Back to Journals » International Journal of Nanomedicine » Volume 21
Recent Advances in Mitochondria-Targeted Nano-Drug Delivery Systems for Cancer Therapy
Authors Huang L, Lei Y ![]() , Zheng P, Chen Q, Wang C, Ma J
, Zheng P, Chen Q, Wang C, Ma J
Received 14 September 2025
Accepted for publication 10 January 2026
Published 2 February 2026 Volume 2026:21 567593
DOI https://doi.org/10.2147/IJN.S567593
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Kamakhya Misra
Li Huang,1,* Yujing Lei,2,* Piao Zheng,3 Qingshan Chen,4 Chenyang Wang,1 Jie Ma5
1Pharmaceutical Preparation Center, The First Hospital of Hunan University of Chinese Medicine, Changsha, People’s Republic of China; 2College of Medicine and Medical Technology, Changsha Cultural Creative and Arts Vocational College, Changsha, People’s Republic of China; 3Department of Discipline Development and Research Management, The Second Hospital of Hunan University of Chinese Medicine, Changsha, People’s Republic of China; 4The Department of Hepatobiliary Pancreatic Hernia Surgery, The First Hospital of Hunan University of Chinese Medicine, Changsha, People’s Republic of China; 5Department of Traditional Chinese Medicine, Hunan Institute for Drug Control, Changsha, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Li Huang, Email [email protected]
Abstract: Cancer remains a major disease that poses a serious threat to human health. Conventional treatments such as radiotherapy, chemotherapy, and surgery are limited by systemic toxicity and tumor recurrence, which hinder the achievement of highly efficient and specific therapy. The development of nanodrug delivery systems has provided new opportunities for cancer treatment. Utilizing mechanisms such as passive targeting (eg, the EPR effect) and active targeting (eg, peptide-based surface modification), these systems can precisely deliver drugs to tumor sites, thereby significantly reducing systemic toxicity. In recent years, research focus has shifted from tissue-level targeting to subcellular organelle targeting, particularly of mitochondria. Functioning as cellular power plants, mitochondria are deeply involved in tumor initiation, progression, and the regulation of apoptosis, making them important targets for cancer therapy. Based on the structural features of mitochondria and their dysfunctional role in cancer, this review systematically explores strategies for mitochondria-targeted nanodrug delivery and summarizes the latest research advances along with future directions in cancer treatment. A unique aspect of this review is its systematic integration of the design principles from mitochondrial substructure characteristics to multi-level targeting strategies, underscoring the innovative potential of nanocarriers in overcoming tumor drug resistance and enabling precise intervention. This work thereby provides a theoretical basis and novel insights for precision oncology.
Keywords: nano-drug delivery carriers, mitochondrial targeting, tumor microenvironment, responsive nanocarriers
Introduction
As a significant threat to human health, cancer has long been a central focus and major challenge in medical research. According to the “Global Cancer Statistics 2022” published by the International Agency for Research on Cancer (IARC) of the World Health Organization, there were close to 20 million new cases of cancer in the year 2022 (including nonmelanoma skin cancers [NMSCs]) alongside 9.7 million deaths from cancer. With demographics-based predictions indicating that the number of new cases of cancer will reach 35 million by 2050.1 Conventional therapeutic modalities, including radiotherapy, chemotherapy, and surgery, are often compromised by dose-limiting systemic toxicity, drug resistance,2 and the risk of tumor recurrence.3 Therefore, there is an urgent need to develop treatment strategies that must not only target tumor cells precisely but also minimize damage to healthy cells.
Mitochondria, often termed the “powerhouse” of the cell, play a central role in tumor initiation and progression. They not only generate adenosine triphosphate (ATP) through oxidative phosphorylation, supplying the energy required for the uncontrolled proliferation of cancer cells, but also regulate key malignant behaviors such as metabolic reprogramming and resistance to apoptosis.4 For instance, the commonly observed “Warburg effect” in tumor cells—characterized by an abnormal reliance on glycolysis for energy production—is closely linked to dysfunctional mitochondrial respiratory chain activity.5 Furthermore, abnormalities such as altered mitochondrial membrane potential and impaired cytochrome c release can diminish the sensitivity of cancer cells to treatments, thereby enhancing their drug resistance.6 Consequently, targeting mitochondrial function to reverse tumor drug resistance has become a major focus of current research. However, existing drug delivery systems face significant challenges: they struggle to penetrate the double-membrane structure of mitochondria and lack the ability to intelligently respond to dynamic changes in the tumor microenvironment, which severely limits their therapeutic efficacy.7
The development of nanodrug delivery systems offers promising strategies to overcome the aforementioned challenges.8 Leveraging the enhanced permeability and retention (EPR) effect in tumor tissues, nanoparticles ranging from 50 to 500 nm can passively accumulate at the tumor site, while active targeting can be achieved through surface modifications—such as ligand conjugation or stimuli-responsive functionalization—thereby significantly enhancing drug enrichment.9 In recent years, research focus has increasingly shifted toward subcellular targeting, particularly mitochondrial-targeted delivery systems. For instance, nanocarriers modified with mitochondrial-targeting peptides (eg, TPP) can achieve precise delivery by leveraging the mitochondrial membrane potential gradient,10 while ROS-responsive carriers can trigger drug release in tumor microenvironments characterized by elevated reactive oxygen species levels.11 Nevertheless, optimizing the penetration efficiency of delivery carriers and enhancing the sensitivity of microenvironment-responsive systems remain critical challenges that require urgent attention.
Based on the relationship between mitochondrial structural characteristics, dysfunction, and tumorigenesis, this review systematically discusses the design strategies of mitochondria-targeted nanodrug delivery carriers and summarizes recent research advances and future prospects in the development of various nanocarriers for mitochondrial targeting in tumor therapy, aiming to provide a theoretical foundation and technical reference for precision cancer treatment.
Mitochondrial Structural Characteristics
Mitochondria are double-membrane-bound organelles whose unique structural features provide a fundamental biological basis for the targeted design of nanodrug delivery carriers (see Figure 1). The following section systematically describes the relationship between mitochondrial structure and targeting strategies across four distinct compartments—inner mitochondrial membrane (OMM), mitochondrial intermembrane space(IMS), the outer mitochondrial membrane (IMM), and mitochondrial matrix—integrated with recent research advances (see Table 1).
 |
Table 1 Key Targets of Each Mitochondrial Substructure |
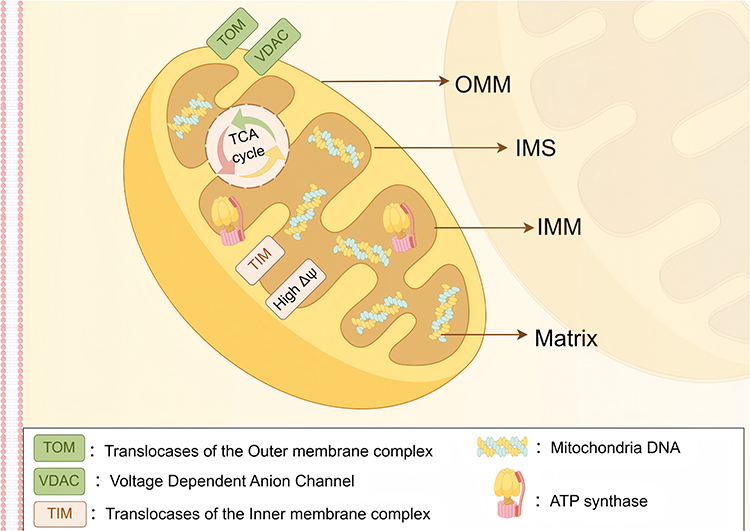 |
Figure 1 Schematic diagram of mitochondrial structure (By Figdraw). Depicted structures include: outer mitochondrial membrane (OMM), inner mitochondrial membrane (IMM), intermembrane space (IMS), and mitochondrial matrix. |
Outer Mitochondrial Membrane (OMM)
OMM is the outermost unit membrane of mitochondria, forming a continuous barrier that separates the organelle from the cytosolic matrix. Specific protein complexes and channels on the OMM can serve as “anchoring sites” for precise drug delivery. The OMM is composed of a phospholipid bilayer and integral proteins. Its surface is rich in voltage-dependent anion channels (VDAC), which adopt a β-barrel structure with a diameter of 2.5–3nm, allowing small molecules (such as ATP and NAD+) or peptides with a molecular weight below 5 kDa to directly translocate.23 In addition, the OMM surface exhibits a high density of negatively charged amino acids, such as glutamic acid and aspartic acid, which enhances its affinity for cationic carriers. This characteristic contributes to the OMM’s high permeability, facilitating the bidirectional exchange of metabolites between mitochondria and the cytosol and enabling efficient nanocarrier penetration. For example, gold nanoparticles with a diameter of approximately 3 nm have been demonstrated to traverse isolated cardiac mitochondria matrix through VDAC.24 Furthermore, the OMM harbors specific protein complexes, such as the TOM translocase complex, which consists of three receptor proteins (Tom20, Tom22, and Tom70), a channel-forming protein (Tom40), and several small Tom proteins. This complex mediates the import of nuclear-encoded mitochondrial precursor proteins into mitochondria, thus representing a promising target for nanocarrier-based therapeutic strategies.25
Mitochondrial Intermembrane Space (IMS)
IMS is the narrow compartment situated between the OMM and the IMM. Due to the high permeability of the OMM, the ion concentration and pH in the IMS are similar to those in the cytosolic matrix, rendering it a transient reservoir and transport zone for metabolites such as ATP/ADP and Ca2+, thereby functioning as a hub for material exchange between the inner and outer membranes. In addition, the IMS serves as a center for redox regulation and a platform for apoptosis signal initiation. On one hand, it harbors a diverse array of redox enzyme systems, including MIA40/ERV1,26 which facilitate the import of disulfide-containing proteins (eg, SAMC, COX19) and maintain an oxidative environment conducive to proper protein folding; On the other hand, IMS stores pro-apoptotic factors such as cytochromeC. Upon activation of apoptotic signals, these factors are released into the cytoplasm through channels in the OMM, thereby triggering the caspase cascade and inducing programmed cell death.27 Therefore, by leveraging the unique biochemical microenvironments of the IMS, surface ligands of nanodrug delivery carriers can be engineered to specifically bind to enzymes or proteins within the intermembrane space, enabling targeted recognition and precise mitochondrial localization.
