Back to Archived Journals » Advances in Genomics and Genetics » Volume 8
Recent advances in computational epigenetics
Received 5 July 2017
Accepted for publication 28 September 2017
Published 28 December 2017 Volume 2018:8 Pages 1—12
DOI https://doi.org/10.2147/AGG.S115524
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr John Martignetti
Heather J Ruskin,1 Ana Barat2
1Advanced Research Computing Centre for Complex Systems Modelling, School of Computing, Dublin City University, Dublin, Ireland; 2Department of Physiology and Medical Physics, Royal College of Surgeons in Ireland, Dublin, Ireland
Abstract: Over the last two decades, the importance of epigenetics for interpretation of diverse genetic and genomic data has become increasingly clear. The need for study of indirect (non-gene) factors determining gene characteristic behavior and organism function, together with analysis of outcomes which are nondeterministic, is now well recognized. Given the increasing availability of large-scale datasets, analysis has inevitably become richer, but also more complex, and the formation of structured hypotheses, together with questions designed to refine experiment, relies heavily on computational tools. In particular, the effort to explore the whole genomic–epigenomic landscape has motivated an interdisciplinary approach towards large-scale multivariable and combinatorial analysis as well as novel database developments. Exploration of heritable changes in phenotype relies not only on newer sophisticated sequencing methods but also on legacy data, revisited for their contribution to understanding of transcriptional regulation and disease. The challenges presented are nontrivial, not least in terms of interpretation across multiple scales from cell to organism, but the field is advancing rapidly. With an early initial focus on cancers, both in development of models and database provision, work is emerging on brain function and neural pathways, while newer targets again are the behavioral sciences, in which interest is now burgeoning. In the following article, key developments and advances are summarized and current methods and tools reviewed.
Keywords: epigenetics, DNA methylation, histone modifications, computational modeling, data analysis, advances, sequencing, databases
Introduction
Following the breakthrough in the typing of the human genome,1,2 the need to understand how chemical modifications can alter gene involvement and function has prompted the study of the control system for gene switching. Developmental traits and differences, as well as disease initiation and progression, are all intrinsically linked to phenotypic plasticity (the degree to which non-genotypic factors determine phenotype form).3,4 Genetic and genomic attributes have been studied in detail, and current knowledge is captured in databases, which include ENCODE, Ensembl, Gene, GEO and GWAS for humans, as well as other organism-specific examples.5–9 The collated data are extensive and draw heavily both on state-of-the-art methods of gene sequencing and on extensive gene expression measurements, providing a basis for investigation of molecular evolution, disease-specific mutations and other. Despite this wealth of data, however, it is now clear that gene factors alone are insufficient to explain the complex mechanisms producing diversity and heritable changes in phenotype. In consequence, the last few decades have seen increased focus on the way in which reprogramming of the transcriptional regulatory network can occur.10–12
Cells control gene expression by wrapping DNA double strands around clusters of core histone proteins in order to form nucleosomes, the building blocks of chromatin. Chromatin structure is known to be affected in the neighborhood of expressed genes, particularly in the case of promoter and enhancer genomic regions.13 Hence, alterations in chromatin structure (caused by chemical modifications of both DNA and histone proteins) influence gene activity, causing speed up, slow down or even suppression of transcriptional initiation. These heritable alterations in the chromatin structure (which regulates transcription through gene expression or activation of protein- and RNA-encoding genes) leave the genetic code unaffected. Epigenesis is thus a second-order effect, which goes beyond the content of the genome to the way in which its message is compiled and implemented during development, cell proliferation and division.14–16
