Back to Archived Journals » Virus Adaptation and Treatment » Volume 7
Recent advances in host–virus interactomics during entry and infection
Authors Bhattacharjee S
Received 30 June 2015
Accepted for publication 29 September 2015
Published 1 December 2015 Volume 2015:7 Pages 57—66
DOI https://doi.org/10.2147/VAAT.S60265
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Jonathan Dinman
Soumen Bhattacharjee
Cell and Molecular Biology Laboratory, Department of Zoology, University of North Bengal, Siliguri, Darjeeling, West Bengal, India
Abstract: Viral infections and pandemics result in millions of deaths worldwide each year. Viruses exploit host cellular processes, not only to gain entry and to deliver their genetic cargo, but also to counteract and use host immune defenses. To this end, a variety of ingenious strategies have evolved in viruses that involve fusion between virus and host membranes, channel formation through the host plasma membranes, disruption of the membrane vesicles, or a combination of these events. The entry and infection pathways of virus are thus largely defined by the interactions between virus particles and their cell surface and cytoplasmic receptors. A thorough analysis of virus–host interactomes may reveal novel mechanisms in virus entry, virus infection, and pathogenic strategies to modulate host metabolic pathways. The study of viral entry, infection, and pathogenesis has evolved over a long period. A host of next-generation technological advancements in this field has been discussed in this review.
Keywords: RNA interference, high-throughput, bioinformatics, viral entry, viral infection, virus–host interactions
Introduction
Viruses, obligate intracellular parasites, are metastable molecular assemblages that should be unlocked systematically during cellular entry by specific molecular and/or cellular environmental cues, with minimal energetic input.1 The transport of genetic material and other essential components through the host cell barriers requires, in most cases, precise attachment of the virions to the cell surface receptor(s) of the permissive host cells. Sometimes, viruses require additional co-receptor(s) on the host cell surface in order to complete successful entry within the cell. Therefore, the presence of cell receptors and co-receptors, where required, determines the fate of viral attachment and entry, especially in animal cells.
The diffusion rates of free virions toward the host cell, as well as the concentrations of virions and host cells, determine the probability and frequency of collision between them, and the initial interaction is guided by electrostatic forces.2,3 The subsequent high-affinity binding depends on hydrophobic and other forces whose strength and specificity are governed primarily by the conformations of the interacting viral and cellular interfaces.2 The avidity of virus binding to cells depends on the engagement of multiple receptor-binding sites on the virion and the fluid nature of the plasma membrane. Studies on the kinetics of virus spread in several viruses (human cytomegalovirus, vesicular stomatitis virus, and T7 bacteriophage) have been undertaken with the help of mathematical modeling.4–6 Investigation on the parameters of viral infection and the patterns of infection spread in oncolytic viruses (eg, modified adenovirus particles or conditionally-replicating adenoviruses [CRAds]) revealed that the probability of an initially infected host cell forming a plaque was 28% and the spread of infection from initially infected cells to the secondarily infected cells was generally inefficient.7
Subsequent to successful binding to specific cell surface receptor(s), the entry and uncoating of virion nucleocapsids through the plasma membranes is mediated by transport pathways specific to each group of viruses. The mechanism of virus entry can be simple fusion of viral membranes with cellular plasma membranes via activation of viral fusion proteins, as in many enveloped viruses (members of family Paramyxoviridae), and via receptor-mediated endocytic pathways (influenza virus, family Orthomyxoviridae).8 The entry of nonenveloped viruses, on the other hand, employs varied mechanisms: from disruption of endosome at low internal pH (adenovirus; family Adenoviridae), through pore formation at the plasma membrane or endosomes (poliovirus; family Picornaviridae), or via caveolae (SV40; family Polyomaviridae).1,9 Recently, macropinocytosis or cellular drinking has come to the focus, as a means of unusual clathrin-independent endocytic pathway of cell entry in a growing list of viruses, which include Ebola virus (EBOV; family Filoviridae), adenovirus 35, influenza A virus, Kaposi’s sarcoma-associated herpesvirus (family Herpesviridae), vaccinia virus (VV) extracellular virions (family Poxviridae), VV mature virions of the IHD-J strain, Nipah virus (family Paramyxoviridae), and Old World Arenaviruses (Lassa virus and Lymphocytic choriomeningitis mammarenavirus; family Arenaviridae).10,11 Viruses that require replication of their genomes within the host nucleus employ a variety of strategies to transport their genomic cargo: disassembly at the nuclear pore complex, through nuclear envelope disruption or both.12
Early studies on host–virus interactomics
Enzyme-based assays provided the initial clues concerning the chemical nature of cell surface components to which virions become attached. Early studies on identification of cell surface receptors depended on enzyme-based characterizations, such as in adenovirus and influenza virus.13,14 The development of technologies such as monoclonal antibody-mediated immunoprecipitation or affinity chromatography, expression of cell surface receptors in non-susceptible cells through gene transformation, and subsequent molecular cloning of cellular receptors from the transformed non-susceptible cells paved the way toward identification of myriad of cell surface receptors/co-receptors required for virus attachment (Table 1).15–17 These discoveries largely depended on the cloning of the receptor genes that allowed detailed mutagenesis analyses and structural studies on host–virus interactions.
