Back to Journals » International Journal of Nanomedicine » Volume 13
Recent advances in "smart" delivery systems for extended drug release in cancer therapy
Authors Kalaydina RV ![]() , Bajwa K, Qorri B
, Bajwa K, Qorri B ![]() , Decarlo A, Szewczuk MR
, Decarlo A, Szewczuk MR ![]()
Received 7 May 2018
Accepted for publication 23 June 2018
Published 20 August 2018 Volume 2018:13 Pages 4727—4745
DOI https://doi.org/10.2147/IJN.S168053
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Regina-Veronicka Kalaydina,1,* Komal Bajwa,2,* Bessi Qorri,1,* Alexandria Decarlo,3 Myron R Szewczuk1
1Department of Biomedical and Molecular Sciences, Queen’s University, 2Postgraduate Medical Education, Graduate Diploma and Professional Master in Medical Sciences, School of Medicine, Queen’s University, 3Department of Biology, Queen’s University, Kingston, ON, Canada
*These authors contributed equally to this work
Abstract: Advances in nanomedicine have become indispensable for targeted drug delivery, early detection, and increasingly personalized approaches to cancer treatment. Nanoparticle-based drug-delivery systems have overcome some of the limitations associated with traditional cancer-therapy administration, such as reduced drug solubility, chemoresistance, systemic toxicity, narrow therapeutic indices, and poor oral bioavailability. Advances in the field of nanomedicine include “smart” drug delivery, or multiple levels of targeting, and extended-release drug-delivery systems that provide additional methods of overcoming these limitations. More recently, the idea of combining smart drug delivery with extended-release has emerged in hopes of developing highly efficient nanoparticles with improved delivery, bioavailability, and safety profiles. Although functionalized and extended-release drug-delivery systems have been studied extensively, there remain gaps in the literature concerning their application in cancer treatment. We aim to provide an overview of smart and extended-release drug-delivery systems for the delivery of cancer therapies, as well as to introduce innovative advancements in nanoparticle design incorporating these principles. With the growing need for increasingly personalized medicine in cancer treatment, smart extended-release nanoparticles have the potential to enhance chemotherapy delivery, patient adherence, and treatment outcomes in cancer patients.
Keywords: nanomedicine, smart delivery systems, extended drug release, personalized medicine
Introduction
Nanoparticles (NPs) have been successfully adopted in electronics, food and agriculture, biosensing, and some areas of nanomedicine; however, their translation to clinical oncology remains limited.1 Although nanomedicine-based drug delivery has been dominating the field of cancer research over the past decade, only a dozen US Food and Drug Administration (FDA)-approved NPs are currently available.2 As such, there is a growing need for novel NPs in oncology to improve drug delivery for cancer treatment, mainly through target-driven design.3 Currently, poor patient outcomes are attributed in part to the low stability, drug solubility and bioavailability, poor pharmacokinetic (PK) and pharmacodynamic (PD) parameters, aspecific distribution, cytotoxicity, and chemoresistance that are characteristic of traditional chemotherapeutic agents.4,5 As a result, nanomedicine-based drug delivery has been of increasing research interest because NPs have been shown to substantially improve the therapeutic efficacy of chemotherapeutic agents by overcoming the various anatomical, physiological, chemical, and clinical barriers associated with intravenous drug administration.4 However, the lack of efficacy in the clinic has made innovative NP-design and -delivery approaches increasingly important in the translation of these promising therapies from bench to bedside.
Recent trends in NP design suggest that there is a focus on multifunctional targeting or “smart” delivery, which incorporates multiple complementary targeting strategies, including passive, active, and stimuli-responsive targeting.6,7 The addition of extended drug-release properties could further improve multifunctional targeting, whereby smart extended-release NPs (SER NPs) can provide additional physiological and clinical benefits, particularly in the treatment of cancer, where drug delivery poses significant challenges.8 SER NPs can be modified with active and stimuli-responsive targeting to take into account the pathophysiological characteristics of the tumor, and can be matched to the patient’s lifestyle by modifying the desired length and duration of extended release. As such, SER NPs represent a promising option in advancing the field of personalized medicine.
Despite the abundance of promising preclinical evidence for several NP formulations, there has been limited clinical advancement. This limitation is attributed to the tendency of NPs to accumulate in the liver and spleen, their low therapeutic efficiency inside tumors, and barriers associated with NP entry into the cell.9,10 The widespread use of NPs in cancer therapy has also been precluded due to traditional NPs exhibiting suboptimal stability in the body, slow intracellular drug release, low accumulation in tumor sites, and low cellular uptake and aspecific targeting.5 Furthermore, the complexity of the multistep process required for NP preparation, safety of the components involved, and stability of the final product contribute to the barriers that limit the use of NPs in clinical practice.1
This review aims to briefly summarize the physiological and clinical relevance and characteristics of SER NPs. We introduce and describe new advancements in nanodelivery systems that have either successfully combined both approaches or display the potential to do so in hopes of improving the efficacy of delivering chemotherapeutic agents to patients. Here, the particular focus is on the recently developed folic acid (FA)-functionalized nanopolymer (FA–diaminobutyric acid [DABA]–styrene-alt-maleic anhydride [SMA]), as well as injectable Pickering emulsions (IPEs), due to their novelty and particular relevance in cancer therapy. We discuss the application of SER NPs in the context of benefits and drawbacks of increasingly complex NP systems, which have the potential to advance the field of cancer treatment and delivery.
Smart nanoparticles
NP drug-delivery systems that can release the drug in response to specific physiological triggers, at the appropriate time, and at the correct target site are referred to as smart NPs.11 For this review, smart NPs refer to those incorporating all three delivery strategies: passive, active, and stimuli-responsive targeting,6 as summarized in Figure 1. The enhanced permeability and retention (EPR) effect, or passive targeting, is the most basic targeting strategy employed by smart NPs.12 The EPR effect and its limitations have been reviewed extensively elsewhere.6,9,13–18 Briefly, the EPR effect is a complex phenomenon dictated by the degree of leaky tumor vascularization and poor lymphatic draining that varies significantly between tumor types, anatomical sites, and patients.14,15,17,19 However, the high intestinal fluid pressure in tumors can prevent successful uptake and homogenous drug distribution.16,18 Long-circulating liposomes, polymers, and micelles are examples of NPs that take advantage of the leaky vasculature of tumors that ultimately allows for the entrapment and accumulation of NPs.19–21
 | Figure 1 Multifunctional targeting employed by “smart” nanoparticles. |
Active targeting
The delivery of NPs can be enhanced by functionalizing NPs with a variety of targeting molecules that are commonly over-expressed on malignant cells, such as carbohydrates, antibodies, and ligands.6,22 Specific examples of targeting moieties used to functionalize NPs are summarized in Table 1. The efficacy of active targeting is restricted to the treatment of tumors expressing a high level of the targeted biomarker, requiring a thorough understanding of the target tumor, or patient stratification.3,23 There are several classes of NPs in development that employ active targeting strategies. However, apart from denileukin diftitox (an engineered protein combining IL2 and diphtheria toxin), which was approved in 2008 but clinically discontinued in 2016, there have not been any FDA-approved NPs using active targeting in the treatment of cancer.20,24 The lack of clinical application may be due to the reported significant dose loss due to lysosomal digestion following receptor-mediated endocytosis, aspecificity of the targeting ligand, and immunogenicity of the targeting ligand, all of which lead to blood clearance.25 Therefore, it is imperative that there is ongoing research exploring methods of overcoming these limitations, as well as incorporating designs that achieve optimal biodistribution and metabolism for maximal drug delivery and efficacy.25
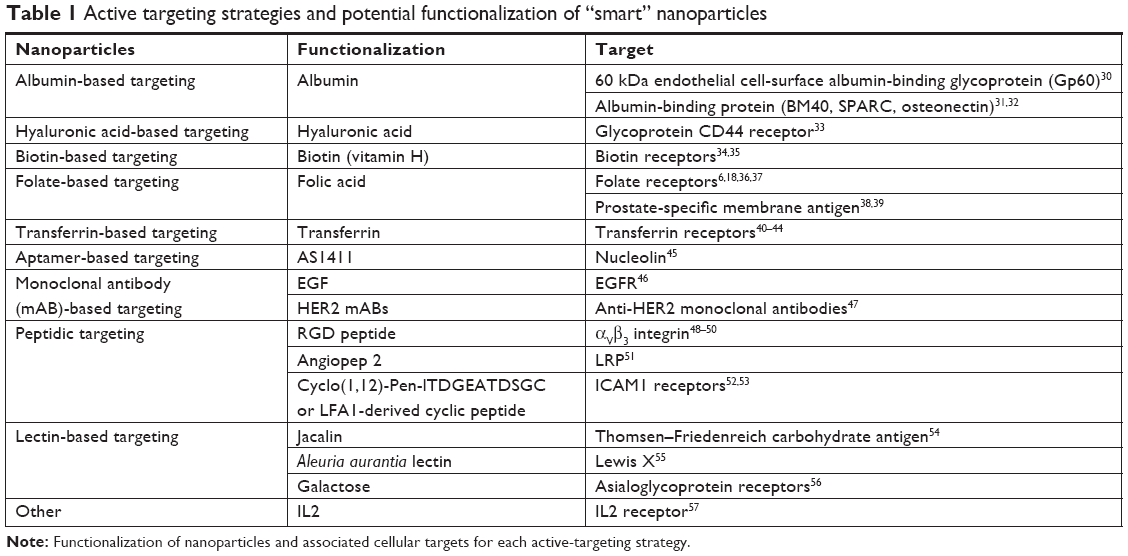 | Table 1 Active targeting strategies and potential functionalization of “smart” nanoparticles |
Stimuli-responsive targeting
Smart NPs may undergo physicochemical structural changes that result in drug release at a particular time and location when exposed to external stimuli, such as heat, pH changes, light, electric/magnetic fields, and ultrasound.1,6,11 These structural changes are a particularly important characteristic of smart NPs, as they allow for the potential to exploit the inherent characteristics of the tumor microenvironment (TME). NPs that contain pendant acidic or basic groups that accept or release protons in response to pH changes are deemed “pH-sensitive” and take advantage of the acidic pH characteristic of tumor cells (6.5–6.8), or endosomes/lysosomes (4.0–6.3).11,26–29 pH-responsive targeting is advantageous due to its suitability for the delivery of thermolabile drugs; however, a lack of toxicity data on pH-sensitive delivery is a drawback of this targeting strategy.11
Physiological importance of smart-nanoparticle drug delivery
Smart NP-delivery systems are advantageous because they have the potential to overcome some of the physiological obstacles faced by traditional chemotherapeutics, summarized in Figure 2. These obstacles include, but are not limited to, renal filtration, hepatic degradation, high tumor-cell density, high interstitial fluid pressure, and drug-efflux pumps.4 As previously discussed, through multiple levels of targeting, smart NPs can preferentially accumulate at the site of the tumor, achieving higher therapeutic indices.58
 | Figure 2 Physiological benefits of “smart” and extended-release nanopolymers. |
Active targeting has been shown to enhance therapeutic efficiency by minimizing off-target effects and reducing the amount of drug that must be administered to achieve a therapeutic response.6 For instance, loaded FA-functionalized, pH-sensitive polymeric micelles and transferrin-conjugated NPs exhibit higher efficacy compared to their respective free drugs in drug-resistant mouse models.59–61 NPs employing active targeting have also been reported to enhance siRNA delivery to tumors compared to traditional NPs that lack active targeting properties.62 FDA-approved nab-paclitaxel, an albumin-based formulation of paclitaxel (Ptx), has significantly enhanced the therapeutic index of solvent-based Ptx and prompted further research into albumin-based targeting strategies for other chemotherapeutics, such as docetaxel.63 These examples of NPs employing active-targeting strategies will be discussed in detail under the section on the clinical importance of smart-NP drug delivery.