Various enzymes are involved in the chemical modification process and are associated with the epigenetic mechanisms, “signatures” or markers of change, which “punctuate” the genetic code.4,17 These fall broadly into chemical and protein groupings, with the former including DNA methylation and the latter various covalent posttranscriptional histone modifications such as methylation, acetylation, phosphorylation, ubiquitylation and sumoylation, with the first two being the more intensively studied to date.18–20 DNA methylation involves the addition of a methyl group to a DNA strand and commonly acts as a mechanism to switch the gene “off” permanently, while histone modifications directly impact chromatin structure and affect gene expression values. These changes are molecule- as well as modification-specific; for example, histone H3 acetylation and deacetylation promote increase and decrease in gene expression, respectively; histone H3 trimethylated at lysine 36 (H3K36me3) or at lysine 4 (H3K4me3) as well as histone H3 dimethylated at lysine 4 (H3K4me2) are marks associated with enhanced gene expression, while H3K27me3 is associated with its repression. An extra level of complexity is added by different histone variants (coded by separate genes) being differentially represented in “open” versus “closed” or “compact” chromatin domains.21,22 Epigenetic marks also reflect imprinting of genes by environmental factors such as diet and lifestyle, with such information also passed on to subsequent generations.23–26
In recent years, heterogeneous micromolecular abnormalities have become increasingly associated with risk, onset and progression of a range of conditions and diseases, such as obesity,11,27 mood disorders and other psychopathologies,28–30 autoimmune and cardiovascular diseases,31,32 as well as cancers33 and ageing.34–36 Moreover, the reversible nature and faster dynamics of epigenetic changes are of major interest in the targeting of intervention, providing key motivation for pharmaceutical development over the last decade.37,38
The need, therefore, to understand epigenetic changes and their influence on disease has stimulated development of numerous computational approaches and tools, for application to data generation, mapping and management, as well as analysis and therapy. While the Human Genome Project (1990–2003) focused on sequencing all genes in human DNA (~20,000, with some three billion base-pairs), a similar large-scale project, the Epigenomics Road Map (2008–date), is exploring specific patterns of epigenetic modifications, with the principal aim of creation of a map of the epigenome for multiple tissue types and cancers.30,39
In particular, the data richness of many biological and medical fields, fueled by new technology and improvements in computing power, has meant that analysis of the patterns of changes, which disrupt normal gene regulation, is now feasible. The challenges presented by the “extra layer” of control have also served to emphasize the importance of interdisciplinary approaches, combining related fields of genomics, such as biochemistry and proteomics, with the hybrid discipline of bioinformatics as well as with traditional aspects of computer science, mathematics and the physical sciences. The formulation of models to explain the biological processes, together with interrogation of diverse data sources and in-depth integrated analysis, is an essential feature of the new paradigm.40–44
In the following sections, modeling epigenetic dynamics from DNA methylation to histone modifications is considered, with achievements and challenges being discussed. The computational resources employed to manage various available data-types are considered, and technology-dependent methods and tools to produce quantitative epigenetic data are discussed next. Attempting illustration of the range of computational epigenetic methods is inevitably selective, due to restrictions of space here as well as to the exponentially growing literature base in different fields. In consequence, this review gives detail on a cancer example, while new directions and developments in other areas are briefly summarized.
Modeling epigenetic dynamics
Epigenetic modifications (both DNA methylation and histone modifications) are characterized by distinctive time scales. Given the natural heterogeneity of epigenetic and epigenomic changes and their combinatorial effect, control is dependent on different signature mechanisms and the multiple interactions between instances of these, which affect gene expression and functionality. Epigenetic regulation corresponds, in consequence, to emergent system behavior, with complex overall dynamics.45–47 Some histone modifications take place much faster than others and the time scale for histone acethylation is much shorter than that for DNA methylation for which the system remains relatively stable,48,49 but rates of change do not apply universally. Epigenetic dynamics have seen heightened interest from the computational modeling community, often in the context of disease initiation and progression. While early work focused on single changes, more recent efforts aim to explore interdependencies and the way in which system evolution occurs.
