover, compound Klf1::Bcl11a mutant mice that carry the human β-globin locus showed further enhanced γ-globin expression compared to single-mutant animals,148 indicating that a strategy targeting both genes together (without affecting non-erythroid functions of BCL11A) might have additional therapeutic benefits in β-thalassemia. In order to move transgene-mediated activation of γ-globin from concept to therapeutic application, shRNA expression from constitutive RNA polymerase III promoters, such as the commonly used U6 promoter (see Figure 3C), needs to be avoided. To this end, Renella et al has surrounded a BCL11A-specific shRNA with the flanking sequences of a naturally occurring miRNA (miR223), allowing its (potentially regulated) expression from RNA polymerase II-driven promoters.149 Using lentiviral vectors for spleen focus-forming virus (SFFV)-promoter-driven BCL11A shRNAmiR expression in murine erythroleukemia cells, approximately 50% of control embryonic εy levels were achieved compared to the equivalent positive U6 shRNA control,150 so that controlled and stable shRNA-mediated HbF induction has achieved an efficiency of potential clinical relevance. Figure 5, in addition to transcription factors negatively regulating the expression of the γ-globin genes,151,152 shows examples of miRNAs validated as regulators of γ-globin gene expression,153–156 either directly155 or through interactions with relevant target transcription-factor mRNA,153,154,156 such as miR-15a and miR-16-1 (targeting MYB),153 miR-23 and miR-27a (targeting KLF3 and Sp1, respectively),156 and miRNA-486-3p (targeting BCL11A).154 Lentiviral vectors carrying sequences of these miRNAs are expected to lead to inhibition of γ-globin gene transcription-factor repressors and induction of HbF.41
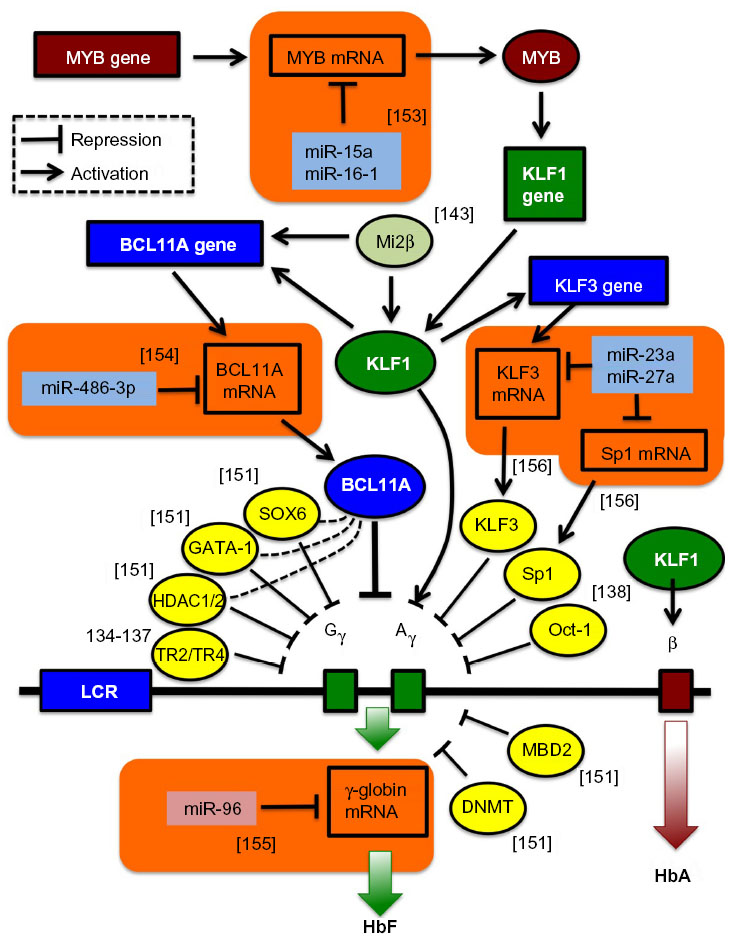 | Figure 5 Novel targets for γ-globin gene transcriptional activation. |
Genome editing
In contrast to gene-augmentation approaches, the direct DNA-level repair of primary mutations would achieve physiological levels of gene expression for each corrected cell and, in the absence of off-target activity, would altogether avoid the risk of insertional mutagenesis inherent to integrating vectors. Genome-editing approaches, however, still suffer from low efficiencies in HSPCs, which, without selection (eg, of iPSC clones) or enrichment steps, mostly precludes their clinical application for gene therapy. However, Genovese et al157 have recently achieved high-efficiency targeted DNA replacement in HSPC from controls and patients with X-linked severe combined immunodeficiency, reaching efficiencies of 3%–11% depending on the subpopulation, thus moving homology-directed gene repair of HSPCs into the realm of clinical application.