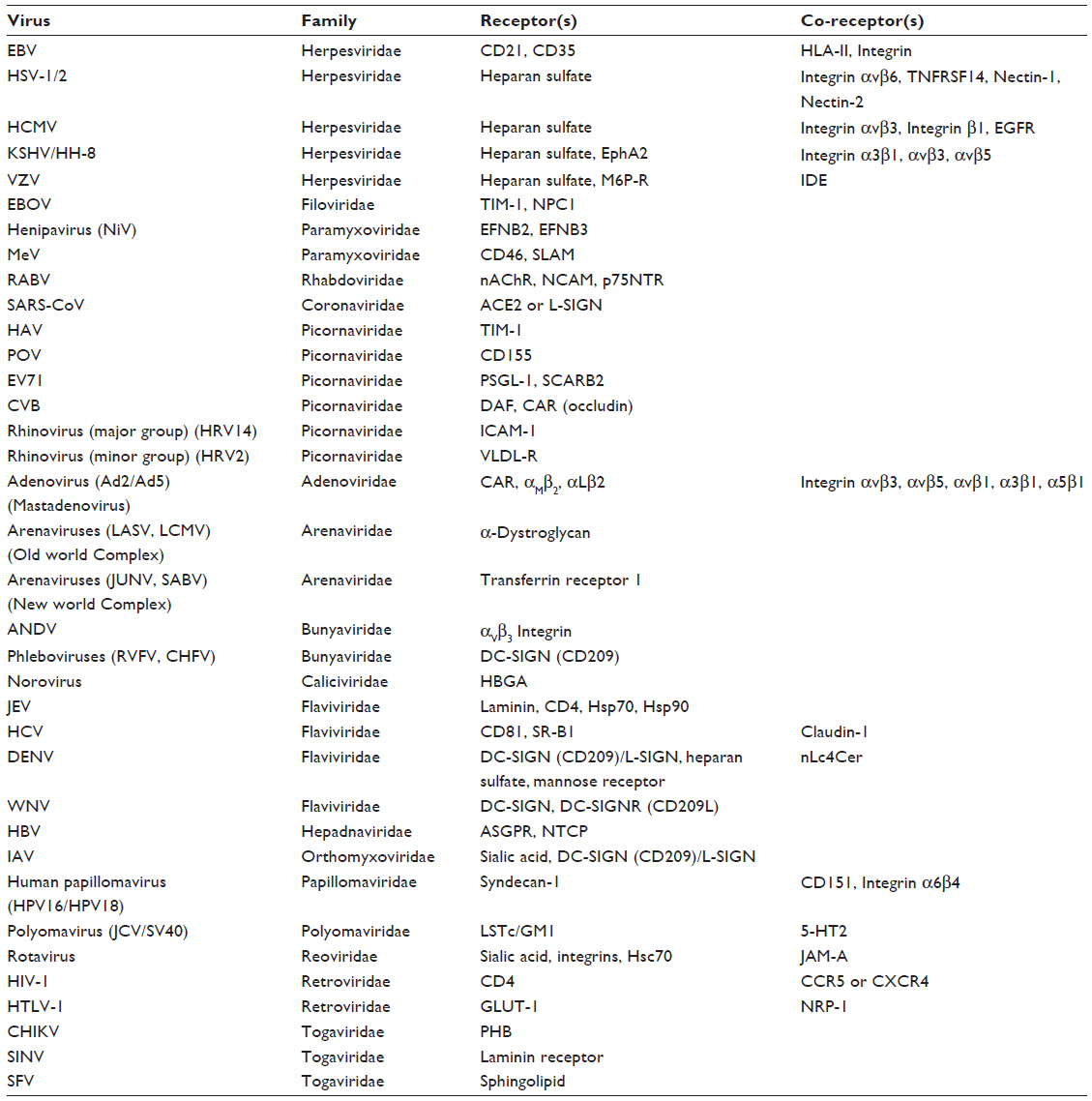 | Table 1 Known host cell receptors and co-receptors involved in attachment and entry of medically important viruses |
In contrast to studies on isolated viral proteins and their interactions with cellular targets, global analyses of virus–host interactions have been studied through a variety of genomics and proteomic tools.18 Numerous proteomic studies have been carried out to study the effect of viral infections on human and other cells. 2D gel electrophoresis of whole-cell lysates taken before and after infection followed by mass spectrometry (MS) identified the proteins detected in a gel.19,20 High-throughput yeast two-hybrid (HT-Y2H) assays have been useful in exploring protein–protein interactions (PPIs) in Saccharomyces cerevisiae using full-length predicted open reading frames.21 The study of virus–host PPIs has been possible through binary Y2H assays or complex affinity purification followed by MS.22 However, the Y2H system of detection of virus–host PPIs has few formidable limitations: frequent false positives; limitation of analysis of hydrophobic membrane proteins, owing to the expression of the reporter system within the nuclei; and lack of mammalian posttranslational modifications in yeast.20 Several strategies have been developed in order to overcome these limitations, such as the use of novel N-terminal bait and C-terminal bait and prey fusion-protein vectors, validation of Y2H data through biochemical and/or cell-based assays, and co-immunoprecipitation studies. In HT-Y2H assays, the limitations discussed above have been addressed through several ingenuous study designs, such as the LUminescence-based Mammalian IntERactome (LUMIER) mapping, and other variations of luminescence-based protein-fragment complementation assays, eg, split-yellow fluorescent protein (YFP) or split-luciferase-based methods.23–25
Virion purification and MS screens have identified several candidate human proteins that putatively interact with human immunodeficiency virus 1 (HIV-1; family Retroviridae).26 Physiological implications of virus–virus protein interaction in VV, hepatitis C virus (HCV), potato virus A, pea seed-borne mosaic virus, and T7 phage have also been studied.27 Systematic Y2H virus–host interaction screens, which are subsequently validated by a variety of methods, have been used to chart several virus–host PPIs, such as in HCV, Epstein–Barr virus (EBV), KSHV and varicella-zoster virus (VZV), dengue virus (DENV), and HIV-1.20,28–32 Chikungunya virus (CHIKV; family Togaviridae)–host-protein interactions have been investigated through HT-Y2H assays that are validated by protein interaction mapping.33 Out of total 30 distinct interactions, 22 high-confidence interaction data sets were generated that contained 21 interactions supported by nsP2 and one by nsP4. The results were further validated in protein complementation assay. In order to identify interacting host proteins, a CHIKV–host-protein interaction map was built up, which indicated several candidate proteins of translational machinery (hnRNP-K), RNA splicing factors (SRSF3), and cytoskeletal proteins (VIM, TACC3, CEP55, and KLC4), that are targeted by CHIKV nsP2. Other cellular proteins identified with nsP2 as bait are regulators of gene transcription and the proteins involved in protein degradation and/or autophagy. A similar study explored putative interacting host factors in severe acute respiratory syndrome-coronavirus (SARS-CoV; family Coronaviridae) replication.34
The Y2H and small interfering RNA (siRNA) screens have been useful in selecting host cofactors interacting with influenza virus, which Shaw classified into several gene clusters, namely, ribosome, coat protein complex I (COPI) vesicle, proton-transporting V-type ATPase complex, spliceosome, nuclear pore/envelope, and kinase/signaling.35 The involvement of ribosome in host mRNA degradation (influenza virus and SARS-CoV), vATPase in pH-dependent endosomal release (SARS-CoV, Semliki Forest virus; family Togaviridae), and nuclear pore-associated protein such as human KPNA1 (Karyopherin alpha 1 or importin alpha 5) and MAPK-signaling pathway in infections of SARS-CoV and HCV has been elucidated through these studies.20
The study of HIV–human protein interaction by Jäger et al employed affinity tagging and purification followed by MS that revealed several interactions of HIV-1 proteins in two human cell lines (HEK293 and Jurkat).36 The authors scored the PPIs using an improvised scoring system (MS interaction statistics) and plotted 497 HIV–human PPIs involving 435 individual human proteins in an interaction map. In addition to several known interactions, the authors discovered novel interactions, such as the interactions of HIV Pol and/or protease (PR) with that of host eIF3 complex subunits, thus indicating a role of this host-protein complex in inhibition of HIV-1 reverse transcriptase.36
In order to model virus infection at the cellular level, we need exhaustive PPI data not only on intravirus–host interactomes but also on virus–host interactomes. Although far from complete, the studies discussed above have generated a huge primary database of intraviral and virus–host PPIs. These results are now being validated by detailed computational and bioinformatic analyses. Such PPI networks and their analyses may prove to be a powerful resource for the development of antiviral therapeutic strategies.
