Back to Archived Journals » Advances in Genomics and Genetics » Volume 5
Recent advances in preimplantation genetic diagnosis
Authors Kahraman S, Beyazyurek C, Tac HA, Pirkevi C, Cetinkaya M, Gulum N
Received 16 July 2014
Accepted for publication 29 August 2014
Published 17 April 2015 Volume 2015:5 Pages 189—203
DOI https://doi.org/10.2147/AGG.S53422
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr John Martignetti
Semra Kahraman, Çağri Beyazyürek, Hüseyin Avni Taç, Caroline Pirkevi, Murat Cetinkaya, Neşe Gülüm
IVF and Reproductive Genetics Center, Istanbul Memorial Hospital, Istanbul, Turkey
Abstract: Preimplantation genetic diagnosis (PGD) is an important method for the identification chromosomal abnormalities and genes responsible for genetic defects in embryos that are created through in vitro fertilization before pregnancy. As the list of conditions and indications for PGD testing is continuing to extend enormously, novel in vitro fertilization techniques and newly established genetic analysis techniques have been implemented in clinical settings in the recent years. Blastocyst-stage biopsy, vitrification techniques, time-lapse imaging, whole-genome amplification, array-based diagnostic techniques, and next-generation sequencing techniques are promising techniques for the accurate diagnosis of diverse genetic conditions and also for the selection of the best embryo that has the highest implantation capacity. The timing and technique used for biopsy, the amplification techniques, the genetic diagnosis techniques, and appropriate genetic counseling play important roles in establishing a successful PGD. In this review, those key points of PGD will be reviewed in detail.
Keywords: preimplantation genetic diagnosis, array comparative genomic hybridization, single-nucleotide polymorphism arrays, next-generation sequencing, monogenic disorders, aneuploidy testing
Introduction
Preimplantation genetic diagnosis (PGD) involves the genetic testing of biopsy material from embryos that are generated through in vitro fertilization (IVF) techniques and involves the transfer of chromosomally normal and disease-free embryos to the uterus. PGD allows couples who have an increased risk of transmitting genetic disorders not only to have healthy children, but also to prevent complications such as health problems and the psychological and financial burdens that may result from the termination of a pregnancy.
PGD was first performed by Handyside et al1 using the polymerase chain reaction (PCR) technique in order to prevent transmission of a sex-linked disorder. Since then, PGD has been applied to many different conditions, and the list of indications for PGD testing has extended enormously. Over those years, it is estimated that PGD has been performed for over 300 monogenic conditions; this treatment has resulted in the births of thousands of healthy children.2–4 The current list of indications for PGD testing could be summarized as follows:
- PGD for monogenic disorders
- Autosomal recessive
- Autosomal dominant
- X-linked
- Y-linked
- Mitochondrial
- Predisposition syndromes
- Preimplantation human leukocyte antigen (HLA) typing for hemopoietic stem cell transplantation
- PGD for numerical and structural chromosomal abnormalities
- Advanced female age
- Previous history of chromosomally abnormal fetus
- Repeated pregnancy losses
- Repetitive implantation failures
- Translocation carriers
- Inversion carriers
- PGD for selecting single euploid embryo transfer.
The biopsy technique and timing, the choice of genetic diagnosis techniques for mutational and chromosomal abnormalities, and appropriate genetic counseling play important roles in establishing a successful PGD.
Developmental stage at time of biopsy and biopsy techniques
There are currently three sources of genetic material available for PGD: polar bodies from oocytes; blastomeres from cleavage-stage embryos; and trophectoderm cells from blastocyst-stage embryos. Choosing the genetic source and the optimal day of biopsy requires reflection on several factors:5
- The timing of the biopsy should allow accurate identification of the genetic errors that need to be screened.
- The identified abnormalities should correspond to an abnormality in the embryo.
- The genetic diagnosis should be obtained to allow the selection of an embryo for transfer.
- The biopsy procedure should not harm the embryos.
Polar body biopsy
Polar body biopsy can be applied either before fertilization or after fertilization. For prefertilization polar body biopsy, the first polar body is removed before fertilization and the second polar body is removed after fertilization. For postfertilization polar body biopsy, both the first and the second polar bodies are removed simultaneously after fertilization. Since the polar bodies have no further role in embryo development, their removal is unlikely to have any negative effects on the subsequent steps of the embryo’s development. Another advantage is the longer time that is available for genetic analysis prior to embryo transfer.6,7 However, polar bodies give no information about the paternal genotype, and therefore cannot be used to diagnose paternal mutations, HLA typing, chromosomal abnormalities caused by paternal meiosis, or mitotic errors, and cannot be used for sex determination.6,7 As a result of these limitations, polar body biopsy is preferred to diagnose mostly aneuploidies in advanced maternal age, maternal inherited disorders such as X-linked anomalies, and maternal translocations. It should also be noted that in some countries, polar body diagnosis is the only option available because of legal restrictions.
With polar body biopsy, the material is obtained very early in the IVF culture period; thus, the genetic laboratory gains precious time for diagnosis and interpretation. Additionally, the biopsies involve only the polar bodies, which are separate and independent from the oocyte, and during the procedure, no portion of the actual embryo is biopsied. However, the capture of embryonic genetic errors with polar body biopsy is low. Even when both polar bodies are biopsied, approximately one error in four is missed.5 A recent publication detailed the results of the serial biopsies of embryos from nine patients. This study confirms that aneuploidy screening of polar bodies provides a relatively low capture rate for errors that are ultimately present at the blastocyst stage.8 In addition, as a technical note, polar bodies may undergo fragmentation, which may complicate the biopsy procedure and lead to misdiagnosis if all the fragments are not retrieved.
Cleavage-stage biopsy
Blastomere biopsy is accomplished at the cleavage stage, which is typically on day 3 of in vitro development. This technique is the oldest and most widely used method for PGD.9 In this method, one or two blastomeres are retrieved from embryos with more than six cells (Figure 1). With cleavage-stage biopsy, both maternal and paternal meiotic errors can be detected. In addition, blastomere biopsy allows enough time for genetic analysis to be performed before the embryo reaches the blastocyst stage and enough time for a fresh transfer with the timely shipment of samples with the use of transport PGD.
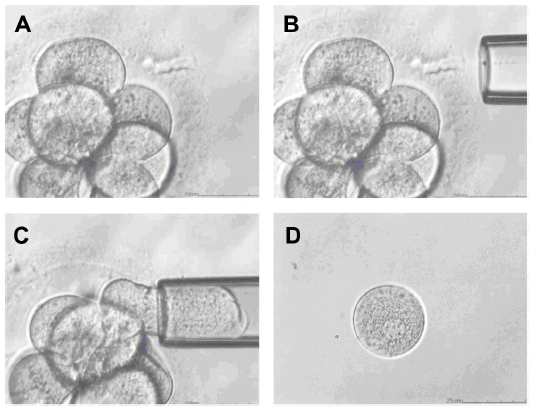 | Figure 1 Blastomere biopsy. |
The choice of embryo transfer day after the blastomere biopsy is critical. Because of the width of the zona opening made for the biopsy procedure itself, the embryo must be manipulated with extra care. The general practice is to transfer compacted embryos or morulas or, in some cases, to wait for blastocyst formation on day 5.10
One of the major concerns at this point is the possible effect of decreasing the cell number on the further development of the biopsied embryo. In addition, an embryo’s further development is negatively affected when the remaining cells are harmed during the biopsy because of the use of an inappropriate technique or an unsuitable medium. The question of the impact of biopsy on developing embryos was studied in a randomized control trial in which a paired design was used.11 The results of the blastomere biopsies demonstrated significant harm, with implantation rates decreasing from 50% to 30%, whereas after the trophectoderm biopsies, equivalent implantation and delivery rates were obtained.11
Although mosaicism has been speculated to be more prevalent in the cleavage-stage embryos compared to the blastocyst-stage ones, according to a detailed analysis, similar degrees of mosaicism have been found irrespective of the developmental stage;12 hence, a cleavage-stage biopsy may be as safe as a blastocyst-stage biopsy.