Owing to higher drug concentrations achieved by active targeting compared to passive targeting, active-targeting strategies have been suggested to show improved therapeutic efficacy due to improved drug penetration.64 However, it has been postulated that coordinating targeting approaches may provide a solution to poor tumor penetration in instances where active targeting does not achieve effective drug accumulation.65 As such, the incorporation of stimuli-responsive targeting is becoming an increasingly popular trend in NP design, as it has been shown to accelerate drug release, improve cellular binding and internalization, and lead to more efficient drug perfusion throughout the tumor volume.66 Additionally, stimuli-responsive delivery is also used to achieve controlled release, which is critical for steady-state drug delivery.18 Controlled drug release has been reported to reduce the toxic side effects of drugs and improve the function of insoluble drugs, such as doxorubicin.67
High tumor-cell density has been an obstacle in the treatment of many cancers, due to their characteristically dense TME. Most notably, pancreatic ductal adenocarcinoma (PDAC) possesses an impenetrable stroma due to its fibrotic nature, in addition to hypovascular and hypoperfused tumor vessels, all of which contribute to increased interstitial pressure.68,69 This limitation has been partially overcome by nab-Ptx, which due to the functionalization of Ptx with albumin, forms a complex with the Gp60 receptor and SPARC, both commonly overexpressed on stromal fibroblasts in PDAC.70 The benefits observed in the PK, PD, and side-effect profiles of currently approved anticancer NPs, such as nab-Ptx, which currently only employs passive targeting, provides a strong rationale for developing NPs with multiple levels of targeting.
Clinical importance of smart-nanoparticle drug delivery
Cancer patients often face severe cytotoxic effects, as most anticancer agents are administered at the maximum tolerated dose, leading to discontinuation of life-saving treatment.71 Intravenous delivery of anticancer agents is also limited due to low drug efficacy, the need for hospitalization, frequent administration, and high cost.4 Smart NPs can allow for the administration of lower doses of drugs while maintaining effective intracellular concentrations, thereby widening the therapeutic window of anticancer agents.72 For instance, nanopolymers possess high drug-loading capacity, which allows them to achieve the same efficacy with smaller doses while minimizing systemic side effects. Therefore, smart-NP formulations that can increase tumor accumulation and specificity for cancer cells through coordinated targeting strategies can provide a therapeutic option that significantly reduces systemic side effects.65
Tran et al73 summarized the recent successes in cancer nanomedicine in the clinic. In particular, the clinical trials of irinotecan liposomal (Onivyde), which employs stimuli-responsive properties, were highlighted as a case study in the clinical success of nanomedicine in cancer therapy. Other NP “success stories” include doxorubicin liposomal (Doxil), a passive targeting FDA-approved liposomal NP that encapsulates doxorubicin for the treatment of ovarian cancer. The half-life of liposomal doxorubicin is approximately 100 times that of free doxorubicin, and the NP formulation reduces cardiotoxicity, which is significant dose-limiting toxicity associated with free doxorubicin.74,75 As a result, liposomal doxorubicin decreases the need for hospitalization and enables continuation of life-saving treatment.
Another pertinent example is albumin-bound Ptx, also known as nab-Ptx (Abraxane), an injectable formulation of Ptx that is used to treat breast, lung, and pancreatic cancers, among others. Nab-Ptx was developed to avoid the toxicities of polyoxyethylated castor-oil solvent (Cremophor) used for Ptx because of its poor aqueous solubility.76 Cremophor has been associated with several toxicities, including hypersensitivity reactions, neutropenia, peripheral neuropathy, and liver toxicity. Nab-Ptx has also been shown to significantly increase progression-free survival of metastatic breast cancer patients compared to solvent-based Ptx.77 Ptx targets metabolically active cancer cells by preventing the typical breakdown of microtubules during cell division. Nab-Ptx is a cell-cycle phase-specific drug, mainly targeting cancer cells in the G2/M phase of the cell cycle, and in combination with gemcitabine (Gem) was FDA-approved as a first-line treatment for PDAC in 2013.70 Chiorean et al78 examined changes in Karnofsky performance status (KPS) for patients’ well-being during treatment with nab-Ptx plus Gem vs Gem alone as first-line therapy for metastatic pancreatic cancer in the Phase III MPACT trial. The study concluded that the two treatment arms had generally comparable time to any KPS deterioration, and while baseline KPS, neutrophil:lymphocyte ratio, age, liver metastases, and region had a significant effect on time to definitive KPS deterioration, treatment arm did not. The limited efficacy of nab-Ptx is attributed to its cell cycle phase-specificity for the G2/M phase, but ongoing research on trapping cancer cells in a sensitive phase of the cell cycle may further enhance the therapeutic effects of nab-Ptx even in treatment-resistant tumors such as pancreatic cancer.79
Preliminary studies from our laboratory using highly aggressive human pancreatic cancer in RAGxCγ double-mutant mice have shown that the tumor xenografts develop not only resistance to nab-Ptx treatment after 66 days, but also metastasize to the liver and lung. Tumor xenografts from the same animal model of human pancreatic cancer also developed resistance to Gem treatment after 30 days, as well as a massive liver metastatic burden. It should be noted that the toxicity profile for Gem and nab-Ptx compared with FOLFIRINOX (a combination of folinic acid, fluorouracil, irinotecan, and oxaliplatin) is similar. However, patients receiving the FOLFIRINOX regimen every 2 weeks with a 46-hour fluorouracil infusion developed higher hematological toxicities and growth-factor usage, while the nab-Ptx plus Gem weekly infusion regimen had higher rates of neuropathy.
The development of chemoresistance is a significant obstacle encountered in oncology, which may be circumvented through the use of smart NPs. One approach is to modify and optimize NPs to avoid cell-surface-pump-mediated multidrug resistance (MDR) involving Pgp-mediated resistance.80 Patients who previously failed drug therapies have demonstrated clinical responses to anticancer agents encapsulated in NPs, bypassing the surface-pump-mediated resistance in humans.81–83 A recent smart-NP formulation incorporating redox-sensitive release of Ptx and an MMP2-triggered mitochondrion-targeting conjugate for mitochondrial delivery of Ptx demonstrated higher cellular uptake and cytotoxicity in MCF7 breast cancer cells.84 A detailed description of how stimuli-responsive targeting strategies may overcome MDR has been eloquently discussed in detail in a recent review by Zhou et al.85
Another approach to overcoming MDR is through the use of gene-silencing drugs. To reverse MDR in human breast cancer cells, Liu et al86 designed a new carrier system loaded with an active siRNA-targeting MDR1 gene. Briefly, phospholipid (PL)-modified cationic polyamidoamine (PAMAM)–siMDR1 complexes were designed to form hybrid nanocomplexes (PL dendriplexes). This new delivery system demonstrated higher gene-silencing efficiency, enhanced cellular uptake of siMDR1 (inhibits mRNA and MDR1 protein expression), decreased Pgp expression, raised cellular accumulation of doxorubicin, and inhibited tumor-cell migration.86 Additionally, when Ptx was introduced into cells after incubation with siMDR1 for 6 hours, either empty or entrapped in dendriplexes/PL dendriplexes and transfected with the only siMDR1, flow-cytometry analyses showed that the PL dendriplex–Ptx combination resulted in a 45.2% induction of cell apoptosis. In contrast, there was 4.15%, 4.97%, and 7.45% induction of apoptosis in control, dendriplexes, and PL-dendriplex cohorts, respectively, the results of which suggest that dendriplexes and PL dendriplexes are not highly cytotoxic, but work synergistically with either siMDR1 or Ptx.
In another study, Li et al87 provided compelling data showing reversal of MDR in MCF7/ADR breast cancer cells using a novel hR3–siMDR1–PAMAM complex (HSPC) in their design of a delivery system. Their complexes were designed and engineered using hR3 anti-EGFR antibody to self-assemble HSPCs via electrostatic interactions for siRNA delivery. The HSPCs were shown to have lower cytotoxicity, higher cellular uptake, and enhanced endosomal escape. It is noteworthy that HSPCs encapsulating siMDR1 reduced MDR1 gene expression by 99.4% with up to sixfold enhancement compared to siMDR1 alone, as well as increased doxorubicin accumulation, downregulated Pgp expression, and suppressed cellular migration in MCF7/ADR breast cancer cells. Furthermore, the combined effect of Ptx with siMDR1-loaded HSPCs showed synergism in reversing MDR, all of which inhibited cell growth and induced cell apoptosis. Other reports have proposed novel NP applications to reverse MDR, such as LAH4L1, an amphipathic cationic polypeptide, to form nanocomplexes via electrostatic interactions with siRNA to have high transfection efficiency in delivering siMDR1 to reverse MDR in ovarian cancer cells.88
Other reports have reversed MDR using codelivery of MDR1-targeting siRNA and doxorubicin using a novel cationic poly(lactide-co-glycolic acid) (PLGA) nanoformulation,89 formulating MDR1 ribozymes with N-(1-[2,3-dileoyloxy]propyl)-N, N, N-trimethylammonium methyl sulfate to form a liposomal complex,90 and constructing a pDNA-iMDR1-shRNA containing a U6-RNA gene-promoter-driven expression vector encoding anti-MDR1/Pgp shRNA molecules.91 Li et al92 gave an essential detailed review on the recent developments on the application in PAMAM dendrimers as useful carriers for drug and genetic material (pDNA, siRNA) delivery in cancer therapy, as well as their use in hybrid NPs, and conjugated or loaded in other NP systems. They also highlighted the efficacy of PAMAM dendrimers in overcoming problems with tumor MDR. Kesharwani et al93 have eloquently highlighted the challenges for effective delivery of siRNAs and oligonucleotides. The report discusses improvements in the design template, with a particular focus on describing hyperbranched PAMAM dendrimers and their unique three-dimensional architecture and nanoscale size.
The cationic surface charge of PAMAM dendrimers not only serve as siRNA-condensing agents and robust nanovectors for targeted delivery, but their functionality also permits conjugation of drugs and genes for the development of hybrid systems for combination therapy. PAMAM-dendrimer toxicity has been reviewed in detail in different models by Naha et al.94 Collectively, the toxic response of PAMAM dendrimers correlated with amine-terminated residue and increased systematically with generation. Replacing the amine groups with hydroxyl (−OH) and carboxylic acid (−COOH) terminated PAMAM dendrimers, resulting in significantly less toxicity. It is noteworthy that the PK parameters, biodistribution, biodegradation, and chronic toxicity of PAMAM dendrimers are not well known.
de Jong and Borm reviewed the safety evaluation of NP formulations for drug delivery, their applications, and hazards.95 The engineering and design of NPs for drug delivery must include the specificity of drug targeting, delivery, and reduced toxicity, while maintaining therapeutic effects, increased safety and biocompatibility, and increased development of new safe medicines. Drug-delivery systems should be designed with the following principles in mind: drug incorporation and release, formulation stability, shelf life, biocompatibility, biodistribution, targeting, and functionality.
One of the drawbacks of administering multiple doses of systemic siRNA carriers is their decreased fluctuation in serum. To overcome this issue, Kim and Song96 developed a targetable, injectable, and noncytotoxic micelleplex hydrogel. The report highlighted the dual function of the micelleplex hydrogel carrier and provided supporting data to demonstrate its active systemic targetable siRNA-delivery carrier with an additive tunable therapeutic time by controlled release of the delivery carrier after only one injection of the hydrogel. Briefly, the micelleplex hydrogel was fabricated by mixing a functionalized folate-linked polyethylenimine-conjugated polyorganophosphazene (FPP), a biodegradable polymer, and amphiphilic forming micelles with siRNA at 4°C. After injection into the body, sol–gel transition in solution as a function of temperature occurred due to hydrophobic interactions. The hydrogel released dissociated micelleplexes, targeting the folate moiety in a time-dependent manner. It is noteworthy that the polyethylenimine moiety in the FPP formulation formed a micelleplex with anionic siRNAs by ionic interactions, which turned into a gel after subcutaneous injection, due to the properties of polyorganophosphazene. These micelleplexes were then released slowly by dissolution and degradation of the gel into the bloodstream directly or through lymphatic vessels. The released micelleplexes in circulation accumulated in the tumor region via the EPR effect, and targeted and enter only tumor cells via FA-receptor-mediated endocytosis. Gene suppression by the siRNA delivered through endocytosis process was caused by cleavage of specifically targeted mRNA only in tumor cells.