The study of virus attachment and entry entered a new era in the last decade with the development of a host of gene-disruption technologies.37 These new technologies include multiplex screenings of small molecule inhibitors, RNA interference (RNAi) screening, haploid screening and genome editing, host–virus interactomics, 3D imaging, and in silico approach (Figure 1). This review explores recent advances and discusses the potential novel applications in the study of virus entry and infection.
 | Figure 1 Advanced tools in the study of host–virus interaction dynamics. |
New approach on virus attachment and infection
Genetic approach to identify host factors involved in viral infection
In an effort to understand the genetic basis of host susceptibility to viral infection/disease, researchers have identified genes that determine the outcome of virus–host interactions. Host proteins involved in susceptibility to viral infections have been explored through genetic mapping and positional cloning techniques in experimental animals. Although limited by the availability of inbred mouse strains and their inherent genetic diversities, these approaches are based on the identification of individuals who differ in susceptibilities to viral infections. Mice that differ in their inherent susceptibilities can be bred and the progenies can be subjected to linkage analyses to identify chromosomal location of candidate genes. Studies in this line have created low-resolution mapping of several mouse genes responsible for susceptibility/resistance to mouse cytomegalovirus (MCMV; reviewed in Webb et al38), West Nile virus39 and mouse adenovirus type 1 (MAV-140).38–40 The pattern of susceptibility/resistance to MCMV infection has identified the Cmv1 locus, a stimulatory member (Ly49H) of Ly49 receptor gene family (homolog of human KIR genes). The importance of Cmv1/Ly49H in modulating MCMV infection in mouse has underlined the role of stimulatory KIRs in controlling viral infections through their interactions with major histocompatibility complex-I (MHC-I) molecules.38 Interferon (IFN)-inducible proteins are important in determining the outcome of many host–virus interactions. Genetics and biochemical studies have established the inhibitory roles of IFN-induced 2′,5′-oligoadenylate synthetase (OAS) family of enzymes and RNAse L in infections with Picornaviruses and with Flaviviruses (West Nile virus) in mouse models. Systematic genetic crosses in inbred mice initially identified a resistance/susceptibility locus (Flv/Wnv) with a chromosomal region. Subsequent positional cloning technique identified the gene cluster encoding the IFN-inducible OAS family of proteins.39 Welton et al used a genome-wide search strategy, quantitative trait locus (QTL) detection, to identify host factors associated with susceptibility of congenic mouse to MAV-1.40 The H-2s haplotype of SJL/J mice was shown not to be associated with susceptibility to MAV-1, and the susceptibility of MAV-1-infected (BALB/cJ × SJL/J) F1 mice was intermediate between that of SJL/J and BALB/cJ mice, indicating that susceptibility is a genetically controlled quantitative trait. Through the analyses of 192 backcross progeny in a genome scan with polymorphic markers, a major QTL was detected on chromosome 15 (logarithm of odds [LOD] score of 21) and MAV-1 susceptibility was shown to be unlinked to the H-2 major histocompatibility locus. Through the development of high-resolution genetic map, a diverse panel of inbred mouse strains was used to identify QTLs associated with regulation of host responses to influenza infection.41 A novel N-ethyl-N-nitrosourea-mediated germline mutagenesis in MCMV-resistant mice and backcrossing led to the identification and positional cloning of host susceptibility genes.42 A similar approach was used to identify a mouse gene, Eif2ak4 (encoding GCN2), involved in susceptibility to MCMV and human adenovirus.43
Antiviral drug screening
Conventional therapy against viral infections has specifically targeted specific viral proteins and enzymes, but this therapy regime is fraught with danger of severe cellular toxicity or the chance of viral targets evolving to resistant varieties, owing to viruses’ intrinsic capacity for rapid genetic changes and evolution of their fitness levels. Therefore, the current need is to develop a combination therapy that increases specificity and efficiency, avoids selection of resistant strains, and enlarges the therapeutic arsenal. This requires alternative therapeutic interventions, such as targeting critical host proteins required for virus entry and replication. Documenting and understanding virus–host PPIs is a prerequisite to identify cellular “druggable” targets. Few promising host-protein antagonistic drugs are ready to be launched commercially, such as Maraviroc (a CCR5 co-receptor antagonist) for the HIV treatment, DAS181 (a recombinant sialidase fusion protein) for influenza virus infection, and TSG101 (human monoclonal antibody against a surface receptor expressed in infected cell) for a variety of viruses.32 Through a process of drug repurposing, the available libraries of small drug molecules can be rescreened for novel interactions with host cell proteins. Cell-based high-throughput screenings of drugs with known biological functions have been useful in identifying host proteins that enhance IFN signaling pathways or regulate replication of viruses, namely, HCV and influenza virus.32