In an early application for gastric cancers,50 computational modeling played a complementary role for in vivo/in vitro experiments in hypothesis testing, providing insight into overall methylation dynamics. Moreover, the role of aberrant promoter methylation in transcription pattern modification, subsequently explored,51 demonstrated the long-term objective for a system model, namely multiple scaling of effects from aberrant cell changes to initiation and progression of disease. Phenomenological models (widely used in the physical and complexity sciences, including systems biology)52 were developed for epigenetic mechanisms, in order to support formulation of hypotheses based on limited data, which could be later refined. Computational micromodels, such as that described,45,53 utilized the Markov Chain Monte Carlo class of algorithms to mimic interdependence of epigenetic events through random sampling of states. Transcription information (as a function of histone modification levels and DNA methylation) permitted movement to new histone states, corresponding to associated transition probabilities, based on empirical data.
The importance of DNA methylation data in genomic stability and cellular plasticity, as well as in genomic imprinting and other normal cell processes, has motivated considerable efforts in modeling, prediction and analysis. Thus, in a study54 on epigenetic leukemia therapy, a dynamic multi-compartmental model of DNA methylation levels based on the activity of the Dnmt methyltransferase and other proteins is described. The numerical solution of the first-order partial differential equation model highlighted the mechanism for CpG island hypo-methylation via local modulation of such proteins. Further, in a discussion of asthma etiology,55 the authors considered epigenome-wide effects and distinguished between the accepted form of “integrator model” (for epigenetic changes leading to disease) and a two-stage model process. In the former, all factors including genetic variants and stochastic events have equal weight in influencing the production of intermediate phenotypes, while, in the latter, initial exposure of methylation quantitative loci leads to genetic variants, which are then modified further by DNA methylation and through additional exposure. (Genetic variants, such as SNP haplotypes, are notably a focus of GWAS.56) More recently still,43 the modeling of DNA methylation dynamics using phylogenetic approaches was proposed, with specific focus on changes in CpG dinucleotides, vital to cell differentiation, as well as the structure of precursor and dependent cell types. A continuous-time Markov chain was used to draw inferences on CpG methylation dynamics.
Identification of intraindividual epigenetic variation with a view to understanding the molecular basis for disease risk has motivated epigenome-wide association studies (EWAS),57,58 and the comparative basis is echoed in development of models such as AgentCrypt.59 Here, the agent-based approach is used to explore intra- and interdependencies in human intestinal crypt structure and dynamics, together with the effect of potential inhibitors on methylation modification of intestinal tissue in disease onset.
Relatively recently, investigation of the interrelationship between histone modifications and DNA methylation has indicated that specific epigenetic combinations determine whether chromatin structure is open or compact.21,22 Specific models for histone modification patterns, the histone code and contributions to the epigenome dynamics have also attracted increased attention in recent years. Thus, in a study,46 the authors explored modifications to the histone code and specialized enzyme recruitment leading to alternative and stable heritable states, which “mark” the DNA sequence and control functions, such as gene expression. The dichotomy of such “marks” whether active or repressive was also considered by Ku et al60 who developed a mathematical model of histone modification dynamics, where bivalent domains are thought to play an important role in stem cell differentiation and are related to known features of chromatin states. Further, a stochastic mathematical model proposed42 describes molecular mechanisms involved in establishing histone modification patterns for a single gene, with non-phenomenological physical parameters.
In efforts to relate histone modifications, DNA methylation and higher-order chromatin structure, a model of transcriptional regulation of epigenetic processes was proposed with a view to reconciling earlier models with experimental data.61 Performance was assessed in terms of stability properties and memory effects, with emphasis placed on experimental validation of theoretical predictions and the need for extension to multi-scale models to explore self-organization of chromatin. Cross talk between DNA methylation pathways and histone modifications has also been considered,20 as have multi-scale effects in a generalized nucleation and looping model for epigenetic memory of histone modifications.44 Cell mechanisms, producing and sustaining these patterns, were investigated as a prerequisite for predicting efficacy of epigenetic drug therapy.