To explore current developments in siRNA-delivery systems using NPs in oncology, particularly those that encapsulate siRNA for the targeted treatment of cancer, Kim et al97 reviewed the current status of clinical trials related to siRNA-based cancer therapy, elucidating the remaining issues that need to be overcome to establish a successful therapy. The report also described various promising design strategies of delivery vehicles for stable and targeted siRNA delivery, including prospects for future design. Additionally, Sarett et al98 reported analyses of complicating factors relevant to the clinical adoption of local siRNA therapeutics, such as polymers, siRNA carriers, and construct types studied in basic research. Among local siRNA therapeutics in the clinic, the authors identified that systems that achieve sustained delivery are mostly absent. Clinical approaches use simple topical treatments or local-injection strategies, due to the ease of use mandated by the regulatory process. Sarett et al98 recommended a balance between complicated design processes and therapeutic efficacy for controlled-release delivery systems to maintain strong gene-silencing activity without repeated doses. To this end, the report outlines the earliest to most recent clinical trials for local siRNA-delivery systems.
Classical methods of smart-drug delivery
There are three generations of delivery system that have been developed based on their degree of specific targeting.99,100 First-generation NPs, defined as those that lack specific targeting, include the successful FDA-approved liposomal daunorubicin, liposomal doxorubicin, and PEGylated liposomal doxorubicin.65 Second-generation NPs have relied on active targeting, while third-generation NPs employ a multistage strategy or smart-drug delivery.99 There is ongoing debate on the cost-versus-benefit analysis of increasingly complex drug-delivery systems, such as smart NPs.1,65
Recent clinical discontinuation of FDA-approved denileukin diftitox, an active-targeting NP for cutaneous T-cell lymphoma, due to production difficulties is evidence of practical limitations of developing increasingly complex NPs.24 Functionalizing NPs increases the complexity of the preparation process, and is associated with increased cost and regulatory barriers that must be overcome.65 To mitigate these challenges, targeting ligands or bioresponsive materials can be coupled directly with NP starting materials before NP formulation, allowing for tighter control over synthesis, uniformity, and target-ligand density.65 Newer formulations of smart-drug delivery must not only improve the PK and PD parameters of free drugs, but also be relatively straightforward to produce to maximize the chances of clinical translation.
Table 2 summarizes the current FDA-approved NPs and describes the type of targeting, cancer indication, advantages, and drawbacks. Other FDA-approved NPs for the treatment of cancer that are not discussed in Table 2 include trastuzumab emtansine (Kadcyla) and NanoTherm (MagForce), as they are beyond the scope of smart NPs. It is noteworthy that the therapeutic efficacy of these NPs is to reduce side effects and selectively accumulate in the disease area for a prolonged period of time with high controllability.101 According to a recent review article by Liu et al,101 the following should be characteristic properties of NPs and the development process: clinically useful and reproducible formulations, high verifiability, precise control over the preparation process, generating nanocarriers with required features, high batch-to-batch reproducibility, design simplicity, and industrial upscaling feasibility. Therefore, future research on smart NPs for controlled drug delivery should focus on more stimulus-sensitive nanomedicine to be clinically utilized.
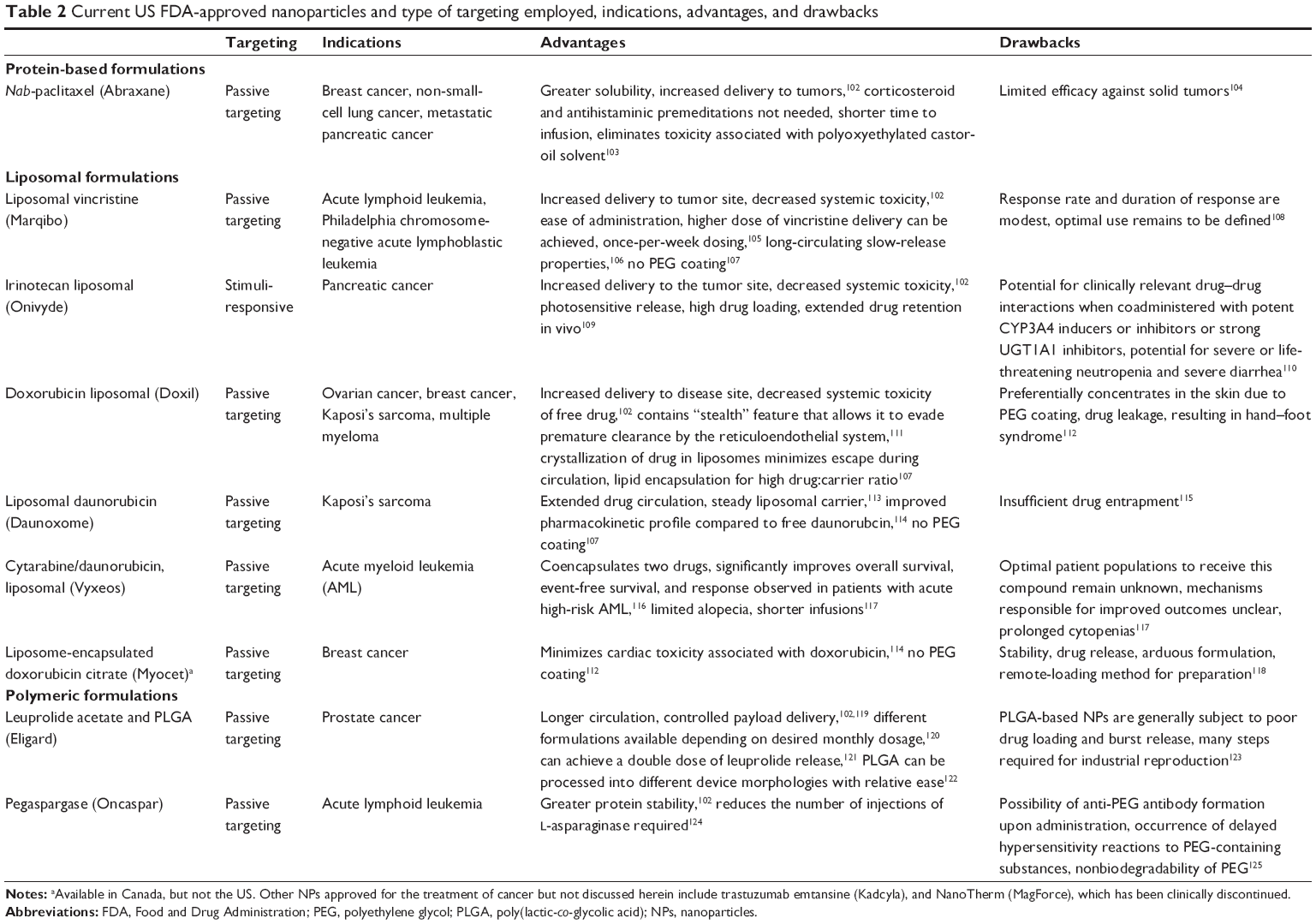 | Table 2 Current US FDA-approved nanoparticles and type of targeting employed, indications, advantages, and drawbacks |
Novel approaches to smart-nanoparticle delivery systems
Advancements in NP design have resulted in multifunctional targeting and multispecificity.65 Recently, the copolymer FA-DABA-SMA was developed, which relies on the EPR effect, FA-receptor targeting, and pH sensitivity.6,18 This smart-delivery system incorporates all three forms of targeting. The amphiphilic polymer is characterized by a hydrophilic outer shell containing a hydrophobic core, which permits encapsulation of hydrophobic chemotherapeutic agents. The polymer exists in cylindrical conformation at neutral pH within the body, and collapses to release its cargo at an acidic pH that is typically characteristic of the TME and lysosomes,18 as depicted in Figure 3. Li et al18 described and summarized the synthesis and characterization of FA-DABA-SMA. In brief, the SMA serves as the template for the NP. The linker, DABA, is used to improve the accessibility of the FA-DABA ligand, which is synthesized by combining FA, dicyclohexylcarbodiimide, and hydroxysuccinimide, resulting in a stable functionalized FA-DABA-SMA polymer at neutral pH and self-assembles into nanostructures in dilute concentrations.18 Due to the reported anticancer effects and fluorescent capabilities of curcumin, it is used in NP studies as a hydrophobic drug load and a fluorescent marker.126,127 Here, the encapsulation process is chemical-free, as curcumin diffuses passively into the core of the NP, thereby minimizing undesired toxicity. This smart-delivery system is particularly advantageous due to its noninvasive delivery and drug release, because the FA-DABA-SMA polymer circulates in the bloodstream and pH changes trigger drug release at the tumor site.5
 | Figure 3 A pH-responsive, “smart” active polymer-delivery system. |
Preclinical evidence of curcumin-loaded FA-DABA-SMA polymer demonstrates significant toxicity and cell death in PANC1 pancreatic cancer cells, with the empty FA-DABA-SMA polymer being nontoxic.6 Approved anticancer drugs currently used in the clinic are being tested in these NPs for their loading capacities and release profiles. Collectively, this smart FA-DABA-SMA NP shows promise for its use in a wide variety of cancer types, due to the specific overexpression of the FA receptor in pancreatic, epithelial, ovarian, cervical, breast, lung, kidney, colorectal, and brain tumors.128
Other recent advances in smart-drug delivery involve the use of multiple types of functionalization and triggering stimuli for drug release. For example, doxorubicin-loaded micelles have recently been developed with reactive oxygen species and dual pH sensitivity.26 Another recent advance in NP design is multispecificity, which is particularly relevant to the dynamic nature of tumor markers and is often a limiting factor of active targeting.65
Stimuli-responsive targeting can be optimized by pairing nanomedicine-based drug delivery with other modalities. For example, magnetic resonance imaging-guided high-intensity focused ultrasound combines different technologies, such as image guidance for radiation treatment and triggering of smart-nanomedicine systems for delivery of drugs. In the case of thermosensitive liposomal doxorubicin, the release is triggered at the tumor site by locally increasing the temperature through high-intensity ultrasound radiation.129
Despite promising advances, some improvements to current smart-NP design could be considered. To circumvent the issue of significant dose loss associated with active targeting, lysosome-escape mechanisms that rely on pH sensitivity have been developed.25 Lysosomal escape aims to minimize inactivation of NPs by acid hydrolases found in endolysosomal compartments, thereby representing a critical design strategy and the focus of ongoing research efforts.130 Additionally, incorporating extended-release design principles into smart NPs would lead to targeted and prolonged-action therapies with unique physiological and clinical advantages, as summarized in Figures 2 and 4.
 | Figure 4 Clinical benefits of “smart” and extended-release NPs. |
Extended-release nanoparticle-delivery systems
Extended-release drug-delivery systems are designed to release drugs over a prolonged period employing steady-rate drug release or controlled release to achieve stable and higher therapeutic potential while minimizing adverse side effects.131 Extended-release NPs used in a clinical setting hold drugs either on their surface or adsorbed in a matrix that attains sustained release.132 Currently, hydrophobic biodegradable polymeric NPs are commonly used for the continuous supply of encapsulated therapeutic agents at the site of the tumour.58
NPs can undergo certain cell-surface modifications to achieve more prolonged circulation. In particular, PEGylation which involves the conjugation of polyethylene glycol (PEG) to a nanopolymer, has been used extensively in nanomedicine.20 It has been shown to increase drug-hydrodynamic radius, prolong plasma-retention time, decrease proteolysis, decrease renal excretion, and shield antigenic determinants from immunodetection.133–137 Etirinotecan pegol is an FDA-approved long-acting topoisomerase 1 inhibitor for breast cancer designed to improve the PK properties and tolerability of irinotecan whose enhanced therapeutic response has been attributed to the more prolonged circulation of PEGylated nanomaterials.138
Physiological importance of extended-release drug delivery
Limiting factors in the physiological success of NPs include size and surface characteristics, as they play roles in circulation time and elimination from the body.139 The optimal NP size that is correlated with more prolonged circulation, increased accumulation, and decreased renal NP clearance is 40–200 nm.140 NPs that are 10–100 nm in size have been shown to increase circulation time of the drug for as long as 160 hours, with modifications in surface area and charge allowing for the ability to reach optimal loading and control of dosing.141 For instance, nanocrystalline cellulose possesses an extensive negatively charged surface area that allows it to bind to large amounts of the drug.142 It can be coated with cationic surfactant (cetyltrimethylammonium bromide), resulting in an altered release profile due to its ability to deliver hydrophobic anticancer agents as a result of its hydrophobic domain.142 High loading capacity in combination with extended-release profiles can reduce the dosage required, thereby minimizing aspecific cytotoxicity.