In vitro cytopathic effects-based high-throughput screening assays to identify novel antiviral drugs against bluetongue virus,44 influenza virus,45–47 SARS-CoV,48 yellow fever virus,49 and Crimean-Congo hemorrhagic fever virus50 have identified unique antivirals. In a novel modification of studying virus–host interactions, a systematic large number of pair-wise drug interactions were explored.51 This multiplex screening method (multiplex screen for interacting compounds) employed a combination of cell culture, immunostaining, fluorescence imaging, and bioinformatics tools and tested approximately 500,000 drug pairs that synergistically inhibited HIV replication in HeLa-based MAGI cells expressing CD4 receptor. The multiplex screen for interacting compounds screening methodology not only identified promising drug pairs but also identified novel targets for HIV therapies that inhibit viral assembly and release.
RNAi screening
RNAi involves genetic modulation of virtually any gene by reducing its mRNA levels and, therefore, subsequent protein expression.52,53 The availability of libraries of siRNAs directed toward almost every human gene enabled several genome-wide high-throughput RNAi (HT-RNAi) screens to identify key host proteins involved in viral infections, such as in influenza virus,47,54 HIV-1,55–57 and HCV.58,59 Although these initial studies generated a wealth of promising leads for drug designing, the outcome of HT-RNAi screens is fraught with many pitfalls: errors in RNAi reagent design, inhomogeneous staining, differences in cell growing properties, transfection/infection efficiencies, signal-detection methodologies, data variations from plate to plate, as well as reliability and reproducibility of readouts. Therefore, bioinformatics and robust statistical approaches may increase the sensitivity of HT-RNAi technology in identifying bonafide hits.60 Integration of HT-RNAi screens with PPI databases may be approached to strengthen the significance and reliability of identification of host dependency factors, reduce false positives and false negatives, and identify novel cellular targets.60
Gene depletion
High-throughput genetic screens can identify not only specific virus–host interactions but also components of cellular pathways that are needed for virus replication. This could lead to the development of multiple drug targets. Novel haploid host–gene disruption through non-lethal antiviral insertional mutagenesis and gene trapping methods have led to the identification of host cell receptors for influenza A and EBOV infections.61,62 Haploid genetic approach has identified host factors required for entry of Lassa virus mediated by the Lassa glycoprotein, in addition to known entry receptor (glycosylated α-dystroglycan), receptor modifiers, and additional genes.63 However, haploid screens may miss biologically significant targets due to induced lethality by mutagenesis.37 More recently, Petersen et al employed insertionally mutagenized haploid human cell line, transcription activator-like effector nuclease-driven gene disruption, and a large-scale siRNA screen to identify members of the major cellular cholesterol regulatory pathway as important targets in entry of Andes virus (family Bunyaviridae).64
CRISPR/Cas9 genome editing
New genetic engineering technologies that specifically target genome loci through DNA double-strand breaks (DSBs) can be classified into three novel platforms: zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and the most powerful clustered regularly interspaced short palindromic repeat (CRISPR)-associated 9 (Cas9) guided silencing.65–67 These new gene-depletion strategies can be used to target putative host cell proteins suspected to be involved in virus interactions.
CRISPR loci evolved in bacteria and archaea as a part of adaptive immune system, which acquire foreign DNA resulting in the production of surveillance complexes consisting of short complementary guide RNAs (gRNAs) and an endonuclease (Cas9). When coexpressed in cells, gRNA and Cas9 can target DNA, namely, putative host-protein gene, for modification/silencing. The gRNAs contain a 20 base-pair guide sequence, which recruits the gRNA/Cas9 complex to its target sequence by base pairing. Successful binding of Cas9 to the target and subsequent endonuclease disruption requires the correct protospacer adjacent motif trinucleotide sequence immediately following the target sequence. Endonuclease Cas9 cuts both strands of DNA causing a DSB, which lies three to four nucleotides upstream of the protospacer adjacent motif sequence. The DSBs may be repaired by an error-prone non-homologous end joining (NHEJ) DNA repair mechanism, which often results in the generation of inserts/deletions at the DSB site. This may lead to disruption of open reading frames in the target genes.
