Back to Journals » Nature and Science of Sleep » Volume 13
Recent Advances in Studies on the Role of Neuroendocrine Disorders in Obstructive Sleep Apnea–Hypopnea Syndrome-Related Atherosclerosis
Authors Wang W, Zheng Y, Li M, Lin S, Lin H
Received 12 April 2021
Accepted for publication 19 July 2021
Published 27 July 2021 Volume 2021:13 Pages 1331—1345
DOI https://doi.org/10.2147/NSS.S315375
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sarah L Appleton
Wanda Wang,1 Yanli Zheng,1 Meimei Li,1 Shu Lin,1– 3 Huili Lin1
1Department of Cardiology, The Second Affiliated Hospital of Fujian Medical University, Quanzhou, Fujian Province, People’s Republic of China; 2Centre of Neurological and Metabolic Research, The Second Affiliated Hospital of Fujian Medical University, Quanzhou, Fujian Province, People’s Republic of China; 3Diabetes and Metabolism Division, Garvan Institute of Medical Research, Sydney, NSW, 2010, Australia
Correspondence: Huili Lin
Department of Cardiology, The Second Affiliated Hospital of Fujian Medical University, Quanzhou, Fujian Province, People’s Republic of China
Email [email protected]
Shu Lin
Centre of Neurological and Metabolic Research, The Second Affiliated Hospital of Fujian Medical University, Quanzhou, Fujian Province, People’s Republic of China
Email [email protected]
Abstract: Cardiovascular disease is a common cause of death worldwide, and atherosclerosis (AS) and obstructive sleep apnea–hypopnea syndrome (OSAHS) critically contribute to the initiation and progression of cardiovascular diseases. OSAHS promotes endothelial injury, vascular smooth muscle cell (VSMC) proliferation, abnormal lipid metabolism, and elevated arterial blood pressure. However, the exact OSAHS mechanism that causes AS remains unclear. The nervous system is widely distributed in the central and peripheral regions. It regulates appetite, energy metabolism, inflammation, oxidative stress, insulin resistance, and vasoconstriction by releasing regulatory factors and participates in the occurrence and development of AS. Studies showed that OSAHS can cause changes in neurophysiological plasticity and affect modulator release, suggesting that neuroendocrine dysfunction may be related to the OSAHS mechanism causing AS. In this article, we review the possible mechanisms of neuroendocrine disorders in the pathogenesis of OSAHS-induced AS and provide a new basis for further research on the development of corresponding effective intervention strategies.
Keywords: obstructive sleep apnea, atherosclerosis, cardiovascular diseases, sympathetic nerve, neuroendocrine disorders
Introduction
Cardiovascular disease is still a major public health problem worldwide and one of the main causes of death, which greatly affects the quality of life and medical expenses of patients.1–3 Atherosclerosis (AS) is a chronic inflammatory disease and the main pathological basis of most cardiovascular diseases.4 Vascular endothelial dysfunction, vascular smooth muscle cell (VSMC) proliferation, leukocyte migration, lipid accumulation, and platelet activation are key links in the process of AS.5–7 Inflammation and oxidative stress are key molecular events in the pathogenesis of AS. The main risk factors for inducing AS include hypertension, obesity, dyslipidemia and diabetes mellitus.8 Previous studies have shown that modulators such as renin-angiotensin II (AngII), glucocorticoids, catecholamines and neuropeptide Y (NPY) have direct or indirect atherogenic effects.9–11 Therefore, neurohumoral dysregulation plays a very important role in the occurrence and development of AS.
Obstructive sleep apnea–hypopnea syndrome (OSAHS) is a highly prevalent disease characterized by repeated episodes of obstruction of the respiratory passages during sleep.12 Its prevalence is positively correlated with age.13 OSAHS may cause a series of pathophysiological changes, such as hypoxemia and hypercapnia.14 By means of immunohistochemistry, fMRI and microneurography observations, the researchers found that under hypoxemia and hypercapnia, the level of discharge of central neurons and peripheral sympathetic neurons changed. Neuronal firing is indirectly involved in the proatherogenic effects, including increased discharge of AngII, glucocorticoids, catecholamine and NPY.
Therefore, the purpose of this review is: first, we collected evidence that OSAHS causes AS from basic and clinical studies; secondly, we reviewed the relationship between OSAHS and central and peripheral neuroendocrine disorders; thirdly, we reviewed the pathophysiological role of related modulators in AS. Finally, we provided existing evidence for the study of effective intervention strategies for OSAHS-related AS and complications.
OSAHS Overview
OSAHS has an increasingly high prevalence globally. An earlier longitudinal study of the Wisconsin Sleep Cohort study estimated that the prevalence of OSAHS was 2% in men and 4% in women aged 30–60 years in the United States.15 A survey conducted 20 years later also in the United Stated estimated that 6% of women and 13% of men aged 30–70 years had moderate to severe OSAHS.16 Obesity is the main risk factor for OSAHS.17,18 The increasing prevalence of obesity may be the main reason for the increased prevalence of OSAHS.19 Moreover, some unalterable factors, such as age, sex, and heredity, are considered as risk factors for OSAHS.20 Patients with OSAHS have intermittent upper respiratory airway obstruction while sleeping, causing apnea, which is the complete airflow cessation, or hypopnea, which is the partial reduction of airflow.21 Significant or complete airway cessation leads to a hypoxic-hypercapnia state, resulting in altered intrathoracic pressure and increased breathing effort. However, subsequent extreme intrathoracic pressure triggers arousal and ends apnea.22 These periodic changes lead to intermittent hypoxia (IH), short awakenings, and sleep fragmentation throughout the night.23,24 Among many related factors, IH is one of the hallmarks of OSAHS. The carotid bodies are bodies located at the bifurcation of common carotid arteries that sense changes in arterial blood oxygen level. IH not only induces enhances carotid body chemosensory reactivity to oxygen, but also carotid baroreceptor attenuation mediated by reactive oxygen species.25 During exposure to IH, the size of the carotid body increases, and this morphological change matches the functional activation.26 After 21 days of IH exposure, the rats underwent bilateral carotid body ablation.27 Carotid body ablation normalizes blood pressure, reduces the ventilatory response to hypoxia, and restores cardiac autonomy and baroreflex function. This indicates that both the autonomic alterations and the progression of cardiovascular pathophysiology induced by IH depend heavily on the heightened carotid bodies chemosensory drive. IH and hypercapnia have synergistic effect in promoting sympathetic outflow and increasing hypertension.28,29 IH and hypercapnia synergistically increase sympathetic nerve activity, which depends not only on the severity of hypercapnic exposures but also on the severity of hypoxia.30 This synergistic effect is most apparent in low lung volume (expiratory) phases of respiration and is inhibited during lung inflation (inspiratory).31 About one-third of adult OSAHS patients had bruxism.32 Most bruxism events occurred close to sleep apnea/hypopnea events, and there is a positive correlation between the frequency of apnea episodes and the frequency of bruxism events.33 The occurrence of bruxism may be closely related to a micro-arousal event that ensued from an obstructive apnea event.34 After the onset of bruxism, upper airway obstruction is reduced, parasympathetic nerve activity is increased, and heart rate is reduced.35 Therefore, bruxism may be a protective response to OSAHS.
OSAHS is conventionally diagnosed using polysomnography in a sleep center, during which sleep and respiratory parameters are monitored.During this examination, abnormal respiratory events are quantified as apneas (airflow absent for >10 seconds) and hypopneas (airflow diminution associated with a fall in arterial oxyhemoglobin saturation of ≥3% or terminated by an electroencephalographic arousal). The number of apnea and hypopnea events per hour during sleep was defined as the apnea–hypopnea index (AHI). OSAHS diagnosis is confirmed by two conditions: 1) an AHI greater than 5; and 2) daytime symptoms, such as daytime sleepiness. Moreover, AHI is used to determine the severity of OSAHS, with AHIs of 5–15, 15–30, and >30 indicating mild, moderate, and severe OSAHS, respectively.36 Daytime sleepiness is a common but not universal accompanying symptom. OSAHS patients with daytime sleepiness have an increased risk of metabolic diseases compared to those with no-daytime sleepiness.37 This may be related to the increased levels of pro-inflammatory cytokines interleukin (IL)-6 and tumor necrosis factor (TNF)-α in patients with disorders of daytime sleepiness.38,39 However, the underlying mechanisms remain unclear, and further research is needed. OSAHS is a chronic disease that can be treated. Continuous positive airway pressure (CPAP) is the main treatment for moderate to severe OSAHS. Effective CPAP treatment can reduce AHI, improve blood pressure, and reduce the biomarkers of metabolic diseases related to OSAHS.40–42 A low-calorie diet combined with an active lifestyle to achieve significant weight loss is a feasible and effective treatment for mild OSAHS.43 For some patients with OSAHS, preventing the patient from sleeping in the supine position can improve daytime sleepiness and AHI.44 Compared with CPAP treatment, this positional therapy may have better adherence.