It has been suggested that inheritance of the “epigenetic code” can be cumulatively summarized in terms of the “Epigenetic Code REplication Machinery” (ECREM), a macromolecular complex, consisting of enzymes such as the DNA methyltransferases, of chromatin organization and noncoding regulatory RNA.32,62 The four mechanisms, identified, include (a) DNA methylation, (b) histone modification, (c) chromatin remodeling and (d) involvement of small (21–26 nt) and noncoding RNAs, whose role in cellular development and protection has been shown to be vital to the epigenetic regulatory network.63 It seems clear that epigenetic events thus closely control gene expression and genomic regulation through multiple generations, with deregulation resulting in phenotype variability and increased susceptibility to disease.64,65 A ideal, comprehensive model, compliant with ECREM principles, would encompass information on all mechanisms involved and dynamically monitor cumulative changes. To date, this goal has not been realized.
Data management – new developments
Data resources and types
While PubMed records over 50,000 papers on epigenetics to date, with more than a third of these appearing since 2013, data generation discussions have been dominated by the intra-generational rather than the inter-generational processes (ie, inheritance of modified phenotype).26 High-level descriptions of biological processes and their concomitant entities are available from the literature and experimental studies, but quantitative data, mainly captured through molecular and epigenetic databases, are increasingly abundant. Such resources range from the small and specialized to extensions of well-established and wide-ranging repositories of nucleotide sequences, transcriptional regulatory sites and transcription factors for human genes and diseases, as well as microarray and gene expression data. Current lists and descriptions are available from Internet hub sites, such as EMBL-EBI (https://www.ebi.ac.uk/training/online/course/bioinformatics-terrified/what-database/relational-databases/primary-and-secondary-databases), NAR (http://www.oxfordjournals.org/nar/database/c/), NGS (https://www.nextgenerationsequencing.info/bioinformatics/genetic-databases/general-genetic-databases), HSLS (https://www.hsls.pitt.edu/obrc/index.php?page=human_genome), TCGA (https://cancergenome.nih.gov) and NCBI (https://www.ncbi.nlm.nih.gov/genbank/).
Nucleotide data,66 generated through next-generation sequencing methods, including RNA, whole genome and exome, as well as targeted technologies,67,68 predominates. Substantial data on gene expression through microarrays and RNA sequencing (including nanopore variants) are also available.69 Specific efforts for epigenetic measures have focused on DNA methylation content and patterns, as well as chromatin-associated proteins and methylated genes in various cancer types and other diseases. The value of the data types has been discussed in articles, including the one by Bock and Lengauer70 which describes inferences on epigenetic states from DNA sequences and the one by Lim et al71 which also reviews contemporary databases and tools, and more recently still through shared internet resources, such as Epigenie72 which incorporates information on current large-scale projects, databases by data type and statistical data analysis and visualization tools.
Databases: graph databases – a new approach
Epigenetic and epigenomic databases have expanded enormously over the last two decades.38,73,74 Of these, the Krembil Family Epigenetics Laboratory captures methylation data on human chromosomes 21 and 22 as well as information on male germ cells and DNA methylation in twins, while MethyLogiX DNA methylation database has similar coverage, targeted to late-onset Alzheimer’s disease. In addition, a number of epigenetic databases have been developed for furthering investigation of different disease types. Small-scale examples include StatEpigen75 (developed to investigate molecular determinants and statistical relationships in colon cancer), while larger-scale relational databases include MethyCancer76 and the broader Catalogue of Somatic Mutations in Cancer (COSMIC).77 Other well-known examples include PubMeth78 and MethInfoText.79 MethDB provides information on DNA methylation content and patterns across a number of species, tissues and phenotypes.80 Other methylation information is contained in MethBank,81 which focuses on integrated next-generation methylation programming data, MethPrimerDB,82 which captures primer sets for human and murine DNA methylation analyses, and REBASE,83 which captures data from GenBank on thousands of DNA methyltransferase genes. Histone sequences (H1, H2 and H2B, H3 and H4) are available in total for nearly 900 species from the Histone Database,84 while HIstome85 contains data on human histone proteins and modifying enzymes. Chromatin-associated protein information and chromatin-remodeling factor sequences in eukaryotes are available from ChromDB86 and CREMOFAC,87 respectively, while CR Cistrome88 contains CHiP-seq data for human and murine histone modification linkages and chromatin regulators.