Repair of causative mutations
Endogenous genomic loci can be altered efficiently and specifically using engineered zinc finger nucleases (ZFN)158–163 and transcription activator-like effector nucleases (TALENs), as recently reported by Voit et al for the human globin locus.164 Moreover, besides ZFN and TALEN, clustered regularly interspaced short palindromic repeats (CRISPR) linked to Cas9 nuclease are now also being investigated for their utility in modifying β-globin.165,166 ZFN (Figure 6A), τALEN (Figure 6B), and CRISPR (Figure 6C) comprise a specifically engineered DNA binding domain fused to a nuclease.31 Binding of a ZFN or TALEN pair at contiguous sequences flanking a target site leads to the dimerization of the FokI (a double-stranded DNA nickase) domain, resulting in a targeted DNA double-strand break, while CRISPR/Cas9 introduces double-strand breaks as a monomer.31 To increase target-site specificity and thus reduce off-target activity, CRISPR linked to nickases and thus requiring dimerization for genome modification are also being investigated.167 The resulting double-strand break (Figure 6D) can be repaired by error-prone nonhomologous end joining or by high-fidelity homology-directed repair in the presence of a homologous DNA donor template (Figure 6E and F). In their study, Voit et al engineered a pair of highly active TALENs that induce modification of about 50% of human β-globin alleles near the site of the sickle mutation. These TALENs stimulate targeted integration of therapeutic, full-length β-globin complementary DNA to the endogenous β-globin locus in about 20% of K562 erythroleukemia cells.164
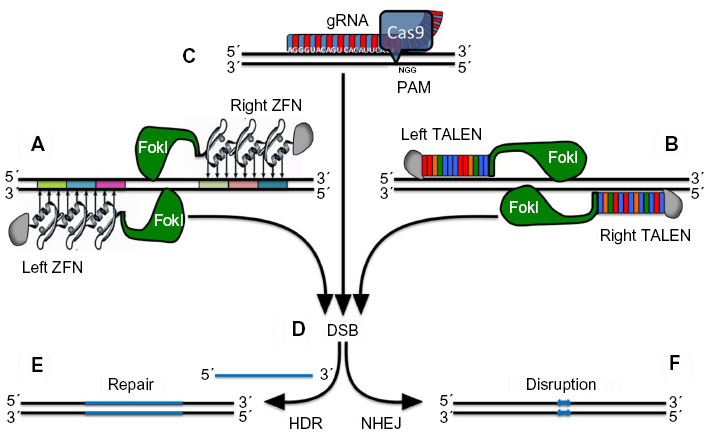 | Figure 6 Alternative strategies for targeted gene repair. |
Using patient-specific iPSCs, Ma et al168 have recently applied this technology to β-thalassemia, with Sun and Zhao169 likewise applying it to SCA patient-specific iPSCs, both groups following the idea that correction of disease-causing mutations offers an ideal therapeutic solution when iPSCs are available. In the β-thalassemia study, Ma et al described a robust process combining efficient generation of integration-free patient-specific β-thalassemia iPSCs and TALEN-based universal correction of HBB mutations in situ. Integration-free and gene-corrected iPSC lines from two patients carrying different types of homozygous mutations were generated. These iPSCs are pluripotent, have normal karyotype, and, more importantly, can be induced to differentiate into hematopoietic progenitor cells and then further to erythroblasts expressing normal β-globin. Interestingly, and of importance for any clinical application of genome-editing tools, the correction process did not generate TALEN-induced off-target mutations.168
HbF activation by genome editing
In contrast to SCA, wherein a single mutation is present in all patients, β-thalassemia is caused by a large variety of mutations, each of which would have to be corrected by an individually validated designer nuclease. The alternative and universally applicable approach of using designer nucleases to induce HbF instead is therefore an attractive option. While results for this strategy as a gene-therapy approach have, to our knowledge, not yet been published in peer-reviewed journals, it is already being employed, as patent applications for corresponding ZFN and TALEN indicate.170,171 Intriguingly, and depending on the target (such as a γ-globin repressor or its binding site), this approach might use nonhomologous end joining to disrupt the target sequence in HSPCs and thus achieve high levels of efficiency that would allow a direct translation to clinical applications for β-thalassemia.
Toward personalized therapy of thalassemia
With hundreds of primary mutations, disease modifiers, and polymorphisms linked to hereditary persistence of HbF,1 β-thalassemia patients can be stratified into clinically distinct subgroups. It is expected, therefore, that the management of β-thalassemia patients will increasingly be customized for stratified classes of β-thalassemia patients, which will also hold for intervention by gene therapy. For instance, with the objective of reaching therapeutic levels of hemoglobins, an optimized gene-therapy protocol might differ between patients with β0 genotypes (without endogenous β-globin expression, such as β039 and β0-IVSI-1 homozygotes or compound heterozygotes) and those with β+ genotypes (with residual β-globin expression, such as β+-IVSI-110 and β+-IVSI-6 homozygotes or compound heterozygotes). In this respect, the response of patients with compound heterozygote β0/β+ genotypes to exogenous β-globin expression might need careful study. Moreover, in the case of mutation-specific genome editing, considerations of personalization are inherent in the approach, while, for other approaches, these considerations might be less obvious but similarly critical. For instance, the efficiency of gene therapy based on exogenous γ-globin gene expression or on the activation of endogenous γ-globin by any of the means discussed above may be in doubt in cases where the patients involved are already expressing high endogenous levels of HbF. These considerations also hold for the combination of gene therapy and pharmacological induction of HbF detailed in the section titled “Combination therapy of gene addition with HbF inducers”, because the individual genetic composition is an important cause of variations in the response and tolerance to drug treatment, as recently reviewed.30 Pharmacogenomic-based studies have clearly demonstrated that several genomic variations (not restricted to the human β-globin gene cluster) are significantly associated with differential responses of β-globinopathy patients to treatment with chemical HbF inducers, such as hydroxyurea.172 This insight renders the use of genomic/transcriptomic analysis to predict the in vivo response and to guide the personalization of any such therapy a logical conclusion.111,112 With the same rationale, the analysis of patient-specific responses in cell culture before therapy, and, in particular, before permanent therapeutic intervention, is strongly recommended83 and will become increasingly common. This trend, combined with the ex vivo approach used for the therapy itself and with an increasing use and creation of patient-specific iPSCs (in particular for gene-correction approaches), is expected to lead to a dramatic increase in biobanking of patient-derived cells, with all the regulatory, management, and ethical issues involved.173
Conclusion
In summary, gene therapy is one of the most promising approaches for the future treatment of β-thalassemia patients and comprises several, at times complementary, strategies. The clinically most advanced approach, that of substituting nonfunctional endogenous β-globin genes with a normal β-globin gene carried by lentiviral vectors, leads to de novo production of HbA. This approach can be enhanced, as in vitro evidence indicates, by additional treatment with inducers of endogenous HbF, which is firmly established as clinically beneficial. In the same vein, numerous gene-therapy approaches also draw on HbF as a positive disease modifier, either by expressing exogenous HbF from a lentiviral vector or by inducing endogenous HbF with a variety of approaches, including the expression of exogenous artificial transcription factors or the disruption of γ-globin repressors or their binding sites. This latter approach has been made possible by an increasingly detailed understanding of globin gene regulation and by the development of rationally designed artificial nucleases for genome editing. Designer nucleases in turn now also allow gene editing of the human globin locus and thus the correction of altered β-globin genes as the most direct gene-therapy approach. As for the cellular targets of gene therapy, human erythropoietic stem cells have been considered in most studies and are still the substrate of choice for clinical applications. However, it can be expected that iPSCs from β-thalassemia patients will play an increasing role in preclinical, and possibly clinical, gene-therapy studies in the future.