Mitochondrial Inner Membrane (IMM)
IMM is situated within OMM and encloses the mitochondrial matrix. It serves as a central site for energy conversion and molecular transport in mitochondria and exhibits highly selective permeability. The structural characteristics and targeting strategies associated with the IMM are as follows: First, the IMM invaginates to form cristae, which significantly increase the surface area of the inner membrane and provide binding sites for key enzyme complexes—such as complexesI-V and ATP synthase—involved in the electron transport chain (ETC), thereby driving oxidative phosphorylation;19,20 Second, the high transmembrane potential (Δψ) provides a critical driving force for mitochondrial targeting. In normal cells, the mitochondrial membrane potential is approximately −157mV, whereas in tumor cells, it is frequently hyperpolarized, reaching values as low as −180mV. This electrochemical gradient facilitates the targeted delivery of nanocarriers. Positively charged nanocarriers, such as nanoparticles functionalized with triphenylphosphonium (TPP), can leverage electrostatic interactions and the membrane potential gradient to achieve selective mitochondrial accumulation;17 Third, cardiolipin is a signature phospholipid of the IMM, and its content and spatial distribution differ between tumor and normal cells, offering a basis for rational design of mitochondria-targeted therapeutics.18
Mitochondrial Matrix
The mitochondrial matrix, as the metabolic hub enclosed by the IMM, provides unique targets for precision cancer therapy due to its rich enzymatic systems and genetic material. It houses key enzymes of the tricarboxylic acid cycle (TCA), including succinate dehydrogenase (SDH) and isocitrate dehydrogenase (IDH), as well as enzymes involved in fatty acid oxidation, collectively regulating tumor energy metabolism. Targeted interventions addressing metabolic dysregulation have made notable progress. For example, studies have demonstrated that shRNA nanocarriers can reverse cisplatin resistance in lung adenocarcinoma by silencing RNA circ0515 and restoring the expression of succinate dehydrogenase subunit B (SDHB).22 Meanwhile, the clinically approved drug AG-120 (ivosidenib) inhibits mutant IDH1 and IDH2, thereby preventing the accumulation of the oncometabolite 2-hydroxyglutarate (2-HG), and has demonstrated significant therapeutic efficacy in the treatment of leukemia and glioma. Another critical target within the mitochondrial matrix is mitochondrial DNA (mtDNA), which, due to the lack of robust DNA repair mechanisms, exhibits a mutation rate exceeding 50% in tumors—particularly in genes such as ND1 and ND6—leading to impairment of the respiratory chain.28 Vitamin K3 induces apoptosis in colorectal cancer cells by competitively inhibiting DNA polymerase γ and effectively halting mitochondrial DNA (mtDNA) replication.21
The Relationship Between Mitochondrial Dysfunction and Tumors
Compared to normal cells, tumor cells exhibit distinct alterations in mitochondrial structure and function, including hyperpolarization of the mitochondrial membrane potential, impaired ATP-generating capacity, and abnormally elevated reactive oxygen species (ROS) production. These functional disparities establish a robust biological foundation for the preferential targeting of tumor cells by mitochondria-directed nanocarriers.
Mitochondrial Membrane Potential (MMP)
Mitochondrial membrane potential (ΔΨm) serves as a critical indicator of normal mitochondrial function, reflecting the electrochemical gradient across the inner mitochondrial membrane between the matrix and the IMS. Under physiological conditions, mitochondria transfer electrons to oxygen via the electron transport chain (ETC) while actively pumping protons from the matrix into the IMS, thereby establishing a proton electrochemical gradient that drives membrane potential generation. In tumor cells, however, respiratory chain dysfunction and glycolytic dominance lead to significant hyperpolarization, with ΔΨm values ranging from −180 to −220mV.29 This hyperpolarization exerts a dual role: on one hand, it promotes tumor proliferation by enhancing nutrient uptake and suppresses apoptosis through inhibition of cytochrome c release; on the other hand, the abnormally elevated negative membrane potential presents a promising targeting opportunity for lipophilic cationic nanocarriers.30 For instance, triphenylphosphonium (TPP+)-modified carriers can selectively accumulate in tumor mitochondria via electrostatic interactions, induce membrane depolarization, and disrupt cellular respiration by inhibiting complexes I and IV of the respiratory chain.31
Mitochondrial pH
Mitochondrial pH is a critical determinant of energy metabolic homeostasis. In normal cells, the extracellular pH (pHe=7.3–7.4) is higher than the intracellular pH (pHi=7.0–7.2). This physiological pH gradient facilitates essential mitochondrial enzymatic reactions, particularly those mediated by key enzymes in the tricarboxylic acid cycle (TCA cycle) and oxidative phosphorylation(OXPHOS), such as citrate synthase and succinate dehydrogenase. In contrast, tumor cells exhibit an inverted pH profile, with pHe ranging from 6.2 to 6.9—lower than pHi (7.12–7.65). This reversal results from the heightened energy demands of rapid tumor proliferation, which drives a metabolic shift from aerobic respiration to glycolysis. This shift leads to the excessive production of lactate, which is actively exported into the extracellular space, thereby contributing to the acidic tumor microenvironment.32 Notably, the mitochondrial matrix maintains a relatively alkaline pH (7.5–8.2), a condition directly maintained by the active translocation of protons from the matrix to the IMS during ATP synthesis.33 Consequently, the development of nanocarriers responsive to mitochondrial pH gradients presents a promising therapeutic strategy for cancer treatment. Such systems enable site-specific drug delivery and release within tumor mitochondria, enhancing therapeutic efficacy while minimizing off-target effects.34
Changes in Reactive Oxygen Species (ROS) Concentration
As the primary site of ROS generation in cells, mitochondria exhibit distinct alterations in ROS concentration dynamics during tumorigenesis.35 Under normal physiological conditions, electron leakage from mitochondrial respiratory chain complexes I and III generates low levels of ROS, with the superoxide anion (O2•−) rapidly converted to hydrogen peroxide (H2O2) by superoxide dismutase (SOD), thereby contributing to the maintenance of redox homeostasis.36 In tumor cells, mitochondrial dysfunction leads to significantly elevated ROS levels (approximately 0.1mM), compared to those in normal cells (approximately 20nM). This increase is primarily attributed to enhanced electron leakage from the electron transport chain(ETC) under hypoxic conditions, coupled with reduced activities of key antioxidant enzymes such as glutathione peroxidase 4 (GPX4) and catalase(CAT), resulting in impaired ROS clearance.37,38 The intracellular ROS concentration exerts a dual role in cancer biology: at moderate levels, ROS promote tumor progression by enhancing proliferative signaling pathways; however, at excessive concentrations, they induce mitochondrial DNA (mtDNA) mutations and lipid peroxidation, ultimately triggering ferroptosis.39 Therefore, precise modulation of ROS levels represents a promising therapeutic strategy for cancer treatment. By leveraging the heightened ROS environment within tumor mitochondria, ROS-responsive nanocarriers can be rationally engineered to enable spatially targeted therapeutic interventions.
Mitochondrial DNA (mtDNA) Mutations
As the unique circular double-stranded genetic material within mitochondria, mtDNA plays a pivotal role in tumor initiation and progression and represents a distinct therapeutic target. Compared to nuclear DNA (nDNA), mtDNA lacks histone protection and has limited DNA repair capacity, making it highly susceptible to oxidative stress and other genotoxic insults, thereby contributing to a significantly higher mutation rate.40 Furthermore, because the 13 proteins encoded by mtDNA constitute essential components of the mitochondrial respiratory chain complexes, mtDNA mutations directly compromise respiratory chain integrity, leading to impaired electron transport and reduced ATP production.41 To meet the increased bioenergetic demands of rapid proliferation, tumor cells are forced to enhance glycolytic flux. This mutation-driven metabolic reprogramming not only ensures sustained energy supply but also promotes the aggressive phenotypes of cancer cells, including enhanced proliferation, invasion, and metastatic capacity. As such, targeting mtDNA and its associated mutational landscape has become a focal point in cancer therapy development, with strategies including nucleic acid-based interference and small-molecule inhibitors showing significant promise.21,42
Mitochondrial Metabolic Pathway Changes
The homeostasis of mitochondrial metabolic pathways is essential for cellular energy production. In normal cells, the mitochondrion-dependent TCA cycle and OXPHOS represent the principal pathways for ATP generation, efficiently harnessing oxygen and nutrients to produce substantial energy output.In contrast, tumor cells exhibit profound metabolic reprogramming, characterized by a heightened dependence on glycolysis for ATP production even under aerobic conditions.43 This metabolic shift confers key survival and growth advantages to cancer cells: First, glycolysis proceeds at a significantly faster rate than OXPHOS, enabling rapid ATP production to meet the elevated energy demands of uncontrolled proliferation. Second, glycolytic intermediates—such as glucose-6-phosphate and fructose-6-phosphate—serve as vital precursors for the biosynthesis of nucleic acids, amino acids, lipids, and other macromolecules, thereby supporting the heightened anabolic activity required for accelerated cell growth and division. Third, lactate produced during glycolysis is actively exported from the cell, leading to acidification of the tumor microenvironment, which not only promotes tumor invasion and metastatic dissemination but also suppresses anti-tumor immune responses.44 Given the central role of glycolysis in both bioenergetics and biosynthetic metabolism in cancer, therapeutic strategies targeting key rate-limiting enzymes—including hexokinase (HK) and phosphofructokinase-1 (PFK-1)—have emerged as promising avenues for cancer intervention.45,46
The Strategy of Targeting Mitochondria with Nano-Drug Delivery Carrier
Mitochondria are the core hub of tumor energy metabolism, and their structural characteristics (OMM permeability, IMM high membrane potential, matrix enzyme system, etc.) and dysfunctions (membrane potential hyperpolarization, ROS accumulation, and mtDNA mutations, etc.) together constitute the biological basis for targeted therapy. Building upon the analysis of key targets within mitochondrial subcompartments presented in Mitochondrial Structural Characteristics and integrating insights from the discussion of mitochondrial dysfunction in tumors detailed in The Relationship Between Mitochondrial Dysfunction and Tumors, this section proposes a systematic and multilevel three-dimensional targeting strategy. First, passive targeting leverages precise modulation of nanocarrier physicochemical properties—such as particle size and surface charge—to exploit the enhanced permeability and retention (EPR) effect in tumor tissues and the mitochondrial membrane potential gradient, thereby enabling selective drug accumulation at the target site. Second, active targeting integrates ligand modifications (eg, TPP, MPP) with microenvironment-responsive mechanisms (including pH, ROS, and enzyme-triggered release) to achieve active recognition and overcome subcellular delivery barriers while facilitating precise organelle-specific targeting. Thirdly, a multimodal combination approach synergistically incorporates photodynamic therapy (PDT) with immunotherapy to significantly amplify mitochondrial damage and enhance overall therapeutic efficacy. These strategies driven by deep integration of interdisciplinary technologies promote a paradigm shift from “tissue-level enrichment” to “precise mitochondrial intervention,” offering a viable solution to address critical limitations of conventional chemotherapy such as drug resistance and systemic toxicity (see Figure 2 and Table 2).
 |
Table 2 Three Major Mitochondrial Targeting Strategies |
 |
Figure 2 Three mitochondrial targeting strategies (By Figdraw). |
Passive Targeting: Precise Regulation of Charge and Particle Size
Passive targeting is an important strategy for nanoparticle targeting of tumor mitochondria, which is mainly based on the physiological characteristics of tumor tissue and the physical properties of mitochondria. For example, the high negative potential of the IMM can attract positively charged nanoparticles, allowing them to be passively enriched in mitochondria to achieve a targeting effect.
Particle Size
Particle size is a key factor affecting the targeting of nanocarriers to mitochondria, and its mechanism of action mainly depends on the cellular uptake pathway and the mitochondrial outer membrane VDAC channel.59,60 Torrano’s team found that when the particle size was <50-100 nm, nanoparticles (such as 46 nm platinum-modified cerium oxide particles) could directly penetrate the cell membrane into the cytoplasm through a non-endocytic pathway and achieve up to 91% mitochondrial colocalization (Manders’ coefficient) within 10 minutes. Transmission electron microscopy confirmed that they were attached to the mitochondrial outer membrane. This rapid targeting relies on transient membrane pore formation induced by surface ultra-small Pt particles (2–5 nm) rather than a simple small-size diffusion effect. In contrast, particles with a particle size >150 nm, such as 143 nm/285 nm Pt-modified cerium oxide, could only enter cells by conventional endocytosis and remained in endocytic vesicles after 8 hours, with less than 6% mitochondrial colocalization. Notably, the particle size dependence of mitochondrial targeting efficiency requires a synergistic action of surface physical-chemical properties—platinum-free 47 nm Ceria particles also failed to target mitochondria.47 Therefore, optimizing the particle size to ~50 nm (in combination with penetration enhancers such as Pt nanoparticles) can maximize the mitochondrial targeting efficiency of nanocellular-based CeOs to achieve precise subcellular delivery.