Evidence on OSAHS-Induced AS
A cross-sectional study described early signs of AS in patients with no obvious comorbidities.45 The independent, validated indicators of early signs of AS include a significant increase in pulse wave velocity, carotid diameter, and intima-media thickness. Moreover, a randomized controlled trial observed that effective CPAP in patients with OSAHS without obvious comorbidities can significantly decrease carotid intima-media thickness and pulse-wave velocity.46 This indicates that OSAHS is an independent risk factor for AS. AS plays a significant role in the pathogenesis of coronary heart disease and ischemic stroke. An intravascular ultrasound showed that the severity of OSAHS was positively correlated with the volume of coronary atherosclerotic plaque.47 Accumulating evidence has shown that coronary heart disease incidence is significantly higher in individuals with OSAHS.48,49 There is a lot of evidence supporting the fact that the risk of ischemic stroke increases in patients with OSAHS compared with that in those who do not have OSAHS, but this risk is not related to confounding factors.50–52 Further studies have shown that mortality risk after an ischemic stroke in patients with OSAHS is significantly increased; however, this increase in risk is not affected by other risk factors.53
Endothelial dysfunction is a key AS mechanism. Normal endothelium regulates vasomotor tension, maintains inflammation and oxidative homeostasis, and can regenerate and be repaired.54 However, in patients with OSAHS, the endothelial regulatory function is destroyed, and endothelial cell (EC) apoptosis occurs.55–57 Circulating endothelial progenitor cells (EPCs) represent endothelial regenerative and repair capacities.58 Some studies have reported that patients with OSAHS had elevated levels of circulating progenitor cells, suggesting endothelial damage.59 However, some studies observed decreased circulating endothelial progenitor cell (EPC) levels in patients with OSAHS.56,60 This may be due to the continuous progression of the disease leading to decreased circulating EPC levels, ultimately depleting endothelial regeneration. Increased arterial stiffness is an early sign of AS and is related to OSAHS. When severe OSAHS coexists with hypertension, OSAHS has an additive effect on arterial stiffness.61 CPAP is considered to be an effective method for treating patients with OSAHS, but whether CPAP can improve endothelial function remains controversial.62–64 This may be related to different CPAP treatment strategies.65
Patients with OSAHS are at an increased risk of metabolic diseases, including hypertension, obesity, dyslipidemia, and diabetes. Such metabolic diseases are main AS risk factors.66 Cross-sectional epidemiological studies have shown that OSAHS is an independent hypertension risk factor.67 OSAHS-induced hypertension is closely related to hyperactivity of the sympathetic nervous system and impairment of endothelial function.68,69 Obesity is a main risk factor for OSAHS, and OSAHS may further increase body weight. OSAHS-induced changes in the levels of leptin, ghrelin and glucocorticoids are related to energy intake and consumption.70,71 Both increase in plasma ghrelin and glucocorticoids stimulate food intake. An increase in plasma leptin suggests that excess energy is associated with resistance to the energy consumption effects of leptin.72 This inadequate balance between energy intake and energy consumption ultimately leads to weight gain. It has been reported that weight loss can reduce inflammation biomarkers in overweight patients with mild OSAHS and improve the sleep patterns of patients with OSAHS.73–75 Serum low-density lipoprotein cholesterol (LDL-C) and high-density lipoprotein cholesterol (HDL-C) proportions are positively correlated with cardiovascular events and independently correlated with OSAHS severity.40 This increase in LDL-C/HDL-C proportions may be due to the down-regulation of lipoprotein lipase following sympathetic nerve outflow expression. OSAHS is a risk factor for the development of type 2 diabetes mellitus and is independent of obesity and other traditional risk factors.76,77 This may be related to an increase in regulators, inflammation, and oxidative stress, which may lead to insulin resistance, impaired sensitivity, and β-cell dysfunction.78 It is worth noting that there may be a two-way relationship between OSAHS and type 2 diabetes mellitus.79 It has been reported that effective CPAP crucially contributes to decreasing the levels of metabolic disease biomarkers, such as leptin, LDL-C/HDL-C ratio, adiponectin, and blood glucose.40,41
The above evidence shows that OSAHS is an important AS risk factor that directly or indirectly promotes AS occurrence and development. OSAHS usually coexists and interacts with metabolic diseases, leading to an increased incidence of cardiovascular events. (Table 1).
 |
Table 1 Evidence on OSAHS-Induced as |
OSAHS and Neuroendocrine Disorders
Sympathetic hyperactivity is an important characteristics of OSAHS. Central neuron groups that control sympathetic outflow include the nucleus tractus solitarius (NTS), median preoptic nucleus (MnPO), paraventricular nucleus (PVN), and rostral ventrolateral medulla (RVLM).80,81 These central neurons regulate sympathetic nerve activity through mutual neural projection; however, they participate in a series of pathophysiological changes by regulating modulator release.
Nucleus Tractus Solitarius
NTS is located in the dorsomedial part of the medulla oblongata and is the first central nucleus that receives peripheral information, including chemoreceptors and baroreceptors. Repeated hypoxia during sleep activates arterial chemoreceptors in patients with OSAHS and releases excitatory amino acid neurotransmitters, that is, glutamate, through the afferent nerve to project information to NTS.27 It should be noted that hypoxia not only affects sympathetic nerve activity through afferent chemical receptors, but also directly affects the neuronal firing of NTS.82 NTS projects information to RVLM via glutamatergic neurons;83 however, NTS indirectly projects information to PVN via A2 noradrenergic neurons, resulting in increased sympathetic nerve activity.84 In addition to receiving chemical information, NTS receives pressure information. Under physiological conditions, elevated arterial pressure can activate NTS and promote the inhibitory output of RVLM parasympathetic neurons by activating GABAergic neurons of the cerebral cortex.85 Studies by Narkiewicz et al28 and Chan et al86 showed that the baroreflex regulation of patients with OSAHS is impaired, consequently increasing sympathetic nerve activity.
Median Preoptic Nucleus
Because of blood-brain barriers, modulators in the plasma are difficult to recognize by the central nervous system. The lamina terminalis is located in the midline anterior wall of the third ventricle. Its structure includes MnPO, subfornical organ (SFO), and organum vasculosum of the lamina terminalis (OVLT). Circulating AngII provides an excitatory drive for MnPO neurons through neural sites that lack a functional blood-brain barrier, including the SFO/OVLT site.87 MnPO drives small cell neurons of PVN through a glutamatergic pathway and participates in the regulation of arterial blood pressure and sympathetic nerve activity;88,89 however, MnPO transmits signals to neuroendocrine cells in PVN to regulate glucocorticoid release. MnPO plays an important role in maintaining the sympathetic excitability of AngII. The feedback between MnPO and AngII seems to be a reason why patients with OSAHS still maintain sympathetic activation when they are awake.
Paraventricular Nucleus
PVN integrates information in an ascending order both from the MnPO and the NTS.90,91 Glutamatergic neurons in PVN transmit information to RVLM and regulate sympathetic outflow.92 Most activated PVN neurons contain corticotropin-releasing hormones (CRHs).93 Previous studies have suggested that the levels of adrenocorticotropic hormone (ACTH) and glucocorticoids in patients with OSAHS increase.94 This suggests that OSAHS activates CRH neurons in PVN, initiates hypothalamic-pituitary adrenergic (HPA) axis hormonal cascades, and contributes to cardiovascular disease occurrence.
Rostral Ventrolateral Medulla
RVLM is an important presympathetic neuron in the central nervous system that regulates sympathetic outflow and arterial blood pressure. It participates in the sympathetic excitatory response stimulated by baroreflex and chemoreceptors. OSAHS-induced hypercapnia may activate RVLM by changing the central acid environment. In human and mouse experiments, hypercapnia combined with hypoxemia can further increase sympathetic outflow.95,96 Hypoxemia may increase central chemoreceptor sensitivity and more frequently activate sympathetic nerves through hypercapnia.
Sympathetic Nerve
The increase in sympathetic nerve outflow is mainly regulated by a variety of OSAHS-induced pathophysiological changes. Reasons behind sympathetic nerve activation may include (As shown in Figure 1): 1) the transmission of hypoxia information of the carotid body to the NTS nucleus, followed by RVLM, and PVN, leading to increased sympathetic nerve activity; 2) increased NTS activity caused by impaired pressure sensitivity, eventually leading to sympathetic nerve activation; 3) increased circulating AngII levels, MnPO activation, information projection to PVN, and finally sympathetic excitatory drive activation due to increased renal sympathetic activity; 4) changes in central acid levels, consequent RVLM stimulation, and subsequent sympathetic nerve excitement, all caused by intra-arterial hypercapnia; and 5) apnea termination by arousal, which is a central nervous event that increases sympathetic activity and suppresses parasympathetic tone.97 Although changes in intrathoracic pressure during apnea increase the frequency of parasympathetic afferent fiber activation,98,99 they are not enough to inhibit the increase of sympathetic nerve outflow. The overall effect of apnea or hypoventilation is an increase in sympathetic outflow.
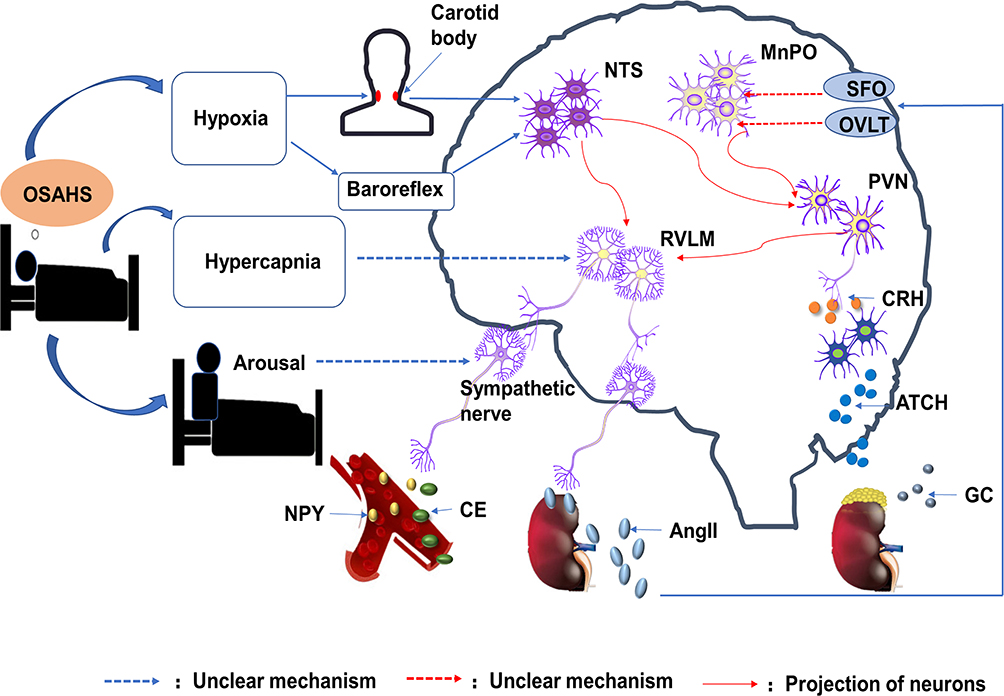 |
Figure 1 Model of central pathways related to obstructive sleep apnea–hypopnea syndrome-induced increased sympathetic outflow. |
When awake during the day, patients with OSAHS breathe normally, and there are no signs of hypoxemia or hypercapnia. Moreover, sympathetic nerve activity is at a high level. The positive feedback relationship between AngII and MnPO may be an important reason for a high sympathetic nerve activity during the day. Central neuroplasticity may be another reason behind persistent sympathetic nerve overactivation.