As a result of all these developments, and after decades in the making, gene therapy of β-thalassemia has reached a critical phase and is beginning to live up to its long-held promise. At this privileged moment in time, the model systems and protocols are in place to test gene-therapy approaches, and the first clinical trials show therapeutic efficiency and guide our decisions for future developments, such as the choice of conditioning regimen (full or mild), the HSPC source (bone-marrow-derived or mobilized), and the inclusion of insulators for gene augmentation. Ongoing optimization of extant gene-augmentation tools and combinatorial approaches with chemical reagents are approaching therapeutic efficiency, even for severe forms of the disease. At the same time, fundamental insights into globin switching and new tools for cellular reprogramming, transcriptional regulation, posttranscriptional silencing, and genome editing have opened up as-yet uncharted territory in what has become a fast-moving and highly competitive field of research. While there is no telling which approach will win out for widespread clinical application in the course of time, vigilance, widespread competence in shared methodology, and the availability of diametrically different treatment strategies will provide the pressure and scope for fast improving efficacy and safety, for the good of the field and for the benefit of the patients.
Acknowledgments
Funded by the EU Seventh Framework Programme for research, technological development, and demonstration under grant agreement number 306201 (THALAMOSS). This work was also supported by grants from MIUR (Italian Ministry of University and Research) (RG and AF), from the Fondazione Cariparo (Cassa di Risparmio di Padova e Rovigo) (RG and AF), by Telethon grant GGP10124 (RG and AF), by the Research Promotion Foundation of Cyprus (ΥΓΕΙΑ/ΒΙΟΣ/0311(BE)/20) (CWL and MK), by grant KL2TR000458 of the Clinical and Translational Science Center at Weill Cornell Medical College (LB), by the NIH-NKLBI 1R01HL102449 grant (SR) and the Daedalus grants (SR). This research was also supported by Associazione Veneta per la Lotta alla Talassemia (AVLT), Rovigo, Italy (RG and AF).
Disclosure
The authors report no conflicts of interest in this work.
References
Kountouris P, Lederer CW, Fanis P, Feleki X, Old J, Kleanthous M. IthaGenes: an interactive database for haemoglobin variations and epidemiology. PLoS One. 2014;9:e103020. | |
Old JM. Screening and genetic diagnosis of haemoglobin disorders. Blood Rev. 2003;17:43–53. | |
Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis. 2010;5:11. | |
Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassaemia. Lancet. 2012;379:373–383. | |
Weatherall DJ. Phenotype-genotype relationships in monogenic disease: lessons from the thalassaemias. Nat Rev Genet. 2001;2:245–255. | |
Nienhuis AW, Nathan DG. Pathophysiology and clinical manifestations of the β-thalassemias. Cold Spring Harb Perspect Med. 2012;2:a011726. | |
Quek L, Thein SL. Molecular therapies in beta-thalassaemia. Br J Haematol. 2007;136:353–365. | |
Rivella S. Ineffective erythropoiesis and thalassemias. Curr Opin Hematol. 2009;16:187–194. | |
Turbpaiboon C, Wilairat P. Alpha-hemoglobin stabilizing protein: molecular function and clinical correlation. Front Biosci (Landmark Ed). 2010;15:1–11. | |
Olivieri NF, Brittenham GM. Management of the thalassemias. Cold Spring Harb Perspect Med. 2013;3. | |
Goss C, Giardina P, Degtyaryova D, Kleinert D, Sheth S, Cushing M. Red blood cell transfusions for thalassemia: results of a survey assessing current practice and proposal of evidence-based guidelines. Transfusion. 2014;54:1773–1781. | |
Poggiali E, Cassinerio E, Zanaboni L, Cappellini MD. An update on iron chelation therapy. Blood Transfus. 2012;10:411–422. | |
King A, Shenoy S. Evidence-based focused review of the status of hematopoietic stem cell transplantation as treatment of sickle cell disease and thalassemia. Blood. 2014;123:3089–3094; quiz 3210. | |
Angelucci E, Matthes-Martin S, Baronciani D, et al; EBMT Inborn Error and EBMT Paediatric Working Parties. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: indications and management recommendations from an international expert panel. Haematologica. 2014;99:811–820. | |
Thein SL, Menzel S, Lathrop M, Garner C. Control of fetal hemoglobin: new insights emerging from genomics and clinical implications. Hum Mol Genet. 2009;18:R216–R223. | |
Fibach E, Bianchi N, Borgatti M, Prus E, Gambari R. Mithramycin induces fetal hemoglobin production in normal and thalassemic human erythroid precursor cells. Blood. 2003;102(4):1276–1281. | |
Lampronti I, Bianchi N, Borgatti M, Fibach E, Prus E, Gambari R. Accumulation of gamma-globin mRNA in human erythroid cells treated with angelicin. Eur J Haematol. 2003;71:189–195. | |
Mischiati C, Sereni A, Lampronti I, et al. Rapamycin-mediated induction of gamma-globin mRNA accumulation in human erythroid cells. Br J Haematol. 2004;126:612–621. | |
Lampronti I, Bianchi N, Zuccato C, Medici A, Bergamini P, Gambari R. Effects on erythroid differentiation of platinum(II) complexes of synthetic bile acid derivatives. Bioorg Med Chem. 2006;14:5204–5210. | |
Zuccato C, Bianchi N, Borgatti M, et al. Everolimus is a potent inducer of erythroid differentiation and gamma-globin gene expression in human erythroid cells. Acta Haematol. 2007;117:168–176. | |
Lampronti I, Bianchi N, Zuccato C, et al. Increase in gamma-globin mRNA content in human erythroid cells treated with angelicin analogs. Int J Hematol. 2009;90:318–327. | |