Representation and querying of these complex systems requires relational statements, linking the multiple interdependencies between genetic and epigenetic modifications. Very recently, however, it has been recognized that structured data management is not the only requirement; the complementary need is 1) for linkage and integration of multiple data types, designed to comply with different data schema, and 2) investigation of advanced hypotheses requiring complex and time-consuming query forms. In consequence, a novel graph database approach, which supports both integration and query speed-up, but also has wide-ranging context, has been proposed.38 Nodes and edges in the graph database, respectively, represent concepts and associations, with the framework readily adaptable to highly interconnected data. Advantages include the use of graphical search algorithms and next-neighbor node-linked traversal searches, which give additional flexibility compared to the more conventional relational databases. Various graph database frameworks exist, including FlockDB, AllegroGraph and Neo4j amongst others,89–91 with the last permitting both multi-relational graphs and directional relationships as well as supporting a flexible declarative query language (Cypher). In particular, the Neo4j framework has found considerable application in the biomedical sciences and can be used to complement data integration as well as exploratory analysis and visualization. Examples of interactive query tools for integration and management of different medical and biological data types are given, together with Neo4j linkages for data management and analysis (Figure 1).38
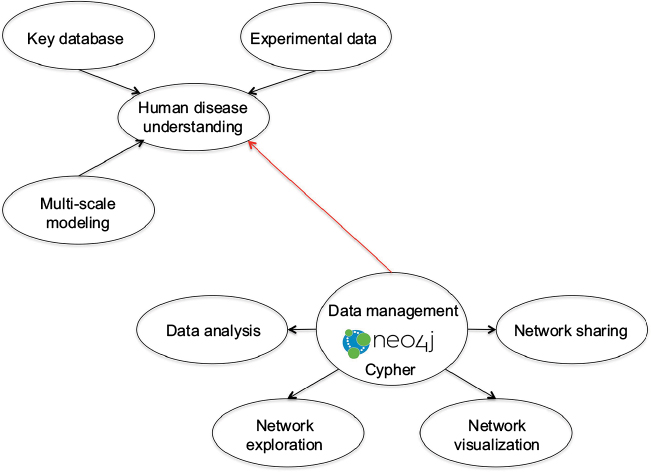 | Figure 1 Schematic diagram of Neo4j. Notes: Important to understanding human diseases, multi-scale computational models link the micromolecular layer and genetic–epigenetic alteration to organism development. Extraction and collation of data describing biomedical systems draws heavily on key publicly available databases (such as UniProt KB, Human Protein Atlas, Reactome, IntAct) as well as project-specific experimental datasets. Graph databases facilitate integration and querying of these heterogeneous datasets. The Neo4j graph database manages and presents incorporated data for analysis (using primarily R, Java and Python), in order to explore and visualize the interconnectivity of the integrated concepts. The Neo4j output graph (available in the JavaScript Object Notation format) can be processed further and linked to network sharing frameworks. Figure courtesy of Dr IA Balaur. |
Neo4j-based frameworks have also been used to assess performance of in silico models of biological systems, notably computational and mathematical models of cancer92,93 (and the BioModels database).94 Moreover, FlockDB supports application to reset rather than traversal searches (based on adjacency graph storage), providing a platform, similar to that of, for example, GraphLab, MapReduce and Scope (amongst others), for scalable execution. A comparative review of these machine learning paradigms for Cloud is provided in the work by Low et al.95 Corbellini et al96 provide an overview of graph databases for graph processing frameworks and for large-scale (predominantly social) networks in their work.
Targeted analysis, methods and tools
The increasing richness of resources for different data types has required corresponding elaboration of algorithms and tools.72,73 Recommendations for the design and analysis of EWAS as well as the interpretation of the complex data generated have been put forward by Michels et al97 and Birney et al98 among others.