Neuroendocrine Disorders and AS
Patients with OSAHS experience sustained sympathetic nerve excitation, which consequently stimulates endocrine system activities, thereby causing neurohumoral disorders. Relevant reports mentioned that levels of modulators, such as AngII, glucocorticoids, catecholamine, and NPY, in patients with OSAHS were significantly higher than those in non-OSAHS patients.70,100–102 Continuous sympathetic nerve excitation promotes inflammatory/immune response and ultimately increases cardiovascular disease-related end-organ damage. Neuroendocrine disorders directly promote AS occurrence and development (Figure 2). AS mechanisms induced by these mediators are summarized in Table 2.
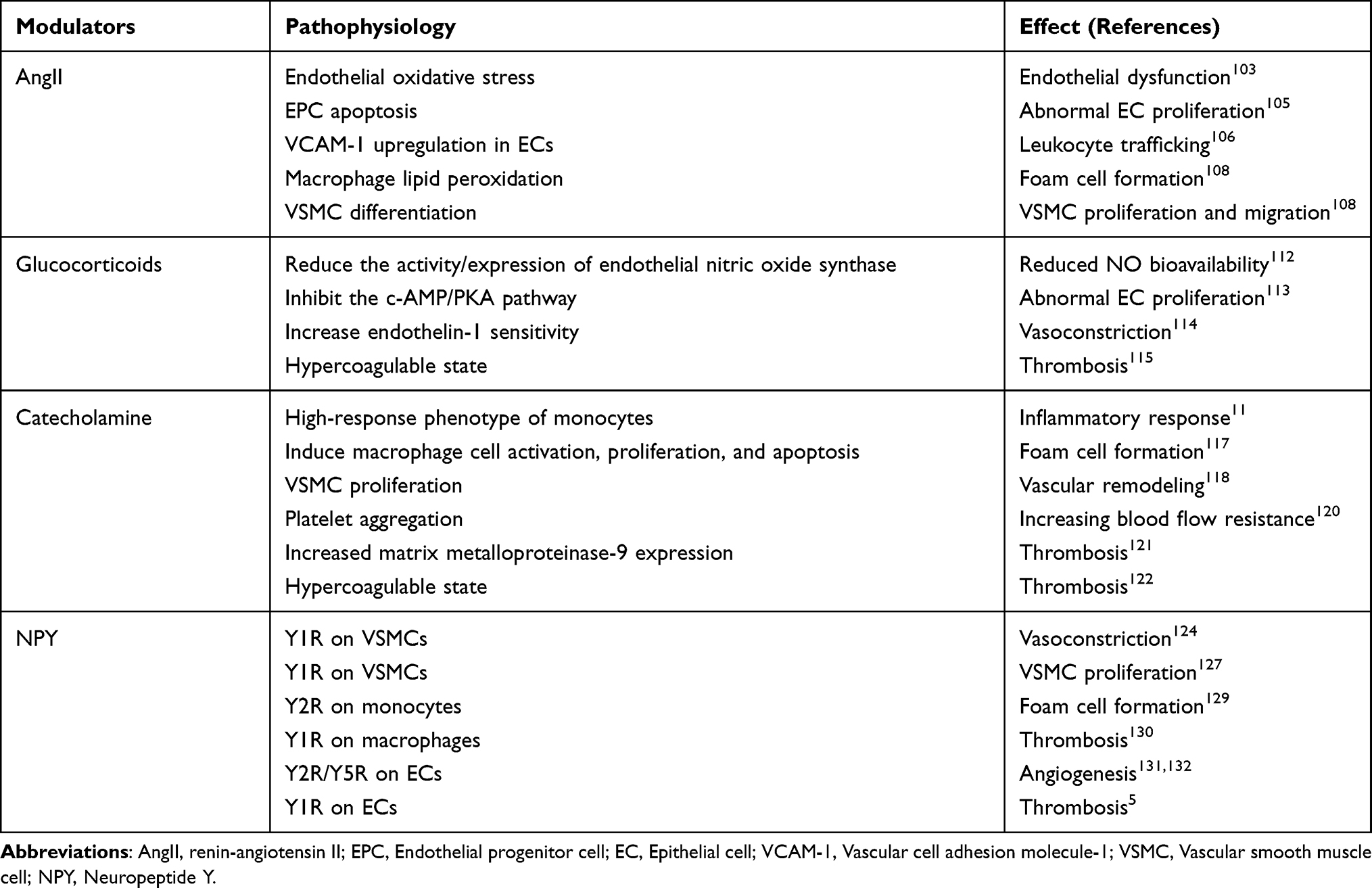 |
Table 2 Mediator-Induced as Mechanisms |
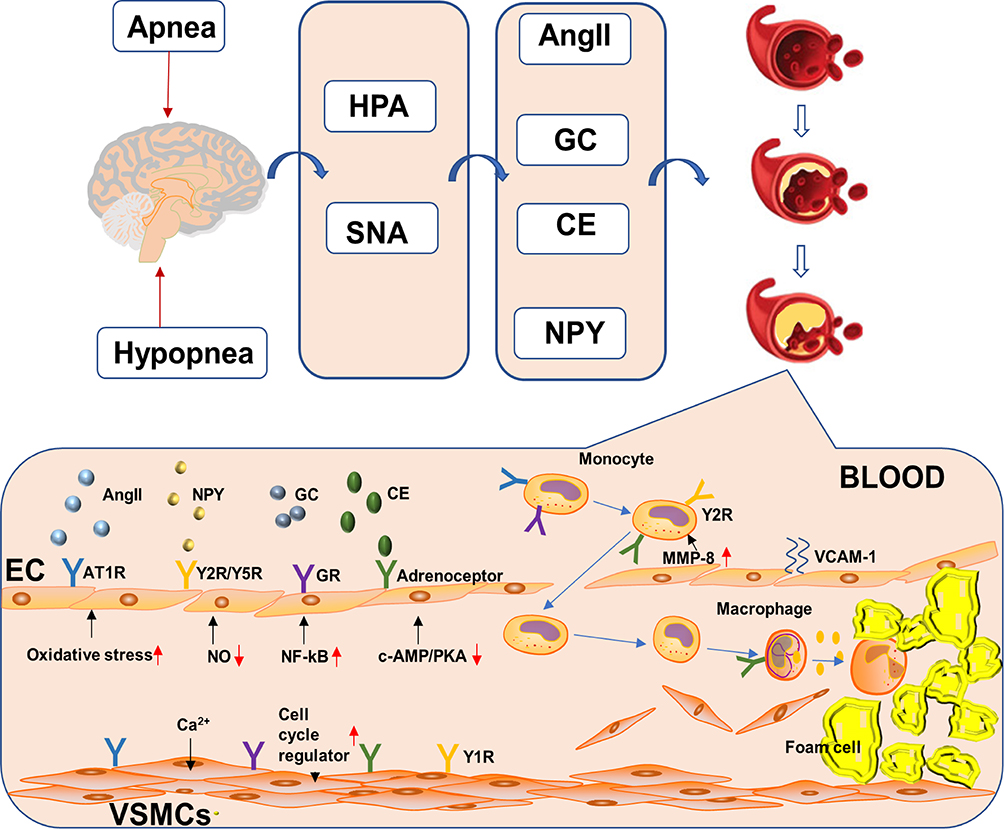 |
Figure 2 Neuroendocrine disorders directly and indirectly promote atherosclerosis occurrence and development. |
AngII and AS
AngII is the renin-angiotensin system (RAS) effector and is an important regulator of cardiovascular function. In patients with OSAHS, sympathetic nerve activation leads to excessive RAS activation, and circulating AngII levels are significantly increased. The atherogenic effect of AngII is mediated by AngII binding to the angiotensin type 1 receptor (AT1R). AngII is an effective vasoconstrictor. It binds to AT1R to increase the concentration of Ca2+ in vascular smooth muscle and increase the activity of vasoconstriction. AngII interacts with endothelial AT1-R and increases endothelial oxidative stress by generating superoxide anions derived from nicotinamide adenine dinucleotide phosphate oxidase.103 Moreover, AngII is a powerful activator of oxidative stress and inflammation. EPCs can differentiate into vascular endothelial cells (ECs) and play a powerful role in EC repair. The depletion and damage of EPCs may cause endothelial dysfunction and cardiovascular disease progression.104 AngII induces EPC apoptosis through the activation of AT1-R, induction of oxidative stress, and activation of redox-sensitive apoptosis signal-regulating kinase 1-dependent proapoptotic pathways.105 The expression of vascular cell adhesion molecule-1 (VCAM-1) in ECs mediates monocyte aggregation. This regulation of VCAM-1 expression occurs at the transcriptional level and is mediated by a redox-sensitive transcription factor nuclear factor-kB (NF-kB). AngII activates transcription factor NF-kB and upregulates VCAM-1 in ECs.106 AngII mediates the leukocyte recruitment cascade through complex redox signaling pathways, which occur in ECs, monocytes, dendritic cells, neutrophils, and lymphocytes.107 AngII can preoxidize lipid macrophage, absorb and oxidize low-density lipoproteins, accelerate foam cell formation, and promote AS occurrence.108 Moreover, AngII can differentiate contractile VSMC, that is, differentiated VSMC from synthetic VSMC, that is, dedifferentiated type and accelerate VSMC proliferation and migration.109 Moreover, AngII causes AS structural remodeling, reduces AS stability, and promotes thrombotic event occurrence.110
Glucocorticoids and AS
In patients with OSAHS, the corticotropin-releasing hormone (CRH) neurons of PVN receive information projections from NTS and MnPO, activate the HPA axis, and cause increased circulating glucocorticoid levels. Under physiological conditions, glucocorticoids have anti-inflammatory, immune regulatory, and metabolic regulation effects. Higher plasma glucocorticoid levels are associated with coronary AS severity. This leads to metabolic and cardiovascular complications.111 Glucocorticoids reduce the bioavailability of NO in blood vessels by reducing the activity/expression of endothelial nitric oxide synthase.112 Glucocorticoids inhibit the c-AMP/PKA pathway and damage EC regeneration and anti-inflammatory and antioxidant functions.113 Glucocorticoids increase the sensitivity of blood vessels to endothelium-derived vasoconstrictors, such as catecholamines and endothelin-1.114 Moreover, glucocorticoids can promote AS development by promoting insulin resistance, dyslipidemia, and hypertension. Glucocorticoids increase the levels of procoagulant factors, mainly including factors VIII, IX, and von Willebrand factor, and impaired fibrinolysis, mainly including plasminogen activator inhibitor-1.115 Glucocorticoids contribute to thrombus formation following AS plaque rupture. Excessive glucocorticoids can promote atherosclerotic lesion calcification.116 It is worth noting that there is a circadian rhythm in the HPA axis, which helps us understand the unique role of glucocorticoids in promoting AS. For example, patients with metastatic breast cancer with flat glucocorticoid circadian rhythms have a higher mortality. This suggests that changes in the circadian rhythm of the HPA axis will affect the progression of diseases. However, there are no reports on the HPA axis circadian rhythm in patients with OSAHS.