Pourfarzad F, von Lindern M, Azarkeivan A, et al. Hydroxyurea responsiveness in β-thalassemic patients is determined by the stress response adaptation of erythroid progenitors and their differentiation propensity. Haematologica. 2013;98:696–704. | |
Finotti A, Gambari R. Recent trends for novel options in experimental biological therapy of β-thalassemia. Expert Opin Biol Ther. 2014;14: 1443–1454. | |
Colah R, Gorakshakar A, Nadkarni A. Global burden, distribution and prevention of β-thalassemias and hemoglobin E disorders. Expert Rev Hematol. 2010;3:103–117. | |
Lederer CW, Basak AN, Aydinok Y, et al. An electronic infrastructure for research and treatment of the thalassemias and other hemoglobinopathies: the Euro-mediterranean ITHANET project. Hemoglobin. 2009;33:163–176. | |
Traeger-Synodinos J, Harteveld CL. Advances in technologies for screening and diagnosis of hemoglobinopathies. Biomark Med. 2014;8:119–131. | |
Ip HW, So CC. Diagnosis and prevention of thalassemia. Crit Rev Clin Lab Sci. 2013;50:125–141. | |
Thein SL. Genetic association studies in β-hemoglobinopathies. Hematology Am Soc Hematol Educ Program. 2013;2013:354–361. | |
Suzuki M, Yamamoto M, Engel JD. Fetal globin gene repressors as drug targets for molecular therapies to treat the β-globinopathies. Mol Cell Biol. 2014;34:3560–3569. | |
Breda L, Rivella S, Zuccato C, Gambari R. Combining gene therapy and fetal hemoglobin induction for treatment of beta-thalassemia. Expert Rev Hematol. 2013;6:255–264. | |
Gaj T, Gersbach CA, Barbas CF 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. | |
Gambari R. Alternative options for DNA-based experimental therapy of β-thalassemia. Expert Opin Biol Ther. 2012;12:443–462. | |
Ryu BY, Persons DA, Evans-Galea MV, Gray JT, Nienhuis AW. A chromatin insulator blocks interactions between globin regulatory elements and cellular promoters in erythroid cells. Blood Cells Mol Dis. 2007;39:221–228. | |
Voon HP, Wardan H, Vadolas J. siRNA-mediated reduction of alpha-globin results in phenotypic improvements in beta-thalassemic cells. Haematologica. 2008;93:1238–1242. | |
Arumugam PI, Scholes J, Perelman N, Xia P, Yee JK, Malik P. Improved human beta-globin expression from self-inactivating lentiviral vectors carrying the chicken hypersensitive site-4 (cHS4) insulator element. Mol Ther. 2007;15:1863–1871. | |
Miccio A, Poletti V, Tiboni F, et al. The GATA1-HS2 enhancer allows persistent and position-independent expression of a β-globin transgene. PLoS One. 2011;6:e27955. | |
Montini E, Cesana D. Genotoxicity assay for gene therapy vectors in tumor prone Cdkn2a-/- mice. Methods Enzymol. 2012;507: 171–185. | |
Cesana D, Ranzani M, Volpin M, et al. Uncovering and dissecting the genotoxicity of self-inactivating lentiviral vectors in vivo. Mol Ther. 2014;22:774–785. | |
Nowrouzi A, Cheung WT, Li T, et al. The fetal mouse is a sensitive genotoxicity model that exposes lentiviral-associated mutagenesis resulting in liver oncogenesis. Mol Ther. 2013;21:324–337. | |
Roselli EA, Mezzadra R, Frittoli MC, et al. Correction of beta-thalassemia major by gene transfer in haematopoietic progenitors of pediatric patients. EMBO Mol Med. 2010;2:315–328. | |
Wilber A, Hargrove PW, Kim YS, et al. Therapeutic levels of fetal hemoglobin in erythroid progeny of β-thalassemic CD34+ cells after lentiviral vector-mediated gene transfer. Blood. 2011;117(10):2817–2826. | |
Boulad F, Wang X, Qu J, et al. Safe mobilization of CD34+ cells in adults with beta-thalassemia and validation of effective globin gene transfer for clinical investigation. Blood. 2014;123:1483–1486. | |
Yannaki E, Karponi G, Zervou F, et al. Hematopoietic stem cell mobilization for gene therapy: superior mobilization by the combination of granulocyte-colony stimulating factor plus plerixafor in patients with β-thalassemia major. Hum Gene Ther. 2013;24:852–860. | |
Fibach E, Bianchi N, Borgatti M, et al. Effects of rapamycin on accumulation of alpha-, beta- and gamma-globin mRNAs in erythroid precursor cells from beta-thalassaemia patients. Eur J Haematol. 2006; 77(5):437–441. | |
Pope SH, Fibach E, Sun J, Chin K, Rodgers GP. Two-phase liquid culture system models normal human adult erythropoiesis at the molecular level. Eur J Haematol. 2000;64:292–303. | |
Breda L, Kleinert DA, Casu C, et al. A preclinical approach for gene therapy of beta-thalassemia. Ann N Y Acad Sci. 2010;1202:134–140. | |
Huber TL. Dissecting hematopoietic differentiation using the embryonic stem cell differentiation model. Int J Dev Biol. 2010;54(6–7):991–1002. | |
Honig GR, Lu SJ, Feng Q, Vida LN, Lee BS, Lanza R. alpha-Thalassemia-like globin gene expression by primitive erythrocytes derived from human embryonic stem cells. Hemoglobin. 2010;34:145–150. | |
Verlinsky Y, Strelchenko N, Kukharenko V, et al. Human embryonic stem cell lines with genetic disorders. Reprod Biomed Online. 2005;10: 105–110. | |
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. | |
Csobonyeiova M, Polak S, Koller J, Danisovic L. Induced pluripotent stem cells and their implication for regenerative medicine. Cell Tissue Bank. Epub July 19, 2014. | |
Kim C. Disease modeling and cell based therapy with iPSC: future therapeutic option with fast and safe application. Blood Res. 2014;49: 7–14. | |
Fan Y, Luo Y, Chen X, Li Q, Sun X. Generation of human β-thalassemia induced pluripotent stem cells from amniotic fluid cells using a single excisable lentiviral stem cell cassette. J Reprod Dev. 2012;58: 404–409. | |