A review on the applications of CRISPR/Cas9 technology in human papillomaviruses (HPV16, HPV18; family Papillomaviridae), hepatitis B virus (family Hepadnaviridae), EBV (family Herpesviridae), and HIV-1 has recently been published.68 These powerful genome editing technologies are sure to delve into many host–virus interactome studies in future.
Novel applications
Protein–protein interactomics and systems biology
Computational prediction of host–pathogen interactions has been developed that infers interactions among domains based on their occurrence in Y2H data. Sprinzak and Margalit demonstrated in S. cerevisiae that characteristic pairs of sequence-signatures can be learned from a database of experimentally determined interacting proteins in which one protein contains one sequence-signature and its interacting partner contains the other sequence-signature.69 The sequence-signatures that appear together in interacting protein pairs more often than expected at random are termed correlated sequence-signatures. Dyer et al combined this domain-based approach with Bayesian statistics to predict interactions among human and Plasmodium falciparum.70 Here, the researchers integrated a number of public intra-species PPI datasets with protein-domain profiles to develop a novel framework for predicting and studying host–pathogen PPI networks. This theme was translated into virus–human interactions where 182 unique human proteins with more than one viral interacting partner were identified.71 Tastan et al used a multitude of information in a learning framework, namely, co-occurrence of functional motifs and their interaction domains and protein classes, gene ontology annotations, posttranslational modifications, tissue distributions and gene expression profiles, and topological properties of the human proteins in the prediction of PPIs between HIV-1 and human proteins.72 The study predicted, among others, interactions of HIV-1 protein Tat and human vitamin D receptor, validating an earlier report that Tat acts with vitamin D receptor in a synergistic manner as a stimulator for HIV-1 LTR activity.72
While protein–protein-interaction databases provide a global view of cellular processes controlled by viruses, analyses of the structural details of individual proteins and their interaction interfaces would provide a dynamics of such interactions. Topological and functional analysis of virus–host interactome has been attempted through domain–domain interactions (DDIs).73 Search for DDIs from PPI dataset by Pfam scan and HMMER 3.0 against Pfam-A models identified 9,598 intrahost DDIs among 2,084 domains, 1,851 intravirus DDIs among 839 domains, and 269 virus–host DDIs between 87 viral domains from 49 viruses and 144 host domains after filtration. Based on this outcome, network distribution, network topology analysis, gene ontology enrichment analysis, and viral disease network were built up. Virus–host interactomes revealed that viruses use unique domains to interact with the same host partners with fundamental functions and conserved DDIs occurring in host interactomes to mediate the interspecies interaction. Thus, viruses seem to perturb the host cellular network by both common and unique strategies.73
In silico studies
Viruses have a strong tendency to directly and indirectly target host proteins that are central to viral infectivity and replication. These proteins are characterized by high values of connectivity and centrality in the cellular network. The study of viral infection at the systems level is now possible through the analyses of virus–host PPIs. These technically challenging, time-consuming, and expensive experiments can be replaced by in silico studies through algorithm-based modeling of “viral infectomes”. Viral proteins seem to interact with highly connected and central proteins within the host infectome network in order to control essential functions of the host cell. In silico simulation of these topological perturbations associated with viral infections can help to identify suitable targets for drug design.74
Posttranslational modifications of multifunctional cholesterol transporter protein Niemann–Pick C1 (NPC1) have been studied in relation to EBOV binding and cellular entry in human.75 NPC1 of nine species, including human, was subjected to multiple sequence analyses, using appropriate similarity matrix in ClustalW2. NetPhos 2.0 program was employed for phosphorylation site assessment of each Thr, Ser, and Tyr residue of the NPC1 protein using artificial neural network framework. Kinase specific phosphorylation sites in NPC1 were then predicted by NetPhosK 1.0 and KinasePhos 2.0. The O-β-GlcNAc modification potential sites in NPC1 were predicted by YinOYang 1.2 and ISOGlyP, which identified potential o-glycosylation sites (Yin Yang sites) and enhancement value product values as an indication of glycosylation rates, respectively. Analyses of solvent accessibility of predicted Ser and Thr residues were done with NetSurfP v1.1, and secondary structure analysis was conducted using POLYVIEW server. Docking of spike glycoprotein of Zaire EBOV was then carried out using structures of the receptor (NPC1), EBOV glycoprotein as ligand, and other active interacting residues for receptor and ligand positions as input with the help of HADDOCK server that used experimental knowledge-driven docking method. A host of revelations emerged from this study, highlighted by the prediction that NPC1-Yin Yang sites may not be important for virus attachment, whereas phosphosite 473 may be important for binding and entry of EBOV.75 In another study, EBOV–human PPIs were investigated through in silico virtual spectroscopy (informational spectrum method) method to decipher key interactions of EBOV glycoprotein and host endothelial extracellular matrix.76