Blastocyst-stage biopsy
Blastocyst-stage embryo biopsy can be defined as the retrieval of a small part of the trophectoderm from a hatching blastocyst (Figure 2). The major advantage of this method is the amount of genetic material available for testing. Having more than one cell to work with prevents most diagnostic errors that are caused by technical problems, such as hybridization failures, amplification failures, or the absence of a nucleus. The allele dropout (ADO) rate is also lower, thereby increasing the reliability and accuracy of the diagnosis.13 A study conducted on both blastomeres and trophectoderm tissue samples demonstrated that the trophectoderm biopsy resulted in a threefold reduction in the number of embryos that remained inadequately evaluated by the use of PGD for single-gene disorders (SGD). The difference was attributed to the larger number of cells obtained via trophectoderm biopsy.13
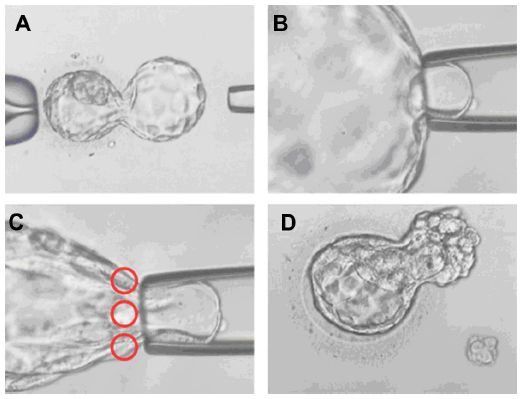 | Figure 2 Day 5 blastocyst-stage embryo biopsy. |
The most common policy with blastocyst-stage biopsy is a day 5 biopsy with a day 6 transfer. However, the time for the analysis is limited by the implantation window of the blastocyst and is thus very short – less than 24 hours. Despite this narrow window, there are two options to overcome the time limitation. One of them is to vitrify biopsied blastocysts and to transfer diagnosed embryos in a subsequent freeze–thaw cycle. The other one is to biopsy very early in the morning and to transfer in the evening. Apart from these options, as long as the rate of embryo development is suitable, trophectoderm biopsy can be applied late in the fourth day and the transfer can occur on the fifth day. With this option, it is still possible to biopsy and then to vitrify the remaining, slower-growing blastocysts on the fifth day. Also, a blastocyst contains over 100 cells, so the removal of 2–10 cells from the trophectoderm is very unlikely to have a detrimental effect either on the blastocyst’s development or on the development of the fetus originating from the inner cell mass (ICM); this provides an advantage over cleavage-stage biopsy.11 It should also be noted that embryonic aneuploidy screening was demonstrated to be beneficial in two randomized controlled trials in which trophectoderm biopsies were used.14–16
Mosaicism, which may cause discordance between ICM and trophectoderm cells, is another issue that has been assessed in blastocysts. Recent reports in which ICM and trophectoderm cells had been evaluated separately by fluorescent in situ hybridization (FISH) and array comparative genomic hybridization (aCGH) techniques demonstrated that aneuploid blastocysts displayed no evidence of preferential segregation of abnormalities to the trophectoderm, and nearly all trophectoderm biopsies derived from the same embryos were concordant; this finding supports the idea that the trophectoderm karyotype is an excellent predictor of the ICM karyotype.17–20
Role of new IVF techniques: time-lapse imaging systems
Culturing embryos until day 5 to select the blastocyst with the best morphology ensures higher implantation rates and decreased miscarriage rates. With the increasing need for cryopreservation and for single-embryo transfer to avoid multiple pregnancies, scoring embryos is a crucial step to select the best embryos for transfer, for cryopreservation, or for PGD to maintain high efficacy of the IVF cycle.
Embryo evaluation is the fundamental of IVF laboratories, but it still remains subjective despite the recent efforts that have been made to find more objective means.21 Continuous monitoring of embryo development from fertilization to blastocyst stage by automated time-lapse imaging has offered the possibility to appraise the timing of embryonic cell division. In a classical morphological evaluation, the embryos have to be removed daily for a few minutes from the incubator to allow for a static observation under the microscope. Conversely, in the time-lapse system, embryos can be continuously monitored without being disturbed. In addition, time-lapse allows for the observation of the exact time points of cell divisions, compaction, blastocyst formation, generation and absorption of fragments, and multinucleation (Figure 3). With this technology, fertile patients referred to IVF for PGD may opt to biopsy only regularly dividing embryos for the first run in order to deselect those with low implantation potential.22 Another option to incorporate time-lapse imaging is to further select among those embryos already identified as euploid.