Flexibility in the functionalization of nanomatrices permits the ability to obtain the desired drug-release profile. Solid-lipid NPs are particularly advantageous due to their combination of polymeric micelles and lipid-based liposomes, which allows them to deliver both hydrophobic and hydrophilic therapeutic agents, demonstrate excellent bioavailability, scalability, and physical stability.131 Premature bursting and rapid removal from circulation reduce the efficacy of the treatment. However, PEGylation coating may offer an alternative option that facilitates drug release over an extended period.143 An essential consideration in optimal extended-release delivery systems is minimal drug burst, with most of the drug released according to a specific release profile.6,18
Clinical importance of extended-release drug delivery
Extended-release NPs are attractive therapeutic options for the long-term treatment of complex chronic diseases, such as cancer, owing to drug release over a prolonged period.144,145 These characteristics of extended-release NPs can achieve improved patient compliance, better life-cycle management of drugs, and extended relief of symptoms, due to a reduction in fluctuating drug levels.146 The clinical advantages of smart NPs are summarized in Figure 4.
Challenges with patient adherence span several stages of treatment, and are primarily attributed to multiple dosing, which is standard in cancer treatment.123,147–149 Patients typically experience challenges when entering a different phase of their treatment cycle, particularly in initiating implementation and treatment persistence.150 Extended-release drug-delivery systems can overcome these challenges in patient adherence by decreasing dosing cycles. Adherence increased from 59% for multiple dosing to 80% with a single daily dose in male patients on long-term antihypertensive medication.151 Conversely, a decline in adherence was observed in patients on a chemotherapy regimen of three doses/day.152
The number of injections required over the course of treatment is an essential consideration in drug delivery for patients and clinicians alike. For instance, PEG–IFNα conjugates have shown clinically superior antiviral activity to free IFNα, and are approved for hepatitis C therapy.153 PEG–IFNα has been useful in the treatment of melanoma and renal-cell carcinoma, and is currently being tested in other solid tumors.154–156
Classical methods of extended-release delivery systems
Controlled drug delivery has been an area of research interest over the past six decades, with early extended-release formulations including oral and transdermal sustained-release systems.100 In oncology, FDA-approved leuprolide for injectable suspensions can achieve slow and sustained drug release for the treatment of prostate cancer.157 Additionally, non-PEGylated liposomal doxorubicin for the treatment of metastatic breast cancer resembles a prolonged infusion that forms a mononuclear phagocyte-system depot capable of slow release into blood circulation.158 Although promising as delivery systems, these methods of extended-release delivery have faced scrutiny, and their limitations are briefly summarized in Table 3.
 | Table 3 Classical methods of extended-release delivery systems and their limitations |
Novel approaches to extended-release drug delivery
A number of novel NP formulations for extended drug release have been developed in recent years. For instance, PLGA polymers have been shown to attain 82% of the cumulative release of dexamethasone in 17 days.130 Clinically, cancer patients undergoing chemotherapy are often given dexamethasone to counteract the adverse side effects of their antitumor treatment. The extended release of dexamethasone from PGLA polymers may overcome the long-term use of dexamethasone, but may result in thrush, bone loss, cataracts, easy bruising, or muscle weakness. PLGA is a copolymer composed of lactic and glycolic acid monomers that are used as a drug-delivery vehicle. PLGA is susceptible to hydrolytic degradation of the ester linkage on the polymer backbone, which results in the release of the encapsulated drug.174 Several PLGA drug-delivery applications, such as Lupron Depot, Risperdal Consta, and Zoladex, have been approved by the FDA, European Medicines Agency, and Health Canada.123 However, therapeutics demonstrating extended-term sustained release (weeks or longer) from PLGA polymers are mostly hydrophobic in nature, with molecular weights of 400–1,000 Da. The design and fabrication of extended-term, sustained release, low-molecular-weight hydrophilic drugs from PLGA has been challenging. One of the most significant challenges in drug delivery is the inherent difficulty in full encapsulation and retention, followed by long-term and targeted delivery of low-molecular-weight hydrophilic therapeutics at the tumor site.
To that end, we have recently reported that oseltamivir phosphate (OP) can prevent the tumor neovascularization, growth, and metastasis of human triple-negative breast175,176 and pancreatic177 cancer cells in heterotopic xenografts of these tumors in RAGxCγ double-mutant mice. Findings have also included a novel signaling paradigm that regulates EGFR,177 insulin receptors,176,178,179 and Toll-like receptors,180 all of which play essential roles in multistage tumorigenesis.181 As such, the controlled release of hydrophilic OP from a biodegradable PLGA cylinder (PLGA-OP) implanted at the tumor site was investigated for its role in limiting tumor neovascularization, growth, and metastasis.182 As shown in Figure 5, PLGA-OP cylinders showed a 20%–25% release profile within 48 hours followed by a continuous metronomic low-dose release of 30%–50% OP for an additional 16 days, with all of the OP released by day 30. To show the therapeutic effect of PLGA-OP, PLGA-OP containing 20 mg OP and empty cylinders surgically implanted at the tumor site of heterotopic xenografts of human pancreatic tumors in RAGxCγ double-mutant mice impeded tumor neovascularization and growth rate and spread to the liver and lungs compared to the untreated cohort over 30 days (Figure 5).182
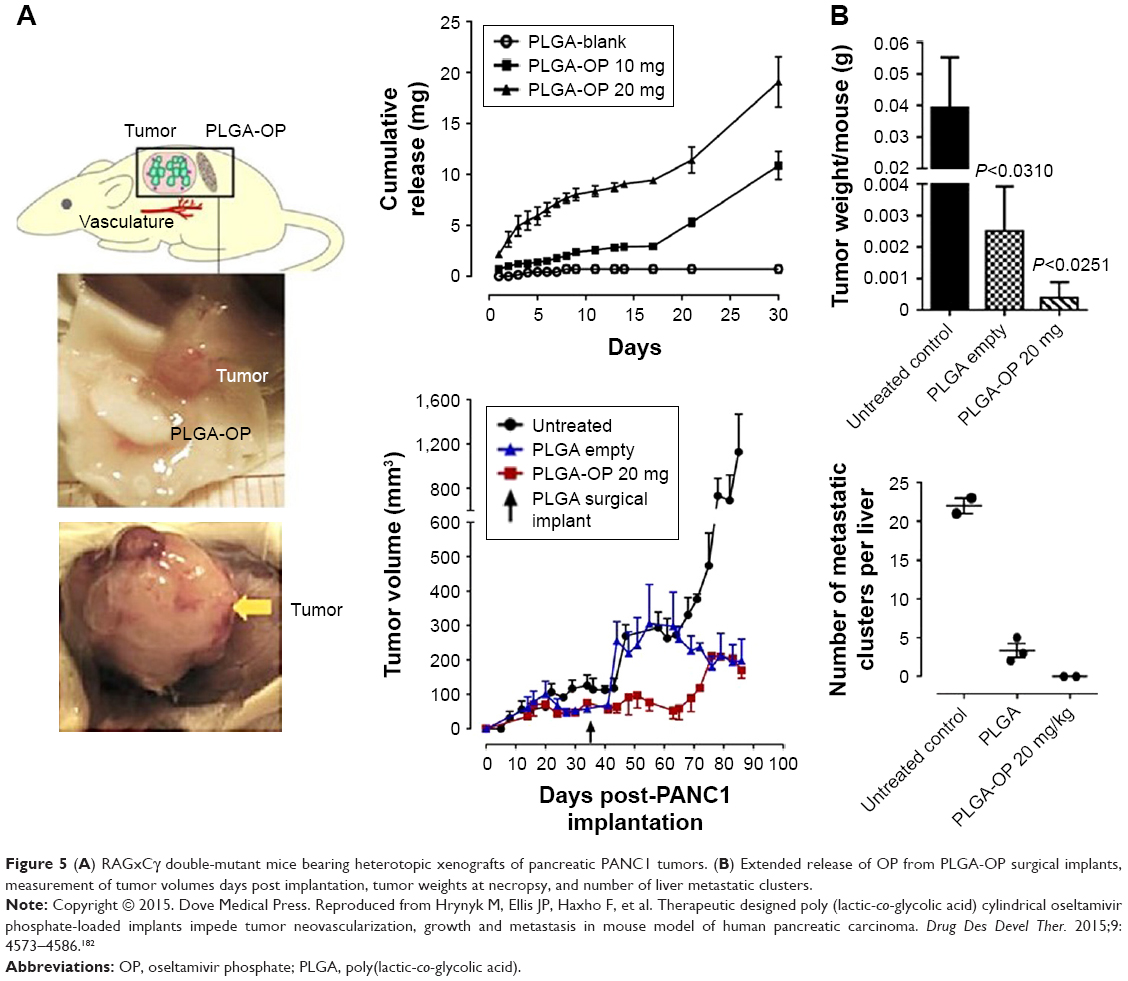 | Figure 5 (A) RAGxCγ double-mutant mice bearing heterotopic xenografts of pancreatic PANC1 tumors. (B) Extended release of OP from PLGA-OP surgical implants, measurement of tumor volumes days post implantation, tumor weights at necropsy, and number of liver metastatic clusters. |
Despite difficulties with the encapsulation of small hydrophilic drugs, OP-particulate encapsulation within polymeric PLGA cylinders using the formulation method described in these studies resulted in full retention of the drug with no loss of therapeutic activity. Collectively, delivery of OP using this delivery system in the treatment of pancreatic cancer cells resulted in disabling the survival mechanism of pancreatic cancer with acquired chemoresistance to Gem.183
Furthermore, the double-layered Gem- and OP-loaded PLGA cylindrical implants were designed and engineered to provide an optimal combinatorial and sequential sustained extended-release of the hydrophilic drugs for 30 days. Here, OP and Gem encapsulated in inner/outer distinct layers of implantable double-layered PLGA cylinders disabled pancreatic cancer cell survival and increased sensitivity to therapy of two low-molecular-weight hydrophilic chemotherapeutics.
The development of amphiphilic polymeric vesicles capable of encapsulating large quantities of a chemotherapeutic agent and improving its targeting efficiency and bioavailability are emerging new delivery platforms for nanomedicine in cancer research. Polymeric micelles have been modified by attaching ligand molecules to the outer shell of the micelle with specificity for biomarkers that are overly expressed on cancer cells. Such ligands as peptides, antibodies, sugars, and aptamers have enabled significant improvement in tumor selectivity and overall therapeutic efficiency of cancer treatments. However, these specific delivery vehicles are limited by low overtime stability, especially with antibodies, triggering immunogenic adverse side effects and high costs. Multiple functionalities using complex hydrophobic/hydrophilic polymeric structures prepared with highly controlled molecular weights and defined architectures are needed to enable self-assembled, stimuli-responsive regions (CO2, pH, and temperature) for triggered drug release and reactive groups for drug conjugation cross-linking and “click” chemistry. To this end, Kapishon et al184 designed a specific smart chemotherapeutic delivery platform for active tumor targeting. They fabricated a new polymeric micelle-delivery system for active tumor targeting followed by micelle–drug internalization via receptor-induced endocytosis. By decorating amphiphilic block copolymeric micelles with OP at the hydrophilic end, oseltamivir–PEG methyl ether methacrylate-block-poly(methyl methacrylate) using reversible addition–fragmentation chain transfer living radical polymerization, the OP micelles had self-assembling properties to give wormlike micellar structures with a molecular weight of 80,000 g/mol. These newly designed micelles for active tumor targeting have triple functionality, such that they exert an antitumor cell effect and at the same time deliver and internalize hydrophobic chemotherapeutic agents.