Although far from complete and still in a developmental flux, huge primary databases of intraviral and virus–host PPIs have been built so far, such as in VirHostNet and VirusMINT.77,78 In combination with literature-derived low-throughput interaction data, Meyniel-Schicklin et al have used these high-quality curated PPI datasets in an integrative and comparative computational analysis of all intraviral and virus–human interactomes through the implementation of graph theory and other computational modelings.79 Their study tried to delineate the intraviral and virus–human PPI network architectures and systems behavior of several human virus networks (DENV, HCV, EBV, herpes simplex virus 1 [HSV-1], SARS-CoV, VV, and VZV). The analyses of virus–human interactomes revealed that viral proteins that target a high number of host proteins are predicted to be more disordered than viral proteins targeting a single host protein. Analysis of viruses, according to the human proteins they target, has identified common and specific viral strategies at the topological as well as the functional levels.79
3D imaging
Visualization of viruses in host cells can reveal finer details of the structures and interactions in virus entry, viral genome replication, assembly, maturation, escape, and further infection. Traditional electron microscopy (EM) methods, complemented by fluorescence microscopy (FM) techniques, now allow us to follow the dynamics of virus–cell interactions. Multistep events of influenza virus entry have been visualized using real-time microscopy, which provided new insights into cellular endocytic pathways.80 Two different host proteins that interact with the Sindbis virus at different stages of infection were identified using a green fluorescent protein-tagged viral protein.81 The interactions between Sindbis virus and host proteins were explored by using a mutant virus expressing the viral nsP3 protein tagged with green fluorescent protein.81 Direct observation of nsP3 localization and isolated nsP3-interacting proteins at various times after infection was recorded via FM. Their results revealed that host factor recruitment to nsP3-containing complexes was time dependent, with a specific early and persistent recruitment of G3BP and a later recruitment of 14-3-3 proteins.81
High structural, as well as temporal, resolution can be obtained through sub-diffraction FM and correlative EM/FM approaches.82 3D reconstruction by transmission electron microscopy serial sections, electron tomography, and focused ion beam scanning electron microscopy plays significant roles. Focused ion beam scanning electron microscopy uses scanning electron beam to generate 3D images from serial section of cells of a resin bed, which captures virus entry in susceptible host cells.83 Dramatic morphological changes in the plasma membrane at the site of entry for VV as well as changes in the vaccinia virion itself have been visualized in several studies.83
Conclusion
Organisms impose multiple barriers to virus entry. However, viruses exploit fundamental cellular structures and processes to gain entry to cells, unpack their genetic cargo, and initiate productive replication. Viruses seem to target common and central host proteins for remodeling the cellular signaling pathways and machinery. Therefore, gaining knowledge on the functions of the individual viral proteins in the host cell and that on their interactions with cellular signaling pathways is of paramount importance in understanding the pathogenesis. Systems virology, a highly integrated interdisciplinary systems biology approach in virology, has been instrumental in the global understanding of hitherto unknown virus–host interactions and especially of the dynamic nature of host responses against viruses. The integration of high-throughput array-based genetic screening, computational systems biology, and in silico strategies has substantiated known interactions and unraveled new interactions between the virus and host proteins. Systems biology approaches may help to identify the impact of virulence factors on the host system based on computational models of signal transduction and pathway analysis. Advances in computational biology in molecular dynamics simulations are required to understand unique features of protein structures and the chain of events that ensue with the initial attachment and entry of virus. The completeness of virus–host PPIs will be cornerstone in the structure-based predictions of virus–host DDIs and virus-induced network perturbations in viral infections. The virus-centric approach in developing antiviral drugs might fail to produce desired effects due to the potential genetic plasticity of the viruses, whereas a host-protein-based approach may identify a broad-range of antiviral drug target candidates. There are already a number of approved drugs designed against human proteins; however, considering the multidimensional nature of PPIs, there is a long way to go before the development of low-risk and low-cost antivirals.
Acknowledgments
The author apologizes to those experts whose valuable works could not be mentioned here due to space limitations. The author also acknowledges editorial help from Ms Sutanuka Chattaraj in preparing the manuscript.
Disclosure
The author declares no conflicts of interest in this work.
References
Marsh M, Helenius A. Virus entry: open sesame. Cell. 2006;124(4):729–740. | |
Puck TT, Garen A, Cline J. The mechanism of virus attachment to host cells. The role of ions in the primary reaction. J Exp Med. 1951;93(1):65–88. | |
Gilbert P-A, Kamen A, Bernier A, Garnier A. A simple macroscopic model for the diffusion and adsorption kinetics of r-Adenovirus. Biotechnol Bioeng. 2007;98:239–251. | |
Lam V, Boehme KW, Compton T, Yin J. Spatial patterns of protein expression in focal infections of human cytomegalovirus. Biotechnol Bioeng. 2006;93(6):1029–1039. | |