 | Figure 3 Time-lapse observations of cleavage and blastulation. |
In a recent study, researchers retrospectively investigated the relationship between morphokinetic variables and ploidy after trophectoderm biopsy and either aCGH or single-nucleotide polymorphism (SNP) array. Multiple aneuploid embryos took longer for the initiation of compaction and for the time to reach full blastocyst stage compared with euploid embryos (Figure 4). Embryos having single or multiple aneuploidy had a later initiation of blastulation compared with euploid embryos. This noninvasive model for ploidy classification may help identify the blastocysts that are considered low risk and allow the remaining ones that score a medium-to-high aneuploidy risk to be vitrified for a later diagnosis and a potential transfer.23,24
 | Figure 4 Time-lapse photos of an embryo dividing irregularly from 6–8 cells and further diagnosed as complex aneuploid at the blastocyst stage (A–C). |
Chromosomally normal and abnormal embryos were also found to have different kinetic behaviors in another study in which embryos were biopsied on day 3 and were analyzed by aCGH. A hierarchical algorithm using the t5–t2 interval and the duration of the third cell cycle (cc3=t5–t3) classified embryos into four categories based on the expected percentage of chromosomally normal embryos and, thus, increased the probability of selecting chromosomally normal embryos by time-lapse morphokinetic analysis.25
Therefore, time-lapse morphological assessments and combined measurements are promising and may improve the ability to predict embryonic aneuploidy. However, invasive diagnostic techniques such as comprehensive chromosomal screening (CCS) still remain the most reliable methods in order to eliminate chromosomally abnormal embryos.26
Recent advances in aneuploidy testing
Although PGD first emerged as a diagnostic tool for the elimination of SGDs,1 it has been more widely used for the purpose of eliminating chromosomal abnormalities in preimplantation embryos and for increasing the take-home baby rates for IVF treatments. Aneuploidy is defined as a numerical chromosomal abnormality that results in a deviation of the number of copies of any of the 23 chromosomes.27 Aneuploidy can originate from either an excess number of chromosomes (eg, trisomy) or from missing chromosomes (eg, monosomy). Haploidy (one set of chromosomes [n]) and triploidy (3n) are associated with the abnormalities of whole chromosome sets. Aneuploidy significantly contributes to IVF failures27 and is the major cause of the first trimester miscarriages.28 PGD for numerical chromosomal abnormalities has been commonly used for couples experiencing recurrent first trimester miscarriages in order to eliminate chromosomally abnormal embryos from transfer. The other common indication for PGD is advanced maternal age. Women of advanced maternal age have a high risk of producing chromosomally abnormal oocytes and, thus, of offspring with chromosomal abnormalities.4 Lastly, balanced translocation carriers (Robertsonian, reciprocal, inversion) may have fertility problems and may experience multiple pregnancy losses due to unbalanced segregation products formed in their gametes in relation to the breakpoints on the chromosomes.4
In 1993, Munné et al29 developed a diagnostic method combining the principles of FISH and the fixation of a single blastomere (Figure 5A) from cleavage-stage human embryos in an attempt to provide an alternative to the PCR technique for the determination of the sex of the embryos. After that, a short FISH procedure was developed in order to screen aneuploidy; this procedure uses fluorochrome- and digoxigenin-labeled DNA probes that are specific for the chromosomes most involved in miscarriages (X, Y, 18, 13, and 21) (Figure 5B).30 The FISH technique was also used to test polar bodies;31,32 this procedure involved either the simultaneous or sequential removal of two polar bodies from a fertilized egg, and it was done with the aim of diagnosing chromosomal nondisjunctions and premature separation of sister chromatids during maternal meiotic divisions. After an improvement of culture media to better support blastocyst-stage embryos, the FISH technique was coupled with a trophectoderm tissue biopsy for the purpose of testing for aneuploidy in blastocysts.33
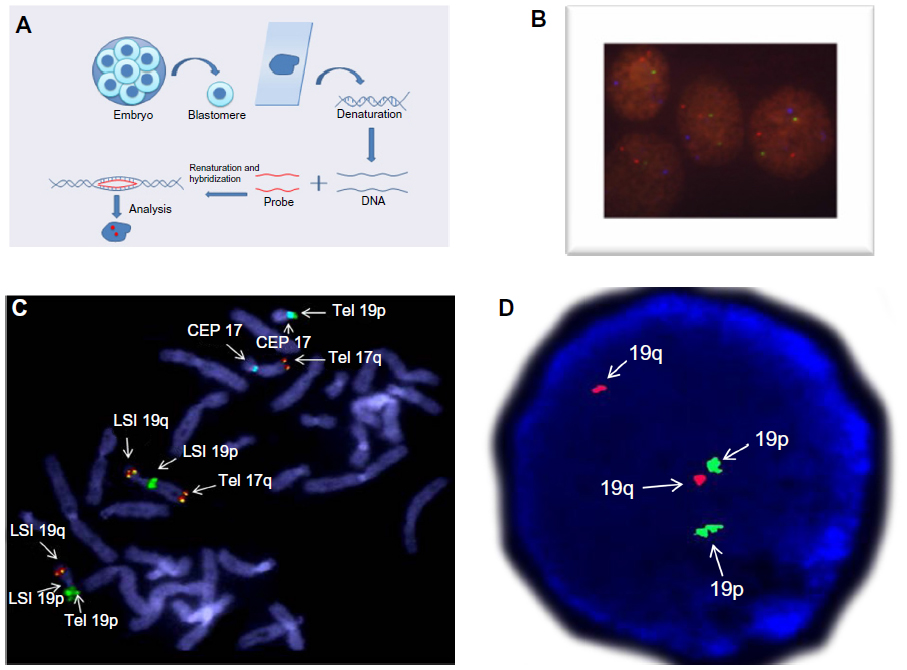 | Figure 5 Overview and applications of the FISH technique. |
The FISH technique has also been used successfully in the diagnosis of unbalanced products and embryos that belong to balanced rearrangement carriers. Because cases for translocation and inversion carriers are different from aneuploidy cases, there is a need for a preclinical workup study in order to both confirm the rearrangement breakpoint regions and to test the efficiency of the probes that will be used for PGD (Figure 5C). For this preclinical study, at least ten metaphase cells and 100 interphase cells should be analyzed with the same probe combination to assess signal specificities and any possibility of polymorphism in the probe-specific regions that may cause difficulties in signal interpretation during PGD (Figure 5D).
Regardless of the embryonic stage during which the biopsy is performed, the FISH technique has many limitations. The number of chromosomes that are screened is limited, and the subjectivity of the method itself leads to inaccurate results when the technique is used suboptimally.34 The FISH method is only available to screen a maximum of 9–12 chromosomes and it is able to detect 60%–80% of all aneuploid embryos.35 Given the fact that aneuploidy could affect any of the chromosomes, FISH still cannot detect a significant proportion of the aneuploidies and segmental abnormalities. The second limitation is suboptimal fixation, since the quality of the FISH results is highly dependent on the quality of the nucleus, which is limited by the experience of the laboratory personnel who performs micromanipulations, such as fixation and biopsy procedures. These issues are considered to be the major reasons that previous randomized controlled trials have failed to show any benefit of PGD in improving live birth rates.36,37
From the beginning of 2010, several CCS techniques have been introduced and have been validated in preclinical studies evaluating accuracy. One technique is aCGH, in which the analysis of all of the chromosomes is done either with multiplex PCR, whole-genome amplification (WGA), or multiple displacement amplification (MDA).38–42 The most widely used techniques are aCGH, SNP arrays, next-generation sequencing (NGS), and real-time quantitative PCR (RT-qPCR).
Comparative genomic hybridization
The first attempts to analyze the comprehensive chromosomal constitutions of embryos started with the implementation of the comparative genomic hybridization (CGH) technique on embryos. CGH provides genome-wide scanning of differences in DNA sequence copy number, unlike FISH, which allows only a limited number of loci to be analyzed. The CGH technique was demonstrated first as a means to screen copy number variations in the entire genome.43,44
CGH is based on the cohybridization of differentially-labeled test and reference DNAs with normal metaphase chromosomes (Figure 6). Measurement of the test-to-reference fluorescence ratios along all chromosomes provides information on the chromosomal regions that are either overrepresented or underrepresented in the test genome. The limitations are as follows: the technique is laborious and time consuming, and the analysis is too lengthy for the limited time frame for human preimplantation embryos to be transplanted into the uterus. For that reason, the technique definitely required freezing during assisted reproductive technique (ART) cycles when blastomere biopsy was used in the study by Wilton.45 Alternatively, polar body biopsy coupled with CGH analysis was demonstrated with success in fresh ART cycles.46 This strategy allowed the freezing of the embryos to be avoided. The viability of these embryos had been significantly affected by freezing techniques in previous decades when advanced vitrification techniques had not yet been implemented in routine clinical usage. The CGH technique has served as a basis to develop array-based CGH (aCGH) technology, which is more robust, accurate, automated, and with higher resolution.