IPEs are another promising novel method of extended-release NPs compared to older formulations.185 In our studies, IPEs were designed and fabricated to replace our surgically implantable PGLA drug-delivery vehicle, as previously discussed, while providing similar dosing and release profiles at the tumor site to the surgical implant.185 The IPE formulation is unique in that compared to previous extended-release drug-delivery systems, it has been developed to adjust the release rate to occur within a target of 30 days.185 The monoglyceride-stabilized PE (MSPE) approach is cost-effective, as it utilizes simple equipment with readily available reagents and has a formulation time of <1 hour.185 In brief, emulsions consist of two liquids that remain insoluble in each other and include an emulsifier that stabilizes the mixture. The emulsion is stabilized by glycerol monostearate containing ≥90% glycerol monostearate (GMS). To produce a smooth emulsion from two separate mixtures, an oil phase and a water phase are prepared and subsequently mixed at a fixed temperature and mixing speed. The oil phase is prepared by mixing GMS with canola oil, resulting in a 4%-by-weight oil phase. The solutes (OP, sodium chloride, and sodium citrate) are added to distilled water to prepare a 4%-by-aqueous-weight phase. These mixtures are then warmed separately to cause melting of GMS, followed by adding the aqueous to the oil phase, producing a coarse water-in-oil emulsion of 20%:80%. This coarse emulsion is then added to a stirrer that carries out stirring and processing to produce an emulsion that is quench-cooled, resulting in a viscous final product.
Preclinical data have shown a decrease in PANC1 pancreatic cancer cell viability after these cells were exposed to MSPE at multiple time points leading up to 30 days.185 This effect was unchanged when comparisons were made between PANC1 exposed to OP only, compared to OP released from the emulsion. A near-linear sustained release of OP from MSPE-encapsulated OP was observed, with <5% of the total drug loading being released over a 30-day period.185 Regarding stability, the viscosity of the emulsion remains unchanged, and both phases remain stable.185 These results suggest that the viscous MSPE emulsion will concentrate at the site of injection.
Future directions: the potential for smart extended-release drug-delivery nanoparticles
Smart NPs that can exhibit sustained, extended-release drug delivery would offer additional advantages from both clinical and physiological standpoints. Studies continue to highlight the importance of localizing treatment and prolonging drug release, both of which achieve therapeutic benefits with limited adverse effects, due to fewer drug interactions and reduced drug-level fluctuations.186 Therefore, modifying NPs to exhibit both extended-release and targeting properties should continue to be an area of intense research focus.
One strategy of developing SER drug-delivery NPs could involve PEGylation of smart NPs to prolong circulation time, as previously discussed. PEGylation of self-assembled hyaluronic acid NPs has been shown to improve active and passive targeting in tumor-bearing mice, reduce liver uptake, and increase circulation time and NP accumulation in tumors.187 Given recent advances in antibody-mediated therapy in cancer research, it is perhaps unsurprising that antibody-targeted NPs represent promising active-targeting strategies that are undergoing rigorous clinical research.65 As such, smart NPs incorporating antibody-mediated targeting that also exhibit extended-release could be of great interest in cancer therapy in light of the benefits of prolonged drug release outlined herein.
Alternatively, current extended-release NPs can be modified to achieve a higher degree of targeting. Dendrimer-based stealth NPs have been used to encapsulate anastrozole, commonly used for the treatment of breast cancer, to improve its water-solubility and extended-release profile.188 Extended release of encapsulated anastrozole was achieved for 40 days, and the authors suggested that the stealth NPs could be further functionalized to construct a targeted therapeutic agent for breast cancer treatment.188 Similarly, IPEs can be functionalized with FA for greater specificity toward malignant cells, due to their overexpression of the FA receptor. Some NPs with targeting properties that display extended release have been in development, such as silk fibroin functionalized with the cyclic arginine–glycine–aspartic acid–phenylalanine–lysine (RGDfK) peptide, and chlorin e6 (a second-generation photosensitizer with antitumor activity when used in conjunction with irradiation), which displays both smart drug delivery and sustained release. Here, active targeting is achieved using the cyclic RGDfK peptide, which targets αVβ3 integrins.50 Silk-fibroin NPs have also displayed extended-release, as only 33% of fluorouracil had been released by the end of the third day in a human gastric cancer model.50 The combination of active targeting and extended-release resulted in a significant reduction in tumor burden in vivo with excellent biocompatibility and safety, with encapsulation of cisplatin, doxorubicin, and Ptx being suggested as future directions to test the efficacy of this formulation.50
Due to the increased focus on patient selection, stratification, and personalized medicine, nanomedicine-based drug-delivery systems in the clinical setting need to take smart and extended-release drug delivery into consideration.21,189 Personalized medicine incorporates a variety of factors, including genetic, genomic, and clinical markers, as well as the patient’s age, race, and other environmental factors.190 Future work looking at additional genetic and epigenetic biomarkers will assist in the development of novel targeting moieties for increasingly effective and customized therapeutic options.191 Potential biomarkers could target solid tumors, tumor vasculature, and supporting cells within the TME.65 Depending on the pathophysiology of the patient’s tumor, appropriate functionalization and triggers for drug release can also significantly improve efficacy.192 For instance, the limitations of the EPR effect may be overcome by selecting patients with angiogenic tumors, using vasodilating agents, such as TNFα, and using imaging strategies to monitor delivery of the NPs.193
Despite the benefits of SER NPs, a significant concern with increasingly complex drug-delivery systems is cost-effective upscaling manufacturing.186 Maintaining the original properties of NPs during the development process is expected to be an obstacle in complex NP formulation.186 The examples explained in detail here, namely FA-DABA-SMA and IPE formulations, involve straightforward preparation processes, safe intermediates, and inexpensive materials.6,18,185 As such, they represent good candidates as potential SER NPs, and future work should explore incorporating both smart and extended-release design principles in these formulations.
Alternatively, self-assembled nanostructures may offer innovative approaches to limitations encountered in the development of NPs. Li et al194 highlighted in an impressive review article cooperation principles in various self-assembled nanostructures based on the principles of molecular cooperation in self-assembled systems. The report focused on the bottom-up chemistry and material-science considerations of nanomedicine with reference to conformation change-induced cooperation in natural self-assembled nanostructures, noncovalent interactions involving the molecular basis of supramolecular cooperation, phase transition-induced cooperation in synthetic self-assembled nanostructures, the fTM molecular mechanism of supramolecular cooperation, and lastly, supramolecular cooperation in addressing the challenges in medicine. Luo et al195 reported a novel nanovaccine, the mechanism of which was dependent on the STING and not on Toll-like receptors or MAVS pathways. The formulation of the nanovaccine is minimalistic, and made by mixing antigen with a synthetic polymeric NP, PC7A, which generated a strong cytotoxic T-cell response with low systemic cytokine expression. PC7A NPs achieved efficient cytosolic delivery of tumor antigens to antigen-presenting cells in draining lymph nodes, which led to increased surface presentation while simultaneously activating type I IFN-stimulated genes. This nanovaccine design produced potent inhibition of tumor growth in melanoma, highlighting nanovaccine formulation as an intense area of research interest.
Conclusion
Recent advances in NP design have led to the development of drug-delivery systems that can overcome several physiological and clinical barriers associated with the traditional administration of chemotherapeutic agents. Smart-drug delivery aims to localize treatment to tumors to reduce cytotoxicity and enhance the therapeutic index by using multifunctional targeting strategies. While older formulations have relied primarily on the EPR effect, recent advances, such as the FA-DABA-SMA polymer, employ three levels of targeting, a chemical-free process of drug encapsulation, noninvasive drug delivery and release, to achieve cytotoxicity against pancreatic cancer cells. Future work should investigate encapsulation of conventional chemotherapeutic drugs into the FA-DABA-SMA polymer, its application toward other cancers that overexpress FA, and improving its extended-release profile. IPEs represent an additional promising NP formulation for extended-drug release. Modifying IPEs to include active targeting properties could successfully combine the concepts of smart delivery with extended release to achieve more significant therapeutic responses, minimal side effects, and improved patient adherence. Therefore, combining different targeting strategies and optimizing the drug-release profile for a coordinated approach toward drug delivery is of particular importance in the drug-delivery field, but the cost and technical difficulties of achieving this complexity must also be considered. This continuously growing field allows for a novel way for clinically implementing personalized medicine in the form of NP-delivery systems, and requires a more profound understanding of the patient’s tumor pathophysiology to be successful.
Author contributions
MRS conceived the review and obtained funding. RVK, KB, BQ, and AD designed the study, carried out the searches, refined and selected the study design, extracted data, and conducted the thematic analyses. RVK, KB, and BQ led the writing of the draft as contributing first authors. MRS finalized the manuscript. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work. All authors approved the final manuscript.
Disclosure
This work was supported in part by grants to MRS from the Natural Sciences and Engineering Research Council of Canada (NSERC) and private-sector cancer funding from the Josefowitz family and Encyt Technologies Inc to MRS. RVK is a recipient of the Queen’s Graduate Award (QGA). BQ is a recipient of the QGA and part of the 2017 Terry Fox Research Institute Transdisciplinary Training Program in Cancer Research. The authors report no other conflicts of interest in this work.