Ochi K, Takayama N, Hirose S, Nakahata T, Nakauchi H, Eto K. Multicolor staining of globin subtypes reveals impaired globin switching during erythropoiesis in human pluripotent stem cells. Stem Cells Transl Med. 2014;3:792–800. | |
Dias J, Gumenyuk M, Kang H, et al. Generation of red blood cells from human induced pluripotent stem cells. Stem Cells Dev. 2011;20: 1639–1647. | |
Tubsuwan A, Abed S, Deichmann A, et al. Parallel assessment of globin lentiviral transfer in induced pluripotent stem cells and adult hematopoietic stem cells derived from the same transplanted β-thalassemia patient. Stem Cells. 2013;31:1785–1794. | |
Kobari L, Yates F, Oudrhiri N, et al. Human induced pluripotent stem cells can reach complete terminal maturation: in vivo and in vitro evidence in the erythropoietic differentiation model. Haematologica. 2012;97:1795–1803. | |
Pászty C. Transgenic and gene knock-out mouse models of sickle cell anemia and the thalassemias. Curr Opin Hematol. 1997;4:88–93. | |
Farrell CM, Grinberg A, Huang SP, et al. A large upstream region is not necessary for gene expression or hypersensitive site formation at the mouse beta-globin locus. Proc Natl Acad Sci U S A. 2000;97: 14554–14559. | |
Rivella S, May C, Chadburn A, Riviere I, Sadelain M. A novel murine model of Cooley anemia and its rescue by lentiviral-mediated human beta-globin gene transfer. Blood. 2003;101:2932–2939. | |
Weizer-Stern O, Adamsky K, Amariglio N, et al. mRNA expression of iron regulatory genes in beta-thalassemia intermedia and beta-thalassemia major mouse models. Am J Hematol. 2006;81:479–483. | |
Vadolas J, Nefedov M, Wardan H, et al. Humanized beta-thalassemia mouse model containing the common IVSI-110 splicing mutation. J Biol Chem. 2006;281:7399–7405. | |
Jamsai D, Zaibak F, Vadolas J, et al. A humanized BAC transgenic/knockout mouse model for HbE/beta-thalassemia. Genomics. 2006;88:309–315. | |
Costa FC, Fedosyuk H, Neades R, de Los Rios JB, Barbas CF 3rd, Peterson KR. Induction of fetal hemoglobin in vivo mediated by a synthetic gamma-globin zinc finger activator. Anemia. 2012;2012:507894. | |
Getman M, England SJ, Malik J, Peterson K, Palis J, Steiner LA. Extensively self-renewing erythroblasts derived from transgenic β-yac mice is a novel model system for studying globin switching and erythroid maturation. Exp Hematol. 2014;42:536–546. e8. | |
McColl B, Kao BR, Lourthai P, et al. An in vivo model for analysis of developmental erythropoiesis and globin gene regulation. FASEB J. 2014;28:2306–2317. | |
Huo Y, McConnell SC, Liu S, et al. Humanized mouse models of Cooley’s anemia: correct fetal-to-adult hemoglobin switching, disease onset, and disease pathology. Ann N Y Acad Sci. 2010;1202:45–51. | |
Huo Y, McConnell SC, Ryan TM. Preclinical transfusion-dependent humanized mouse model of beta thalassemia major. Blood. 2009;113: 4763–4770. | |
Huo Y, McConnell SC, Liu SR, et al. Humanized mouse model of Cooley’s anemia. J Biol Chem. 2009;284:4889–4896. | |
Arumugam P, Malik P. Genetic therapy for beta-thalassemia: from the bench to the bedside. Hematology Am Soc Hematol Educ Program. 2010;2010:445–450. | |
May C, Rivella S, Callegari J, et al. Therapeutic haemoglobin synthesis in beta-thalassaemic mice expressing lentivirus-encoded human beta-globin. Nature. 2000;406:82–86. | |
Breda L, Casu C, Gardenghi S, et al. Therapeutic hemoglobin levels after gene transfer in β-thalassemia mice and in hematopoietic cells of β-thalassemia and sickle cells disease patients. PLoS One. 2012;7:e32345. | |
Imren S, Fabry ME, Westerman KA, et al. High-level beta-globin expression and preferred intragenic integration after lentiviral transduction of human cord blood stem cells. J Clin Invest. 2004;114:953–962. | |
Puthenveetil G, Scholes J, Carbonell D, et al. Successful correction of the human beta-thalassemia major phenotype using a lentiviral vector. Blood. 2004;104:3445–3453. | |
Miccio A, Cesari R, Lotti F, et al. In vivo selection of genetically modified erythroblastic progenitors leads to long-term correction of beta-thalassemia. Proc Natl Acad Sci U S A. 2008;105:10547–10552. | |
Wang CX, Sather BD, Wang X, et al. Rapamycin relieves lentiviral vector transduction resistance in human and mouse hematopoietic stem cells. Blood. 2014;124:913–923. | |
Phaltane R, Lachmann N, Brennig S, Ackermann M, Modlich U, Moritz T. Lentiviral MGMT(P140K)-mediated in vivo selection employing a ubiquitous chromatin opening element (A2UCOE) linked to a cellular promoter. Biomaterials. 2014;35:7204–7213. | |
Ackermann M, Lachmann N, Hartung S, et al. Promoter and lineage independent anti-silencing activity of the A2 ubiquitous chromatin opening element for optimized human pluripotent stem cell-based gene therapy. Biomaterials. 2014;35:1531–1542. | |
Dighe N, Khoury M, Mattar C, et al. Long-term reproducible expression in human fetal liver hematopoietic stem cells with a UCOE-based lentiviral vector. PLoS One. 2014;9:e104805. | |
Groth AC, Liu M, Wang H, Lovelett E, Emery DW. Identification and characterization of enhancer-blocking insulators to reduce retroviral vector genotoxicity. PLoS One. 2013;8:e76528. | |
Arumugam PI, Urbinati F, Velu CS, Higashimoto T, Grimes HL, Malik P. The 3′ region of the chicken hypersensitive site-4 insulator has properties similar to its core and is required for full insulator activity. PLoS One. 2009;4:e6995. | |
Papapetrou EP, Lee G, Malani N, et al. Genomic safe harbors permit high β-globin transgene expression in thalassemia induced pluripotent stem cells. Nat Biotechnol. 2011;29:73–78. | |