 | Figure 6 Comparative genomic hybridization technique. |
aCGH
The technique of aCGH is similar to CGH, but instead of hybridization on metaphase chromosomes, the hybridization is either on chips (usually bacterial artificial chromosomes) or on arrays, and the results are analyzed with specialized software.47 This technique includes the following steps: extraction of genomic DNA; WGA; fluorescent labeling of samples and references; mixing of differentially-labeled probes; hybridization on arrays; washing; and then scanning and analysis by the computer program (Figure 7A–C). This method is fully automated, and the whole procedure is completed within 12 hours. The software analyzes the ratio of red and green intensity information of fluorescent colors in each position, and then compares these ratios with those of the male and female reference DNA. The decision of chromosomal status is made by the software by calculating the standard deviation ratios after smoothing and normalizing the data; if the ratio for a specific chromosome is higher than +0.3 (>0.3), the software reports this abnormality as a “gain” (usually a trisomy), and if this ratio is below -0.3, it reports as a “loss” (usually a monosomy). In addition to the numerical abnormalities, this technique is capable of the detection of segmental aneuploidies as small as 2 Mb and imbalances formed by abnormal segregations due to rearrangements (Figure 7D).48–50
 | Figure 7 Overview and applications of the aCGH technique. |
The aCGH system can diagnose unbalanced products formed from any Robertsonian carrier and the vast majority of reciprocal translocations carriers. However, the diagnosis of reciprocal translocations smaller than 2 Mb in length has not yet been validated either by aCGH or by any other array systems. This remains an indication for FISH-based PGD for translocations. However, as an alternative over FISH, aCGH has the ability to screen not only translocated chromosomes, but also the ones that are not involved in the rearrangements. The high rates of aneuploidies in nontranslocated chromosomes have shown the importance of aneuploidy screening by the aCGH technique in both young and old female age groups.51 Furthermore, a possibility of an interchromosomal effect (ICE) that could be responsible for the increased incidence of aneuploidies in embryos belonging to Robertsonian translocation carriers has been proposed by the authors of a retrospective study in which FISH was used.52 More recently, Alfarawati et al53 reported ICE in Robertsonian translocation carriers, but not in reciprocal translocation carriers. In that study, in which the aCGH technique was used for comprehensive chromosomal assessments, the authors claimed that ICE is more pronounced in cleavage-stage embryos than in blastocysts; this difference might be attributed to mitotic instability.53 Regardless of whether ICE is responsible for aneuploidies, these studies show the benefit of CCS in the translocation carrier’s embryos.
Although aCGH is a widely used array-based detection technique, there are some pitfalls. This technique cannot detect polyploidies, such as triploidy and tetraploidy. Balanced chromosomal rearrangements, such as translocations, inversions, and uniparental disomies, cannot be detected because the total amount of DNA is the same as that of the control DNA. Also, this method cannot be used to diagnose SGDs.
The aCGH method has been applied successfully in all three embryonic developmental stages, including prefertilization and postfertilization (polar body), cleavage, and blastocyst.6,7,14,54 According to some studies in which implantation rates were examined as a primary outcome, preimplantation genetic screening with aCGH following day 3 blastomere biopsy markedly improved IVF outcomes while lowering multiple pregnancy rates and miscarriages.54,55 Since blastocyst-stage transfers are being used more often by clinics, the risk of multiple gestations and the selection of the best embryo that has the highest implantation capability has gained more importance. In order to determine whether performing CCS on blastocysts can reduce the risk of multiple gestation, Yang et al14 conducted a randomized controlled study. The researchers assessed clinical outcomes after single-blastocyst transfers by comparing morphological assessment alone and in combination with aCGH on good-prognosis patients. According to their results, blastocyst transfer after aCGH testing significantly increased ongoing pregnancy rates in the aCGH group (P=0.009).
RTqPCR
In 2012, Treff et al56,57 developed a RTqPCR-based methodology that provides simultaneous assessment of 24 chromosomes in less than 4 hours. This technique was based on multiplex amplification of 96 loci (four for each chromosome) and was performed with the use of a commercial assay and master mixes.56,57 This new 4-hour method was validated with a consistency of 100% both in cell lines with previously well-characterized karyotypes and in discarded human embryos, with a consistency of 100%.57 The authors demonstrated that this method is superior because it has the required accuracy and it is a fast method that may provide both same-day trophectoderm biopsy coupled with 24-chromosome aneuploidy screening and fresh blastocyst-stage transfers. With a similar strategy of using CCS with qPCR, Forman et al15 performed a controlled trial in which they compared the clinical outcomes of the transfer of a single euploid blastocyst with the transfers of two untested blastocysts. According to their results, they found similar ongoing pregnancy rates (60.7% after the single euploid blastocyst transfer versus 65.1% after the two untested blastocyst transfers), but significantly lower rates of twins in the single euploid transfer group (0% versus 53.4%). Although this method has a reduced resolution compared to aCGH and SNP-array systems, it has been demonstrated that this technique was successful in diagnosing unbalanced products derived from embryos that belonged to three rearrangement carriers whose karyotypes were unknown before the initiation of experiments.58
Other techniques
Newer platforms such as SNP arrays and karyomapping techniques are showing promise for more detailed chromosomal analysis than previous array-based techniques.11,17,59–61 In general, the system relies on the detection of variations in the number of copies and of loss of heterozygosity. SNPs among alleles are used for heterozygosity information that then can be used to identify the copy numbers of each chromosome or chromosomal region. With this platform, aneuploidies, segmental imbalances, recombinations, uniparental disomies, single-nucleotide mutations, and HLA compatibility could be detected.41 While some platforms use only copy-number variation information,60 some others use an algorithm called parental support,62 which explicitly computes the confidence in each copy-number call and is able to determine the parental origin of the detected abnormalities. The disadvantages of techniques based on parental support are the length, the cost, and the complexity of the protocol, all of which make these techniques less applicable in clinical laboratories.59 Although some successful research studies and randomized controlled trials have been conducted,11,17,60,61,63,64 which have already revolutionized the knowledge about the origins and mechanisms of aneuploidy in both cleavage-stage and blastocyst-stage embryos, this technique still needs improvements on applicability, and the costs still need to be reduced for routine use.
Recently, the NGS technique has been applied in a clinical setting.65 In this study, samples from 68 cycles were biopsied at cleavage stage and were analyzed both by aCGH and by the NGS technique. In this study, the same WGA samples were tested by both techniques; NGS showed 100% sensitivity and consistency with aCGH. This finding demonstrates that a robust, high-throughput technology may replace array-based techniques in the near future because of its cost effectiveness. The NGS technique involves library preparation, bar coding, sequencing, and analysis. It has numerous advantages, such as allowing the opportunity to evaluate multiple samples on the same sequencing chip with the use of a bar-coding system. This reduces the time and cost. Moreover, it gives the opportunity to evaluate more than one type of condition; SGDs, HLA compatibility, translocations, aneuploidy, and mitochondrial mutations can be analyzed at the same time. This breakthrough may start a new era in the scientific history of reproductive genetics.