Surface Charge
The surface charge of nanocarriers plays a crucial role in their targeting to mitochondria. The significantly negative environment of mitochondria makes positively charged nanocarriers more likely to interact with them through strong electrostatic attraction and achieve effective targeting. Mitchell et al demonstrated that the mitochondrial-targeting peptide SS-31 utilizes its positive surface charge to interact electrostatically with the negatively charged mitochondrial lipid bilayer, which is a key mechanism for its function.48 Similarly, Yang et al found that compared with negatively charged or neutral gold nanoparticles, positively charged gold nanoparticles can more effectively target tumor cell mitochondria and significantly enhance photodynamic therapy outcomes.49 However, surface charge not only affects targeting efficiency but also significantly regulates the circulation time of nanocarriers in vivo, cellular uptake efficiency, and biomolecular interactions. Zhang et al found that although positively charged nanocarriers are beneficial for mitochondrial targeting, they tend to non-specifically bind to a large number of negatively charged biomolecules (such as serum proteins and cell membrane proteoglycans) in vivo. This leads to rapid clearance by the immune system and shortens their circulation half-life in vivo.50 Therefore, designing efficient mitochondrial-targeting nanocarriers requires balancing in vivo stability with targeting ability.
Active Targeting: Dual Drive of Ligand Modification and Microenvironment Response
The active targeting mechanism takes advantage of the specific biological characteristics of tumor mitochondria, such as the unique pH, high levels of ROS, and special membrane potential, to design nanoparticles that can actively recognize and bind to tumor mitochondria for precise targeted delivery. This targeting method can significantly improve the enrichment efficiency of nanocarriers in tumor mitochondria, enhance the therapeutic effect of drugs, and reduce damage to normal tissues and cells.
Ligand Modification
Ligand modification is one of the important strategies to achieve mitochondrial targeting of nano-drug delivery carriers. By attaching ligands with specific recognition abilities to the surface of nanocarriers, these carriers can accurately target tumor mitochondria and improve the therapeutic effect of drugs. Common ligands for targeting mitochondria include triphenylphosphine (TPP) and mitochondrial-penetrating peptides (MPPs).
TPP is the most widely used mitochondrial targeting ligand. Its chemical structure is (C6H5)3P. The three groups are covalently linked to the central phosphorus atom, which gives it good lipid solubility and enables it to effectively penetrate the lipid bilayer of both the cell membrane and mitochondrial membrane. Due to the high negative membrane potential (Δψm) maintained by the inner mitochondrial membrane of tumors, the strong positive charge (P+) of TPP can specifically cross the mitochondrial membrane and accumulate in the mitochondrial matrix under electrostatic driving forces. This provides a new design concept for targeting mitochondria. Combining TPP+ molecules with anti-tumor drugs can create new agents with mitochondrial targeting functions. Song et al demonstrated the feasibility of this idea by using TPP+ as a carrier to deliver triptolide (TP) to mitochondria, significantly enhancing its anti-tumor activity while reducing systemic toxicity.61 Lu’s team developed and constructed a self-assembled nano-targeted drug delivery system LND-SS-Pt-TPP+/HA-CD. β-cyclodextrin-conjugated aqueous acid (HA-CD)-encapsulated prodrug nanoparticles can target CD44 on tumor surfaces and further deliver prodrugs to intracellular mitochondria through TPP to inhibit metabolic reprogramming, which is used for treating cisplatin-resistant lung cancer.52 In addition to using TPP ligand modification vectors alone, they can be combined with other methods to further enhance their anti-tumor effects.53,62 These studies collectively demonstrate that TPP-modified nanocapsules can overcome the limitations of conventional delivery systems and significantly increase effective drug concentrations in mitochondria.
MPPs are a class of mitochondria-targeting peptides with high efficiency in membrane penetration. The structure of MPPs is rich in cationic amino acids (eg, arginine, lysine) and hydrophobic residues (eg, phenylalanine). This structure endows MPPs with two key properties: on the one hand, the cations in the structure can be driven by electrostatic interactions for cellular uptake, which takes advantage of the high negative potential of the mitochondrial inner membrane to achieve specific targeting; on the other hand, hydrophobic residues facilitate their penetration through both the phospholipid bilayer and OMM and IMM.63 Kelly’s team reported for the first time a synthetic peptide that exhibits efficient cellular uptake and specific mitochondrial localization. The peptide sequence contains positively charged lysine and arginine; these cations promote charge-driven intracellular uptake, while D-arginine increases cellular stability. At the same time, this polypeptide sequence includes lipophilic phenylalanine, which helps MPPs better enter mitochondria through phospholipid bilayer structures and cross the hydrophobic IMM while achieving high cell permeability and mitochondrial localization.64,65 At an application level, Yang et al developed smart nanosystems functionalized with MPP: an MPP-modified N-(2-hydroxypropyl) methacrylamide (HPMA) copolymer loaded with doxorubicin (PM), along with nuclear-accumulated HPMA copolymer Dox conjugate (PN). After co-delivery of these two copolymers(PMN), PM promoted cell apoptosis and inhibited tumor metastasis by destroying mitochondria, while PN inhibited cell proliferation and promoted apoptosis by destroying nuclei.66
Responsive Modification
Responsive modification is the use of environmentally responsive materials to modify nano-drug delivery carriers, enabling them to achieve targeted delivery to mitochondria and drug release under the stimulation of specific tumor microenvironments or intracellular environments, thereby improving therapeutic effects. There are many specific stimulating factors in the tumor microenvironment and intracellular environment, such as pH value, temperature, REDOX potential, enzyme concentration, etc. These factors provide a basis for the design of responsive nano-drug delivery carriers.
As mentioned above, the mitochondrial matrix is weakly alkaline (pH ~8.0). Based on this property, the incorporation of weakly acidic drugs into nanocarriers can be considered to target mitochondria because the solubility of weakly acidic drugs in an alkaline environment is significantly improved. Tan et al designed (4-carboxy-butyl) triphenylphosphine bromide (CTPP) conjugated with a glycolipid-like conjugate (CSOSA) and loaded it with the weakly acidic drug Celastrol (Cela) micelles to verify this mechanism. In a neutral environment (pH 7.4), Cela is negatively charged, forming electrostatic equilibrium with weak cations, so the drug is stably encapsulated in the hydrophobic CSOSA core in a hydrophobic state. After exposure to the alkaline mitochondrial environment (pH 8.0), the electrostatic equilibrium between Cela and weak cations breaks down, and the release rate of Cela from the micelles increases. Additionally, alkaline pH increases water solubility of Cela in micelles, which enhances drug solubility.67 Furthermore, pH values vary across different regions of tumor cells: The extracellular environment is weakly acidic (pH 6.5–7.2), lysosomes are strongly acidic (pH 4.5–5.5), and the mitochondrial matrix has a pH of approximately 8.0. These pH gradient differences provide a foundation for hierarchical targeting in designing pH-responsive nanocarriers.54
The cytosolic concentration of glutathione (GSH) in tumor cells (2–10 mM) is significantly higher than that in the extracellular matrix (2–20 μM) and normal cells (> 4-fold), while the level of ROS is markedly elevated.68 This difference provides a specific activation basis for redox-responsive carriers. The GSH-responsive system can achieve targeted drug release by triggering disulfide bond cleavage through high concentrations of GSH in tumor cells. For example, the lipid-polymer hybrid nanoparticle developed by Zhou et al has a triple-functional design: a PLGA core encapsulating paclitaxel (PTX), a TPP-amphiphilic polymer (C18-PEG2000-TPP) for mitochondrial targeting, and a reduction-responsive polymer (DLPE-S-S-mPEG4000) serving as a GSH-sensitive switch. After the carrier enters the tumor cells, intracellular GSH cleaves the disulfide bond, leading to mPEG4000 falling off and exposing the TPP-targeted groups. This directs the carrier to accumulate in mitochondria, ultimately inducing apoptosis.69 The ROS-responsive system can take advantage of abnormal ROS levels in tumor cells to trigger functional group transformation. The N-alkylamino-ferrocene system designed by Reshetnikov et al exploits the lipophilic conversion of ferrocene (log P value from −0.5 to 2.1) in the presence of ROS to enrich ferrocene in mitochondrial membranes and induce membrane potential collapse, achieving selective killing of cancer cells.70
Tumor-specific enzymes, such as hyaluronidase, esterase, alkaline phosphatase, and matrix metalloproteinases, are highly expressed, providing a precise trigger switch for mitochondrial targeted delivery.71,72 Typical multistage enzyme response strategies include the mesoporous silica system designed by Naz et al (MSN-DPH), which blocks TPP-modified drug-loaded pores through hyaluronic acid (HA). When HA is degraded by hyaluronidase overexpressed in the tumor microenvironment, the pore opens to release doxorubicin (DOX) and exposes the mitochondrial targeting group TPP, significantly increasing mitochondrial drug accumulation. This induces apoptosis in MGC-803 gastric cancer cells.55 For example, Song et al developed a hyaluronidase-responsive nanosystem that innovatively uses 9-O-octadecyl-substituted berberine derivatives(BD) for self-assembly kernel formation. DSPE-PEG2000 modification improves stability by masking positive charges with negatively charged HA coating. When HAase in the tumor microenvironment degrades HA, positively charged PEG/BD nanoparticles (drug loading >70%) are exposed, enhancing cell uptake and lysosomal escape while facilitating mitochondrial targeting through charge-mediated enhancement. This process dissipates mitochondrial membrane potential and releases cytochrome C while regulating the balance of Bcl-2 family proteins and up-regulating ROS levels. Finally, it shows significant anti-tumor effects in an A549 xenograft model.56
Combination of Multimodal Technologies: Interdisciplinary Breakthrough for Synergy
Combination Therapy with Photodynamic Therapy
Photodynamic Therapy (PDT) functions on the principle of using photosensitizers to generate reactive oxygen species (ROS) under light of a specific wavelength to kill tumor cells. However, the clinical application of traditional PDT has long been constrained by limitations such as insufficient light penetration depth (<3 mm), hypoxia in the tumor microenvironment (oxygen partial pressure <10 mmHg), and the nonspecific distribution of photosensitizers.73 In recent years, the integration of nanocarrier technology with mitochondrial targeting strategies has brought significant breakthroughs to this field. Through nanocarrier delivery, photosensitizers can not only better overcome physiological barriers but can also achieve precise subcellular localization at the organelle level using mitochondrial-targeting ligands (eg, TPP),74 thereby significantly amplifying the cytotoxic effects of PDT and activating programmed cell death pathways. For example, a TSPO-targeted photosensitizer, IR700DX-6T, developed by Zhou et al, induces a mitochondrial ROS burst under 690 nm light irradiation. This activates the ROS/p38/CASP3/GSDME signaling axis, simultaneously triggering GSDME-dependent pyroptosis and Caspase-3-mediated apoptosis. In a MSS-type colorectal cancer model, this approach significantly enhanced the efficacy of anti-PD-1 immunotherapy, achieving an 82% tumor regression rate in the combination therapy group.75 To further improve targeting and synergistic effects, Lv et al constructed a mitochondria-targeted multifunctional nanoplateform (HCuS-TH302@PDA-Ce6/TPP NPs). This platform uses hollow copper sulfide nanoparticles (HCuS NPs) as the carrier, loaded with the hypoxia-activated prodrug TH302, and surface-modified with the photosensitizer Ce6 and the mitochondrial-targeting moiety TPP. Under dual-wavelength laser irradiation (660 nm to excite Ce6 for ROS generation and exacerbate hypoxia, and 808 nm to produce a photothermal effect), the system successfully achieved spatiotemporally cascaded synergy among PDT, photothermal therapy (PTT), and hypoxia-activated chemotherapy. This resulted in a remarkably high tumor growth inhibition rate of 95.53% in a B16F10 tumor-bearing mouse model.57 These advances fully demonstrate the great potential of mitochondrial targeting strategies in overcoming the limitations of traditional PDT and enhancing therapeutic efficacy.