In epigenome mapping initiatives, computational modeling has motivated use of a range of new technologies in an attempt to correlate characteristic behavior and explore joint methylation profiles for multiple targets. Thus, DNA methylation arrays (ChIP-Chip), ChIP-seq, methylation-targeted sequencing (eg, methylated DNA immunoprecipitation sequencing), bisulfite sequencing and others all feature widely, while various tools have been developed. These include ACME for identification of ChIP enrichment sites99 and aids for mapping both short and long bisulfite sequence to the reference genome,100,101 as well as tools for quantitative measurement of cytosine methylation levels, with examples including Bismark102 and MOABS.103 The latter is based on a beta-binomial hierarchical model for differential methylation, while a similar regression basis is used to model whole-genome bisulfite data in detection of differentially methylated sites in RADMeth.104 Bisulfite sequence-mapped data of count form have a complicated variance–covariance structure, but recently MACAU,105 a tool to identify differential DNA methylation, based on a binomial mixed model which takes account of both over-dispersion and genetic relatedness, has been described. Bayesian-based model tools include Bis-SNP,41 which identifies allele-specific epigenetic events, as well as a faster version BS-SNPer.106 Novel high-throughput nanopore sequencing variants, as well as diversity in technological platforms and in required sequencing depth, have also stimulated contributions to the recent literature.107,108
Increasingly, sophisticated bioinformatic methods are required for downstream analyses as researchers attempt to interpret multi-locus methylation information from multiple samples, for example, methods such as model-based clustering described by Houseman et al,109 tailored for data obtained with methylation-specific microarrays. Multivariate statistical methods, particularly for both supervised and unsupervised clustering, principal component analysis, regression and visualization tools, such as heatmaps, have proved vital to interpretation of outcomes for these complex data,110 which are generated by combinations of epigenetic changes and molecular events.111,112
A major challenge faced by EWAS is intra-sample cell-type heterogeneity (different fractions of component cell types in the sample), and a number of statistical algorithms have been developed to address this issue. These algorithms can be classified as reference-based (with defined a priori DNA methylation profile for the tissue of interest) and reference-free (with a tissue-specific DNA methylation profile unavailable).113–115
Text- and data-mining examples for extraction of epigenetic information from the literature, together with appropriate computational, mathematical and statistical methods, are widely reported.116–118 Pooling summary-level genome-wide and epigenome-wide studies may provide powerful new insights,119 with combined query criteria highlighting multiple levels of control, which can apply to even a single change, as noted.38 Furthermore, the previous focus of the epi-informatic approach, on DNA methylation and histone modification and the patterns that apply in various disease manifestations, has now extended to the integration of data within a scaffold network such as that for protein interaction, which specifies correlation between methylation and gene expression. Abnormal epigenetic marks may appear in cells of different types, with increased phenotypic plasticity associated with these anomalies and crucially linked to network properties, which can offer insight for diagnostics and therapy.120,121 In this context also, genetic tools, such as genome-scale libraries, are attracting epi-informatic efforts, with other posttranscriptional modifications also used to investigate modulators of protein stability and mediate loss-of-function screening, for example for cancers.