Catecholamine and AS
Compared with individuals who do not have OSAHS, circulating catecholamine, including norepinephrine (NA) and epinephrine, levels in patients with OSAHS were significantly higher, and this is due to sympathetic hyperactivity. Physiological NA concentrations are sufficient to cause vasoconstriction. Monocytes bind to catecholamines through β-adrenergic receptors to form a continuous high-response phenotype, which is characterized by an enhanced pro-inflammatory response.11 This phenotype promotes plaque inflammation and accelerates AS. Catecholamine signal transduction in macrophages can induce cell activation, proliferation, and apoptosis and promote plaque core formation.117 It was found that NA stimulated VSMC cell cycle regulator expression, induced VSMC proliferation, and accelerated vascular remodeling.118 Interestingly, NA stimulates to reduce hydraulic conductivity,119 and this may be a reason behind the patchy distribution of AS in arteries. Catecholamine can induce platelet aggregation in a dose-dependent manner, thereby increasing blood flow resistance and promoting plaque formation.120 NA enhances the expression of matrix metalloproteinase-9 in monocytes released by lipopolysaccharide through the ERK/JNK-c-Fos pathway,121 which causes plaque instability and rupture and leads to adverse outcomes. Catecholamines promote hypercoagulability and increase thrombosis risk after plaque rupture.122
NPY and AS
NPY is a peptide neurotransmitter that is stored together with NA in presynaptic vesicles and large and dense core vesicles. Circulating NPY levels in patients with OSAHS increases when hyperactive sympathetic nerves are pathologic.70 Additionally, endothelial dysfunction further stimulates NPY secretion.123 NPY mainly participates in pathophysiology by binding to Y1R/Y2R/Y5R. NPY binds to Y1R on Vascular smooth muscle cells (VSMCs) and exerts a vasoconstriction effect,124 helping enhance the vasoconstriction of AngII and NA and promote the discontinuity of ECs.125,126 Animal experiments have found that NPY promotes the proliferation of VSMCs by binding to Y1R on VSMCs, leading to intimal thickening.127 Our study further confirmed that NPY affects the proliferation of VSMCs by binding to Y1R to regulate geminin.128 NPY binding to Y2R can drive monocyte migration and adhesion, possibly leading to foam cell formation.129 Our recent study further confirmed that NPY binds to macrophage Y1R to regulate metalloproteinase-8 expression and promote macrophage migration to the vascular injury site.130 Combinations of Y2R/Y5R can stimulate EC proliferation, adhesion, and migration and promote angiogenesis.131,132 Neovascularization may destroy plaque stability, increase bleeding risk, and lead to thrombosis. NPY promotes platelet activation and aggregation by binding to Y1R on ECs, thus promoting thrombosis.5
OSAHS, AS, and Metabolic Diseases
AS plays an important role in the pathogenesis of arteriosclerotic cardiovascular diseases. This can be caused by multiple independent or combined risk factors, including hypertension, obesity, dyslipidemia, and diabetes. OSAHS is widely associated with these risk factors through neuroendocrine mechanisms. Research on the pathogenesis of neuroendocrine diseases and understanding the relationship between these risk factors and AS is helpful to develop better treatment strategies.
Hypertension
OSAHS is independently associated with hypertension. Hypoxemia and hypercapnia also affect local vasodilation.133 However, due to the vasoconstrictor effect of sympathetic nerves on hypoxia and hypercapnia, blood pressure gradually rises during apnea.134 Consistent with sympathetic nerve activity, daytime blood pressure in patients with OSAHS still increases. This abnormality may originate from the elements of the nervous system, such as chemoreceptors and baroreceptors, and may be secondary to increased renal sympathetic activity. This background activity is set by the core neuron network located in NTS, MnPO, PVN, and RVLM. Increases in sympathetic activity caused by the central neuron plasticity may be related to increases in daytime blood pressure in these patients.69 The ultimate neuronal activity manifestation is changes in circulatory media, causing vasoconstriction and increased circulatory resistance. The positive feedback between AngII and MnPO may be another hypertension cause. Impaired baroreflex sensitivity further increases blood pressure.
Hypertension can impair vasomotor activity, cause permanent endothelial injury, and increase cell lipid permeability.135 Elevated sVCAM-1 levels are considered a biomarker of endothelial injury induced by hypertension.136 Oxidative stress and inflammation are major factors allowing hypertension to promote endothelial dysfunction, vascular aging, and AS.137 Moreover, AS can promote hypertension occurrence.
Obesity
Obesity is caused by an imbalance between energy intake and expenditure. The deterioration of sleep disordered breathing is believed to be mainly due to obesity.138 Following this deterioration, the weight of patients with non-mild OSAHS increased by 10%, and moderate to severe OSAHS increased by six-fold.17 Excessive fat accumulation in the neck can lead to airway narrowing and decreased diastolic function. Adipose tissue is not only a place where the body stores energy, but is also an important endocrine organ. Adipose tissue secretes a variety of cytokines and inflammatory factors, such as leptin, adiponectin, TNF-α, and IL-6, that are involved in the pathogenesis of OSAHS.139
Sympathetic nerve hyperactivity is thought to be associated with obesity.140 Glucocorticoids can trigger feeding behavior by activating AMP-activated protein kinase (AMPK) in the hypothalamus. Chronic glucocorticoid exposure promotes adipogenesis through the differentiation of preadipocytes.141 Glucocorticoids can promote fat decomposition and visceral fat deposition, thus promoting fat redistribution, leading to centripetal obesity. NPY binds to Y1R on adipocytes and downregulates glucose transporter 4, leading to insulin resistance and metabolic abnormalities in adipocytes.142 Moreover, NPY greatly promotes adipocyte proliferation and differentiation, and fat accumulation.143,144
Dyslipidemia
Previous studies have shown that OSAHS can promote dyslipidemia.145,146 Dyslipidemia is a recognized risk factor for AS. These abnormalities include increased plasma cholesterol, low-density lipoprotein (LDL), very LDL, and triglyceride levels, and decreased plasma high density lipoprotein (HDL) levels. The concentration of glucocorticoids is closely related to elevated blood lipid levels.147 Glucocorticoids play a “permissive action” on lipolysis mediated by catecholamines and growth hormones, promoting lipolysis and increasing blood lipid concentration. LDL-C/HDL-C is considered to be positively correlated with AS progression.148 HDL has anti-atherosclerotic and antioxidant effects. Increases in LDL-C/HDL-C levels are directly proportional to the severity of OSAHS. AMPK plays a key role in regulating lipid metabolism. Activated AMPK stimulates catabolic pathways and inhibits anabolic pathways. Glucocorticoids can promote the inflow of liver free fatty acids by activating liver AMPK,149 leading to very LDL formation and promoting AS occurrence. It has been observed in animal experiments that treatment with AngII receptor blockers can reduce the overproduction of triglycerides, suggesting that AngII stimulates triglyceride production in the liver.150 NPY increases the expression of cholesterol producing protein and cholesterol synthesis through Y1 and Y5 receptors. This effect is mediated by activating the ERK1/2 signaling pathway.151 Dyslipidemia upregulates and activates AT1R, promotes ox-LDL into ECs, and leads to endothelial dysfunction.152 Hyperlipidemia produces excessive reactive oxygen species, damages ECs, and leads to AS.153
Diabetes
OSAHS has a causal relationship with diabetes and is unrelated to obesity.154 A ganglionic blockade reduces insulin resistance, suggesting a causal relationship between sympathetic nerve excitement and diabetes.155 Insufficient insulin secretion and insulin resistance are key factors in diabetes development. The severity of OSAHS is positively correlated with insulin resistance.156 Glucocorticoids have an anti-insulin effect. Glucocorticoids inhibit AMPK activity in the periphery of adipose tissue, leading to lipolysis and accumulation of plasma free fatty acids (FFAs).157 FFAs may mediate insulin resistance by inhibiting insulin signal transduction and activating the pro-inflammatory NF-kB pathway through toll-like receptors, secreting many pro-inflammatory and pro-atherosclerotic cytokines and chemokines.158 Insulin resistance leads to impaired insulin signal transduction and reduces nitric oxide activation in ECs and VSMCs.159 NPY inhibits insulin secretion by binding to the pancreatic β-cell Y1R.160 AngII stimulation leads to increased oxidative stress, possibly contributing to insulin resistance.161 Catecholamines mediate the inhibition of insulin secretion by activating α-2 adrenoceptors in β-cells.162 Hyperglycemia caused by insulin resistance and hyposecretion is cytotoxic. Hyperglycemia promotes inflammatory cytokine production through a variety of cellular effects, such as the activation of transcription factor NF-kB.163 The imbalance between nitric oxide bioavailability and reactive oxygen species caused by hyperglycemia can lead to endothelial dysfunction.164 High glucose levels may induce the expression of lipoxygenase in ECs and stimulate the adhesion of monocytes to ECs.165 Elevated glucose levels can lead to LDL oxidation.166 Autonomic neuropathy in diabetic patients may affect the innervation and collapse of the upper airway and ventilation drive, contributing to the pathogenesis of OSAHS.