Recent advances in mutation testing
Since its first application in 1990, PGD for SGD has become an effective alternative to prenatal diagnosis.1 Moreover, PGD is an excellent option for families who have an affected child who is in need of bone marrow transplantation. Preimplantation HLA typing can facilitate the birth of an HLA-matched donor infant,66,67 and hematopoietic progenitor cells from this donor could then be transplanted into the affected child with a successful cure as a result.68
PGD by multiplex PCR method
Currently, the most common method of PGD of SGD involves the use of a single-cell multiplex PCR for the amplification of short tandem repeat (STR) polymorphic markers that are located in close proximity to the mutation site. Those polymorphic STR markers are repeats of DNA that are mostly heterozygous and whose sizes vary greatly among individuals. STR length values that are linked to the mutated allele can be determined via fragment analysis using paternal and maternal genomic DNA prior to PGD, and the genotypes of the generated embryos can be diagnosed by linkage analysis during PGD (Figures 8 and 9). In addition to direct mutation techniques via restriction length polymorphism or minisequencing, the use of multiple STR markers that are linked to the mutation site is useful to overcome the diagnostic problems of ADO, which is the amplification failure of one of two alleles in a locus and is one of the major causes of misdiagnosis during PGD.4
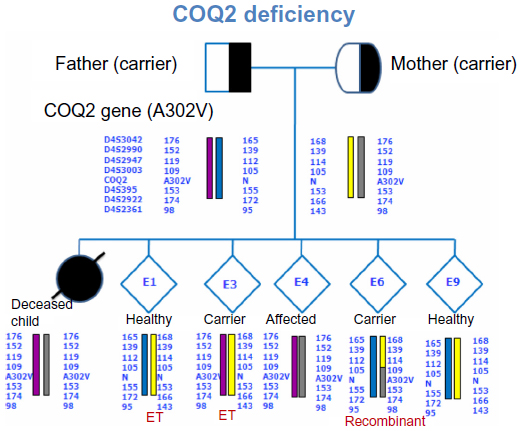 | Figure 8 Actual case sample of linked STR marker analysis for the COQ2 gene. |
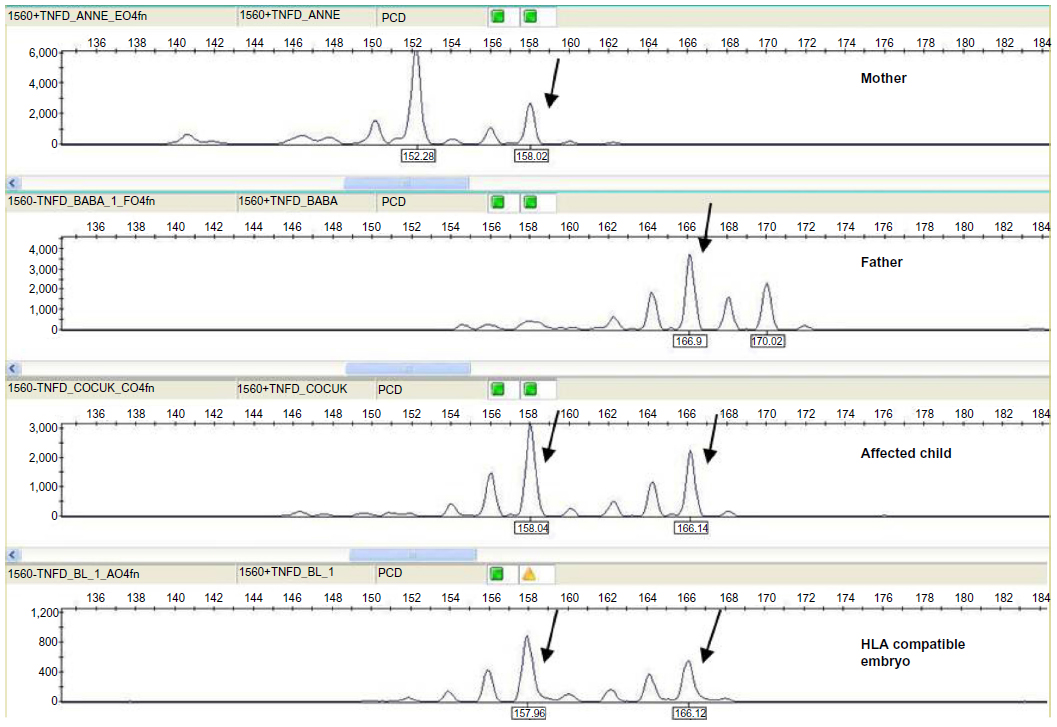 | Figure 9 An informative STR analysis in the HLA class II region in order to determine HLA compatibility of the resulting embryo. |
WGA (Whole genome amplification)
The major disadvantages of PGD by multiplex PCR are the amount of time and the amount of labor required to optimize the process of the PCR protocol so that the interaction of the PCR primers does not result in the failure of the PCR reaction. An alternative approach to overcome those problems is the amplification of the single-cell genome by WGA.
Some of the WGA protocols are PCR-based, such as primer extension PCR (PEP) and degenerated oligonoucleotide primed PCR (DOP-PCR). PEP was initially used on single sperms,69 and the protocol was adapted for the amplification of single blastomeres in PGD.70 Incomplete genome coverage and the amplification bias between the genomic loci of the amplified products were main disadvantages of PEP. In DOP-PCR, a partially degenerate primer that binds at many sites throughout the genome, is used during several low-temperature annealing cycles. More specific priming at the fragments will be generated by the increased annealing temperature. DOP-PCR is mainly used for aCGH applications.
MDA71,72 is a non-PCR, isothermal method for DNA amplification. MDA, during its early use to amplify single blastomeres for PGD,71,73 was a long method (over 16 hours), and the ADO rates of the amplification products were as high as 34%.
The preimplantation haplotyping approach was developed as an alternative to the previous methods, which had relied completely on PCR. Renwick et al introduced the single-cell haplotyping approach by using WGA by MDA.74 In this technique, DNA from single cells was amplified using MDA; the resulting products were then tested by using disease-specific PCR multiplexes applied under standard laboratory conditions to determine the haplotypes in the embryo. With this method, 12 different monogenic disorders with 38 different mutations were diagnosed successfully; these results demonstrate that preimplantation genetic haplotyping provides a robust, efficient, and successful alternative to single-cell PCR for monogenic diseases.75 Despite the high ADO rates (average 27%) in this study, the method did not cause any problems with diagnosis; the percentage of embryos with no diagnosis due to ADO or recombination was 0.82%. Later, MDA was modified to give results in a 4-hour period with approximately 10% ADO rates, and it was used for the detection of monogenic disorders.72 The MDA method was also successfully used for array-based preimplantation testing.76
SNP arrays
An SNP is a variation at a single position (single DNA base pair) in a DNA sequence. An SNP microarray contains immobilized DNA sequences (Figure 10). It detects SNPs within a genome at a high resolution; this detection enables karyomapping, a process in which the SNP genotype of a person is determined. For SGDs, the SNP genotypes of the parents and a reference (an affected child or another affected relative) can be analyzed at a gene of interest, and linkage information can be used to select unaffected embryos during PGD.41,77
The main advantage of karyomapping over the traditional, targeted approach is that karyomapping is applicable to any inherited SGD within the informative SNP loci without the development of costly, time-consuming, and laborious patient- or disease-specific designs. In addition, SNP data enable the detection of chromosomal abnormalities (meiotic trisomies, monosomies, and deletions);77 the elimination of those embryos may improve implantation rates and lower miscarriage rates. Karyomapping data can also be used for HLA typing via the use of SNP data over the HLA region located on chromosome 6.41
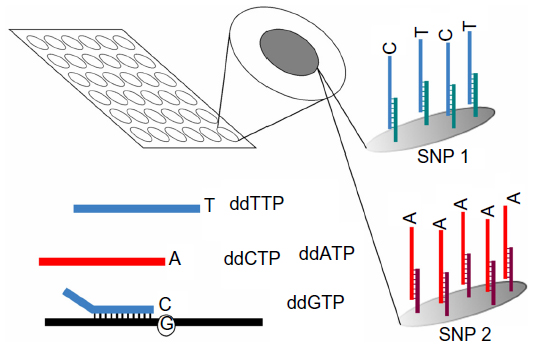 | Figure 10 The working principle of SNP arrays. |
However, the major disadvantage in karyomapping is that it does not include a mutation detection method; therefore, a reference (an affected child or another affected relative) is always needed to establish linkage information, which is not always available for every couple. Similarly, it is not possible to apply karyomapping to de novo mutation cases without additional work with direct mutation testing.