The case for cancer and data integration
Many epigenetic and epigenomic studies have focused on cancer, but distinction between cancerous and healthy states is not straightforward as cancer is neither a single disease nor uniform in progression or markers.64,122 Epigenetic variability is intrinsic in normal tissue, so that achieving reliable targets for diagnosis and treatment of malignancies is heavily dependent on this and on the molecular properties that distinguish cell classes.123,124 Major disruption in cell-cycle mechanisms of molecular adhesion and regulation results in abnormal gene expression and mutation of tumor-suppressor genes in tumors and neighbor tissue.40,65 Transcriptional states and gene mutations are some of the many properties, operating at the genome level, that characterize cancer phenotypes, but refinement of these classifications requires additional epigenetic information. Core relationships between DNA and histone proteins contribute to de-regulation of nuclear events in cells, including DNA damage repair as well as replication and transcription, and are prominent in disease initiation and progression.19,125 In a recent review of an earlier model, its developers suggest that some genes are epigenetically disrupted even before occurrence of mutations leading to malignancies, causing altered differentiation throughout tumor evolution.33
Many correlations between DNA-dependent events and histones also occur at the level of histone posttranslational modifications, leading to recruitment of non-histone proteins via specialized binding domains, rather than to alterations in nucleosome structure (the Histone Code hypothesis). Changes to chromatin conformation, effected through these histone modifications and binding of methyl residues on DNA cytosines from CpG dinucleotides, lead to “closure” and impedance of transcription. Considerable emphasis is also given in the literature to abnormal DNA methylation and hypermethylation of CpG islands situated close to promoter regions, as well as concomitant methylation of multiple loci, with strong indications that downregulation of expression of core genes results, together with distinctive phenotypes.12,16,126,127
In the particular case of cancer, molecular subtypes (or stratification) reflect both disease etiology (with different molecular mechanisms disrupted in distinct subtypes) and different cell compositions. The former is evidenced, for example, by levels of differential gene expression and different sets of somatic mutations, and the latter by cell fractions; for example, in colon cancer, the mesenchymal128 subtype contains a larger fraction of stromal cells than other subtypes. The characterization of subtypes is important as these can respond differentially to various treatments, but the importance of methylation data in terms of cancer molecular subtyping has been recognized only recently. Current computational methods for determining neoplastic disease subtypes are based on identifying groups of differentially expressed genes (ie, biomarkers) that can best discriminate between these. However, these methods can be unreliable since they yield different biomarker sets when applied to data from different studies.129 Thus, in addition to using network approaches128–130 to refine and better characterize existing subtype signatures, integrating -omics data of different types can enhance molecular subtyping of malignant neoplastic disease. In an analysis on colon cancer data, for example, consideration of genome-wide methylation in the context of expression-based subtype data derived from different datasets revealed that two molecular subtypes, little differentiated by expression, were distinguishable with respect to locus-specific methylation131 (illustrated in Figure 2 for subtypes: Goblet-like/C2 and Inflammatory/C3 cells), and confirmed for a larger set of samples.128