Briefly, neuroendocrine disorders caused by OSAHS directly damage endothelial function or indirectly promote the occurrence and development of metabolic diseases. Moreover, those metabolic diseases are interrelated. For example, there is a causal relationship between hypertension and dyslipidemia, which eventually leads to a wider range of cardiovascular diseases.
Conclusion and Prospect
This review highlights the widespread prevalence of OSAHS and its relation to AS. OSAHS is a multi-system disorder that plays an important role in AS development by causing neuroendocrine disorders. Strong evidence indicates that global OSAHS complications are not only limited to sleep disorders, but also include major extra respiratory complications. In the long term, OSAHS increases the risk of cardiovascular and cerebrovascular mortality. Hypertension, obesity, dyslipidemia, and diabetes are the strongest clinical risk factors related to the progression of OSHAS. The underlying mechanisms of OSAHS-mediated AS causing neuroendocrine disorders are characterized by complex correlations and interactions. Further studies on these potential mechanisms will help determine future targets for clinical intervention and treatment strategies for OSAHS related to cardiovascular diseases.
Mice exposed to chronic IH and a high-cholesterol diet developed AS.167 Hypoxia or a high-fat diet alone did not promote AS. It is suggested that OSAHS causes AS not only by one single mechanism, but by hypoxia, central neuron activity, and sympathetic overactivity. “Syndrome Z” refers to a combination of metabolic syndromes (X syndrome) and OSAHS.168 This reflects the close relationship between OSAHS and metabolic diseases and emphasizes that OSAHS is related to cardiovascular risk under the joint effect of metabolic diseases. Moreover, CPAP combined with weight loss has a greater impact on certain cardiovascular risk factors than CPAP alone.169,170 This further proves that OSAHS is a multi-system disorder, and OSAHS treatment should be comprehensive. Sympathetic nerve outflow is regulated by the central autonomic neural network. The activation of AT1R can induce an increase in reactive oxygen species in cells, which is an important mechanism of central autonomic neural network regulation.171 Hence, AT1R receptor blocker treatment may be a more effective OSAHS-related hypertension treatment strategy.
Most changes in central neuron activity caused by OSAHS are based on animal experiments, which require more human experiments to be confirmed. Because the fact that the sympathetic nervous system accelerates the pathological pro-inflammatory immune process has attracted increasing attention, how hypoxemia/hypercapnia sensitizes specific parts of the central nervous system is particularly important. Currently, there is no unified theory to explain the relationship between OSAHS and AS. More basic and clinical research is needed to describe this relationship more comprehensively.
Abbreviations
AS, Atherosclerosis; AHI, Apnea–hypopnea index; AMPK, AMP-activated protein kinase; AngII, Angiotensin II; AT1R, Angiotensin type 1 receptor; ATCH, Adrenocorticotropic hormone; CPAP, Continuous positive airway pressure; CRH, Corticotropin-releasing hormone; CRHs, Corticotropin-releasing hormones; EC, Endothelial cell; ECs, Endothelial cells; EPC, Endothelial progenitor cell; EPCs, Endothelial progenitor cells; HDL, High density lipoprotein; HDL-C, High-density lipoprotein cholesterol; HPA, Hypothalamic-pituitary adrenergic; IH, Intermittent hypoxia; IL, Interleukin; LDL, Low-density lipoprotein; LDL-C, Low-density lipoprotein cholesterol; MnPO, Median preoptic nucleus; NA, Norepinephrine; NF-kB, Nuclear factor-kB; NPY, Neuropeptide Y; NTS, Nucleus tractus solitarius; OSAHS, Obstructive sleep apnea–hypopnea syndrome; OVLT, Organum vasculosum of the lamina terminalis; PVN, Paraventricular nucleus; RAS, Renin-angiotensin system; RVLM, Rostral ventrolateral medulla; SFO, Subfornical organ; TNF, Tumor necrosis factor; VCAM-1, Vascular cell adhesion molecule-1; VSMC, Vascular smooth muscle cell; VSMCs, Vascular smooth muscle cells.
Funding
This work was supported by Science and Technology Bureau of Quanzhou [grant number 2020CT003], Natural Science Foundation of Fujian Province [grant numbers 2018J01199] and Science and Technology Planning Project of Quanzhou [grant numbers 2018C053R].
Disclosure
The authors report no conflicts of interest in this work.
References
1. Goradel N, Hour F, Negahdari B, et al. Stem cell therapy: a new therapeutic option for cardiovascular diseases. J Cell Biochem. 2018;119(1):95–104. doi:10.1002/jcb.26169
2. Soler-Botija C, Gálvez-Montón C, Bayés-Genís A. Epigenetic biomarkers in cardiovascular diseases. Front Genet. 2019;10:950.
3. Pardali E, Dimmeler S, Zeiher A, Rieger M. Clonal hematopoiesis, aging, and cardiovascular diseases. Exp Hematol. 2020;83:95–104.
4. Libby P, Buring J, Badimon L, et al. Atherosclerosis. Nat Rev Disprimers. 2019;5(1):56.
5. Zhu P, Sun W, Zhang C, Song Z, Lin S. The role of neuropeptide Y in the pathophysiology of atherosclerotic cardiovascular disease. Int J Cardiol. 2016;220:235–241.
6. Zhu Y, Xian X, Wang Z, et al. Research progress on the relationship between atherosclerosis and inflammation. Biomolecules. 2018;8:3.
7. Bäck M, Yurdagul A, Tabas I, Öörni K, Kovanen P. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol. 2019;16(7):389–406.
8. Herrington W, Lacey B, Sherliker P, Armitage J, Lewington S. Epidemiology of atherosclerosis and the potential to reduce the global burden of atherothrombotic disease. Circ Res. 2016;118(4):535–546.
9. Ruiz-Ortega M, Lorenzo O, Rupérez M, et al. Role of the renin-angiotensin system in vascular diseases: expanding the field. Hypertension. 2001;38(6):1382–1387.
10. Walker B. Glucocorticoids and cardiovascular disease. Eur J Endocrinol. 2007;157(5):545–559.
11. van der Heijden C, Groh L, Keating S, et al. Catecholamines induce trained immunity in monocytes in vitro and in vivo. Circ Res. 2020;127(2):269–283.
12. Punjabi N, Caffo B, Goodwin J, et al. Sleep-disordered breathing and mortality: a prospective cohort study. PLoS Med. 2009;6(8):e1000132.
13. Senaratna C, Perret J, Lodge C, et al. Prevalence of obstructive sleep apnea in the general population: a systematic review. Sleep Med Rev. 2017;34:70–81.
14. Tripathi A, Melnik A, Xue J, et al. Intermittent hypoxia and hypercapnia, a hallmark of obstructive sleep apnea, alters the gut microbiome and metabolome. mSystems. 2018;3:3.
15. Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med. 1993;328(17):1230–1235.
16. Peppard P, Young T, Barnet J, Palta M, Hagen E, Hla K. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol. 2013;177(9):1006–1014.
17. Peppard P, Young T, Palta M, Dempsey J, Skatrud J. Longitudinal study of moderate weight change and sleep-disordered breathing. JAMA. 2000;284(23):3015–3021.
18. Anandam A, Akinnusi M, Kufel T, Porhomayon J, El-Solh A. Effects of dietary weight loss on obstructive sleep apnea: a meta-analysis. Sleep & Breathing = Schlaf & Atmung. 2013;17(1):227–234.
19. Flegal K, Carroll M, Ogden C, Curtin L. Prevalence and trends in obesity among US adults, 1999–2008. JAMA. 2010;303(3):235–241.
20. Eckert D, Malhotra A. Pathophysiology of adult obstructive sleep apnea. Proc Am Thorac Soc. 2008;5(2):144–153.
21. Gottlieb D, Punjabi N. Diagnosis and management of obstructive sleep apnea: a review. JAMA. 2020;323(14):1389–1400.
22. Tahrani A. Obstructive sleep apnoea in diabetes: does it matter? Diabetes Vasc Dis Res. 2017;14(5):454–462.
23. Dempsey J, Veasey S, Morgan B, O’Donnell C. Pathophysiology of sleep apnea. Physiol Rev. 2010;90(1):47–112.
24. Gleeson K, Zwillich C, White D. The influence of increasing ventilatory effort on arousal from sleep. Am Rev Respir Dis. 1990;142(2):295–300.
25. Peng Y, Nanduri J, Zhang X, et al. Endothelin-1 mediates attenuated carotid baroreceptor activity by intermittent hypoxia. J Appl Physiol. 2012;112(1):187–196.
26. López-Barneo J. Oxygen sensing and stem cell activation in the hypoxic carotid body. Cell Tissue Res. 2018;372(2):417–425.
27. Del Rio R, Andrade D, Lucero C, Arias P, Iturriaga R. Carotid body ablation abrogates hypertension and autonomic alterations induced by intermittent hypoxia in rats. Hypertension. 2016;68(2):436–445.
28. Narkiewicz K, Pesek C, Kato M, Phillips B, Davison D, Somers V. Baroreflex control of sympathetic nerve activity and heart rate in obstructive sleep apnea. Hypertension. 1998;32(6):1039–1043.
29. Moreira T, Takakura A, Colombari E, Guyenet P. Central chemoreceptors and sympathetic vasomotor outflow. J Physiol. 2006;577:369–386.
30. Madirazza K, Pecotic R, Pavlinac Dodig I, Valic M, Dogas Z. Hyperoxia blunts renal sympathetic nerve activity response to acute intermittent hypercapnia in rats. J Physiol Pharmacol. 2019;70:5.