Since array technology is used in the karyomapping technique, the cost of the test per embryo is currently higher than that of the traditional multiplex PCR technique. But it should be noted that the preclinical workup cost, which is necessary for the existing STR method, is not required in karyomapping; this saving may result in the reduction of costs overall.
NGS
Although recent advances in NGS have created opportunities for possible PGD applications (Figure 11), the experience with NGS on single cells is limited. NGS has been performed successfully on single tumor cells to quantify genomic copy number78 and on sperm cells to determine recombination hot spots.79 Recently, NGS was performed on trophectoderm cells80 and on blastomeres65 for the detection of aneuploidies and of unbalanced chromosomal rearrangements; the results of these experiments were successful.
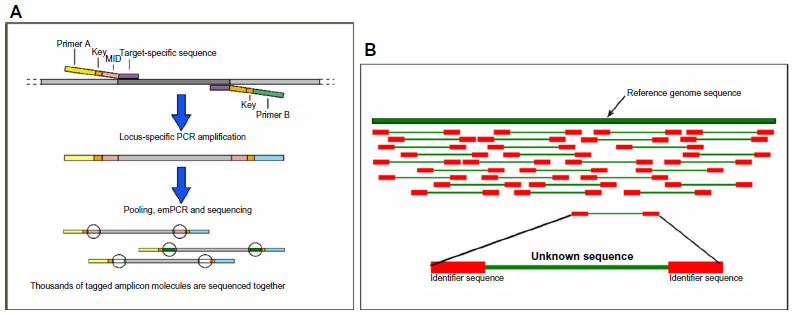 | Figure 11 Next-generation sequencing and preimplantation genetic diagnosis. |
The sensitivity and specificity of this method depends on the sequencing depth and the coverage of the regions of interest. Complex bioinformatic analyses are necessary to provide a sequence analysis since there is a large quantity of data obtained through massive parallel sequencing. First applications of the NGS technique on PGD for monogenic disorders81 involved a targeted, semiconductor technology-based NGS method, which was used with a bar-coding protocol that gave results in less than 24 hours. Genotype results from the NGS method were consistent with the results of the conventional STR method. However, further studies are necessary to establish the accuracy and reliability of this technique, as well as its applicability for single blastomeres.
Conclusion
Although noninvasive techniques, such as time-lapse analysis and noninvasive assessments of embryos and oocytes, are being developed in order to generate an alternative to PGD, invasive diagnostic techniques still remain the most reliable methods to eliminate affected and chromosomally abnormal embryos. In the last couple of decades, preventive medicine has gained more attention worldwide. This, together with the implementation of more powerful, comprehensive, and cost-effective techniques, such as NGS, will strengthen the place of PGD in ART and increase the demand for PGD.
Currently, the choice of technique depends mostly on the indication (whether the purpose is either mutation testing or chromosomal analysis) and on the cost, the availability, and the applicability of the technique. As the list of conditions and indications for PGD testing is continuing to extend enormously, the techniques have been evolving toward a universal method for the simultaneous diagnosis of multiple types of genetic conditions such as monogenic disorders, HLA typing, rearrangements, aneuploidy screening, and the selection of the best embryo that has the highest implantation capacity. The development of such a method will be possible in the near future.
Disclosure
The authors report no conflicts of interest in this work.
References
Handyside AH, Kontogianni EH, Hardy K, Winston RM. Pregnancies from biopsied human preimplantation embryos sexed by Y-specific DNA amplification. Nature. 1990;344(6268):768–770. | |
Harton GL, Magli MC, Lundin K, Montag M, Lemmen J, Harper JC. European Society for Human Reproduction and Embryology (ESHRE) PGD Consortium/Embryology Special Interest Group. ESHRE PGD Consortium/Embryology Special Interest Group - best practice guidelines for polar body and embryo biopsy for preimplantation genetic diagnosis/screening (PGD/PGS). Hum Reprod. 2011 Jan;26(1):41–46. | |
Preimplantation Genetic Diagnosis International Society (PGDIS). Guidelines for good practice in PGD: programme requirements and laboratory quality assurance. Reprod Biomed Online. 2008; 16(1):134–147. | |
Harper JC, Wilton L, Traeger-Synodinos J, et al. The ESHRE PGD Consortium: 10 years of data collection. Hum Reprod Update. 2008; 18(3):234–247. | |
Scott KL, Hong KH, Scott RT. Selecting the optimal time to perform biopsy for preimplantation genetic testing. Fertil Steril. 2013;100(3):608–614. | |
Christopikou D, Tsorva E, Economou K, et al. Polar body analysis by array comparative genomic hybridization accurately predicts aneuploidies of maternal meiotic origin in cleavage stage embryos of women of advanced maternal age. Hum Reprod. 2013;28(5):1426–1434. | |
Geraedts J, Montag M, Magli MC, et al. Polar body array CGH for prediction of the status of the corresponding oocyte. Part I: clinical results. Hum Reprod. 2011;26(11):3173–3180. | |
Capalbo A, Bono S, Spizzichino L, et al. Sequential comprehensive chromosome analysis on polar bodies, blastomeres and trophoblast: insights into female meiotic errors and chromosomal segregation in the preimplantation window of embryo development. Hum Reprod. 2013; 28(2):509–518. | |
Xu K, Montag M. New perspectives on embryo biopsy: not how, but when and why? Semin Reprod Med. 2012;30(4):259–266. | |
Grifo J, Talebian S, Keegan D, Krey L, Adler A, Berkeley A. Ten-year experience with preimplantation genetic diagnosis (PGD) at the New York University School of Medicine Fertility Center. Fertil Steril. 2007;88(4):978–981. | |
Scott RT Jr, Upham KM, Forman EJ, Zhao T, Treff NR. Cleavage-stage biopsy significantly impairs human embryonic implantation potential while blastocyst biopsy does not: a randomized and paired clinical trial. Fertil Steril. 2013;100(3):624–630. | |
Taylor TH, Gitlin SA, Patrick JL, Crain JL, Wilson JM, Griffin DK. The origin, mechanisms, incidence and clinical consequences of chromosomal mosaicism in humans. Hum Reprod Update. 2014;20(4):571–581. | |
Forman EJ, Ferry KM, Gueye NA, Smith RD, Stevens J, Scott RT Jr. Trophectoderm biopsy for single-gene disorder preimplantation genetic diagnosis (PGD) is significantly more reliable than day 3 blastomere biopsy. Fertil Steril. 2011;96(3):S222. | |
Yang Z, Liu J, Collins GS, et al. Selection of single blastocysts for fresh transfer via standard morphology assessment alone and with array CGH for good prognosis IVF patients: results from a randomized pilot study. Mol Cytogenet. 2012;5(1):24. | |
Forman EJ, Hong KH, Ferry KM, et al. In vitro fertilization with single euploid blastocyst transfer: a randomized controlled trial. Fertil Steril. 2013;100(1):100–107. e1. | |
Scott RT, Upham KM, Forman EJ, et al. Blastocyst biopsy with comprehensive chromosome screening and fresh embryo transfer significantly increases in vitro fertilization implantation and delivery rates: a randomized controlled trial. Fertil Steril. 2013;100(3):697–703. | |