Nonetheless, combined PDT/PTT modalities face potential side effects and challenges of drug resistance on the path to clinical translation. Photothermal therapy may damage surrounding normal tissues due to local overheating, triggering inflammatory responses. The efficacy of PDT is strictly dependent on oxygen supply, and the intrinsically hypoxic tumor microenvironment may limit its effects. Furthermore, tumor cells can upregulate endogenous antioxidant systems (eg, glutathione, superoxide dismutase) to scavenge excess ROS, thereby developing resistance to ROS-induced apoptosis. More complexly, subtle interactions exist between different treatment modalities. For instance, while PTT-induced vascular disruption can enhance drug retention at the tumor site, it may also exacerbate hypoxia in the tumor core, potentially weakening the potency of certain oxygen-dependent chemotherapeutic agents. However, this treatment-aggravated hypoxic microenvironment can also be strategically exploited. By designing and delivering hypoxia-activated prodrugs (eg, TH302), the challenge can be transformed into a therapeutic advantage through cascaded killing. Therefore, a deep understanding and careful balancing of these synergistic and antagonistic relationships are crucial for designing safe and highly effective multimodal combination treatment regimens.
Combination Therapy with Immunotherapy
Mitochondrial targeting strategies are emerging as a pivotal breakthrough for enhancing the efficacy of immunotherapy. The core rationale lies in precisely modulating mitochondrial function to reshape anti-tumor immune responses across multiple levels. First, regulating mitochondrial apoptotic pathways can directly enhance the killing efficiency of immune cells. For instance, research by Pan et al demonstrated that the mitochondrial apoptosis (mtApoptosis) initiation status in tumor cells directly influences their susceptibility to Natural Killer (NK) cells. Combining BH3 mimetics with NK cell therapy synergistically induced tumor regression at low effector-to-target ratios and significantly prolonged survival.76 This mechanism also provides a theoretical basis for improving T cell function. However, this apoptosis-based synergy has a dual nature: while excessive mitochondrial damage releases tumor antigens, it may also lead to the substantial release of ATP and potassium ions, inadvertently recruiting immunosuppressive regulatory T cells (Tregs) or myeloid-derived suppressor cells (MDSCs), thereby suppressing effector T cell function and creating a potential antagonistic effect. Moreover, prolonged stimulation may induce acquired resistance in tumor cells via the upregulation of anti-apoptotic proteins (eg, Bcl-2, Mcl-1).
Consequently, research has further focused on immune activation mechanisms that go beyond apoptosis. An ultrasound-responsive liposome system (LID) developed by Wang et al innovatively utilizes locally generated ROS to oxidize mitochondrial DNA (mtDNA) within tumor cells. The oxidized mtDNA, upon transfer to dendritic cells (DCs), potently activates the cGAS-STING pathway, leading to a substantial increase in IFN-β secretion. Combining this strategy with PD-L1 blockade achieved significant tumor complete remission.58 This approach ingeniously converts mitochondrial damage into a potent innate immune signal.
On the other hand, immune evasion mechanisms themselves involve mitochondria. Saha et al revealed a novel mechanism whereby cancer cells unidirectionally hijack mitochondria from immune cells (eg, T cells) via tunneling nanotubes (TNTs). This directly leads to metabolic exhaustion in immune cells and enhanced metabolism in tumor cells. Targeting key regulators of this process (eg, Miro1) or disrupting TNT assembly using inhibitors like L-778123 can effectively block mitochondrial transfer, thereby reversing immune exhaustion. In animal models, such targeting strategies combined with PD-1 inhibitors demonstrated remarkable synergistic tumor suppression and survival benefits.77
In summary, the combination of mitochondrial targeting and immunotherapy represents a multi-layered regulatory system, encompassing mechanisms ranging from directly enhancing cytotoxicity and activating innate immune signaling to blocking immune evasion. However, their interactions are complex, presenting possibilities from synergy to antagonism, and may induce resistance. Therefore, the key to future clinical translation lies in a deep understanding of the dynamic balance between these mechanisms and the subsequent optimization of administration strategies (eg, timing, dosage, and sequential regimens) to maximize therapeutic synergy and circumvent potential risks.
From Theory to Practice: Technical Hurdles and Translational Considerations in Targeting Strategies
While passive, active, and stimuli-responsive targeting strategies have demonstrated significant potential in preclinical research, their clinical translation faces numerous technical hurdles that require careful assessment early in the development process.
Limitations and Heterogeneity of Passive Targeting
Passive targeting primarily relies on the EPR effect. However, this strategy suffers from inherent theoretical drawbacks and clinical uncertainties. Its foremost limitation stems from the physical barriers presented by solid tumors: elevated interstitial fluid pressure, a dense extracellular matrix, and inadequate lymphatic drainage. These factors collectively impede the uniform penetration of nanoparticles, leading to drug accumulation at the tumor periphery and insufficient delivery to the central region—a phenomenon known as the “perfusion-limited penetration barrier”.78
A more critical challenge lies in the intrinsic heterogeneity of the EPR effect itself. It varies substantially among individual patients, across different tumor types, and at various disease stages.79 This high degree of unpredictability renders the therapeutic efficacy of passive targeting strategies—which depend solely on size control—highly inconsistent in clinical applications.
The “Ligand Dilemma” in Active Targeting
Active targeting, based on ligand modification, aims to achieve precise “lock-and-key” recognition. However, its practical application is constrained by a multifaceted “ligand dilemma,” where several critical factors interact in a limiting manner.
The first challenge lies in optimizing ligand surface decoration. An optimal density window exists for ligands on the carrier surface. Too low a density results in insufficient targeting, while excessive density can impair binding to the target due to steric hindrance and may accelerate immune clearance due to over-modification.
Secondly, the risk of off-target effects is difficult to eliminate. Many highly effective mitochondrial-targeting ligands (eg, the cationic peptide TPP) carry a strong positive charge, which confers a universal affinity for negatively charged cell membranes. Consequently, while targeting tumor cells, they are also prone to non-specific uptake by normal tissues (particularly the liver and kidneys), raising potential toxicity concerns.80
A more intractable core bottleneck is the low efficiency of intracellular delivery. Even after successful targeting and internalization, most carriers remain trapped within the endolysosomal pathway. Without an efficient endosomal escape mechanism, the drug cannot reach the cytosol, let alone the final mitochondrial target. Studies indicate that from intravenous injection to exerting a pharmacological effect within tumor cells, nanocarriers suffer significant attrition at each physiological step, resulting in extremely low final delivery efficiency. This constitutes a fundamental barrier for ligand-based targeting strategies on their path to clinical translation.81
Design Bottlenecks and Translational Hurdles in Stimuli-Responsive Systems
Stimuli-responsive nanosystems are designed to achieve controlled drug release within the tumor microenvironment. However, their practical application in living organisms often confronts challenges related to precision and reliability, constituting major obstacles to clinical translation.
First, the triggering signals lack sufficient specificity. Typical features of the tumor microenvironment, such as mild acidity and elevated reactive oxygen species (ROS) levels, significantly overlap with conditions found in other bodily sites (eg, inflamed tissues) or intracellular compartments (eg, endosomes). This overlap makes it difficult to achieve strict tumor-“specific” triggering, often leading to premature drug release at non-target sites. This not only diminishes therapeutic efficacy but may also increase systemic toxicity.82
Second, the response kinetics often fail to align with therapeutic requirements. Even when carriers are successfully triggered, their response speed, drug release profile, and the effective duration required to damage mitochondria are frequently mismatched. Excessively rapid release may cause a local drug spike followed by quick clearance, while overly slow release may prevent the attainment of therapeutic concentrations at the target site. Both scenarios compromise the ultimate therapeutic outcome.
Furthermore, the responsive materials themselves may introduce new safety concerns. Chemical bonds (eg, disulfide bonds, hydrazone bonds) introduced to confer responsiveness can alter the material’s degradation behavior in vivo. The long-term biosafety of the resulting metabolic intermediates or final products remains unclear and must be evaluated as an independent potential risk.83
Therefore, overcoming these bottlenecks requires a shift in future design paradigms: moving from reliance on a single stimulus toward integrated systems capable of distinguishing multiple biological signals, and combining controlled release with efficient sub-organelle targeting capabilities (eg, endosomal escape). This approach is crucial for advancing such systems from conceptual designs to clinically effective therapies.
Mitochondrion-Targeted Nanocarriers for Cancer Therapy
Based on the structural characteristics of mitochondria and their functional abnormalities in tumor cells, researchers have developed various types of nanomedicine delivery systems aimed at achieving precise targeting and efficient drug delivery to mitochondria. Such carriers not only need to possess excellent biocompatibility and high drug loading capacity, but also need to integrate passive or active targeting mechanisms to effectively overcome the delivery challenges posed by the double-layer membrane barrier of mitochondria and the complex tumor microenvironment. Currently, the most notable mitochondrial-targeted nanocarriers mainly include polymer-based nanocarriers, lipid-based nanocarriers, and inorganic nanomaterials, which have shown promising application prospects in anti-tumor therapy (see Table 3).