References
Torchilin VP. Multifunctional, stimuli-sensitive nanoparticulate systems for drug delivery. Nat Rev Drug Discov. 2014;13(11):813–827. | ||
Fornaguera C, García-Celma M. Personalized nanomedicine: a revolution at the nanoscale. J Pers Med. 2017;7(4):E12. | ||
Hare JI, Lammers T, Ashford MB, Puri S, Storm G, Barry ST. Challenges and strategies in anti-cancer nanomedicine development: an industry perspective. Adv Drug Deliv Rev. 2017;108:25–38. | ||
Lammers T. Improving the efficacy of combined modality anticancer therapy using HPMA copolymer-based nanomedicine formulations. Adv Drug Deliv Rev. 2010;62(2):203–230. | ||
Yang M, Yu L, Guo R, Dong A, Lin C, Zhang J. A modular coassembly approach to all-in-one multifunctional nanoplatform for synergistic codelivery of doxorubicin and curcumin. Nanomaterials. 2018;8(3):E167. | ||
Li X, Szewczuk M, Malardier-Jugroot C. Folic acid-conjugated amphiphilic alternating copolymer as a new active tumor targeting drug delivery platform. Drug Des Devel Ther. 2016;10:4101–4110. | ||
Gu FX, Karnik R, Wang AZ, et al. Targeted nanoparticles for cancer therapy. Nano Today. 2007;2(3):14–21. | ||
Rodzinski A, Guduru R, Liang P, et al. Targeted and controlled anticancer drug delivery and release with magnetoelectric nanoparticles. Sci Rep. 2016;6:20867. | ||
Pérez-Herrero E, Fernández-Medarde A. Advanced targeted therapies in cancer: drug nanocarriers, the future of chemotherapy. Eur J Pharm Biopharm. 2015;93:52–79. | ||
Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33(9):941–951. | ||
Honey PJ, Rijo J, Anju A, Anoop KR. Smart polymers for the controlled delivery of drugs: a concise overview. Acta Pharm Sin B. 2014;4(2):120–127. | ||
Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–6392. | ||
Danhier F, Feron O, Préat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148(2):135–146. | ||
Bae YH. Drug targeting and tumor heterogeneity. J Control Release. 2009;133(1):2–3. | ||
Bae YH, Park K. Targeted drug delivery to tumors: myths, reality and possibility. J Control Release. 2011;153(3):198–205. | ||
Heldin CH, Rubin K, Pietras K, Östman A. High interstitial fluid pressure: an obstacle in cancer therapy. Nat Rev Cancer. 2004;4(10):806–813. | ||
Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7(11):653–664. | ||
Li X, Mctaggart M, Malardier-Jugroot C. Synthesis and characterization of a pH responsive folic acid functionalized polymeric drug delivery system. Biophys Chem. 2016;214–215:17–26. | ||
Wang M, Thanou M. Targeting nanoparticles to cancer. Pharmacol Res. 2010;62(2):90–99. | ||
Bobo D, Robinson KJ, Islam J, Thurecht KJ, Corrie SR. Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials to date. Pharm Res. 2016;33(10):2373–2387. | ||
Lammers T, Rizzo LY, Storm G, Kiessling F. Personalized nanomedicine. Clin Cancer Res. 2012;18(18):4889–4894. | ||
Byrne JD, Betancourt T, Brannon-Peppas L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv Drug Deliv Rev. 2008;60(15):1615–1626. | ||
Renoux B, Raes F, Legigan T, et al. Targeting the tumour microenvironment with an enzyme-responsive drug delivery system for the efficient therapy of breast and pancreatic cancers. Chem Sci. 2017;8(5):3427–3433. | ||
Wang Z, Zheng Q, Zhang H, et al. Ontak-like human IL-2 fusion toxin. J Immunol Methods. 2017;448:51–58. | ||
Chen WC, Zhang AX, Li SD. Limitations and niches of the active targeting approach for nanoparticle drug delivery. Eur J Nanomed. 2012;4(2–4):89–93. | ||
Qu CX, Li JZ, Zhou YJ, et al. Targeted delivery of doxorubicin via CD147-mediated ROS/pH dual-sensitive nanomicelles for the efficient therapy of hepatocellular carcinoma. AAPS J. 2018;20(2):34. | ||
Adochite RC, Moshnikova A, Carlin SD, et al. Targeting breast tumors with pH (low) insertion peptides. Mol Pharm. 2014;11(8):2896–2905. | ||
Adochite RC, Moshnikova A, Golijanin J, Andreev OA, Katenka NV, Reshetnyak YK. Comparative study of tumor targeting and biodistribution of pH (low) insertion peptides (pHLIP peptides) conjugated with different fluorescent dyes. Mol Imaging Biol. 2016;18(5):686–696. | ||
Weerakkody D, Moshnikova A, el-Sayed NS, et al. Novel pH-sensitive cyclic peptides. Sci Rep. 2016;6:31322. | ||
Vogel SM, Minshall RD, Pilipović M, Tiruppathi C, Malik AB. Albumin uptake and transcytosis in endothelial cells in vivo induced by albumin-binding protein. Am J Physiol Lung Cell Mol Physiol. 2001;281(6):L1512–L1522. | ||
Frei E. Albumin binding ligands and albumin conjugate uptake by cancer cells. Diabetol Metab Syndr. 2011;3(1):11. | ||
Desai NP, Trieu V, Hwang LY, Wu R, Soon-Shiong P, Gradishar WJ. Improved effectiveness of nanoparticle albumin-bound (nab) paclitaxel versus polysorbate-based docetaxel in multiple xenografts as a function of HER2 and SPARC status. Anticancer Drugs. 2008;19(9):899–909. | ||
Choi KY, Saravanakumar G, Park JH, Park K. Hyaluronic acid-based nanocarriers for intracellular targeting: Interfacial interactions with proteins in cancer. Colloids Surf B Biointerfaces. 2012;99:82–94. | ||
Taheri A, Dinarvand R, Atyabi F, Nouri F, et al. Targeted delivery of methotrexate to tumor cells using biotin functionalized methotrexate-human serum albumin conjugated nanoparticles. J Biomed Nanotechnol. 2011;7(6):743–753. | ||
Taheri A, Dinarvand R, Nouri FS, et al. Use of biotin targeted methotrexate-human serum albumin conjugated nanoparticles to enhance methotrexate antitumor efficacy. Int J Nanomedicine. 2011;6:1863–1874. | ||
Patil YB, Toti US, Khdair A, Ma L, Panyam J. Single-step surface functionalization of polymeric nanoparticles for targeted drug delivery. Biomaterials. 2009;30(5):859–866. | ||
Parveen S, Sahoo SK. Evaluation of cytotoxicity and mechanism of apoptosis of doxorubicin using folate-decorated chitosan nanoparticles for targeted delivery to retinoblastoma. Cancer Nanotechnol. 2010;1(1–6):47–62. | ||
Hrkach J, von Hoff D, Ali MM, et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med. 2012;4128(128):ra139. | ||
von Hoff DD, Mita MM, Ramanathan RK, et al. Phase I study of PSMA-targeted docetaxel-containing nanoparticle BIND-014 in patients with advanced solid tumors. Clin Cancer Res. 2016;22(13):3157–3163. | ||
Hong M, Zhu S, Jiang Y, et al. Novel anti-tumor strategy: PEG-hydroxycamptothecin conjugate loaded transferrin-PEG-nanoparticles. J Control Release. 2010;141(1):22–29. | ||
Visser CC, Stevanović S, Voorwinden LH, et al. Validation of the transferrin receptor for drug targeting to brain capillary endothelial cells in vitro. J Drug Target. 2004;12(3):145–150. | ||
Gan CW, Feng SS. Transferrin-conjugated nanoparticles of poly(lactide)-d-α-tocopheryl polyethylene glycol succinate diblock copolymer for targeted drug delivery across the blood–brain barrier. Biomaterials. 2010;31(30):7748–7757. | ||
Jain A, Chasoo G, Singh SK, Saxena AK, Jain SK. Transferrin-appended PEGylated nanoparticles for temozolomide delivery to brain: in vitro characterisation. J Microencapsul. 2011;28(1):21–28. | ||
Camp ER, Wang C, Little EC, et al. Transferrin receptor targeting nanomedicine delivering wild-type p53 gene sensitizes pancreatic cancer to gemcitabine therapy. Cancer Gene Ther. 2013;20(4):222–228. | ||
Liu J, Wei T, Zhao J, et al. Multifunctional aptamer-based nanoparticles for targeted drug delivery to circumvent cancer resistance. Biomaterials. 2016;91:44–56. | ||
Silva CO, Petersen SB, Reis CP, et al. EGF functionalized polymer-coated gold nanoparticles promote EGF photostability and EGFR internalization for photothermal therapy. PLoS One. 2016;11(10):e0165419. | ||
Cirstoiu-Hapca A, Buchegger F, Bossy L, Kosinski M, Gurny R, Delie F. Nanomedicines for active targeting: physico-chemical characterization of paclitaxel-loaded anti-HER2 immunonanoparticles and in vitro functional studies on target cells. Eur J Pharm Sci. 2009;38(3):230–237. | ||
Wang Z, Chui WK, Ho PC, Ho PC. Design of a multifunctional PLGA nanoparticulate drug delivery system: evaluation of its physicochemical properties and anticancer activity to malignant cancer cells. Pharm Res. 2009;26(5):1162–1171. | ||
Danhier F, Vroman B, Lecouturier N, et al. Targeting of tumor endothelium by RGD-grafted PLGA-nanoparticles loaded with paclitaxel. J Control Release. 2009;140(2):166–173. | ||
Mao B, Liu C, Zheng W, et al. Cyclic cRGDfk peptide and chlorin e6 functionalized silk fibroin nanoparticles for targeted drug delivery and photodynamic therapy. Biomaterials. 2018;161:306–320. | ||
Xin H, Jiang X, Gu J, et al. Angiopep-conjugated poly(ethylene glycol)-co-poly(ε-caprolactone) nanoparticles as dual-targeting drug delivery system for brain glioma. Biomaterials. 2011;32:4293–4305. | ||
Yusuf-Makagiansar H, Siahaan TJ. Binding and internalization of an LFA-1-derived cyclic peptide by ICAM receptors on activated lymphocyte: a potential ligand for drug targeting to ICAM-1-expressing cells. Pharm Res. 2001;18(3):329–335. | ||
Chittasupho C, Xie SX, Baoum A, Yakovleva T, Siahaan TJ, Berkland CJ. ICAM-1 targeting of doxorubicin-loaded PLGA nanoparticles to lung epithelial cells. Eur J Pharm Sci. 2009;37(2):141–150. | ||
Obaid G, Chambrier I, Cook MJ, Russell DA. Cancer targeting with biomolecules: a comparative study of photodynamic therapy efficacy using antibody or lectin conjugated phthalocyanine-PEG gold nanoparticles. Photochem Photobiol Sci. 2015;14(4):737–747. | ||
Bhat R, García I, Aznar E, et al. Lectin-gated and glycan functionalized mesoporous silica nanocontainers for targeting cancer cells overexpressing Lewis X antigen. Nanoscale. 2018;10(1):239–249. | ||
Wu X, Tan YJ, Toh HT, et al. Stimuli-responsive multifunctional glyconanoparticle platforms for targeted drug delivery and cancer cell imaging. Chem Sci. 2017;8(5):3980–3988. | ||
Frick SU, Domogalla MP, Baier G, et al. Interleukin-2 functionalized nanocapsules for T cell-based immunotherapy. ACS Nano. 2016;10:9216–9226. | ||
Singh R, Lillard JW. Nanoparticle-based targeted drug delivery. Exp Mol Pathol. 2009;86(3):215–223. | ||
Lee ES, Na K, Bae YH. Doxorubicin loaded pH-sensitive polymeric micelles for reversal of resistant MCF-7 tumor. J Control Release. 2005;103(2):405–418. | ||
Suzuki R, Takizawa T, Kuwata Y, et al. Effective anti-tumor activity of oxaliplatin encapsulated in transferrin–PEG-liposome. Int J Pharm. 2008;346(1–2):143–150. | ||
Sahoo SK, Ma W, Labhasetwar V. Efficacy of transferrin-conjugated paclitaxel-loaded nanoparticles in a murine model of prostate cancer. Int J Cancer. 2004;112(2):335–340. | ||
Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME. Impact of tumor-specific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging. Proc Natl Acad Sci U S A. 2007;104(39):15549–15554. | ||
Gradishar WJ. Albumin-bound paclitaxel: a next-generation taxane. Expert Opin Pharmacother. 2006;7(8):1041–1053. | ||
Pietersz GA, Wang X, Yap ML, Lim B, Peter K. Therapeutic targeting in nanomedicine: the future lies in recombinant antibodies. Nanomedicine (Lond). 2017;12(15):1873–1889. | ||
Cheng Z, al Zaki A, Hui JZ, Muzykantov VR, Tsourkas A. Multifunctional nanoparticles: cost versus benefit of adding targeting and imaging capabilities. Science. 2012;338(6109):903–910. | ||
Du J, Lane LA, Nie S. Stimuli-responsive nanoparticles for targeting the tumor microenvironment. J Control Release. 2015;219:205–214. | ||