Haseltine EL, Lam V, Yin J, Rawlings JB. Image-guided modeling of virus growth and spread. Bull Math Biol. 2008;70(6):1730–1748. | |
Fort J, Mendez V. Time-delayed spread of viruses in growing plaques. Phys Rev Lett. 2002;89(17):178101. | |
Hofacre A, Wodarz D, Komarova NL, Fan H. Early infection and spread of a conditionally replicating adenovirus under conditions of plaque formation. Virol. 2012;423:89–96. | |
Kielian M. Mechanisms of virus membrane fusion proteins. Ann Rev Virol. 2014;1:171–189. | |
Mudhakir D, Harashima H. Learning from the viral journey: how to enter cells and how to overcome intracellular barriers to reach the nucleus. AAPS J. 2009;11(1):65–77. doi: 10.1208/s12248-009-9080-9. | |
Kunz S. Receptor binding and cell entry of old world arenaviruses reveal novel aspects of virus–host interaction. Virology. 2009;387(2):245–249. | |
Mercer J, Helenius A. Gulping rather than sipping: macropinocytosis as a way of virus entry. Curr Opin Microbiol. 2012;15:1–10. | |
Fay N, Pante N. Old foes, new understandings: nuclear entry of small non-enveloped DNA viruses. Curr Opin Virol. 2015;12:59–65. | |
Philipson L, Lonberg-Holm K, Pettersson U. Virus-receptor interaction in an adenovirus system. J Virol. 1968;2(10):1064–1075. | |
Bergelson LD, Bukrinskaya AG, Prokazova NV, et al. Role of gangliosides in reception of influenza virus. Eur J Biochem. 1982;128(2–3):467–474. | |
Flint SJ, Enquist LW, Racaniello VR, Skalka AM. Principles of Virology: Molecular Biology, Pathogenesis and Control of Animal Viruses. second ed. ASM Press; 2004. | |
Grove J, Marsh M. The cell biology of receptor-mediated virus entry. J Cell Biol. 2011;195(7):1071–1082. | |
viralzone.expasy.org [homepage on the Internet]. ViralZone. SIB, Swiss Institute of Bioinformatics. PMID:20947564. Available from http://viralzone.expasy.org/. Accessed November 23, 2015. | |
Bhattacharjee S. Role of genomic and proteomic tools in the study of host–virus interactions and virus evolution. Ind J Virol. 2013;24(3):306–311. | |
Maxwell KL, Frappier L. Viral proteomics. Microbiol Mol Biol Rev. 2007;71(2):398–411. | |
Ma-Lauer Y, Lei J, Hilgenfeld R, von Brunn A. Virus–host interactomes- antiviral drug discovery. Curr Opin Virol. 2012;2:614–621. | |
Uetz P, Giot L, Cagney G, et al. A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. | |
Bailer SM, Haas J. Connecting viral with cellular interactomes. Curr Opin Microbiol. 2009;12:453–459. | |
Barrios-Rodiles M, Brown KR, Ozdamar B, et al. Highthroughput mapping of a dynamic signaling network in mammalian cells. Science. 2005;307:1621–1625. | |
Nyfeler B, Michnick SW, Hauri HP. Capturing protein interactions in the secretory pathway of living cells. Proc Natl Acad Sci U S A. 2005;102:6350–6355. | |
Cassonnet P, Rolloy C, Neveu G, et al. Benchmarking a luciferase complementation assay for detecting protein complexes. Nat Methods. 2011;8:990–992. | |
Chertova E, Chertov O, Coren LV, et al. Proteomic and biochemical analysis of purified human immunodeficiency virus type 1 produced from infected monocyte-derived macrophages. J Virol. 2006;80(18):9039–9052. | |
Uetz P, Rajagopala SV, Dong YA, Haas J. From ORFeomes to protein interaction maps in viruses. Genome Res. 2004;14(10B):2029–2033. | |
Flajolet M, Rotondo G, Daviet L, et al. A genomic approach of the hepatitis C virus generates a protein interaction map. Gene. 2000; 242(1–2):369–379. | |
Calderwood MA, Venkatesan K, Xing L, et al. Epstein-Barr virus and virus human protein interaction maps. Proc Natl Acad Sci U S A. 2007;104:7606–7611. | |
Uetz P, Dong YA, Zeretzke C, et al. Herpesviral protein networks and their interaction with the human proteome. Science. 2006;311(5758):239–242. | |
Mairiang D, Zhang H, Sodja A, et al. Identification of new protein interactions between dengue fever virus and its hosts, human and mosquito. PLoS One. 2013;8(1):e53535. doi: 10.1371/journal.pone.0053535. | |
de Chassey B, Meyniel-Schicklin L, Aublin-Gex A, Andre P, Vincent Lotteau V. New horizons for antiviral drug discovery from virus–host protein interaction networks. Curr Opin Virol. 2012;2:606–613. | |
Bouraï M, Lucas-Hourani M, Gad HH, et al. Mapping of chikungunya virus interactions with host proteins identified nsp2 as a highly connected viral component. J Virol. 2012;86(6):3121–3134. | |
Pfefferle S, Schöpf J, Kögl M, et al. The SARS-coronavirus–host interactome: identification of cyclophilins as target for pan-coronavirus inhibitors. PLoS Pathog. 2011;7(10):e1002331. | |
Shaw ML. The host interactome of influenza virus presents new potential targets for antiviral drugs. Rev Med Virol. 2011;21:358–369. | |
Jäger S, Cimermancic P, Gulbahce N, et al. Global landscape of HIV-human protein complexes. Nature. 2011;481:365–370. | |
Kilcher S, Mercer J. Next generation approaches to study virus entry and infection. Curr Opin Virol. 2014;4:8–14. | |
Webb JR, Lee SH, Vidal SM. Genetic control of innate immune responses against cytomegalovirus: MCMV meets its match. Genes Immun. 2002;3:250–262. | |
Samuel CE. Host genetic variability and West Nile virus susceptibility. Proc Natl Acad Sci U S A. 2002;99:11555–11557. | |
Welton AR, Chesler EJ, Sturkie C, Jackson AU, Hirsch GN, Spindler KR. Identification of quantitative trait loci for susceptibility to mouse adenovirus type 1. J Virol. 2005;79:11517–11522. | |
Bottomly D, Ferris MT, Aicher LD, et al. Expression quantitative trait Loci for extreme host response to influenza a in precollaborative cross mice. G3 (Bethesda). 2012;2:213–221. | |
Crozat K, Georgel P, Rutschmann S, et al. Analysis of the MCMV resistome by ENU mutagenesis. Mamm Genome. 2006;17:398–406. | |
Won S, Eidenschenk C, Arnold CN, et al. Increased susceptibility to DNA virus infection in mice with a GCN2 mutation. J Virol. 2012;86:1802–1808. | |
Li Q, Maddox C, Rasmussen L, Hobrath JV, White LE. Assay development and high throughput antiviral drug screening against bluetongue virus. Antiviral Res. 2009;83(3):267–273. | |