31. Jouett N, Watenpaugh D, Dunlap M, Smith M. Interactive effects of hypoxia, hypercapnia and lung volume on sympathetic nerve activity in humans. Exp Physiol. 2015;100(9):1018–1029.
32. Tan M, Yap A, Chua A, Wong J, Parot M, Tan K. Prevalence of sleep bruxism and its association with obstructive sleep apnea in adult patients: a retrospective polysomnographic investigation. J Oral Facial Pain Headache. 2019;33(3):269–277.
33. Kostrzewa-Janicka J, Jurkowski P, Zycinska K, Przybyłowska D, Mierzwińska-Nastalska E. Sleep-related breathing disorders and bruxism. Adv Exp Med Biol. 2015;873:9–14.
34. Hosoya H, Kitaura H, Hashimoto T, et al. Relationship between sleep bruxism and sleep respiratory events in patients with obstructive sleep apnea syndrome. Sleep & Breathing = Schlaf & Atmung. 2014;18(4):837–844.
35. Lavigne G, Huynh N, Kato T, et al. Genesis of sleep bruxism: motor and autonomic-cardiac interactions. Arch Oral Biol. 2007;52(4):381–384.
36. Quan SF, Gillin JC, Littner MR, Shepard JW. Sleep-related breathing disorders in adults: recommendations for syndrome definition and measurement techniques in clinical research. The Report of an American Academy of Sleep Medicine Task Force. Sleep. 1999;22(5):667–689.
37. Garbarino S, Scoditti E, Lanteri P, Conte L, Magnavita N, Toraldo D. Obstructive sleep apnea with or without excessive daytime sleepiness: clinical and experimental data-driven phenotyping. Front Neurol. 2018;9:505.
38. Vgontzas A, Bixler E, Chrousos G. Metabolic disturbances in obesity versus sleep apnoea: the importance of visceral obesity and insulin resistance. J Intern Med. 2003;254(1):32–44.
39. Li Y, Vgontzas A, Fernandez-Mendoza J, et al. Objective, but not subjective, sleepiness is associated with inflammation in sleep apnea. Sleep. 2017;40:2.
40. Kawano Y, Tamura A, Kadota J. Association between the severity of obstructive sleep apnea and the ratio of low-density lipoprotein cholesterol to high-density lipoprotein cholesterol. Metabolism. 2012;61(2):186–192.
41. Cuhadaroğlu C, Utkusavaş A, Oztürk L, Salman S, Ece T. Effects of nasal CPAP treatment on insulin resistance, lipid profile, and plasma leptin in sleep apnea. Lung. 2009;187(2):75–81.
42. West S, Nicoll D, Wallace T, Matthews D, Stradling J. Effect of CPAP on insulin resistance and HbA1c in men with obstructive sleep apnoea and type 2 diabetes. Thorax. 2007;62(11):969–974.
43. Tuomilehto H, Seppä J, Partinen M, et al. Lifestyle intervention with weight reduction: first-line treatment in mild obstructive sleep apnea. Am J Respir Crit Care Med. 2009;179(4):320–327.
44. Srijithesh P, Aghoram R, Goel A, Dhanya J. Positional therapy for obstructive sleep apnoea. Cochrane Database Syst Rev. 2019;5:CD010990.
45. Drager L, Bortolotto L, Lorenzi M, Figueiredo A, Krieger E, Lorenzi-Filho G. Early signs of atherosclerosis in obstructive sleep apnea. Am J Respir Crit Care Med. 2005;172(5):613–618.
46. Drager L, Bortolotto L, Figueiredo A, Krieger E, Lorenzi G. Effects of continuous positive airway pressure on early signs of atherosclerosis in obstructive sleep apnea. Am J Respir Crit Care Med. 2007;176(7):706–712.
47. Turmel J, Sériès F, Boulet L, et al. Relationship between atherosclerosis and the sleep apnea syndrome: an intravascular ultrasound study. Int J Cardiol. 2009;132(2):203–209.
48. Hla K, Young T, Hagen E, et al. Coronary heart disease incidence in sleep disordered breathing: the Wisconsin Sleep Cohort Study. Sleep. 2015;38(5):677–684.
49. Zhang J, Guo Q, Peng L, et al. The association of neck circumference with incident congestive heart failure and coronary heart disease mortality in a community-based population with or without sleep-disordered breathing. BMC Cardiovasc Disord. 2018;18(1):108.
50. Munoz R, Duran-Cantolla J, Martínez-Vila E, et al. Severe sleep apnea and risk of ischemic stroke in the elderly. Stroke. 2006;37(9):2317–2321.
51. Arzt M, Young T, Finn L, Skatrud J, Bradley T. Association of sleep-disordered breathing and the occurrence of stroke. Am J Respir Crit Care Med. 2005;172(11):1447–1451.
52. Valenza M, Baranchuk A, Valenza-Demet G, Muñoz-Casaubon T, Martin-Navajas J, Healey J. Prevalence of risk factors for atrial fibrillation and stroke among 1210 patients with sleep disordered breathing. Int J Cardiol. 2014;174(1):73–76.
53. Yaggi H, Concato J, Kernan W, Lichtman J, Brass L, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med. 2005;353(19):2034–2041.
54. Shantsila E, Watson T, Lip G. Endothelial progenitor cells in cardiovascular disorders. J Am Coll Cardiol. 2007; 49(7):741–752.
55. Kato M, Roberts-Thomson P, Phillips B, et al. Impairment of endothelium-dependent vasodilation of resistance vessels in patients with obstructive sleep apnea. Circulation. 2000;102(21):2607–2610.
56. Jelic S, Padeletti M, Kawut S, et al. Inflammation, oxidative stress, and repair capacity of the vascular endothelium in obstructive sleep apnea. Circulation. 2008;117(17):2270–2278.
57. El Solh A, Akinnusi M, Baddoura F, Mankowski C. Endothelial cell apoptosis in obstructive sleep apnea: a link to endothelial dysfunction. Am J Respir Crit Care Med. 2007;175(11):1186–1191.
58. Fadini G, Agostini C, Sartore S, Avogaro A. Endothelial progenitor cells in the natural history of atherosclerosis. Atherosclerosis. 2007;194(1):46–54.
59. Kizawa T, Nakamura Y, Takahashi S, Sakurai S, Yamauchi K, Inoue H. Pathogenic role of angiotensin II and oxidised LDL in obstructive sleep apnoea. Eur Respir J. 2009;34(6):1390–1398.
60. de la Peña M, Barceló A, Barbe F, et al. Endothelial function and circulating endothelial progenitor cells in patients with sleep apnea syndrome. Respir Int Rev Thoracic Dis. 2008;76(1):28–32.
61. Drager L, Bortolotto L, Figueiredo A, Silva B, Krieger E, Lorenzi-Filho G. Obstructive sleep apnea, hypertension, and their interaction on arterial stiffness and heart remodeling. Chest. 2007;131(5):1379–1386.
62. Ip M, Tse H, Lam B, Tsang K, Lam W. Endothelial function in obstructive sleep apnea and response to treatment. Am J Respir Crit Care Med. 2004;169(3):348–353.
63. Kohler M, Craig S, Pepperell J, et al. CPAP improves endothelial function in patients with minimally symptomatic OSA: results from a subset study of the MOSAIC trial. Chest. 2013;144(3):896–902.
64. Simpson P, Hoyos C, Celermajer D, Liu P, Ng M. Effects of continuous positive airway pressure on endothelial function and circulating progenitor cells in obstructive sleep apnoea: a randomised sham-controlled study. Int J Cardiol. 2013;168(3):2042–2048.
65. Peker Y, Glantz H, Eulenburg C, Wegscheider K, Herlitz J, Thunström E. Effect of positive airway pressure on cardiovascular outcomes in coronary artery disease patients with nonsleepy obstructive sleep apnea. The RICCADSA randomized controlled trial. Am J Respir Crit Care Med. 2016;194(5):613–620.
66. Grundy S, Cleeman J, Daniels S, et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112(17):2735–2752.
67. Peppard P, Young T, Palta M, Skatrud J. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med. 2000;342(19):1378–1384.
68. Tamisier R, Pépin J, Rémy J, et al. 14 nights of intermittent hypoxia elevate daytime blood pressure and sympathetic activity in healthy humans. Eur Respir J. 2011;37(1):119–128.
69. Gilmartin G, Lynch M, Tamisier R, Weiss J. Chronic intermittent hypoxia in humans during 28 nights results in blood pressure elevation and increased muscle sympathetic nerve activity. Am J Physiol Heart Circ Physiol. 2010;299(3):H925–931.
70. Barceló A, Barbé F, Llompart E, et al. Neuropeptide Y and leptin in patients with obstructive sleep apnea syndrome: role of obesity. Am J Respir Crit Care Med. 2005;171(2):183–187.
71. Harsch I, Konturek P, Koebnick C, et al. Leptin and ghrelin levels in patients with obstructive sleep apnoea: effect of CPAP treatment. Eur Respir J. 2003;22(2):251–257.
72. Phillips B, Kato M, Narkiewicz K, Choe I, Somers V. Increases in leptin levels, sympathetic drive, and weight gain in obstructive sleep apnea. Am J Physiol Heart Circ Physiol. 2000;279(1):H234–237.
73. Sahlman J, Seppä J, Herder C, et al. Effect of weight loss on inflammation in patients with mild obstructive sleep apnea. Nutri Metabol Cardiovasc Dis. 2012;22(7):583–590.
74. Smith P, Gold A, Meyers D, Haponik E, Bleecker E. Weight loss in mildly to moderately obese patients with obstructive sleep apnea. Ann Intern Med. 1985;103:850–855.
75. Johansson K, Neovius M, Lagerros Y, et al. Effect of a very low energy diet on moderate and severe obstructive sleep apnoea in obese men: a randomised controlled trial. BMJ (Clinical Research Ed). 2009;339:b4609.