 |
Table 3 Mitochondrion-Targeted Nanocarriers for Cancer Therapy |
Polymeric Nanocarriers (NPs)
Polymeric nanocarriers are usually composed of biodegradable polymers (polylactic acid, polyglycolic acid, polycaprolactone, and polylactic acid-glycolic acid copolymer (PLGA), etc). Compared with liposomes or inorganic nanocarriers, the core advantages of polymeric nanocarriers are as follows: first, their precise size and charge can maximize electrostatic targeting efficiency. The mitochondrial membrane potential (ΔΨm≈-180 mV) strongly drives positively charged nanoparticles, and polymer carriers can be precisely regulated through molecular design. For example, Dhar studied the relationship between the size and surface charge of PLGA nanoparticles and their mitochondrial uptake efficiency. The particle size (80–140 nm) and surface charge (−30 to +30 mV) were controlled using a nanoprecipitation method. The results showed that 80–100 nm-sized positively charged NPs (+30 mV) had the highest mitochondrial uptake rate, while negatively charged NPs could hardly be detected.84
Secondly, polymeric carriers can simultaneously load targeting ligands, stimuli-responsive units, and multi-mechanism therapeutic agents, significantly enhancing synergistic treatment efficacy while resolving delivery conflicts. A representative example is the glucose-PEG-azo-IR808-S-S-PTX polymer-drug conjugate (self-assembled into micelles) designed by Ma et al’s team, which exemplifies this advantage: specifically, the polymer integrates via covalent bonding glucose (targeting GLUT1 transporters), an azo bond (hypoxia-responsive unit), a disulfide bond (reduction/GSH-responsive unit), along with the chemotherapeutic drug PTX (microtubule-disrupting agent) and the photothermal agent IR808 (mitochondria-targeting photothermal agent). Notably, this simultaneous loading is not merely a physical mixture but involves the tight integration of originally incompatible components (such as hydrophilic glucose and hydrophobic IR808) into a single molecular carrier through chemical bonding along the polymer backbone. During delivery and activation, these co-loaded elements enable precise cascade responses: first, the glucose ligand guides the carrier to achieve active targeting and accumulation at the tumor site via GLUT1. Subsequently, under stimulation by the tumor-specific hypoxic microenvironment (under simulated conditions), the incorporated azo bond cleaves at a rate exceeding 90% within 12 hours, resulting in exposure of the hydrophobic core due to PEG detachment. Following cellular internalization in a reducing environment with high glutathione concentration (GSH~10mM), the integrated disulfide bond breaks synchronously releasing both encapsulated chemotherapeutic drug PTX and photothermal agent IR808 (cumulative drug release reaches 86.6%, far exceeding 15.4% under physiological conditions effectively preventing premature leakage). Finally, under near-infrared light irradiation at 808 nm, co-loaded and released IR808 exerts photothermal conversion rapidly elevating local temperature by 30°C within 5 minutes. Beyond directly disrupting mitochondria, it synergizes with PTX’s microtubule inhibition to exert potent tumor cell killing effects that reduce IC50 value significantly to 1.3 nM. Compared to burst release typical of conventional carriers, this cascade design achieves spatiotemporally precise coordination of targeting→deshielding→drug release→therapy effectively addressing core challenges in multi-mechanism treatments such as delivery compatibility spatiotemporal controllability and synergistic efficacy enhancement.85
It is noteworthy that certain degradable polymers have demonstrated promising biocompatibility potential in specific experimental models. Their safety advantages are typically manifested in two aspects: controllable metabolic clearance and reduced off-target effects. For example, take the cationic amphiphilic AIE polymer PTD designed by Zhou et al: Mechanistically, PTD can self-assemble into micelles with a particle size of approximately 4.3 nm. Its small-molecule degradation products are expected to be cleared via metabolic pathways, thereby potentially avoiding long-term organ accumulation. Experimental data further support its low toxicity: after treating mouse 3T3 fibroblasts for 24 hours with either blank PTD or DOX-loaded PTD at a concentration of 2.0 μg/mL, cell viability remained above 80%, indicating low baseline toxicity to normal cells. Furthermore, its pH-responsive release properties enhance targeting safety by enabling precise DOX release in the weakly acidic tumor microenvironment while maintaining structural stability under neutral physiological conditions. This mechanism may significantly minimize off-target damage to healthy tissues.97 Such studies provide proof of concept for utilizing degradable polymers to circumvent long-term toxicity risks. However, it is crucial to emphasize that the safety window of these carriers is highly sensitive to their specific chemical structure, molecular weight, and surface modifications. Whether the favorable outcomes observed in simplified models (eg, single cell lines, short-term experiments) can be replicated in more complex in vivo physiological environments or chronic disease states, and their complete metabolic fate and long-term effects, require validation through more systematic preclinical studies.
Lipid Nanocarriers
Lipid-based nanocarriers (such as conventional liposomes, MITO-Porter systems, and biomimetic exosomes) have become one of the preferred platforms for mitochondrial-targeted delivery due to their natural biomimetic properties that closely resemble the mitochondrial membrane. Compared with polymer carriers, their core advantage lies in a significant breakthrough in the transmembrane delivery mechanism: the outer mitochondrial membrane is rich in cardiolipin (CL), and liposomes can simulate the compositional characteristics of the mitochondrial membrane by incorporating CL or cationic lipids (such as dodecyltrimethylammonium bromide DQA and triphenylphosphine TPP).89,90 Besides electrostatic adsorption mediated by the mitochondrial membrane potential (ΔΨm≈-180 mV), they can also directly deliver therapeutic molecules to the mitochondrial matrix through membrane fusion, significantly enhancing delivery efficiency. The DF-MITO-Porter system developed by Harashima’s team is a representative example of this innovative mechanism. This system adopts a unique double-layer functionalized structural design: the outer lipid membrane (DOPE/PA) endows the carrier with an ability to escape from endosomes, effectively avoiding lysosomal degradation; while the inner lipid membrane (DOPE/SM or DOPE/PA) utilizes specific recognition capabilities of sphingomyelin (SM) or phosphatidic acid (PA) to achieve efficient fusion with the mitochondrial membrane. Crucially, this system has successfully delivered biologically active macromolecule DNase I with high efficiency and precisely targeted it to HeLa cells’ mitochondrial matrix. Functional experiments further confirmed that delivered DNase I could selectively degrade mtDNA causing significant dysfunction in mitochondria, with a half-maximal effective dose (ED50) as low as 0.33 μg; moreover, its delivery efficiency was 15 times higher than that of traditional single-layer MITO-Porter systems.91 In contrast, positively charged polymer carriers—although they can adsorb onto the surface of mitochondria via electrostatic interactions—lack unique membrane fusion abilities inherent to liposomes and thus are unable to effectively deliver drugs into matrices, fundamentally restricting their delivery efficiency and application potential.
Liposomes exhibit significant advantages in drug loading compatibility, with their unique structural design being particularly prominent: the vesicular structure composed of phospholipid bilayers can efficiently integrate drug molecules with diverse properties through physical interactions without relying on chemical bonding. Specifically, the hydrophilic inner cavity can encapsulate water-soluble drugs (such as the photosensitizer ICG), while the hydrophobic bilayer can accommodate lipophilic drugs (such as DOX). Additionally, the surface can load specific functional molecules through anchoring strategies (such as phospholipid-drug complexes). This “domain-specific loading” feature effectively avoids the need for complex chemical modifications, significantly simplifying the construction process of the delivery system. The LID liposomes developed by Wang’s team fully demonstrate this advantage: first, the photosensitizer ICG is covalently linked to phospholipid DOPE to form DOPE-ICG, successfully anchored on the liposome surface; second, DOX is efficiently encapsulated in the hydrophilic core of the liposome and forms crystals driven by a transmembrane gradient. This strategy combines physical encapsulation and surface anchoring mechanisms, achieving efficient co-loading of two therapeutic agents without complex covalent modification or structural changes to either drug. More importantly, this excellent drug loading compatibility directly translates into a powerful synergistic therapeutic effect: under ultrasound stimulation, LID liposomes not only efficiently generate ROS but also significantly promote transport of DOX to the cell nucleus, thereby achieving efficient killing of tumor cells. Crucially, this system can induce oxidative damage to tumor mtDNA and promote transfer of oxidized mtDNA to dendritic cells, thereby activating cGAS-STING immune signaling pathway. After combined immune checkpoint blockade therapy, complete regression of 87.5% of orthotopic MC38 tumors and 100% of distant tumors was achieved.58 Thus,the outstanding drug loading compatibility of liposomes serves as a key structural basis for achieving multi-mechanism synergistic anti-tumor effects.
Current research indicates that through rational formulation design, liposomes can exhibit promising biocompatibility. Evidence supporting their safety primarily derives from assessments of in vitro cellular tolerance and in vivo organ toxicity. For instance, a study by Sun et al demonstrated that blank liposomes maintained cell viability close to 100% even at concentrations as high as 500 μg/mL, suggesting a wide in vitro safety window.92 In a more complex in vivo model, alkyl glucoside-modified AGCL liposomes developed by Luo et al, when loaded with celastrol, demonstrated potential for reducing systemic toxicity. In treated mice, key serum parameters for liver and kidney function (ALT, AST, UREA, CRE, LDH) remained within normal ranges, and hematoxylin and eosin (H&E)-stained sections of major organs (heart, liver, spleen, lungs, kidneys) showed no significant pathological damage.93 These collective findings suggest that through sophisticated molecular design, liposomes hold promise for achieving low acute toxicity and favorable in vivo tolerance. However, translating these preclinical findings into clinical application necessitates a cautious evaluation of their long-term safety upon repeated administration. Key variables requiring systematic clarification in future translational research include, for example, potential immunogenicity triggered by cationic lipids, the cumulative effects of different phospholipid compositions after repeated dosing, and the long-term impact of their final metabolic products.
Inorganic Nanocarriers
Inorganic nanocarriers (such as gold nanoparticles, mesoporous silica, and graphene oxide) have demonstrated unique advantages in mitochondrial-targeted drug delivery due to their tunable physicochemical properties (such as excellent photothermal conversion performance and high specific surface area) and precise surface functionalization capabilities. The core breakthrough lies in the synergistic effect of physical targeting and stimulus-responsive mechanisms: by modifying the surface with mitochondrial-targeting ligands (such as TPP) and leveraging the inherent near-infrared photothermal effect of inorganic materials, spatiotemporally controlled drug release and precise subcellular intervention at the organelle level can be achieved. Taking the gold nanorod composite system (Au@DOX/CQ@TPP-DCD) developed by Chen’s team as an example, the carrier’s surface is covalently linked with TPP ligands, enabling selective enrichment in the mitochondrial region driven by mitochondrial membrane potential. Meanwhile, the gold nanorod core generates significant local heating (ΔT≈25-30 °C) under 808 nm laser irradiation, which not only triggers drug release but also cooperatively induces massive generation of ROS through the photothermal effect, directly disrupting mitochondrial structure and function. Experimental results show that this carrier exhibits significant mitochondrial colocalization in 4T1 cancer cells (verified by confocal laser scanning microscopy [CLSM]). The combination of autophagy inhibitor chloroquine (CQ) with near-infrared light (1.0 W/cm2 for 3 min) enables a synergistic effect involving photodynamic therapy, photothermal therapy, and chemotherapy triple modalities, achieving precise functional regulation of mitochondria. In vivo anti-tumor experiments further confirm that this strategy can achieve a 75% complete tumor regression without obvious systemic toxicity, demonstrating good safety.94
In terms of drug compatibility, inorganic nanocarriers have significantly surpassed the limitations of traditional delivery systems through a multi-loading strategy, which is specifically reflected in the following two aspects: Firstly, mesoporous silica nanoparticles (MSN) with a high specific surface area (>500 m2/g, measured value of 553 m2/g) and controllable pore size (approximately 3 nm) can achieve highly efficient and high-capacity loading of small molecule chemotherapeutic drugs (Dox), with a drug loading capacity as high as 110 mg/g. Their surface can be further modified with targeting ligands (HA) and mitochondrial-targeting molecules (TPP), thereby enabling enzyme-responsive drug release (triggered by HAase) and precise subcellular targeting at the mitochondrial level (selective accumulation in mitochondria).55 Secondly, graphene oxide (GO), with its two-dimensional planar structure and sp2 hybridized carbon skeleton, can efficiently load hydrophobic photosensitizers (ICG) through π–π stacking and hydrophobic interactions. Meanwhile, the abundant carboxyl groups on its surface provide sufficient reaction sites for covalent coupling of targeting molecules (TPP). The TPP-PPG@ICG nanosystem developed by Zeng et al is a typical example: ICG is embedded between the PEI-PEG modified GO layers through π–π interactions, achieving a drug loading rate as high as 30.4% (w/w); TPP is covalently linked through stable amide bonds to achieve efficient mitochondrial-targeted delivery. Under 808 nm laser irradiation, this system can simultaneously generate significant photothermal heating effects (ΔT>23°C; rising from 25°C to 48°C within 5 minutes) and photodynamic effects (singlet oxygen generation), synergistically disrupting mitochondrial membrane integrity and inhibiting ATP synthesis while significantly suppressing the growth of drug-resistant osteosarcoma both in vitro and in vivo without obvious systemic toxicity—verified by blood biochemical indicators and histopathological examination.95 This “inorganic core-functional shell-drug reservoir” integrated design strategy effectively overcomes the inherent limitations of traditional carriers in thermosensitive drug encapsulation, long-term stability, and multimodal synergistic therapy while demonstrating strong comprehensive performance advantages.