Dong A, Rivella S, Breda L. Gene therapy for hemoglobinopathies: progress and challenges. Transl Res. 2013;161:293–306. | |
Yannaki E, Emery DW, Stamatoyannopoulos G. Gene therapy for β-thalassaemia: the continuing challenge. Expert Rev Mol Med. 2010;12:e31. | |
Bank A, Dorazio R, Leboulch P. A phase I/II clinical trial of beta-globin gene therapy for beta-thalassemia. Ann N Y Acad Sci. 2005; 1054:308–316. | |
Bluebird bio I. Bluebird bio reports rapid transfusion independence in beta-thalassemia major patients treated with its lentiglobin product candidate [press release]. Cambridge, MA: bluebird bio Headquarters; June 14, 2014. Available from: http://investor.bluebirdbio.com/phoenix.zhtml?c=251820&p=irol-newsArticle&ID=1939867&highlight=. Accessed November 26, 2014. | |
Cavazzana-Calvo M, Payen E, Negre O, et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467:318–322. | |
Cavazzana M, Ribeil J-A, Payen E, et al. Outcomes of gene therapy for beta-thalassemia major via transplantation of autologous hematopoietic stem cells transduced ex vivo with a lentiviral beta-globin vector (Abstract submission 3287, Abstract number S742). Presented at: European Hematology Association, 19th Annual Congress, Milan, Italy. June 14, 2014. | |
Bluebird Bio Inc. Bluebird bio Announces First Patient with Sickle Cell Disease Transplanted with LentiGlobin Gene Therapy. In, 2014. | |
Nagel RL, Bookchin RM, Johnson J, et al. Structural bases of the inhibitory effects of hemoglobin F and hemoglobin A2 on the polymerization of hemoglobin S. Proc Natl Acad Sci U S A. 1979;76:670–672. | |
McCune SL, Reilly MP, Chomo MJ, Asakura T, Townes TM. Recombinant human hemoglobins designed for gene therapy of sickle cell disease. Proc Natl Acad Sci U S A. 1994;91:9852–9856. | |
Levasseur DN, Ryan TM, Reilly MP, McCune SL, Asakura T, Townes TM. A recombinant human hemoglobin with anti-sickling properties greater than fetal hemoglobin. J Biol Chem. 2004;279: 27518–27524. | |
Romero Z, Urbinati F, Geiger S, et al. β-globin gene transfer to human bone marrow for sickle cell disease. J Clin Invest. 2013;123:3317–3330. | |
Nishino T, Tubb J, Emery DW. Partial correction of murine beta-thalassemia with a gammaretrovirus vector for human gamma-globin. Blood Cells Mol Dis. 2006;37:1–7. | |
Nishino T, Cao H, Stamatoyannopoulos G, Emery DW. Effects of human gamma-globin in murine beta-thalassaemia. Br J Haematol. 2006;134:100–108. | |
Pestina TI, Hargrove PW, Jay D, Gray JT, Boyd KM, Persons DA. Correction of murine sickle cell disease using gamma-globin lentiviral vectors to mediate high-level expression of fetal hemoglobin. Mol Ther. 2009;17:245–252. | |
Samakoglu S, Lisowski L, Budak-Alpdogan T, et al. A genetic strategy to treat sickle cell anemia by coregulating globin transgene expression and RNA interference. Nat Biotechnol. 2006;24:89–94. | |
Persons DA, Allay ER, Sawai N, et al. Successful treatment of murine beta-thalassemia using in vivo selection of genetically modified, drug-resistant hematopoietic stem cells. Blood. 2003;102:506–513. | |
Hanawa H, Hargrove PW, Kepes S, Srivastava DK, Nienhuis AW, Persons DA. Extended beta-globin locus control region elements promote consistent therapeutic expression of a gamma-globin lentiviral vector in murine beta-thalassemia. Blood. 2004;104:2281–2290. | |
Perumbeti A, Higashimoto T, Urbinati F, et al. A novel human gamma-globin gene vector for genetic correction of sickle cell anemia in a humanized sickle mouse model: critical determinants for successful correction. Blood. 2009;114:1174–1185. | |
Zhao H, Pestina TI, Nasimuzzaman M, Mehta P, Hargrove PW, Persons DA. Amelioration of murine beta-thalassemia through drug selection of hematopoietic stem cells transduced with a lentiviral vector encoding both gamma-globin and the MGMT drug-resistance gene. Blood. 2009;113:5747–5756. | |
Reid ME, El Beshlawy A, Inati A, et al. A double-blind, placebo-controlled phase II study of the efficacy and safety of 2,2-dimethylbutyrate (HQK-1001), an oral fetal globin inducer, in sickle cell disease. Am J Hematol. 2014;89:709–713. | |
Perrine SP, Pace BS, Faller DV. Targeted fetal hemoglobin induction for treatment of beta hemoglobinopathies. Hematol Oncol Clin North Am. 2014;28:233–248. | |
Ahmadvand M, Noruzinia M, Fard AD, et al. The role of epigenetics in the induction of fetal hemoglobin: a combination therapy approach. Int J Hematol Oncol Stem Cell Res. 2014;8:9–14. | |
Fard AD, Hosseini SA, Shahjahani M, Salari F, Jaseb K. Evaluation of novel fetal hemoglobin inducer drugs in treatment of β-hemoglobinopathy disorders. Int J Hematol Oncol Stem Cell Res. 2013;7:47–54. | |
Rahim F, Allahmoradi H, Salari F, et al. Evaluation of signaling pathways involved in γ-globin gene induction using fetal hemoglobin inducer drugs. Int J Hematol Oncol Stem Cell Res. 2013;7:41–46. | |
Qian X, Chen J, Zhao D, Guo L. Plastrum testudinis induces γ-globin gene expression through epigenetic histone modifications within the γ-globin gene promoter via activation of the p38 MAPK signaling pathway. Int J Mol Med. 2013;31:1418–1428. | |
Fibach E, Prus E, Bianchi N, et al. Resveratrol: antioxidant activity and induction of fetal hemoglobin in erythroid cells from normal donors and β-thalassemia patients. Int J Mol Med. 2012;29:974–982. | |
Franco SS, De Falco L, Ghaffari S, et al. Resveratrol accelerates erythroid maturation by activation of FoxO3 and ameliorates anemia in beta-thalassemic mice. Haematologica. 2014;99:267–275. | |