76. Anothaisintawee T, Reutrakul S, Van Cauter E, Thakkinstian A. Sleep disturbances compared to traditional risk factors for diabetes development: systematic review and meta-analysis. Sleep Med Rev. 2016;30:11–24.
77. Nagayoshi M, Punjabi N, Selvin E, et al. Obstructive sleep apnea and incident type 2 diabetes. Sleep Med. 2016;25:156–161.
78. Punjabi N, Beamer B. Alterations in glucose disposal in sleep-disordered breathing. Am J Respir Crit Care Med. 2009;179(3):235–240.
79. Labarca G, Dreyse J, Salas C, et al. Risk of mortality among patients with moderate to severe obstructive sleep apnea and diabetes mellitus: results from the SantOSA cohort. Sleep & Breathing = Schlaf & Atmung. 2021;4:1–9.
80. Dampney R. Functional organization of central pathways regulating the cardiovascular system. Physiol Rev. 1994;74(2):323–364.
81. Knight W, Little J, Carreno F, Toney G, Mifflin S, Cunningham J. Chronic intermittent hypoxia increases blood pressure and expression of FosB/DeltaFosB in central autonomic regions. Am J Physiol Regul Integr Comp Physiol. 2011;301(1):R131–139.
82. Mifflin S, Cunningham J, Toney G. Neurogenic mechanisms underlying the rapid onset of sympathetic responses to intermittent hypoxia. J Appl Physiol. 2015;119(12):1441–1448.
83. Guyenet P. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7(5):335–346.
84. Bathina C, Rajulapati A, Franzke M, Yamamoto K, Cunningham J, Mifflin S. Knockdown of tyrosine hydroxylase in the nucleus of the solitary tract reduces elevated blood pressure during chronic intermittent hypoxia. Am J Physiol Regul Integr Comp Physiol. 2013;305(9):R1031–1039.
85. Zoccal D, Furuya W, Bassi M, Colombari D, Colombari E. The nucleus of the solitary tract and the coordination of respiratory and sympathetic activities. Front Physiol. 2014;5:238.
86. Chan J, Chen W, Lee H, Chan S. Elevated Fos expression in the nucleus tractus solitarii is associated with reduced baroreflex response in spontaneously hypertensive rats. Hypertension. 1998;32(5):939–944.
87. Johnson A, Cunningham J, Thunhorst R. Integrative role of the lamina terminalis in the regulation of cardiovascular and body fluid homeostasis. Clin Exp Pharmacol Physiol. 1996;23(2):183–191.
88. Mourão A, de Mello A, Dos Santos Moreira Met al. Median preoptic nucleus excitatory neurotransmitters in the maintenance of hypertensive state. Brain Res Bull. 2018;142:207–215.
89. McKinley M, Yao S, Uschakov A, McAllen R, Rundgren M, Martelli D. The median preoptic nucleus: front and centre for the regulation of body fluid, sodium, temperature, sleep and cardiovascular homeostasis. Acta Physiologica. 2015;214(1):8–32.
90. Marciante A, Wang L, Little J, Cunningham J. Caspase lesions of PVN-projecting MnPO neurons block the sustained component of CIH-induced hypertension in adult male rats. Am J Physiol Heart Circ Physiol. 2020;318(1):H34–H48.
91. King T, Heesch C, Clark C, Kline D, Hasser E. Hypoxia activates nucleus tractus solitarii neurons projecting to the paraventricular nucleus of the hypothalamus. Am J Physiol Regul Integr Comp Physiol. 2012;302(10):R1219–1232.
92. Koba S, Hanai E, Kumada N, Kataoka N, Nakamura K, Watanabe T. Sympathoexcitation by hypothalamic paraventricular nucleus neurons projecting to the rostral ventrolateral medulla. J Physiol. 2018;596(19):4581–4595.
93. Maruyama N, Mitchell N, Truong T, Toney G. Activation of the hypothalamic paraventricular nucleus by acute intermittent hypoxia: implications for sympathetic long-term facilitation neuroplasticity. Exp Neurol. 2019;314:1–8.
94. Henley D, Russell G, Douthwaite J, et al. Hypothalamic-pituitary-adrenal axis activation in obstructive sleep apnea: the effect of continuous positive airway pressure therapy. J Clin Endocrinol Metab. 2009;94(11):4234–4242.
95. Somers V, Mark A, Zavala D, Abboud F. Contrasting effects of hypoxia and hypercapnia on ventilation and sympathetic activity in humans. J Appl Physiol. 1989;67(5):2101–2106.
96. Molkov Y, Zoccal D, Moraes D, Paton J, Machado B, Rybak I. Intermittent hypoxia-induced sensitization of central chemoreceptors contributes to sympathetic nerve activity during late expiration in rats. J Neurophysiol. 2011;105(6):3080–3091.
97. Horner R, Brooks D, Kozar L, Tse S, Phillipson E. Immediate effects of arousal from sleep on cardiac autonomic outflow in the absence of breathing in dogs. J Appl Physiol. 1995;79(1):151–162.
98. Somers V, Dyken M, Skinner J. Autonomic and hemodynamic responses and interactions during the Mueller maneuver in humans. J Auton Nerv Syst. 1993;44:253–259.
99. Zwillich C, Devlin T, White D, Douglas N, Weil J, Martin R. Bradycardia during sleep apnea. Characteristics and mechanism. J Clin Invest. 1982;69(6):1286–1292.
100. Møller D, Lind P, Strunge B, Pedersen E. Abnormal vasoactive hormones and 24-hour blood pressure in obstructive sleep apnea. Am J Hypertens. 2003;16(4):274–280.
101. Tomfohr L, Edwards K, Dimsdale J. Is obstructive sleep apnea associated with cortisol levels? A systematic review of the research evidence. Sleep Med Rev. 2012;16(3):243–249.
102. Clark R, Boudoulas H, Schaal S, Schmidt H. Adrenergic hyperactivity and cardiac abnormality in primary disorders of sleep. Neurology. 1980;30(2):113–119.
103. Ryan M, Didion S, Mathur S, Faraci F, Sigmund C. Angiotensin II-induced vascular dysfunction is mediated by the AT1A receptor in mice. Hypertension. 2004;43(5):1074–1079.
104. Hill J, Zalos G, Halcox J, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348(7):593–600.
105. Endtmann C, Ebrahimian T, Czech T, et al. Angiotensin II impairs endothelial progenitor cell number and function in vitro and in vivo: implications for vascular regeneration. Hypertension. 2011;58(3):394–403.
106. Pueyo M, Gonzalez W, Nicoletti A, Savoie F, Arnal J, Michel J. Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via nuclear factor-kappaB activation induced by intracellular oxidative stress. Arterioscler Thromb Vasc Biol. 2000;20(3):645–651.
107. Piqueras L, Sanz M. Angiotensin II and leukocyte trafficking: new insights for an old vascular mediator. Role of redox-signaling pathways. Free Radic Biol Med. 2020;157:38–54.
108. Keidar S, Kaplan M, Hoffman A, Aviram M. Angiotensin II stimulates macrophage-mediated oxidation of low density lipoproteins. Atherosclerosis. 1995;115(2):201–215.
109. Rudijanto A. The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis. Acta Medica Indonesiana. 2007;39(2):86–93.
110. Eguchi S, Frank G, Mifune M, Inagami T. Metalloprotease-dependent ErbB ligand shedding in mediating EGFR transactivation and vascular remodelling. Biochem Soc Trans. 2003;31:1198–1202.
111. Alevizaki M, Cimponeriu A, Lekakis J, Papamichael C, Chrousos G. High anticipatory stress plasma cortisol levels and sensitivity to glucocorticoids predict severity of coronary artery disease in subjects undergoing coronary angiography. Metabolism. 2007;56(2):222–226.
112. Schäfer S, Wallerath T, Closs E, et al. Dexamethasone suppresses eNOS and CAT-1 and induces oxidative stress in mouse resistance arterioles. Am J Physiol Heart Circ Physiol. 2005;288(1):H436–444.
113. Golbidi S, Frisbee J, Laher I. Chronic stress impacts the cardiovascular system: animal models and clinical outcomes. Am J Physiol Heart Circ Physiol. 2015;308(12):H1476–1498.
114. Lee J, Zhang J, Massmann G, Figueroa J. Antenatal betamethasone increases vascular reactivity to endothelin-1 by upregulation of CD38/cADPR signaling. J Dev Orig Health Dis. 2014;5(1):56–62.
115. Coelho M, Santos C, Vieira Neto L, Gadelha M. Adverse effects of glucocorticoids: coagulopathy. Eur J Endocrinol. 2015;173(4):M11–21.
116. Kirton J, Wilkinson F, Canfield A, Alexander M. Dexamethasone downregulates calcification-inhibitor molecules and accelerates osteogenic differentiation of vascular pericytes: implications for vascular calcification. Circ Res. 2006;98(10):1264–1272.
117. Barnes M, Carson M, Nair M. Non-traditional cytokines: how catecholamines and adipokines influence macrophages in immunity, metabolism and the central nervous system. Cytokine. 2015;72(2):210–219.
118. Guo J, Sun N. Cell cycle regulator geminin is dispensable for the proliferation of vascular smooth muscle cells. Sci China Life Sci. 2013;56(8):731–738.
119. Chooi K, Comerford A, Sherwin S, Weinberg P. Noradrenaline has opposing effects on the hydraulic conductance of arterial intima and media. J Biomech. 2017;54:4–10.
120. Matsukawa T, Hikasa Y. Effects of imidazoline and nonimidazoline α-adrenoceptor agonists and antagonists, including xylazine, medetomidine, dexmedetomidine, yohimbine, and atipamezole, on aggregation of feline platelets. Am J Vet Res. 2020;81(2):159–171.
121. Yin X, Zhou L, Han F, et al. Beta-adrenoceptor activation by norepinephrine enhances lipopolysaccharide-induced matrix metalloproteinase-9 expression through the ERK/JNK-c-Fos pathway in human THP-1 cells. J Atheroscler Thromb. 2017;24(1):55–67.
122. Preckel D, von Känel R. Regulation of hemostasis by the sympathetic nervous system: any contribution to coronary artery disease? Heartdrug. 2004;4(3):123–130.