In current research models, certain inorganic nanocarriers have demonstrated considerable biocompatibility potential under specific administration protocols, with evidence primarily derived from assessments of in vivo biodistribution and short-term toxicity. For instance, following intravenous injection of a graphene oxide-based carrier (eg, TPP-PPG@ICG) at a high dose of 10 mg/kg, no significant pathological alterations were observed in the major organs (heart, liver, spleen, lungs, kidneys) of experimental mice, and relevant hematological and biochemical parameters remained within normal ranges. This suggests low systemic toxicity under the tested conditions.95 Similarly, PEGylated gold nanorods (GSNR-TPPs), after intravenous administration, accumulated at tumor sites via the EPR effect. Comprehensive blood biochemical analysis (AST, ALT, ALP, BUN) and histopathological examination (H&E staining) detected no marked impairment of liver or kidney function, nor evidence of organ inflammation or necrosis, further supporting the favorable biocompatibility of such materials within the specific experimental window.96 Collectively, these studies provide preliminary safety data supporting the potential future clinical application of inorganic nanocarriers.
However, distinct from their organic counterparts, the inherent physicochemical properties of inorganic materials present unique challenges for clinical translation. These mainly include: a non-negligible tendency for long-term in vivo retention; the sustained release of metal ions resulting from material degradation or corrosion; and the potentially significant impact that subtle batch-to-batch variations in physicochemical properties (eg, size, morphology, surface charge) arising from synthesis processes may have on toxicity profiles. These factors collectively necessitate a more prospective and systematic strategy for assessing their long-term safety.
In summary, leveraging their distinct physicochemical properties, polymeric, liposomal, and inorganic nanocarriers have demonstrated unique advantages for mitochondrial-targeted delivery and have accumulated preliminary biocompatibility data under controlled experimental conditions. However, the safety evidence scattered across these disparate research systems remains insufficient to construct a complete and predictable clinical safety profile. The true safety of a nanocarrier is a dynamic outcome resulting from the interactions among the material itself, the complex in vivo environment, and individual variability—far more complex than what can be captured by isolated toxicity reports. Therefore, building a comprehensive clinical safety profile necessitates a systematic deconstruction from the mechanistic level. The following sections will elaborate on these aspects from three key perspectives: toxicity mechanisms, immunogenicity, and evaluation paradigms.
Safety Challenges for Clinical Translation: Mechanisms, Immunogenicity, and Evaluation in Combination Therapy
The safety of nanocarriers is a dynamic outcome resulting from the interaction between their physicochemical properties and complex biological systems.98 This multifaceted interplay is primarily manifested at two levels: first, the fundamental mechanisms of toxicity, referring to direct damage potentially caused by the carrier material itself; and second, immunogenic responses, denoting the potential for excessive or aberrant immune activation triggered by the carrier’s interaction with the immune system. To comprehensively address the unique challenges posed by nanocarriers, particularly in light of their increasingly prevalent use within combination therapies, safety evaluation systems must evolve towards more systematic and predictive new paradigms. The following analysis will proceed from these three interconnected dimensions.
Firstly, at the level of fundamental toxicity, the dominant mechanisms differ significantly among carrier materials. The risks of polymeric nanocarriers (especially cationic ones) mainly stem from the electrostatic interaction between their positively charged surfaces and cell membranes, potentially leading to membrane damage and intracellular stress. The hemolytic risk of liposomes is highly dependent on their composition and surface charge. For inorganic nanomaterials (eg, silica), concerns center on their potential organ-specific retention, which may lead to chronic inflammation and fibrosis.99 Notably, when these carriers are employed in combination therapies, their toxicity profiles may change significantly and introduce new complexities. For instance, photosensitive components in carriers can increase the risk of cutaneous phototoxicity when combined with phototherapy; co-administration with chemotherapeutic agents possessing specific organ toxicities may exacerbate hepatic or renal burden due to competition for metabolic or excretion pathways. Therefore, while understanding the toxicology of individual materials is fundamental, evaluating combination regimens necessitates systematic investigation of the interactive toxicity between treatment modalities, with a focus on potential synergistic adverse effects.
Secondly, regarding the interaction with the immune system, the complexity far exceeds a simple “foreign body” response. The carrier itself can act as an “adjuvant” to activate innate immunity, while the “protein corona” formed by the adsorption of plasma proteins onto its surface directly dictates immune cell recognition and subsequent fate.100 In the context of combination therapy, the risks associated with this immune interaction are further amplified. For example, when mitochondrial-targeted nanocarriers are combined with immune agonists (eg, STING agonists) or immune checkpoint inhibitors, there is a risk of hyperactivating both innate and adaptive immune systems, potentially inducing severe immune-related adverse events such as cytokine release syndrome. Consequently, finely tuning the composition of the protein corona through surface engineering to balance immune activation and suppression becomes particularly critical in combination immunotherapy regimens.
Given the multifaceted complexity of the aforementioned toxicity mechanisms and immune responses, particularly the new challenges introduced by combination therapies, traditional toxicological evaluation systems are proving insufficient. To reliably support clinical translation, the establishment of more predictive evaluation paradigms is imperative. This requires: 1) Integrating advanced immunotoxicity assessments to systematically investigate the potential impact of nanoformulations, both alone and in combination, on the immune system; 2) Conducting interaction-toxicity studies oriented towards combination therapies, developing long-term toxicology models aligned with clinical dosing regimens (eg, timing, sequence, dosage) that go beyond the safety assessment scope of monotherapies; 3) Employing more relevant and complex models, such as validating safety in patient-derived organoids or humanized mouse models, to more accurately predict human responses. Ultimately, establishing standardized safety databases and evaluation benchmarks for nanomedicines will facilitate a paradigm shift in safety from “reactive testing” to “proactive design,” accelerating the development of safe and effective nanomedicine products.
Summary and Prospect
Mitochondrial-targeted therapy utilizing nanodrug delivery systems offers novel strategies to overcome tumor resistance, induce apoptosis, and activate antitumor immunity. By precisely controlling carrier size and surface charge, and modifying them with targeting ligands such as TPP and mitochondrial-penetrating peptides, the enrichment and release of drugs within mitochondria can be significantly enhanced. Currently, various carriers, including polymers, liposomes, and inorganic nanomaterials, demonstrate distinct advantages in mitochondrial-targeted delivery. Liposomes can efficiently deliver drugs to the mitochondrial matrix via membrane fusion, while inorganic nanomaterials possess both high drug-loading capacity and unique photothermal conversion capabilities. Furthermore, the incorporation of stimuli-responsive designs (eg, triggered by pH, ROS, or enzymes) further improves treatment precision and spatiotemporal controllability.
Although mitochondrial-targeted nanocarriers have demonstrated significant potential in preclinical studies, their clinical translation faces multiple challenges. First, the large-scale production of complex carriers requires stringent control to ensure high batch-to-batch consistency and stability, imposing demanding requirements on manufacturing processes. Second, the in vivo metabolic pathways, biodistribution, and long-term safety of these carriers necessitate systematic validation. For instance, certain inorganic nanomaterials may pose risks of long-term retention, while cationic polymers require careful evaluation of their potential immunogenicity. Furthermore, tumor heterogeneity leads to functional diversity in mitochondria across different patients and even among distinct lesions within the same individual. This variability complicates the development of universal targeting strategies. Future research should focus on elucidating the heterogeneous characteristics of mitochondrial function to advance the development of personalized and precision therapeutic approaches.
Future research should prioritize the following key directions: (1) the development of novel biomimetic carriers—such as engineered exosomes or cell membrane-coated nanoparticles—that leverage their intrinsic targeting capabilities and biocompatibility to improve deep tumor penetration and enhance mitochondrial delivery efficiency; (2) the design of intelligent, stimuli-responsive systems integrated with artificial intelligence and real-time imaging technologies, enabling dynamic monitoring of biodistribution and precise, adaptive drug release; (3) the advancement of multimodal combination therapies—including photothermal, photodynamic, and immunotherapeutic strategies—to simultaneously disrupt mitochondrial function and elicit robust systemic anti-tumor immune responses through spatiotemporal control; and (4) comprehensive evaluation of long-term toxicity and carrier metabolic fate, together with the establishment of standardized manufacturing protocols and quality control frameworks, to accelerate the clinical translation of these promising therapeutic platforms.
Data Sharing Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Research Program of Hunan Provincial Health Commission (202213014042 to L.Huang), the Outstanding Youth Project of Hunan Provincial Education Department (22B0375 to L.Huang), the Natural Science Foundation of Hunan Province (2026JJ80930 to L.Huang), the Natural Science Foundation of China (No. 82505324 to P.Zheng), the Outstanding Innovative Youth Training Program of Changsha (kq2306029 to Q.S. Chen), and the Scientific Research Fund of Hunan Provincial Education Department (No.25C1747 to Y.J. Lei).