Noah JW, Severson W, Noah DL, Rasmussen L, White EL, Jonsson CB. A cell-based luminescence assay is effective for high-throughput screening of potential influenza antivirals. Antiviral Res. 2007;73:50–59. | |
Severson WE, McDowell M, Ananthan S, et al. High-throughput screening of a 100,000-compound library for inhibitors of influenza A virus (H3N2). J Biomol Screen. 2008;13:879–887. | |
König R, Stertz S, Zhou Y, et al. Human host factors required for influenza virus replication. Nature. 2010;463:813–817. | |
Severson WE, Shindo N, Sosa M, et al. Development and validation of a high-throughput screen for inhibitors of SARS CoV and its application in screening of a 100,000-compound library. J Biomol Screen. 2007;12:33–40. | |
Gong E, Ivens T, van den Eynde C, Hallenberger S, Hertogs K. Development of robust antiviral assays for profiling compounds against a panel of positive-strand RNA viruses using ATP/luminescence readout. J Virol Methods. 2008;151:121–125. | |
Paragas J, Whitehouse CA, Endy TP, Bray M. A simple assay for determining antiviral activity against Crimean-Congo hemorrhagic fever virus. Antiviral Res. 2004;62:21–25. | |
Tan X, Hu L, Luquette LJ 3rd, et al. Systematic identification of synergistic drug pairs targeting HIV. Nat Biotechnol. 2012;30(11):1125–1130. | |
Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. | |
Timmons L, Fire A. Specific interference by ingested dsRNA. Nature. 1998;395:854. | |
Karlas A, Machuy N, Shin Y, et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature. 2010;463:818–822. | |
Brass AL, Dykxhoorn DM, Benita Y, et al. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–926. | |
König R, Zhou Y, Elleder D, et al. Global analysis of host–pathogen interactions that regulate early-stage HIV-1 replication. Cell. 2008;135:49–60. | |
Zhou H, Xu M, Huang Q, et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe. 2008;4:495–504. | |
Li Q, Brass AL, Ng A, et al. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc Natl Acad Sci U S A. 2009;106(38):16410–16415. | |
Tai AW, Benita Y, Peng LF, et al. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe. 2009;5:298–307. | |
Amberkar S, Kiani NA, Bartenschlager R, Alvisi G, Kaderali L. High-throughput RNA interference screens integrative analysis: towards a comprehensive understanding of the virus-host interplay. World J Virol. 2013;2(2):18–31. | |
Carette JE, Guimaraes CP, Varadarajan M, et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science. 2009;326(5957):1231–1235. | |
Carette JE, Raaben M, Wong AC, et al. Ebola virus entry requires the cholesterol transporter Niemann–Pick C1. Nature. 2011;477:340–343. | |
Jae LT, Raaben M, Riemersma M, et al. Deciphering the glycosylome of dystroglycanopathies using haploid screens for lassa virus entry. Science. 2013;340(6131):479–483. | |
Petersen J, Drake MJ, Bruce EA, et al. The major cellular sterol regulatory pathway is required for andes virus infection. PLoS Pathog. 2014;10(2):e1003911. | |
Carroll D. Genome engineering with zinc-finger nucleases. Genetics. 2011;188:773–782. | |
Miller JC, Tan S, Qiao G, et al. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol. 2011;29:143–148. | |
Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–338. | |
White MK, Hu W, Khalili K. The CRISPR/Cas9 genome editing methodology as a weapon against human viruses. Discov Med. 2015;19(105):255–262. | |
Sprinzak E, Margalit H. Correlated sequence-signatures as markers of protein–protein interaction. J Mol Biol. 2001;311:681–692. | |
Dyer MD, Murali TM, Sobral BW. Computational prediction of host–pathogen protein–protein interactions. Bioinformatics. 2007;23(13):I159–I166. | |
Dyer MD, Murali TM, Sobral BW. The landscape of human proteins interacting with viruses and other pathogens. PLoS Pathog. 2008; 4(2):e32. | |
Tastan O, Qi Y, Carbonell JG, Klein-Seetharaman J. Prediction of interactions between HIV-1 and human proteins by information integration. Pac Symp Biocomput. 2009:516–527. | |
Zheng L-L, Li C, Ping J, Zhou Y, Li Y, Hao P. The domain landscape of virus–host interactomes. BioMed Res Int. 2014;2014:13. Article ID 867235. | |
Navratil V, de Chassey B, Combe CR, Lotteau V. When the human viral infectome and diseasome networks collide: towards a systems biology platform for the aetiology of human diseases. BMC Systems Biol. 2011;5:13. | |
Basharat Z, Yasmin A. In silico assessment of phosphorylation and O-b-GlcNAcylation sites in human NPC1 protein critical for Ebola virus entry. Infect Genet Evol. 2015;34:326–338. | |
Veljkovic V, Glisic S, Muller CP, et al. In silico analysis suggests interaction between Ebola virus and the extracellular matrix. Front Microbiol. 2015;6:135. | |
Navratil V, de Chassey B, Meyniel L, et al. VirHostNet: a knowledge base for the management and the analysis of proteome-wide virus–host interaction networks. Nucleic Acids Res. 2009;37:D661–D668. | |
Chatr-aryamontri A, Ceol A, Peluso D, et al. VirusMINT: a viral protein interaction database. Nucleic Acids Res. 2009;37:D669–D673. | |
Meyniel-Schicklin L, de Chassey B, Andre P, Lotteau V. Viruses and interactomes in translation. Mol Cell Proteomics. 2012;11: M111014738. | |
Lakadamyali M, Rust MJ, Babcock HP, Xiaowei Zhuang X. Visualizing infection of individual influenza viruses. Proc Natl Acad Sci U S A. 2003;100(16):9280–9285. | |
Cristea IM, Carroll J-WN, Rout MP, Rice CM, Chait BT, MacDonald MR. Tracking and elucidating Alphavirus-Host protein interactions. J Biol Chem. 2006;281(40):30269–30278. | |
Chojnacki J, Muller B. Investigation of HIV-1 assembly and release using modern fluorescence imaging techniques. Traffic. 2013;14:15–24. | |
Risco C, de Castro IF, Sanz-Sanchez L, Narayan K, Grandinetti G, Sriram Subramaniam S. Three-dimensional imaging of viral infections. Ann Rev Virol. 2014;1:453–473. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.