123. Choi B, Shin M, Kim E, et al. Elevated Neuropeptide Y in endothelial dysfunction promotes macrophage infiltration and smooth muscle foam cell formation. Front Immunol. 2019;10:1701.
124. Hubers S, Wilson J, Yu C, et al. DPP (Dipeptidyl Peptidase)-4 inhibition potentiates the vasoconstrictor response to NPY (Neuropeptide Y) in humans during renin-angiotensin-aldosterone system inhibition. Hypertension. 2018;72(3):712–719.
125. Thulin T, Erlinge D. Neuropeptide Y and hypertension. Nutrition. 1995; 11:495–497.
126. Saraf R, Mahmood F, Amir R, Matyal R. Neuropeptide Y is an angiogenic factor in cardiovascular regeneration. Eur J Pharmacol. 2016;776:64–70.
127. Zukowska-Grojec Z, Pruszczyk P, Colton C, et al. Mitogenic effect of neuropeptide Y in rat vascular smooth muscle cells. Peptides. 1993;14(2):263–268.
128. Jiang Z, Zhou Y, Chen X, et al. Different effects of neuropeptide Y on proliferation of vascular smooth muscle cells via regulation of Geminin. Mol Cell Biochem. 2017;433:205–211.
129. Nave H, Bedoui S, Moenter F, et al. Reduced tissue immigration of monocytes by neuropeptide Y during endotoxemia is associated with Y2 receptor activation. J Neuroimmunol. 2004;155:1–12.
130. Wu W, Peng S, Shi Y, Li L, Song Z, Lin S. NPY promotes macrophage migration by upregulating matrix metalloproteinase-8 expression. J Cell Physiol. 2021;236(3):1903–1912.
131. Zukowska-Grojec Z, Karwatowska-Prokopczuk E, Rose W, et al. Neuropeptide Y: a novel angiogenic factor from the sympathetic nerves and endothelium. Circ Res. 1998;83(2):187–195.
132. Li L, Najafi A, Kitlinska J, et al. Of mice and men: neuropeptide Y and its receptors are associated with atherosclerotic lesion burden and vulnerability. J Cardiovasc Transl Res. 2011;4(3):351–362.
133. Daugherty R, Scott J, Dabney J, Haddy F. Local effects of O2 and CO2 on limb, renal, and coronary vascular resistances. Am J Physiol. 1967;213(5):1102–1110.
134. Imadojemu V, Gleeson K, Gray K, Sinoway L, Leuenberger U. Obstructive apnea during sleep is associated with peripheral vasoconstriction. Am J Respir Crit Care Med. 2002;165(1):61–66.
135. Kassan M, Galán M, Partyka M, et al. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler Thromb Vasc Biol. 2012;32(7):1652–1661.
136. Tchalla A, Wellenius G, Travison T, et al. Circulating vascular cell adhesion molecule-1 is associated with cerebral blood flow dysregulation, mobility impairment, and falls in older adults. Hypertension. 2015;66(2):340–346.
137. Guzik T, Touyz R. Oxidative stress, inflammation, and vascular aging in hypertension. Hypertension. 2017;70(4):660–667.
138. Pretto J, Gyulay S, Hensley M. Trends in anthropometry and severity of sleep-disordered breathing over two decades of diagnostic sleep studies in an Australian adult sleep laboratory. Med J Aust. 2010;193(4):213–216.
139. Li M, Li X, Lu Y. Obstructive sleep apnea syndrome and metabolic diseases. Endocrinology. 2018;159(7):2670–2675.
140. Seematter G, Guenat E, Schneiter P, Cayeux C, Jéquier E, Tappy L. Effects of mental stress on insulin-mediated glucose metabolism and energy expenditure in lean and obese women. Am J Physiol Endocrinol Metab. 2000;279(4):E799–805.
141. Campbell J, Peckett A, D’souza A, Hawke T, Riddell M. Adipogenic and lipolytic effects of chronic glucocorticoid exposure. Am J Physiol Cell Physiol. 2011;300(1):C198–209.
142. Gericke M, Schröder T, Kosacka J, Nowicki M, Klöting N, Spanel-Borowski K. Neuropeptide Y impairs insulin-stimulated translocation of glucose transporter 4 in 3T3-L1 adipocytes through the Y1 receptor. Mol Cell Endocrinol. 2012;348(1):27–32.
143. Shin M, Choi B, Kim E, et al. Elevated Pentraxin 3 in obese adipose tissue promotes adipogenic differentiation by activating Neuropeptide Y signaling. Front Immunol. 2018;9:1790.
144. Zhang L, Macia L, Turner N, et al. Peripheral neuropeptide Y Y1 receptors regulate lipid oxidation and fat accretion. Int J Obesity (2005). 2010;34(2):357–373.
145. Davies R, Turner R, Crosby J, Stradling J. Plasma insulin and lipid levels in untreated obstructive sleep apnoea and snoring; their comparison with matched controls and response to treatment. J Sleep Res. 1994;3(3):180–185.
146. Iesato K, Tatsumi K, Saibara T, et al. Decreased lipoprotein lipase in obstructive sleep apnea syndrome. Circu J. 2007;71(8):1293–1298.
147. Vukomanovic V, Ignjatovic V, Mihaljevic O, Vuleta K, Matovic M. Glucose and lipid abnormalities in patients with adrenal incidentalomas. Hell J Nucl Med. 2019;22:7–14.
148. Nicholls S, Tuzcu E, Sipahi I, et al. Statins, high-density lipoprotein cholesterol, and regression of coronary atherosclerosis. JAMA. 2007;297(5):499–508.
149. Christ-Crain M, Kola B, Lolli F, et al. AMP-activated protein kinase mediates glucocorticoid-induced metabolic changes: a novel mechanism in Cushing’s syndrome. FASEB J. 2008;22(6):1672–1683.
150. Ran J, Hirano T, Adachi M. Chronic ANG II infusion increases plasma triglyceride level by stimulating hepatic triglyceride production in rats. Am J Physiol Endocrinol Metab. 2004;287(5):E955–961.
151. Chen F, Zhou Y, Yang K, Shen M, Wang Y. NPY stimulates cholesterol synthesis acutely by activating the SREBP2-HMGCR pathway through the Y1 and Y5 receptors in murine hepatocytes. Life Sci. 2020;262:118478.
152. Wang X, Phillips M, Mehta J. LOX-1 and angiotensin receptors, and their interplay. Cardiovasc Drugs Ther. 2011;25(5):401–417.
153. Huang Q, Qin L, Dai S, et al. AIP1 suppresses atherosclerosis by limiting hyperlipidemia-induced inflammation and vascular endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2013;33(4):795–804.
154. Floras J. Sleep apnea and cardiovascular disease: an enigmatic risk factor. Circ Res. 2018;122(12):1741–1764.
155. Gamboa A, Okamoto L, Arnold A, et al. Autonomic blockade improves insulin sensitivity in obese subjects. Hypertension. 2014;64(4):867–874.
156. de Lima F, Mazzotti D, Tufik S, Bittencourt L. The role inflammatory response genes in obstructive sleep apnea syndrome: a review. Sleep & Breathing = Schlaf & Atmung. 2016;20(1):331–338.
157. Lim C, Kola B, Korbonits M. AMPK as a mediator of hormonal signalling. J Mol Endocrinol. 2010;44(2):87–97.
158. Boden G. Obesity, insulin resistance and free fatty acids. Curr Opin Endocrinol Diabetes Obes. 2011;18(2):139–143.
159. DeFronzo R. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: the missing links. The Claude Bernard Lecture 2009. Diabetologia. 2010;53(7):1270–1287.
160. Cho Y, Kim C. Neuropeptide Y promotes beta-cell replication via extracellular signal-regulated kinase activation. Biochem Biophys Res Commun. 2004;314(3):773–780.
161. Ohki K, Wakui H, Kishio N, et al. Angiotensin II Type 1 receptor-associated protein inhibits angiotensin II-induced insulin resistance with suppression of oxidative stress in skeletal muscle tissue. Sci Rep. 2018;8(1):2846.
162. Chan S, Perrett C, Morgan N. Differential expression of alpha 2-adrenoceptor subtypes in purified rat pancreatic islet A- and B-cells. Cell Signal. 1997;9(1):71–78.
163. La Sala L, Prattichizzo F, Ceriello A. The link between diabetes and atherosclerosis. Eur J Prev Cardiol. 2019;26:15–24.
164. Sun W, Zhu P, Shi Y, et al. Current views on neuropeptide Y and diabetes-related atherosclerosis. Diabetes Vasc Dis Res. 2017;14(4):277–284.
165. Kanter J, Johansson F, LeBoeuf R, Bornfeldt K. Do glucose and lipids exert independent effects on atherosclerotic lesion initiation or progression to advanced plaques? Circ Res. 2007;100(6):769–781.
166. Baynes J, Thorpe S. Glycoxidation and lipoxidation in atherogenesis. Free Radic Biol Med. 2000;28(12):1708–1716.
167. Savransky V, Nanayakkara A, Li J, et al. Chronic intermittent hypoxia induces atherosclerosis. Am J Respir Crit Care Med. 2007;175(12):1290–1297.
168. Drager L, Togeiro S, Polotsky V, Lorenzi-Filho G. Obstructive sleep apnea: a cardiometabolic risk in obesity and the metabolic syndrome. J Am Coll Cardiol. 2013;62(7):569–576.
169. Hudgel D. Critical review: CPAP and weight management of obstructive sleep apnea cardiovascular co-morbidities. Sleep Med Rev. 2018;37:14–23.
170. Chirinos J, Gurubhagavatula I, Teff K, et al. CPAP, weight loss, or both for obstructive sleep apnea. N Engl J Med. 2014;370(24):2265–2275.
171. Cruz J, Flôr A, França-Silva M, Balarini C, Braga V. Reactive oxygen species in the paraventricular nucleus of the hypothalamus alter sympathetic activity during metabolic syndrome. Front Physiol. 2015;6:384.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.