Zhao X, Liu P. Reduction-responsive core–shell–corona micelles based on triblock copolymers: novel synthetic strategy, characterization, and application as a tumor microenvironment-responsive drug delivery system. ACS Appl Mater Interfaces. 2015;7(1):166–174. | ||
Adiseshaiah PP, Crist RM, Hook SS, Mcneil SE. Nanomedicine strategies to overcome the pathophysiological barriers of pancreatic cancer. Nat Rev Clin Oncol. 2016;13(12):750–765. | ||
Nielsen MF, Mortensen MB, Detlefsen S. Key players in pancreatic cancer-stroma interaction: cancer-associated fibroblasts, endothelial and inflammatory cells. World J Gastroenterol. 2016;22(9):2678–2700. | ||
Bhaw-Luximon A, Jhurry D. New avenues for improving pancreatic ductal adenocarcinoma (PDAC) treatment: selective stroma depletion combined with nano drug delivery. Cancer Lett. 2015;369(2):266–273. | ||
Kennedy EJ. Biological drug products: development and strategies. ChemMedChem. 2014;9(12):2814–2815. | ||
Kalimuthu K, Lubin BC, Bazylevich A, et al. Gold nanoparticles stabilize peptide-drug-conjugates for sustained targeted drug delivery to cancer cells. J Nanobiotechnology. 2018;16(1):34. | ||
Tran S, Degiovanni PJ, Piel B, Rai P. Cancer nanomedicine: a review of recent success in drug delivery. Clin Transl Med. 2017;6:44. | ||
Rahman AM, Yusuf SW, Ewer MS. Anthracycline-induced cardiotoxicity and the cardiac-sparing effect of liposomal formulation. Int J Nanomedicine. 2007;2(4):567–583. | ||
Batist G. Cardiac safety of liposomal anthracyclines. Cardiovasc Toxicol. 2007;7(2):72–74. | ||
Kundranda M, Niu J. Albumin-bound paclitaxel in solid tumors: clinical development and future directions. Drug Des Devel Ther. 2015;9:3767–3777. | ||
Untch M, Jackisch C, Schneeweiss A, et al. Nab-paclitaxel versus solvent-based paclitaxel in neoadjuvant chemotherapy for early breast cancer (GeparSepto – GBG 69): a randomised, phase 3 trial. Lancet Oncol. 2016;17(3):345–356. | ||
Chiorean EG, von Hoff D, Wan Y, Margunato-Debay S, Botteman M, Goldstein D. Performance status dynamics during treatment with nab-paclitaxel plus gemcitabine versus gemcitabine alone for metastatic pancreatic cancer. Cancer Manag Res. 2018;10:1389–1396. | ||
Hoffman RM, Bouvet M. Nanoparticle albumin-bound-paclitaxel: a limited improvement under the current therapeutic paradigm of pancreatic cancer. Expert Opin Pharmacother. 2015;16(7):943–947. | ||
Madhavan S, Gusev Y, Harris M, et al. G-DOC: a systems medicine platform for personalized oncology. Neoplasia. 2011;13(9):771–783. | ||
Lee K, Chung H, Im S. Multicenter phase II study of a Cremophor-free polymeric micelle-formulated paclitaxel in patients (pts) with metastatic breast cancer (MBC). J Clin Oncol. 2006;24(18 Suppl):10520. | ||
Nemunaitis J, Cunningham C, Senzer N, et al. Phase I study of CT-2103, a polymer-conjugated paclitaxel, and carboplatin in patients with advanced solid tumors. Cancer Invest. 2005;23(8):671–676. | ||
Northfelt DW, Dezube BJ, Thommes JA, et al. Efficacy of PEGylated-liposomal doxorubicin in the treatment of AIDS-related Kaposi’s sarcoma after failure of standard chemotherapy. J Clin Oncol. 1997;15(2):653–659. | ||
Ma P, Chen J, Bi X, et al. Overcoming multidrug resistance through the GLUT1-mediated and enzyme-triggered mitochondrial targeting conjugate with redox-sensitive paclitaxel release. ACS Appl Mater Interfaces. 2018;10(15):12351–12363. | ||
Zhou L, Wang H, Li Y. Stimuli-responsive nanomedicines for overcoming cancer multidrug resistance. Theranostics. 2018;8(4):1059–1074. | ||
Liu J, Li J, Liu N, et al. In vitro studies of phospholipid-modified PAMAM-siMDR1 complexes for the reversal of multidrug resistance in human breast cancer cells. Int J Pharm. 2017;530(1–2):291–299. | ||
Li J, Liu J, Guo N, Zhang X. Reversal of multidrug resistance in breast cancer MCF-7/ADR cells by h-R3-siMDR1-PAMAM complexes. Int J Pharm. 2016;511(1):436–445. | ||
Guo N, Gao C, Liu J, et al. Reversal of ovarian cancer multidrug resistance by a combination of LAH4-L1-siMDR1 nanocomplexes with chemotherapeutics. Mol Pharm. 2018;15(5):1853–1861. | ||
Misra R, Das M, Sahoo BS, Sahoo SK. Reversal of multidrug resistance in vitro by co-delivery of MDR1 targeting siRNA and doxorubicin using a novel cationic poly(lactide-co-glycolide) nanoformulation. Int J Pharm. 2014;475(1–2):372–384. | ||
Masuda Y, Kobayashi H, Holland JF, Ohnuma T. Reversal of multidrug resistance by a liposome-MDR1 ribozyme complex. Cancer Chemother Pharmacol. 1998;42(1):9–16. | ||
Gu J, Fang X, Hao J, Sha X. Reversal of P-glycoprotein-mediated multidrug resistance by CD44 antibody-targeted nanocomplexes for short hairpin RNA-encoding plasmid DNA delivery. Biomaterials. 2015;45:99–114. | ||
Li J, Liang H, Liu J, Wang Z. Poly(amidoamine) (PAMAM) dendrimer mediated delivery of drug and pDNA/siRNA for cancer therapy. Int J Pharm. 2018;546(1–2):215–225. | ||
Kesharwani P, Banerjee S, Gupta U, et al. PAMAM dendrimers as promising nanocarriers for RNAi therapeutics. Mater Today. 2015;18(10):565–572. | ||
Naha P, Mukherjee S, Byrne H. Toxicology of engineered nanoparticles: focus on poly(amidoamine) dendrimers. Int J Environ Res Public Health. 2018;15(2):E338. | ||
de Jong WH, Borm PJ. Drug delivery and nanoparticles: applications and hazards. Int J Nanomedicine. 2008;3(2):133–149. | ||
Kim YM, Song SC. Targetable micelleplex hydrogel for long-term, effective, and systemic siRNA delivery. Biomaterials. 2014;35(27):7970–7977. | ||
Kim HJ, Kim A, Miyata K, Kataoka K. Recent progress in development of siRNA delivery vehicles for cancer therapy. Adv Drug Deliv Rev. 2016;104:61–77. | ||
Sarett SM, Nelson CE, Duvall CL. Technologies for controlled, local delivery of siRNA. J Control Release. 2015;218:94–113. | ||
Miele E, Spinelli GP, Miele E, Tomao F, Tomao S. Albumin-bound formulation of paclitaxel (Abraxane ABI-007) in the treatment of breast cancer. Int J Nanomedicine. 2009;4:99–105. | ||
Park K. Controlled drug delivery systems: past forward and future back. J Control Release. 2014;190:3–8. | ||
Liu D, Yang F, Xiong F, Gu N. The smart drug delivery system and its clinical potential. Theranostics. 2016;6(9):1306–1323. | ||
Ventola CL. Progress in nanomedicine: approved and investigational nanodrugs. Pharm Ther. 2017;42(12):742–755. | ||
Montana M, Ducros C, Verhaeghe P, Terme T, Vanelle P, Rathelot P. Albumin-bound paclitaxel: the benefit of this new formulation in the treatment of various cancers. J Chemother. 2011;23(2):59–66. | ||
Yano S, Zhang Y, Miwa S, et al. Spatial–temporal FUCCI imaging of each cell in a tumor demonstrates locational dependence of cell cycle dynamics and chemoresponsiveness. Cell Cycle. 2014;13(13):2110–2119. | ||
Douer D. Efficacy and Safety of Vincristine Sulfate Liposome Injection in the Treatment of Adult Acute Lymphocytic Leukemia. Oncologist. 2016;21(7):840–847. | ||
Silverman JA, Deitcher SR. Marqibo (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother Pharmacol. 2013;71(3):555–564. | ||
Dawidczyk CM, Russell LM, Searson PC. Nanomedicines for cancer therapy: state-of-the-art and limitations to pre-clinical studies that hinder future developments. Front Chem. 2014;2:69. | ||
Davis T, Farag SS. Treating relapsed or refractory Philadelphia chromosome-negative acute lymphoblastic leukemia: liposome-encapsulated vincristine. Int J Nanomedicine. 2013;8:3479–3488. | ||
Drummond DC, Noble CO, Guo Z, Hong K, Park JW, Kirpotin DB. Development of a highly active nanoliposomal irinotecan using a novel intraliposomal stabilization strategy. Cancer Res. 2006;66(6):3271–3277. | ||
Lamb YN, Scott LJ. Liposomal Irinotecan: a review in metastatic pancreatic adenocarcinoma. Drugs. 2017;77(7):785–792. | ||
Shah N, Mohammad AS, Saralkar P, et al. Investigational chemotherapy and novel pharmacokinetic mechanisms for the treatment of breast cancer brain metastases. Pharmacol Res. 2018;132:47–68. | ||
Rafiyath SM, Rasul M, Lee B, Wei G, Lamba G, Liu D. Comparison of safety and toxicity of liposomal doxorubicin vs. conventional anthracyclines: a meta-analysis. Exp Hematol Oncol. 2012;1(1):10. | ||
Forssen EA, Coulter DM, Proffitt RT. Selective in vivo localization of daunorubicin small unilamellar vesicles in solid tumors. Cancer Res. 1992;52(12):3255–3261. | ||
Bulbake U, Doppalapudi S, Kommineni N, Khan W. Liposomal formulations in clinical use: an updated review. Pharmaceutics. 2017;9(4):E12. | ||
Mufamadi MS, Pillay V, Choonara YE, et al. A review on composite liposomal technologies for specialized drug delivery. J Drug Deliv. 2011;2011:939851. | ||
Lancet JE, Gl U, Cortes JE, et al. Final results of a phase III randomized trial of CPX-351 versus 7+3 in older patients with newly diagnosed high risk (secondary) AML. J Clin Oncol. 2016;34(15 Suppl):7000. | ||
Chen EC, Fathi AT, Brunner AM. Reformulating acute myeloid leukemia: liposomal cytarabine and daunorubicin (CPX-351) as an emerging therapy for secondary AML. Onco Targets Ther. 2018;11:3425–3434. | ||
Collier MA, Bachelder EM, Ainslie KM. Electrosprayed myocet-like liposomes: an alternative to traditional liposome production. Pharm Res. 2017;34(2):419–426. | ||
Agrahari V, Agrahari V. Facilitating the translation of nanomedicines to a clinical product: challenges and opportunities. Drug Discov Today. 2018;23(5):974–991. | ||
Wex J, Sidhu M, Odeyemi I, Abou-Setta AM, Retsa P, Tombal B. Leuprolide acetate 1-, 3- and 6-monthly depot formulations in androgen deprivation therapy for prostate cancer in nine European countries: evidence review and economic evaluation. Clinicoecon Outcomes Res. 2013;5:257–269. | ||
Tombal B, Berges R. Eligard: advantages for optimal testosterone control. Eur Urol Suppl. 2006;5(18):900–904. | ||
Jain RA. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials. 2000;21(23):2475–2490. | ||
Danhier F, Ansorena E, Silva JM, Coco R, le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. | ||
Pillai G, Ceballos-Coronel ML. Science and technology of the emerging nanomedicines in cancer therapy: a primer for physicians and pharmacists. Sage Open Med. 2013;1(3):2050312113513759. | ||
Zhang F, Liu MR, Wan HT. Discussion about several potential drawbacks of PEGylated therapeutic proteins. Biol Pharm Bull. 2014;37(3):335–339. | ||
Shehzad A, Lee J, Huh TL, Lee YS. Curcumin induces apoptosis in human colorectal carcinoma (HCT-15) cells by regulating expression of Prp4 and p53. Mol Cells. 2013;35(6):526–532. | ||
Kunwar A, Barik A, Mishra B, Rathinasamy K, Pandey R, Priyadarsini KI. Quantitative cellular uptake, localization and cytotoxicity of curcumin in normal and tumor cells. Biochim Biophys Acta. 2008;1780(4):673–679. | ||
Cheung A, Bax HJ, Josephs DH, et al. Targeting folate receptor alpha for cancer treatment. Oncotarget. 2016;7(32):52553–52574. | ||
Lyon PC, Griffiths LF, Lee J, et al. Clinical trial protocol for TARDOX: a phase I study to investigate the feasibility of targeted release of lyso-thermosensitive liposomal doxorubicin (ThermoDox) using focused ultrasound in patients with liver tumours. J Ther Ultrasound. 2017;5:28. | ||
Panyam J, Zhou WZ, Prabha S, Sahoo SK, Labhasetwar V. Rapid endo-lysosomal escape of poly(dl-lactide-co-glycolide) nanoparticles: implications for drug and gene delivery. FASEB J. 2002;16(10):1217–1226. | ||
Ramasamy T, Tran TH, Choi JY, et al. Layer-by-layer coated lipid–polymer hybrid nanoparticles designed for use in anticancer drug delivery. Carbohydr Polym. 2014;102:653–661. | ||