Ma YN, Chen MT, Wu ZK, et al. Emodin can induce K562 cells to erythroid differentiation and improve the expression of globin genes. Mol Cell Biochem. 2013;382:127–136. | |
Italia K, Jijina F, Merchant R, et al. Comparison of in-vitro and in-vivo response to fetal hemoglobin production and γ-mRNA expression by hydroxyurea in hemoglobinopathies. Indian J Hum Genet. 2013;19:251–258. | |
Pecoraro A, Rigano P, Troia A, et al. Quantification of HBG mRNA in primary erythroid cultures: prediction of the response to hydroxyurea in sickle cell and beta-thalassemia. Eur J Haematol. 2014;92:66–72. | |
Bianchi N, Zuccato C, Lampronti I, Borgatti M, Gambari R. Fetal hemoglobin inducers from the natural world: a novel approach for identification of drugs for the treatment of β-thalassemia and sickle-cell anemia. Evid Based Complement Alternat Med. 2009;6:141–151. | |
Zuccato C, Breda L, Salvatori F, et al. A combined approach for β-thalassemia based on gene therapy-mediated adult hemoglobin (HbA) production and fetal hemoglobin (HbF) induction. Ann Hematol. 2012;91:1201–1213. | |
Kaiser J. Gene therapy. Beta-thalassemia treatment succeeds, with a caveat. Science. 2009;326:1468–1469. | |
Ikeda K, Mason PJ, Bessler M. 3′UTR-truncated Hmga2 cDNA causes MPN-like hematopoiesis by conferring a clonal growth advantage at the level of HSC in mice. Blood. 2011;117:5860–5869. | |
Morishita A, Zaidi MR, Mitoro A, et al. HMGA2 is a driver of tumor metastasis. Cancer Res. 2013;73:4289–4299. | |
Sadelain M. Globin gene transfer for the treatment of β-thalassemia and Sickle Cell Disease. Presented at: American Society for Gene and Cell Therapy 19th Annual Congress, Washington, DC, May 24, 2014. | |
Sadelain M, Rivière I, Wang X, et al. Strategy for a multicenter phase I clinical trial to evaluate globin gene transfer in beta-thalassemia. Ann N Y Acad Sci. 2010;1202:52–58. | |
Deng W, Lee J, Wang H, et al. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell. 2012;149:1233–1244. | |
Deng W, Rupon JW, Krivega I, et al. Reactivation of developmentally silenced globin genes by forced chromatin looping. Cell. 2014;158(4):849–860. | |
Rupon JW, Deng W, Wang H, et al. Using forced chromatin looping to overcome developmental silencing of embryonic and fetal β-type globin genes in adult erythroid cells. Blood. 2013;122:433. | |
Gräslund T, Li X, Magnenat L, Popkov M, Barbas CF 3rd. Exploring strategies for the design of artificial transcription factors: targeting sites proximal to known regulatory regions for the induction of gamma-globin expression and the treatment of sickle cell disease. J Biol Chem. 2005;280:3707–3714. | |
Wilber A, Tschulena U, Hargrove PW, et al. A zinc-finger transcriptional activator designed to interact with the gamma-globin gene promoters enhances fetal hemoglobin production in primary human adult erythroblasts. Blood. 2010;115:3033–3041. | |
Voon HP, Vadolas J. Controlling alpha-globin: a review of alpha-globin expression and its impact on beta-thalassemia. Haematologica. 2008;93:1868–1876. | |
Xie SY, Ren ZR, Zhang JZ, et al. Restoration of the balanced alpha/beta-globin gene expression in beta654-thalassemia mice using combined RNAi and antisense RNA approach. Hum Mol Genet. 2007;16: 2616–2625. | |
Voon HP, Wardan H, Vadolas J. Co-inheritance of alpha- and beta-thalassaemia in mice ameliorates thalassaemic phenotype. Blood Cells Mol Dis. 2007;39:184–188. | |
Nasimuzzaman M, Khandros E, Wang X, et al. Analysis of alpha hemoglobin stabilizing protein overexpression in murine beta-thalassemia. Am J Hematol. 2010;85:820–822. | |
Roosjen M, McColl B, Kao B, Gearing LJ, Blewitt ME, Vadolas J. Transcriptional regulators Myb and BCL11A interplay with DNA methyltransferase 1 in developmental silencing of embryonic and fetal β-like globin genes. FASEB J. 2014;28:1610–1620. | |
Forget BG. Progress in understanding the hemoglobin switch. N Engl J Med. 2011;365:852–854. | |
Sankaran VG, Xu J, Byron R, et al. A functional element necessary for fetal hemoglobin silencing. N Engl J Med. 2011;365:807–814. | |
Zhou D, Liu K, Sun CW, Pawlik KM, Townes TM. KLF1 regulates BCL11A expression and gamma- to beta-globin gene switching. Nat Genet. 2010;42:742–744. | |
Sankaran VG, Menne TF, Xu J, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322:1839–1842. | |
Tanabe O, McPhee D, Kobayashi S, et al. Embryonic and fetal beta-globin gene repression by the orphan nuclear receptors, TR2 and TR4. EMBO J. 2007;26:2295–2306. | |
Tanabe O, Shen Y, Liu Q, et al. The TR2 and TR4 orphan nuclear receptors repress Gata1 transcription. Genes Dev. 2007;21:2832–2844. | |
Cui S, Kolodziej KE, Obara N, et al. Nuclear receptors TR2 and TR4 recruit multiple epigenetic transcriptional corepressors that associate specifically with the embryonic β-type globin promoters in differentiated adult erythroid cells. Mol Cell Biol. 2011;31:3298–3311. | |
Campbell AD, Cui S, Shi L, et al. Forced TR2/TR4 expression in sickle cell disease mice confers enhanced fetal hemoglobin synthesis and alleviated disease phenotypes. Proc Natl Acad Sci U S A. 2011;108: 18808–18813. | |
Xu XS, Hong X, Wang G. Induction of endogenous gamma-globin gene expression with decoy oligonucleotide targeting Oct-1 transcription factor consensus sequence. J Hematol Oncol. 2009;2:15. | |
Jiang J, Best S, Menzel S, et al. cMYB is involved in the regulation of fetal hemoglobin production in adults. Blood. 2006;108:1077–1083. | |
Sankaran VG, Xu J, Orkin SH. Transcriptional silencing of fetal hemoglobin by BCL11A. Ann N Y Acad Sci. 2010;1202:64–68. | |