Disclosure
Dr. Li Huang and Ms. Yujing Lei are co-first authors for this study. The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74(3):229–23. doi:10.3322/caac.21834
2. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12(1):31–46. doi:10.1158/2159-8290.CD-21-1059
3. Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25(3):486–541. doi:10.1038/s41418-017-0012-4
4. Vasan K, Werner M, Chandel NS. Mitochondrial metabolism as a target for cancer therapy. Cell Metab. 2020;32(3):341–352. doi:10.1016/j.cmet.2020.06.019
5. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23(1):27–47. doi:10.1016/j.cmet.2015.12.006
6. Kalkavan H, Green DR. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018;25(1):46–55. doi:10.1038/cdd.2017.179
7. Buss CG, Bhatia SN. Nanoparticle delivery of immunostimulatory oligonucleotides enhances response to checkpoint inhibitor therapeutics. Proc Natl Acad Sci U S A. 2020;117(24):13428–13436. doi:10.1073/pnas.2001569117
8. Tian Y, Zhang Y, Yao S, et al. Room temperature exciton polariton condensation in high-quality Sn-doped CdS microsheet. Nano Res. 2025;18(8):94907525. doi:10.26599/NR.2025.94907525
9. Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33(9):941–951. doi:10.1038/nbt.3330
10. Butler LM, Perone Y, Dehairs J, et al. Lipids and cancer: emerging roles in pathogenesis, diagnosis and therapeutic intervention. Adv Drug Deliv Rev. 2020;159:245–293. doi:10.1016/j.addr.2020.07.013
11. Li MY, Naik TS, Siu L, et al. Lyn kinase regulates egress of flaviviruses in autophagosome-derived organelles. Nat Commun. 2020;11(1):5189. doi:10.1038/s41467-020-19028-w
12. Liu Y, Zhang H, Liu Y, et al. Hypoxia-induced GPCPD1 depalmitoylation triggers mitophagy via regulating PRKN-mediated ubiquitination of VDAC1. Autophagy. 2023;19(9):2443–2463. doi:10.1080/15548627.2023.2182482
13. Li Z, Bao Z, Tan J, et al. Neobractatin induces pyroptosis of esophageal cancer cells by TOM20/BAX signaling pathway. Phytomedicine. 2024;128:155547. doi:10.1016/j.phymed.2024.155547
14. Liu J, Liu Y, Li H, et al. Polyphyllin D induces apoptosis and protective autophagy in breast cancer cells through JNK1-Bcl-2 pathway. J Ethnopharmacol. 2022;282:114591. doi:10.1016/j.jep.2021.114591
15. Dmello RS, Palmieri M, Thilakasiri PS, et al. Combination of bazedoxifene with chemotherapy and SMAC-mimetics for the treatment of colorectal cancer. Cell Death Dis. 2024;15(4):255. doi:10.1038/s41419-024-06631-8
16. Zhang P, Zhou C, Ren X, et al. Inhibiting the compensatory elevation of xCT collaborates with disulfiram/copper-induced GSH consumption for cascade ferroptosis and cuproptosis. Redox Biol. 2024;69:103007. doi:10.1016/j.redox.2023.103007
17. Yue C, Yang Y, Song J, et al. Mitochondria-targeting near-infrared light-triggered thermosensitive liposomes for localized photothermal and photodynamic ablation of tumors combined with chemotherapy. Nanoscale. 2017;9(31):11103–11118. doi:10.1039/C7NR02193C
18. Chien PY, Wang J, Carbonaro D, et al. Novel cationic cardiolipin analogue-based liposome for efficient DNA and small interfering RNA delivery in vitro and in vivo. Cancer Gene Ther. 2005;12(3):321–328. doi:10.1038/sj.cgt.7700793
19. Andrzejewski S, Gravel SP, Pollak M, St-Pierre J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014;2:12. doi:10.1186/2049-3002-2-12
20. Zhang C, Liu Z, Bunker E, et al. Sorafenib targets the mitochondrial electron transport chain complexes and ATP synthase to activate the PINK1-Parkin pathway and modulate cellular drug response. J Biol Chem. 2017;292(36):15105–15120. doi:10.1074/jbc.M117.783175
21. Sasaki R, Suzuki Y, Yonezawa Y, et al. DNA polymerase gamma inhibition by vitamin K3 induces mitochondria-mediated cytotoxicity in human cancer cells. Cancer Sci. 2008;99(5):1040–1048. doi:10.1111/j.1349-7006.2008.00771.x
22. Yuan Y, Wu Y, Li C, et al. Circ0515 reprogramming mitochondrial succinate metabolism and promotes lung adenocarcinoma progression through regulating SDHB. Cell Death Dis. 2025;16(1):497. doi:10.1038/s41419-025-07830-7
23. Ruprecht JJ, Kunji E. The SLC25 mitochondrial carrier family: structure and mechanism. Trends Biochem Sci. 2020;45(3):244–258. doi:10.1016/j.tibs.2019.11.001
24. Salnikov V, Lukyánenko YO, Frederick CA, Lederer WJ, Lukyánenko V. Probing the outer mitochondrial membrane in cardiac mitochondria with nanoparticles. Biophys J. 2007;92(3):1058–1071. doi:10.1529/biophysj.106.094318
25. Kiebler M, Pfaller R, Söllner T, et al. Identification of a mitochondrial receptor complex required for recognition and membrane insertion of precursor proteins. Nature. 1990;348(6302):610–616. doi:10.1038/348610a0
26. Thomas LW, Stephen JM, Ashcroft M. CHCHD4 regulates the expression of mitochondrial genes that are essential for tumour cell growth. Biochim Biophys Acta Mol Basis Dis. 2024;1870(7):167282. doi:10.1016/j.bbadis.2024.167282
27. Bello NF, Lamsoul I, Heuzé ML, et al. The E3 ubiquitin ligase specificity subunit ASB2beta is a novel regulator of muscle differentiation that targets filamin B to proteasomal degradation. Cell Death Differ. 2009;16(6):921–932. doi:10.1038/cdd.2009.27
28. Gao Y, Tong H, Li J, et al. Mitochondria-targeted nanomedicine for enhanced efficacy of cancer therapy. Front Bioeng Biotechnol. 2021;9:720508. doi:10.3389/fbioe.2021.720508
29. Shirmanova MV, Druzhkova IN, Lukina MM, et al. Intracellular pH imaging in cancer cells in vitro and tumors in vivo using the new genetically encoded sensor SypHer2. Biochim Biophys Acta. 2015;1850(9):1905–1911. doi:10.1016/j.bbagen.2015.05.001
30. Kafkova A, Trnka J. Mitochondria-targeted compounds in the treatment of cancer. Neoplasma. 2020;67(3):450–460. doi:10.4149/neo_2020_190725N671
31. Fan XY, Liu YJ, Cai YM, et al. A mitochondria-targeted organic arsenical accelerates mitochondrial metabolic disorder and function injury. Bioorg Med Chem. 2019;27(5):760–768. doi:10.1016/j.bmc.2019.01.008
32. Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer. 2011;11(9):671–677. doi:10.1038/nrc3110
33. van der Bliek AM, Sedensky MM, Morgan PG. Cell biology of the mitochondrion. Genetics. 2017;207(3):843–871. doi:10.1534/genetics.117.300262
34. Chen E, Wang T, Zhang J, et al. Mitochondrial targeting and pH-Responsive nanogels for Co-Delivery of lonidamine and paclitaxel to conquer drug resistance. Front Bioeng Biotechnol. 2021;9:787320. doi:10.3389/fbioe.2021.787320
35. Serrano MC, Feito MJ, González-Mayorga A, Diez-Orejas R, Matesanz MC, Portolés MT. Response of macrophages and neural cells in contact with reduced graphene oxide microfibers. Biomater Sci. 2018;6(11):2987–2997. doi:10.1039/C8BM00902C
36. Oyewole AO, Birch-Machin MA. Mitochondria-targeted antioxidants. FASEB J. 2015;29(12):4766–4771. doi:10.1096/fj.15-275404
37. Waypa GB, Marks JD, Guzy R, et al. Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ Res. 2010;106(3):526–535. doi:10.1161/CIRCRESAHA.109.206334
38. Cho H, Cho YY, Shim MS, Lee JY, Lee HS, Kang HC. Mitochondria-targeted drug delivery in cancers. Biochim Biophys Acta Mol Basis Dis. 2020;1866(8):165808. doi:10.1016/j.bbadis.2020.165808
39. Porporato PE, Filigheddu N, Pedro J, Kroemer G, Galluzzi L. Mitochondrial metabolism and cancer. Cell Res. 2018;28(3):265–280. doi:10.1038/cr.2017.155
40. Shokolenko IN, Wilson GL, Alexeyev MF. Aging: a mitochondrial DNA perspective, critical analysis and an update. World J Exp Med. 2014;4(4):46–57. doi:10.5493/wjem.v4.i4.46
41. Gammage PA, Frezza C. Mitochondrial DNA: the overlooked oncogenome. BMC Biol. 2019;17(1):53. doi:10.1186/s12915-019-0668-y
42. Peng L, Gu S, Hou M, Hou X. DNA hydrogels for cancer diagnosis and therapy. Chembiochem. 2024;25(24):e202400494. doi:10.1002/cbic.202400494
43. Zhang Y, Li W, Niu J, Fan Z, Li X, Zhang H. Reprogramming of glucose metabolism in pancreatic cancer: mechanisms, implications, and therapeutic perspectives. Front Immunol. 2025;16:1586959. doi:10.3389/fimmu.2025.1586959
44. Rojas-Pirela M, Andrade-Alviárez D, Rojas V, et al. Exploring glycolytic enzymes in disease: potential biomarkers and therapeutic targets in neurodegeneration, cancer and parasitic infections. Open Biol. 2025;15(2):240239. doi:10.1098/rsob.240239
45. Roberts DJ, Miyamoto S. Hexokinase II integrates energy metabolism and cellular protection: akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015;22(2):248–257. doi:10.1038/cdd.2014.173
46. Cui Y, Li C, Sang F, Cao W, Qin Z, Zhang P. Natural products targeting glycolytic signaling pathways-an updated review on anti-cancer therapy. Front Pharmacol. 2022;13:1035882. doi:10.3389/fphar.2022.1035882
47. Torrano AA, Herrmann R, Strobel C, et al. Cell membrane penetration and mitochondrial targeting by platinum-decorated ceria nanoparticles. Nanoscale. 2016;8(27):13352–13367. doi:10.1039/C5NR08419A
48. Mitchell W, Ng EA, Tamucci JD, et al. The mitochondria-targeted peptide SS-31 binds lipid bilayers and modulates surface electrostatics as a key component of its mechanism of action. J Biol Chem. 2020;295(21):7452–7469. doi:10.1074/jbc.RA119.012094
49. Yang Y, Gao N, Hu Y, et al. Gold nanoparticle-enhanced photodynamic therapy: effects of surface charge and mitochondrial targeting. Ther Deliv. 2015;6(3):307–321. doi:10.4155/tde.14.115
50. Zhang J, Li M, Wang M, et al. Effects of the surface charge of polyamidoamine dendrimers on cellular exocytosis and the exocytosis mechanism in multidrug-resistant breast cancer cells. J Nanobiotechnology. 2021;19(1):135. doi:10.1186/s12951-021-00881-w
51. Halimu G, Zhang Q, Liu L, et al. Toxic effects of nanoplastics with different sizes and surface charges on epithelial-to-mesenchymal transition in A549 cells and the potential toxicological mechanism. J Hazard Mater. 2022;430:128485. doi:10.1016/j.jhazmat.2022.128485
52. Lu H, Tong W, Jiang M, et al. Mitochondria-Targeted multifunctional nanoprodrugs by inhibiting metabolic reprogramming for combating cisplatin-resistant lung cancer. ACS Nano. 2024;18(32):21156–21170. doi:10.1021/acsnano.4c04024
53. Meng Z, Wang B, Liu Y, et al. Mitochondria-targeting polydopamine-coated nanodrugs for effective photothermal- and chemo-synergistic therapies against lung cancer. Regen Biomater. 2022;9:rbac051. doi:10.1093/rb/rbac051