Westesen K, Bunjes H, Koch MH. Physicochemical characterization of lipid nanoparticles and evaluation of their drug loading capacity and sustained release potential. J Control Release. 1997;48(2–3):223–236. | ||
Molineux G. PEGylation: engineering improved biopharmaceuticals for oncology. Pharmacotherapy. 2003;23(8 Pt 2):3S–8S. | ||
Wang M, Xie F, Wen X, et al. Therapeutic PEG-ceramide nanomicelles synergize with salinomycin to target both liver cancer cells and cancer stem cells. Nanomedicine (Lond). 2017;12(9):1025–1042. | ||
Yao XL, Yoshioka Y, Ruan GX, et al. Optimization and internalization mechanisms of PEGylated adenovirus vector with targeting peptide for cancer gene therapy. Biomacromolecules. 2012;13(8):2402–2409. | ||
Balalaeva IV, Zdobnova TA, Krutova IV, Brilkina AA, Lebedenko EN, Deyev SM. Passive and active targeting of quantum dots for whole-body fluorescence imaging of breast cancer xenografts. J Biophotonics. 2012;5(11–12):860–867. | ||
Sykes EA, Chen J, Zheng G, Chan WC. Investigating the impact of nanoparticle size on active and passive tumor targeting efficiency. ACS Nano. 2014;8(6):5696–5706. | ||
Cortés J, Rugo HS, Twelves C, et al. Safety and tolerability of etirinotecan pegol in advanced breast cancer: analysis of the randomized, phase 3 BEACON trial. Springerplus. 2016;5:1033. | ||
Cho K, Wang X, Nie S, Chen Z, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14(5):1310–1316. | ||
Liechty WB, Peppas NA. Expert opinion: Responsive polymer nanoparticles in cancer therapy. Eur J Pharm Biopharm. 2012;80(2):241–246. | ||
Wang B, Jiang W, Yan H, et al. Novel PEG-graft-PLA nanoparticles with the potential for encapsulation and controlled release of hydrophobic and hydrophilic medications in aqueous medium. Int J Nanomedicine. 2011;6:1443–1451. | ||
Jackson JK, Letchford K, Wasserman BZ, Ye L, Hamad WY, Burt HM. The use of nanocrystalline cellulose for the binding and controlled release of drugs. Int J Nanomedicine. 2011;6:321–330. | ||
Amoozgar Z, Wang L, Brandstoetter T, Wallis SS, Wilson EM, Goldberg MS. Dual-layer surface coating of PLGA-based nanoparticles provides slow-release drug delivery to achieve metronomic therapy in a paclitaxel-resistant murine ovarian cancer model. Biomacromolecules. 2014;15(11):4187–4194. | ||
Prosperi D, Colombo M, Zanoni I, Granucci F. Drug nanocarriers to treat autoimmunity and chronic inflammatory diseases. Semin Immunol. 2017;34:61–67. | ||
Owen A, Rannard S. Considerations for clinically-relevant nanomedicine therapies for chronic diseases. Nanomedicine (Lond). 2015;10(20):3103–3107. | ||
Hoffman A. Pharmacodynamic aspects of sustained release preparations. Adv Drug Deliv Rev. 1998;33(3):185–199. | ||
Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces. 2010;75(1):1–18. | ||
Sklar JJ, OH VM, Li SC. Factors affecting therapeutic compliance: a review from the patient’s perspective. Ther Clin Risk Manag. 2008;4(1):269–286. | ||
Claxton AJ, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther. 2001;23(8):1296–1310. | ||
Vrijens B, de Geest S, Hughes DA, et al. A new taxonomy for describing and defining adherence to medications. Br J Clin Pharmacol. 2012;73(5):691–705. | ||
Eisen SA, Miller DK, Woodward RS, Spitznagel E, Przybeck TR. The effect of prescribed daily dose frequency on patient medication compliance. Arch Intern Med. 1990;150(9):1881–1884. | ||
Noens L, van Lierde MA, de Bock R, et al. Prevalence, determinants, and outcomes of nonadherence to imatinib therapy in patients with chronic myeloid leukemia: the ADAGIO study. Blood. 2009;113(22):5401–5411. | ||
Wang YS, Youngster S, Grace M, Bausch J, Bordens R, Wyss DF. Structural and biological characterization of PEGylated recombinant interferon alpha-2b and its therapeutic implications. Adv Drug Deliv Rev. 2002;54(4):547–570. | ||
Bukowski R, Ernstoff MS, Gore ME, et al. PEGylated interferon alfa-2b treatment for patients with solid tumors: a phase I/II study. J Clin Oncol. 2002;20(18):3841–3949. | ||
Atkins MB, Hodi FS, Thompson JA, et al. Pembrolizumab (Pembro) plus ipilimumab (Ipi) or PEGylated interferon alfa-2b (PEG-IFN) for advanced melanoma or renal cell carcinoma (RCC). J Clin Oncol. 2016;34(15 Suppl):3013. | ||
Nunes AT, Green D, Ekwede I, Zoon K, Annunziata CM. Phase 1 study of intraperitoneal infusion of autologous monocytes with PEGinterferon alfa-2b and interferon gamma-1b in women with recurrent or refractory ovarian cancer, fallopian tube cancer, or primary peritoneal cancer. J Clin Oncol. 2016;35(15 Suppl):3092. | ||
Katsuki S, Matoba T, Koga J, Nakano K, Egashira K. Anti-inflammatory nanomedicine for cardiovascular disease. Front Cardiovasc Med. 2017;4:87. | ||
Batist G, Barton J, Chaikin P, Swenson C, Welles L. Myocet (liposome-encapsulated doxorubicin citrate): a new approach in breast cancer therapy. Expert Opin Pharmacother. 2002;3(12):1739–1751. | ||
Dixit N, Maurya SD, Sagar BP. Sustained release drug delivery system. Indian J Res Pharm Biotechnol. 2013;1(3):305–310. | ||
Tarun P, Vishal S, Gaurav S, Satyanand T, Chirag P, Anil G. Novel oral sustained release technology: a concise review sustained release drug disadvantages. Int J Res Dev Pharm Life Sci. 2013;2(2):262–269. | ||
Park K. Controlled drug delivery systems: past forward and future back. J Control Release. 2014;190:3–8. | ||
Müllertz A, Perrie Y, Rades T. Analytical Techniques in the Pharmaceutical Sciences. Heidelberg: Springer; 2016. | ||
Xu M, Liew CV, Heng PW. Evaluation of the coat quality of sustained release pellets by individual pellet dissolution methodology. Int J Pharm. 2015;478(1):318–327. | ||
Davidson RS, Kehoe G, inventors; Cure Pharmaceutical, assignee. Methods for modulating dissolution, bioavailability, bioequivalence and drug delivery profile of thin film drug delivery systems, controlled-release thin film dosage formats, and methods for their manufacture and use. US patent 8999372B2. 2015 Apr 7. | ||
Mccarthy CA, Ahern RJ, Dontireddy R, Ryan KB, Crean AM. Mesoporous silica formulation strategies for drug dissolution enhancement: a review. Expert Opin Drug Deliv. 2016;13(1):93–108. | ||
Kidane A, Bhatt PP, inventors; Supernus Pharmaceuticals, assignee. Osmotic drug delivery system. US patent 15162829. 2016 Feb 4. | ||
Chen J, Pan H, Ye T, et al. Recent aspects of osmotic pump systems: functionalization, clinical use and advanced imaging technology. Curr Drug Metab. 2016;17(3):279–291. | ||
Zaman M, Qureshi J, Ejaz H, et al. Oral controlled release drug delivery system and characterization of oral tablets: a review. Pak J Pharm Sci. 2016;2(1):67–76. | ||
Auiler JF, Liu K, Lynch JM, Gelotte CK. Effect of Food on early drug exposure from extended-release stimulants: results from the Concerta, Adderall XR Food Evaluation (CAFE) study. Curr Med Res Opin. 2002;18(5):311–316. | ||
Chaudhry NC, Saunders L. Sustained release of drugs from ion exchange resins. J Pharm Pharmacol. 1956;8(11):975–986. | ||
Singh BN, Kim KH. Floating drug delivery systems: an approach to oral controlled drug delivery via gastric retention. J Control Release. 2000;63(3):235–259. | ||
Kumar V, Prajapati S, Soni GC, Singh M, Kumar N. Sustained release matrix type drug delivery system: a review. World J Pharm Pharm Sci. 2012;1(3):934–960. | ||
Pundir S, Badola A, Sharma D. Sustained release matrix technology and recent advance in matrix drug delivery system: a review. Int J Drug Res Technol. 2017;3(1):12–20. | ||
Nair LS, Laurencin CT. Biodegradable polymers as biomaterials. Prog Polym Sci. 2007;32(8–9):762–798. | ||
Haxho F, Allison S, Alghamdi F, et al. Oseltamivir phosphate monotherapy ablates tumor neovascularization, growth, and metastasis in mouse model of human triple-negative breast adenocarcinoma. Breast Cancer (Dove Med Press). 2014;6:191–203. | ||
Haxho FA, Neufeld RJ, Szewczuk MR. Novel insulin receptor-signaling platform. Int J Diabetes Clin Res. 2014;1:1–10. | ||
Gilmour AM, Abdulkhalek S, Cheng TS, et al. A novel epidermal growth factor receptor-signaling platform and its targeted translation in pancreatic cancer. Cell Signal. 2013;25(12):2587–2603. | ||
Alghamdi F, Guo M, Abdulkhalek S, Crawford N, Amith SR, Szewczuk MR. A novel insulin receptor-signaling platform and its link to insulin resistance and type 2 diabetes. Cell Signal. 2014;26(6):1355–1368. | ||
Haxho F, Haq S, Szewczuk MR. Biased G protein-coupled receptor agonism mediates Neu1 sialidase and matrix metalloproteinase-9 crosstalk to induce transactivation of insulin receptor signaling. Cell Signal. 2018;43:71–84. | ||
Abdulkhalek S, Amith SR, Franchuk SL, et al. Neu1 sialidase and matrix metalloproteinase-9 cross-talk is essential for Toll-like receptor activation and cellular signaling. J Biol Chem. 2011;286(42):36532–36549. | ||
Haxho F, Neufeld RJ, Szewczuk MR. Neuraminidase-1: a novel therapeutic target in multistage tumorigenesis. Oncotarget. 2016;7(26):40860–40881. | ||
Hrynyk M, Ellis JP, Haxho F, et al. Therapeutic designed poly (lactic-co-glycolic acid) cylindrical oseltamivir phosphate-loaded implants impede tumor neovascularization, growth and metastasis in mouse model of human pancreatic carcinoma. Drug Des Devel Ther. 2015;9:4573–4586. | ||
O’Shea LK, Abdulkhalek S, Allison S, Neufeld RJ, Szewczuk MR. Therapeutic targeting of Neu1 sialidase with oseltamivir phosphate (Tamiflu) disables cancer cell survival in human pancreatic cancer with acquired chemoresistance. Onco Targets Ther. 2014;7:117–134. | ||
Kapishon V, Allison S, Whitney RA, Cunningham MF, Szewczuk MR, Neufeld RJ. Oseltamivir-conjugated polymeric micelles prepared by RAFT living radical polymerization as a new active tumor targeting drug delivery platform. Biomater Sci. 2016;4(3):511–521. | ||
Wood K, Szewczuk MR, Rousseau D, Neufeld RJ. Oseltamivir phosphate released from injectable Pickering emulsions over an extended term disables human pancreatic cancer cell survival. Oncotarget. 2018;9(16):12754–12768. | ||
Wakaskar RR. Promising effects of nanomedicine in cancer drug delivery. J Drug Target. 2018;26(4):319–324. | ||
Choi KY, Yoon HY, Kim JH, et al. Smart nanocarrier based on PEGylated hyaluronic acid for cancer therapy. ACS Nano. 2011;5(11):8591–8599. | ||
Sarkar K, Yang H. Encapsulation and extended release of anti-cancer anastrozole by stealth nanoparticles. Drug Deliv. 2008;15(5):343–346. | ||
Tietjen GT, Saltzman WM. Nanomedicine gets personal. Sci Transl Med. 2015;7(314):314fs47. | ||
Maitland ML, Schilsky RL. Clinical trials in the era of personalized oncology. CA Cancer J Clin. 2011;61(6):365–381. | ||
Nicolini C, Bragazzi N, Pechkova E. Nanoproteomics enabling personalized nanomedicine. Adv Drug Deliv Rev. 2012;64(13):1522–1531. | ||
Kydd J, Jadia R, Velpurisiva P, Gad A, Paliwal S, Rai P. Targeting strategies for the combination treatment of cancer using drug delivery systems. Pharmaceutics. 2017;9(4):E46. | ||
Lammers T, Kiessling F, Hennink WE, Storm G. Drug targeting to tumors: principles, pitfalls and (pre-)clinical progress. J Control Release. 2012;161(2):175–187. | ||
Li Y, Wang Y, Huang G, Gao J. Cooperativity principles in self-assembled nanomedicine. Chem Rev. 2018;118(11):5359–5391. | ||
Luo M, Wang H, Wang Z, et al. A STING-activating nanovaccine for cancer immunotherapy. Nat Nanotechnol. 2017;12(7):648–654. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.