Borg J, Papadopoulos P, Georgitsi M, et al. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat Genet. 2010;42:801–805. | |
Borg J, Phylactides M, Bartsakoulia M, et al. KLF10 gene expression is associated with high fetal hemoglobin levels and with response to hydroxyurea treatment in β-hemoglobinopathy patients. Pharmacogenomics. 2012;13:1487–1500. | |
Amaya M, Desai M, Gnanapragasam MN, et al. Mi2β-mediated silencing of the fetal γ-globin gene in adult erythroid cells. Blood. 2013;121:3493–3501. | |
Sankaran VG. Targeted therapeutic strategies for fetal hemoglobin induction. Hematology Am Soc Hematol Educ Program. 2011;2011: 459–465. | |
Tallack MR, Perkins AC. Three fingers on the switch: Krüppel-like factor 1 regulation of γ-globin to β-globin gene switching. Curr Opin Hematol. 2013;20:193–200. | |
Manchinu MF, Marongiu MF, Poddie D, et al. In vivo activation of the human δ-globin gene: the therapeutic potential in β-thalassemic mice. Haematologica. 2014;99:76–84. | |
Xu J, Peng C, Sankaran VG, et al. Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science. 2011;334:993–996. | |
Esteghamat F, Gillemans N, Bilic I, et al. Erythropoiesis and globin switching in compound Klf1::Bcl11a mutant mice. Blood. 2013;121: 2553–2562. | |
Renella R, Perlov A, Harris CE, et al. Hematopoietic SIN lentiviral micro RNA-mediated silencing of BCL11A: pre-clinical evidence for a sickle cell disease gene-therapy trial. Presented at: American Society of Hematology 54th Annual Meeting, Atlanta, GA, December 10, 2012. | |
Guda S, Peng D, Bauer DE, et al. Optimization of lentivirus vector RNA polymerase II driven microRNA embedded shRNAs for enhanced processing and efficient knockdown of Bcl11a for induction of fetal hemoglobin in erythroid cells. Presented at: American Society for Gene and Cell Therapy 19th Annual Congress, Washington, DC, May 23, 2014. | |
Xu J, Bauer DE, Orkin SH. Targeting regulators of hemoglobin F. The Hematologist. 2011; 1 Sept. | |
Xu J, Bauer DE, Kerenyi MA, et al. Corepressor-dependent silencing of fetal hemoglobin expression by BCL11A. Proc Natl Acad Sci U S A. 2013;110(16):6518–23. | |
Sankaran VG, Menne TF, Šćepanović D, et al. MicroRNA-15a and -16–1 act via MYB to elevate fetal hemoglobin expression in human trisomy 13. Proc Natl Acad Sci U S A. 2001;108:1519–1524. | |
Lulli V, Romania P, Morsilli O, et al. MicroRNA-486–3p regulates γ-globin expression in human erythroid cells by directly modulating BCL11A. PLoS One. 2013;8(4):e60436. | |
Azzouzi I, Moest H, Winkler J, et al. MicroRNA-96 directly inhibits γ-globin expression in human erythropoiesis. PLoS One. 2011;6:e22838. | |
Ma Y, Wang B, Jiang F, et al. A feedback loop consisting of microRNA 23a/27a and the β-like globin suppressors KLF3 and SP1 regulates globin gene expression. Mol Cell Biol. 2013;33(20):3994–4007. | |
Genovese P, Schiroli G, Escobar G, et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510: 235–240. | |
Zou J, Mali P, Huang X, Dowey SN, Cheng L. Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood. 2011;118:4599–4608. | |
Katada H, Komiyama M. Artificial restriction DNA cutters to promote homologous recombination in human cells. Curr Gene Ther. 2011;11:38–45. | |
Hockemeyer D, Soldner F, Beard C, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 2009;27:851–857. | |
Perez EE, Wang J, Miller JC, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26:808–816. | |
Urnov FD, Miller JC, Lee YL, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–651. | |
Voit RA, McMahon MA, Sawyer SL, Porteus MH. Generation of an HIV resistant T-cell line by targeted “stacking” of restriction factors. Mol Ther. 2013;21:786–795. | |
Voit RA, Hendel A, Pruett-Miller SM, Porteus MH. Nuclease-mediated gene editing by homologous recombination of the human globin locus. Nucleic Acids Res. 2014;42:1365–1378. | |
Patsali P, Mussolino C, Stephanou C, et al. Towards personalized gene therapy for β-thalassemia in Cyprus. Presented at: American Society for Gene and Cell Therapy 19th Annual Congress, Washington, DC, May 23, 2014. | |
Cradick TJ, Fine EJ, Antico CJ, Bao G. CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013;41:9584–9592. | |
Tsai SQ, Wyvekens N, Khayter C, et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol. 2014;32:569–576. | |
Ma N, Liao B, Zhang H, et al. Transcription activator-like effector nuclease (TALEN)-mediated gene correction in integration-free beta-thalassemia induced pluripotent stem cells. J Biol Chem. 2013;288:34671–34679. | |
Sun N, Zhao H. Seamless correction of the sickle cell disease mutation of the HBB gene in human induced pluripotent stem cells using TALENs. Biotechnol Bioeng. 2014;111:1048–1053. | |
Cost GJ, Gregory PD, Guschin D, Holmes MC, Miller JC, Paschon D et al. Methods and compositions for treatment of a genetic condition (US20100093913). In: Sangamo Biosciences, Inc, 2014. | |
Cost GJ, Gregory PD, Guschin D, Holmes MC, Miller JC, Paschon D et al. Methods and compositions for treatment of a genetic condition (US20140080216). In: Sangamo Biosciences, Inc., 2014. | |
Gravia A, Chondrou V, Sgourou A, et al. Individualizing fetal hemoglobin augmenting therapy for β-type hemoglobinopathies patients. Pharmacogenomics. 2014;15(10):1355–1364. | |
Patel A. Tissue banking for research – bench to bedside and back – myth, reality or fast fading reality at the dawn of a personalised healthcare era. Cell Tissue Bank. 2011;12:19–21. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.