Northrop LE, Treff NR, Levy B, Scott RT. SNP microarray-based 24 chromosome aneuploidy screening demonstrates that cleavage-stage FISH poorly predicts aneuploidy in embryos that develop to morphologically normal blastocysts. Mol Hum Reprod. 2010;16(8):590–600. | |
Capalbo A, Wright G, Elliott T, Ubaldi FM, Rienzi L, Nagy ZP. FISH reanalysis of inner cell mass and trophectoderm samples of previously array-CGH screened blastocysts shows high accuracy of diagnosis and no major diagnostic impact of mosaicism at the blastocyst stage. Hum Reprod. 2013;28(8):2298–2307. | |
Johnson DS, Cinnioglu C, Ross R, et al. Comprehensive analysis of karyotypic mosaicism between trophectoderm and inner cell mass. Mol Hum Reprod. 2010;16(12):944–949. | |
Fragouli E, Lenzi M, Ross R, Katz-Jaffe M, Schoolcraft WB, Wells D. Comprehensive molecular cytogenetic analysis of the human blastocyst stage. Hum Reprod. 2008;23(11):2596–2608. | |
Alpha Scientists in Reproductive Medicine and ESHRE Special Interest Group of Embryology. The Istanbul consensus workshop on embryo assessment: proceedings of an expert meeting. Hum Reprod. 2011;26(6):1270–1283. | |
Rubio I, Kuhlmann R, Agerholm I, et al. Limited implantation success of direct-cleaved human zygotes: a time-lapse study. Fertil Steril. 2012;98(6):1458–1463. | |
Campbell A, Fishel S, Bowman N, Duffy S, Sedler M, Hickman CF. Modelling a risk classification of aneuploidy in human embryos using non-invasive morphokinetics. Reprod Biomed Online. 2013;26(5):477–485. | |
Montag M. Morphokinetics and embryo aneuploidy: has time come or not yet? Reprod Biomed Online. 2013;26(6):528–530. | |
Basile N, Morbeck D, García-Velasco J, Bronet F, Meseguer M. Type of culture media does not affect embryo kinetics: a time-lapse analysis of sibling oocytes. Hum Reprod. 2013;28(3):634–641. | |
Swain JE. Could time-lapse embryo imaging reduce the need for biopsy and PGS? J Assist Reprod Genet. 2013;30(8):1081–1090. | |
Munné S, Bahçe M, Sandalinas M, et al. Differences in chromosome susceptibility to aneuploidy and survival to first trimester. Reprod Biomed Online. 2004;8(1):81–90. | |
Benkhalifa M, Kasakyan S, Clement P, et al. Array comparative genomic hybridization profiling of first-trimester spontaneous abortions that fail to grow in vitro. Prenat Diagn. 2005;25(10):894–900. | |
Munné S, Weier HU, Stein J, Grifo J, Cohen J. A fast and efficient method for simultaneous X and Y in situ hybridization of human blastomeres. J Assist Reprod Genet. 1993;10(1):82–90. | |
Munné S, Lee A, Rosenwaks Z, Grifo J, Cohen J. Diagnosis of major chromosome aneuploidies in human preimplantation embryos. Hum Reprod. 1993;8(12):2185–2191. | |
Verlinsky Y, Cieslak J, Freidine M, et al. Polar body diagnosis of common aneuploidies by FISH. J Assist Reprod Genet. 1996;13(2):157–162. | |
Kuliev A, Cieslak J, Ilkevitch Y, Verlinsky Y. Chromosomal abnormalities in a series of 6,733 human oocytes in preimplantation diagnosis for age-related aneuploidies. Reprod Biomed Online. 2003;6(1):54–59. | |
McArthur SJ, Leigh D, Marshall JT, de Boer KA, Jansen RP. Pregnancies and live births after trophectoderm biopsy and preimplantation genetic testing of human blastocysts. Fertil Steril. 2005;84(6):1628–1636. | |
Munné S, Gianaroli L, Tur-Kaspa I, et al. Substandard application of preimplantation genetic screening may interfere with its clinical success. Fertil Steril. 2007;88(4):781–784. | |
Munné S, Fragoulie E, Colls P, Katz-Jaffe M, Schoolcraft W, Wells D. Improved detection of aneuploid blastocysts using a new 12- chromosome FISH test. Reprod Biomed Online. 2010;20(1):92–97. | |
Twisk M, Mastenbroek S, Hoek A, et al. No beneficial effect of preimplantation genetic screening in women of advanced maternal age with a high risk for embryonic aneuploidy. Hum Reprod. 2008;23(12):2813–2817. | |
Mastenbroek S, Twisk M, van der Veen F, Repping S. Preimplantation genetic screening: a systematic review and meta-analysis of RCTs. Hum Reprod Update. 2011;17(4):454–466. | |
Gutiérrez-Mateo C, Colls P, Sánchez-García J, et al. Validation of microarray comparative genomic hybridization for comprehensive chromosome analysis of embryos. Fertil Steril. 2011;95(3):953–958. | |
Colls P, Escudero T, Fischer J, et al. Validation of array comparative genome hybridization for diagnosis of translocations in preimplantation human embryos. Reprod Biomed Online. 2012;24(6):621–629. | |
Fiorentino F, Biricik A, Bono S, et al. Development and validation of a next-generation sequencing-based protocol for 24-chromosome aneuploidy screening of embryos. Fertil Steril. 2014;101(5):1375–1382. | |
Natesan SA, Bladon AJ, Coskun S, et al. Genome-wide karyomapping accurately identifies the inheritance of single-gene defects in human preimplantation embryos in vitro. Genet Med. Epub May 8, 2014. | |
Treff NR, Scott RT. Four-hour quantitative real-time polymerase chain reaction-based comprehensive chromosome screening and accumulating evidence of accuracy, safety, predictive value, and clinical efficacy. Fertil Steril. 2013;99(4):1049–1053. | |
Kallioniemi A, Kallioniemi OP, Sudar D, et al. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science. 1992;258(5083):818–821. | |
Kallioniemi A, Visakorpi T, Karhu R, Pinkel D, Kallioniemi OP. Gene copy number analysis by fluorescence in situ hybridization and comparative genomic hybridization. Methods. 1996;9(1):113–121. | |
Wilton L. Preimplantation genetic diagnosis and chromosome analysis of blastomeres using comparative genomic hybridization. Hum Reprod Update. 2005;11(1):33–41. | |
Wells D, Escudero T, Levy B, Hirschhorn K, Delhanty JD, Munné S. First clinical application of comparative genomic hybridization and polar body testing for preimplantation genetic diagnosis of aneuploidy. Fertil Steril. 2002;78(3):543–549. | |
Le Caignec C, Spits C, Sermon K, et al. Single-cell chromosomal imbalances detection by array CGH. Nucleic Acids Res. 2006;34(9):e68. | |
Fiorentino F, Spizzichino L, Bono S, et al. PGD for reciprocal and Robertsonian translocations using array comparative genomic hybridization. Hum Reprod. 2011;26(7):1925–1935. | |
Viaggi CD, Cavani S, Pierluigi M, et al. Characterization of a complex rearrangement involving chromosomes 1, 4 and 8 by FISH and array-CGH. J Appl Genet. 2012;53(3):285–288. | |
Alfarawati S, Fragouli E, Colls P, Wells D. First births after preimplantation genetic diagnosis of structural chromosome abnormalities using comparative genomic hybridization and microarray analysis. Hum Reprod. 2011;26(6):1560–1574. | |