54. Zhang J, Yang C, Pan S, et al. Eph A10-modified pH-sensitive liposomes loaded with novel triphenylphosphine-docetaxel conjugate possess hierarchical targetability and sufficient antitumor effect both in vitro and in vivo. Drug Deliv. 2018;25(1):723–737. doi:10.1080/10717544.2018.1446475
55. Naz S, Wang M, Han Y, et al. Enzyme-responsive mesoporous silica nanoparticles for tumor cells and mitochondria multistage-targeted drug delivery. Int J Nanomed. 2019;14:2533–2542. doi:10.2147/IJN.S202210
56. Song J, Lin C, Yang X, et al. Mitochondrial targeting nanodrugs self-assembled from 9-O-octadecyl substituted berberine derivative for cancer treatment by inducing mitochondrial apoptosis pathways. J Control Release. 2019;294:27–42. doi:10.1016/j.jconrel.2018.11.014
57. Lv J, Wang S, Qiao D, Lin Y, Hu S, Li M. Mitochondria-targeting multifunctional nanoplatform for cascade phototherapy and hypoxia-activated chemotherapy. J Nanobiotechnology. 2022;20(1):42. doi:10.1186/s12951-022-01244-9
58. Wang C, Zhang R, He J, et al. Ultrasound-responsive low-dose doxorubicin liposomes trigger mitochondrial DNA release and activate cGAS-STING-mediated antitumour immunity. Nat Commun. 2023;14(1):3877. doi:10.1038/s41467-023-39607-x
59. Colombini M. The VDAC channel: molecular basis for selectivity. Biochim Biophys Acta. 2016;1863(10):2498–2502. doi:10.1016/j.bbamcr.2016.01.019
60. Aksoyoglu MA, Podgornik R, Bezrukov SM, Gurnev PA, Muthukumar M, Parsegian VA. Size-dependent forced PEG partitioning into channels: VDAC, OmpC, and α-hemolysin. Proc Natl Acad Sci U S A. 2016;113(32):9003–9008. doi:10.1073/pnas.1602716113
61. Song H, Xing W, Shi X, Zhang T, Lou H, Fan P. Antitumor and toxicity study of mitochondria-targeted triptolide derivatives using triphenylphosphine (TPP+) as a carrier. Bioorg Med Chem. 2021;50:116466. doi:10.1016/j.bmc.2021.116466
62. Wang H, Shi W, Zeng D, et al. pH-activated, mitochondria-targeted, and redox-responsive delivery of paclitaxel nanomicelles to overcome drug resistance and suppress metastasis in lung cancer. J Nanobiotechnology. 2021;19(1):152. doi:10.1186/s12951-021-00895-4
63. Zhao T, Liu X, Singh S, et al. Mitochondria penetrating peptide-conjugated TAMRA for live-cell long-term tracking. Bioconjug Chem. 2019;30(9):2312–2316. doi:10.1021/acs.bioconjchem.9b00465
64. Jeena MT, Kim S, Jin S, Ryu JH. Recent progress in mitochondria-targeted drug and drug-free agents for cancer therapy. Cancers. 2019;12(1):4. doi:10.3390/cancers12010004
65. Selmin F, Magri G, Gennari CG, Marchianò S, Ferri N, Pellegrino S. Development of poly(lactide-co-glycolide) nanoparticles functionalized with a mitochondria penetrating peptide. J Pept Sci. 2017;23(2):182–188. doi:10.1002/psc.2952
66. Yang J, Li Q, Zhou M, et al. Concurrent impairment of nucleus and mitochondria for synergistic inhibition of cancer metastasis. Int J Pharm. 2021;608:121077. doi:10.1016/j.ijpharm.2021.121077
67. Tan Y, Zhu Y, Zhao Y, et al. Mitochondrial alkaline pH-responsive drug release mediated by Celastrol loaded glycolipid-like micelles for cancer therapy. Biomaterials. 2018;154:169–181. doi:10.1016/j.biomaterials.2017.07.036
68. Huang Y, Wang T, Tan Q, et al. Smart Stimuli-Responsive and mitochondria targeting delivery in cancer therapy. Int J Nanomed. 2021;16:4117–4146. doi:10.2147/IJN.S315368
69. Zhou W, Yu H, Zhang LJ, et al. Redox-triggered activation of nanocarriers for mitochondria-targeting cancer chemotherapy. Nanoscale. 2017;9(43):17044–17053. doi:10.1039/C7NR06130G
70. Reshetnikov V, Daum S, Janko C, et al. ROS-Responsive N-Alkylaminoferrocenes for cancer-cell-specific targeting of mitochondria. Angew Chem Int Ed Engl. 2018;57(37):11943–11946. doi:10.1002/anie.201805955
71. Pramanik SK, Sreedharan S, Singh H, et al. Mitochondria targeting Non-Isocyanate-Based polyurethane nanocapsules for enzyme-triggered drug release. Bioconjug Chem. 2018;29(11):3532–3543. doi:10.1021/acs.bioconjchem.8b00460
72. Liu S, Jiang Y, Cheng X, et al. Mitochondria-targeting nanozyme for catalytical therapy and radiotherapy with activation of cGAS-STING. Colloids Surf B Biointerfaces. 2024;244:114137. doi:10.1016/j.colsurfb.2024.114137
73. Hare JI, Lammers T, Ashford MB, Puri S, Storm G, Barry ST. Challenges and strategies in anti-cancer nanomedicine development: an industry perspective. Adv Drug Deliv Rev. 2017;108:25–38. doi:10.1016/j.addr.2016.04.025
74. Pan WL, Tan Y, Meng W, et al. Microenvironment-driven sequential ferroptosis, photodynamic therapy, and chemotherapy for targeted breast cancer therapy by a cancer-cell-membrane-coated nanoscale metal-organic framework. Biomaterials. 2022;283:121449. doi:10.1016/j.biomaterials.2022.121449
75. Zhou Y, Zhang W, Wang B, et al. Mitochondria-targeted photodynamic therapy triggers GSDME-mediated pyroptosis and sensitizes anti-PD-1 therapy in colorectal cancer. J Immunother Cancer. 2024;12(3):e008054. doi:10.1136/jitc-2023-008054
76. Pan R, Ryan J, Pan D, Wucherpfennig KW, Letai A. Augmenting NK cell-based immunotherapy by targeting mitochondrial apoptosis. Cell. 2022;185(9):1521–1538.e18. doi:10.1016/j.cell.2022.03.030
77. Saha T, Dash C, Jayabalan R, et al. Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat Nanotechnol. 2022;17(1):98–106. doi:10.1038/s41565-021-01000-4
78. Sindhwani S, Syed AM, Ngai J, et al. The entry of nanoparticles into solid tumours. Nat Mater. 2020;19(5):566–575. doi:10.1038/s41563-019-0566-2
79. Li Z, Xiao C, Yang X, Li Z. Progress in the mechanical properties of nanoparticles for tumor-targeting delivery. Chem Soc Rev. 2025;54(11):5698–5734. doi:10.1039/D3CS00912B
80. Zhao Z, Ukidve A, Krishnan V, Mitragotri S. Effect of physicochemical and surface properties on in vivo fate of drug nanocarriers. Adv Drug Deliv Rev. 2019;143:3–21. doi:10.1016/j.addr.2019.01.002
81. Lapin NA, Vergara LA, Mackeyev Y, et al. Biotransport kinetics and intratumoral biodistribution of malonodiserinolamide-derivatized [60]fullerene in a murine model of breast adenocarcinoma. Int J Nanomed. 2017;12:8289–8307. doi:10.2147/IJN.S138641
82. Yang Q, Wu Q, Liu H, Wu J, Ma F, Tian X. Tumor microenvironment-responsive and modulatory manganese-based nanoenzyme for enhanced tumor immunotherapy. Front Pharmacol. 2024;15:1518983. doi:10.3389/fphar.2024.1518983
83. Xu C, Nam J, Hong H, Xu Y, Moon JJ. Positron emission tomography-guided photodynamic therapy with biodegradable mesoporous silica nanoparticles for personalized cancer immunotherapy. ACS Nano. 2019;13(10):12148–12161. doi:10.1021/acsnano.9b06691
84. Marrache S, Dhar S. Engineering of blended nanoparticle platform for delivery of mitochondria-acting therapeutics. Proc Natl Acad Sci U S A. 2012;109(40):16288–16293. doi:10.1073/pnas.1210096109
85. Ma P, Wei G, Chen J, Jing Z, Wang X, Wang Z. GLUT1 targeting and hypoxia-activating polymer-drug conjugate-based micelle for tumor chemo-thermal therapy. Drug Deliv. 2021;28(1):2256–2267. doi:10.1080/10717544.2021.1992039
86. Zou Y, Sun Y, Wang Y, et al. Cancer cell-mitochondria hybrid membrane coated Gboxin loaded nanomedicines for glioblastoma treatment. Nat Commun. 2023;14(1):4557. doi:10.1038/s41467-023-40280-3
87. Zhang S, Zheng F, Liu K, et al. Mitochondria-targeting polymer micelles in stepwise response releasing gemcitabine and destroying the mitochondria and nucleus for combined antitumor chemotherapy. Int J Mol Sci. 2022;23(20).
88. Shueng PW, Yu LY, Hou HH, Chiu HC, Lo CL. Charge conversion polymer-liposome complexes to overcome the limitations of cationic liposomes in mitochondrial-targeting drug delivery. Int J Mol Sci. 2022;23(6):3080. doi:10.3390/ijms23063080
89. Birk AV, Chao WM, Bracken C, Warren JD, Szeto HH. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol. 2014;171(8):2017–2028. doi:10.1111/bph.12468
90. Shi M, Zhang J, Li X, et al. Mitochondria-targeted delivery of doxorubicin to enhance antitumor activity with HER-2 peptide-mediated multifunctional pH-sensitive DQAsomes. Int J Nanomed. 2018;13:4209–4226. doi:10.2147/IJN.S163858
91. Yamada Y, Harashima H. Delivery of bioactive molecules to the mitochondrial genome using a membrane-fusing, liposome-based carrier, DF-MITO-Porter. Biomaterials. 2012;33(5):1589–1595. doi:10.1016/j.biomaterials.2011.10.082
92. Sun J, Jiang L, Lin Y, et al. Enhanced anticancer efficacy of paclitaxel through multistage tumor-targeting liposomes modified with RGD and KLA peptides. Int J Nanomed. 2017;12:1517–1537. doi:10.2147/IJN.S122859
93. Luo P, Zhang Q, Shen S, et al. Mechanistic engineering of celastrol liposomes induces ferroptosis and apoptosis by directly targeting VDAC2 in hepatocellular carcinoma. Asian J Pharm Sci. 2023;18(6):100874. doi:10.1016/j.ajps.2023.100874
94. Chen S, Gao W, Chang S, et al. Mitochondria-targeting nanomedicines with autophagy inhibitor to enhance cancer photothermal-chemotherapy. Regen Biomater. 2025;12:rbae141. doi:10.1093/rb/rbae141
95. Zeng WN, Yu QP, Wang D, et al. Mitochondria-targeting graphene oxide nanocomposites for fluorescence imaging-guided synergistic phototherapy of drug-resistant osteosarcoma. J Nanobiotechnology. 2021;19(1):79. doi:10.1186/s12951-021-00831-6
96. Liu P, Wang Y, Liu Y, Tan F, Li J, Li N. S-nitrosothiols loaded mini-sized Au@silica nanorod elicits collagen depletion and mitochondrial damage in solid tumor treatment. Theranostics. 2020;10(15):6774–6789. doi:10.7150/thno.42661
97. Zhou J, Wang H, Wang W, Ma Z, Chi Z, Liu S. A cationic amphiphilic AIE polymer for mitochondrial targeting and imaging. Pharmaceutics. 2022;15(1):103. doi:10.3390/pharmaceutics15010103
98. Havelikar U, Ghorpade KB, Kumar A, et al. Comprehensive insights into mechanism of nanotoxicity, assessment methods and regulatory challenges of nanomedicines. Discov Nano. 2024;19(1):165. doi:10.1186/s11671-024-04118-1
99. Lazareva PI, Stupin VA, Lazarev KA, Litvitskiy PF, Manturova NE, Silina EV. Biodistribution and toxicological impact assessment of cerium dioxide nanoparticles in murine models: a systematic review of in vivo and ex vivo studies. Pharmaceutics. 2025;17(11):1475. doi:10.3390/pharmaceutics17111475
100. Ren T, Ma L, Fu P, Zhang C. Nanoparticles used for the delivery of RNAi-Based therapeutics. Pharmaceutics. 2025;17(11):1502. doi:10.3390/pharmaceutics17111502
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.