Beyazyurek C, Yapan C, Ekmekci CG, et al. O3 aneuploidy screening reveals high incidence of abnormalities of chromosomes which are not involved in the rearrangements. Reproductive BioMedicine Online. 2012;24 Suppl 2:S40. | |
Gianaroli L, Magli MC, Ferraretti AP, et al. Possible interchromosomal effect in embryos generated by gametes from translocation carriers. Hum Reprod. 2002;17(12):3201–3207. | |
Alfarawati S, Fragouli E, Colls P, Wells D. Embryos of robertsonian translocation carriers exhibit a mitotic interchromosomal effect that enhances genetic instability during early development. PLoS Genet. 2012;8(10):e1003025. | |
Keltz MD, Vega M, Sirota I, et al. Preimplantation genetic screening (PGS) with Comparative genomic hybridization (CGH) following day 3 single cell blastomere biopsy markedly improves IVF outcomes while lowering multiple pregnancies and miscarriages. J Assist Reprod Genet. 2013;30(10):1333–1339. | |
Rodrigo L, Mateu E, Mercader A, et al. New tools for embryo selection: comprehensive chromosome screening by array comparative genomic hybridization. Biomed Res Int. 2014;2014:517125. | |
Treff NR, Tao X, Su J, et al. Tracking embryo implantation using cell-free fetal DNA enriched from maternal circulation at 9 weeks gestation. Mol Hum Reprod. 2011;17(7):434–438. | |
Treff NR, Tao X, Ferry KM, Su J, Taylor D, Scott RT. Development and validation of an accurate quantitative real-time polymerase chain reaction-based assay for human blastocyst comprehensive chromosomal aneuploidy screening. Fertil Steril. 2012;97(4):819–824. | |
Treff NR, Forman EJ, Katz-Jaffe MG, Schoolcraft WB, Levy B, Scott RT. Incidental identification of balanced translocation carrier patients through comprehensive chromosome screening of IVF-derived blastocysts. J Assist Reprod Genet. 2013;30(6):787–791. | |
Handyside AH. PGD and aneuploidy screening for 24 chromosomes by genome-wide SNP analysis: seeing the wood and the trees. Reprod Biomed Online. 2011;23(6):686–691. | |
Treff NR, Su J, Tao X, Levy B, Scott RT. Accurate single cell 24 chromosome aneuploidy screening using whole genome amplification and single nucleotide polymorphism microarrays. Fertil Steril. 2010;94(6):2017–2021. | |
Scott RT Jr, Ferry K, Su J, Tao X, Scott K, Treff NR. Comprehensive chromosome screening is highly predictive of the reproductive potential of human embryos: a prospective, blinded, nonselection study. Fertil Steril. 2012;97(4):870–875. | |
Johnson DS, Gemelos G, Baner J, et al. Preclinical validation of a microarray method for full molecular karyotyping of blastomeres in a 24-h protocol. Hum Reprod. 2010;25(4):1066–1075. | |
Ling J, Zhuang G, Tazon-Vega B, et al. Evaluation of genome coverage and fidelity of multiple displacement amplification from single cells by SNP array. Mol Hum Reprod. 2009;15(11):739–747. | |
Rabinowitz M, Ryan A, Gemelos G, et al. Origins and rates of aneuploidy in human blastomeres. Fertil Steril. 2012;97(2):395–401. | |
Fiorentino F, Biricik A, Bono S, et al. Development and validation of a next-generation sequencing-based protocol for 24-chromosome aneuploidy screening of embryos. Fertil Steril. 2014;101(5):1375–1382. | |
Verlinsky Y, Rechitsky S, Schoolcraft W, Strom C, Kuliev A. Preimplantation diagnosis for Fanconi anemia combined with HLA matching. JAMA. 2001;285(24):3130–3133. | |
Kahraman S, Beyazyurek C, Ekmekci CG. Seven years of experience of preimplantation HLA typing: a clinical overview of 327 cycles. Reprod Biomed Online. 2011;23(3):363–371. | |
Kahraman S, Beyazyurek C, Yesilipek MA, et al. Successful haematopoietic stem cell transplantation in 44 children from healthy siblings conceived after preimplantation HLA matching. Reprod Biomed Online. 2014;29(3):340–351. | |
Zhang L, Cui X, Schmitt K, Hubert R, Navidi W, Amheim N. Whole genome amplification from a single cell: implications for genetic analysis. Proc Natl Acad Sci U S A. 1992;89(13):5847–5851. | |
Sermon K, Lissens W, Joris H, Van Steirteghem A, Liebaers I. Adaptation of the primer extension preamplification (PEP) reaction for preimplantation diagnosis: single blastomere analysis using short PEP protocols. Mol Hum Reprod. 1996;2(3):209–212. | |
Handyside AH, Robinson MD, Simpson RJ, et al. Isothermal whole genome amplification from single and small numbers of cells: a new era for preimplantation genetic diagnosis of inherited disease. Mol Hum Reprod. 2004;10(10):767–772. | |
Lau EC, Janson MM, Roesler MR, Avner ED, Strawn EY, Bick DP. Birth of a healthy infant following preimplantation PKHD1 haplotyping for autosomal recessive polycystic kidney disease using multiple displacement amplification. J Assist Reprod Genet. 2010;27(7):397–407. | |
Hellani A, Coskun S, Benkhalifa M, et al. Multiple displacement amplification on single cell and possible PGD applications. Mol Hum Reprod. 2004;10(11):847–852. | |
Renwick PJ, Trussler J, Ostad-Saffari E, et al. Proof of principle and first cases using preimplantation genetic haplotyping – a paradigm shift for embryo diagnosis. Reprod Biomed Online. 2006;13(1):110–119. | |
Renwick P, Trussler J, Lashwood A, Braude P, Ogilvie CM. Preimplantation genetic haplotyping: 127 diagnostic cycles demonstrating a robust, efficient alternative to direct mutation testing on single cells. Reprod Biomed Online. 2010;20(4):470–476. | |
Hellani A, Abu-Amero K, Azouri J, El-Akoum S. Successful pregnancies after application of array-comparative genomic hybridization in PGS-aneuploidy screening. Reprod Biomed Online. 2008;17(6):841–847. | |
Handyside AH, Harton GL, Mariani B, et al. Karyomapping: a universal method for genome wide analysis of genetic disease based on mapping crossovers between parental haplotypes. J Med Genet. 2010;47(10):651–658. | |
Navin N, Kendall J, Troge J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472(7341):90–94. | |
Wang J, Fan HC, Behr B, Quake SR. Genome-wide single-cell analysis of recombination activity and de novo mutation rates in human sperm. Cell. 2012;150(2):402–412. | |
Yin X, Tan K, Vajta G, et al. Massively parallel sequencing for chromosomal abnormality testing in trophectoderm cells of human blastocysts. Biol Reprod. 2013;88(3):69. | |
Treff NR, Fedick A, Tao X, Devkota B, Taylor D, Scott RT. Evaluation of targeted next-generation sequencing-based preimplantation genetic diagnosis of monogenic disease. Fertil Steril. 2013;99(5):1377–1384. e6. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.