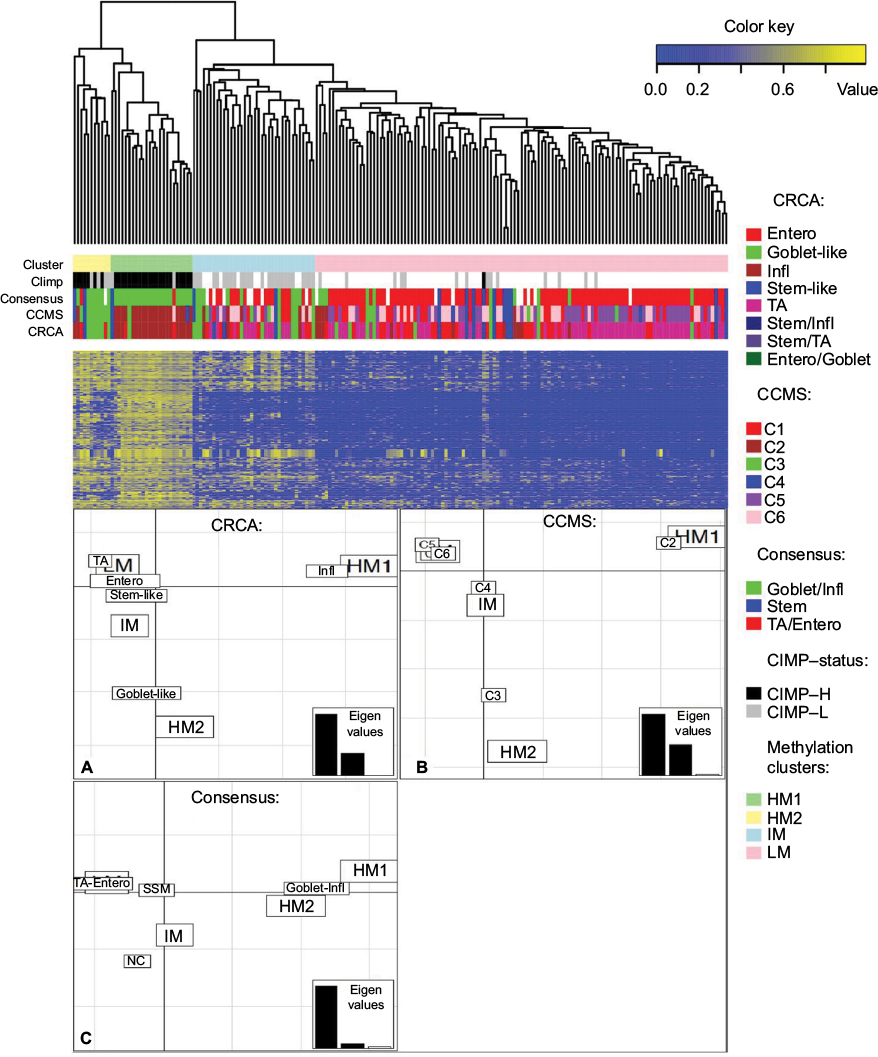 | Figure 2 Clusters of colon cancer methylation data from the NIH TCGA portal obtained with average linkage clustering (methylation clusters HM1, HM2, IM and LM) are shown with corresponding matched expression-based subtypes obtained from three different subtyping schemes, namely CRCA (CRCassigner-786),132 CCMS (Colon Cancer Molecular Subtypes)133 and consensus134 (involving 5, 6 and 3 subtypes, respectively). Notes: The clustering analysis on the upper part of the figure shows that the first two subtyping schemes classify most samples from the two highly methylated clusters HM1 and HM2 in two different expression-based subtypes: CRCA stratifies the samples to Infl and Goblet-like subtypes and CCMS to C2 and C3 respectively. For the third signature (consensus),134 these samples are classified to one single subtype (Goblet/Infl). The lower part of the figure illustrates FCA for the methylation clusters and these three expression-based subtyping signatures (panels A–C), spatial proximity between two labels on the factorial plane illustrating closeness/correspondence of the labeled modalities. FCA shows how subtypes Infl and C2 are very close to HM1 but clearly distinct from Goblet-like and C3 subtypes, which are in turn very close to HM2 (panels A and B), demonstrating how these subtypes can be distinguished by their respective methylation profiles. Note how for the third ‘consensus’ signature (panel C) the HM1 and HM2 labels do not appear separated any more, but are brought close together by their correspondence to the fused subtype ‘Goblet-Infl’. SSM, stem/serrated/mesenchymal134 – a subtype belonging to the consensus subtyping scheme.134 TA, transit-amplifying (in the CRCA subtyping Scheme,133). Figure adapted from Barat A, Ruskin HJ, Byrne AT, Prehn JH. Integrating colon cancer microarray data: associating locus-specific methylation groups to gene expression-based classifications. Microarrays (Basel). 2015;4(4):630–646. © 2015 by the authors; licensee MDPI, Basel, Switzerland. Creative Commons license available at: https://creativecommons.org/licenses/by/4.0/legalcode.131 Abbreviations: CIMP, CpG island methylator phenotype; CIMP-H, CIMP high; CIMP-L, CIMP low; Entero, Enterocyte; FCA, factorial correspondence analysis; HM, high methylation; IM, intermediate methylation; Infl, inflammatory; LM, low methylation; NC, not belonging to any subtype; NIH, [US] National Institutes of Health; TCGA, The Cancer Genome Atlas. |
It is clearly important to ensure quality of methylation data in integrative analyses. A large proportion of cancer archival data (with extensive histological and clinical–pathological records and other -omics data linkage) is available as formalin-fixed paraffin-embedded (FFPE) samples, and concerns have been raised as to the impact of this preservation method on quality of sequencing.135 Nevertheless, recent research135,136 has shown that targeted sequencing can be successfully used to assess genome-wide methylation from FFPE samples, with an investigation of methylation calling in matched fresh-frozen and FFPE samples. Genome-scale DNA methylation assessment has led to mixed results, however, in terms of establishment of methylation prognostic profiles, for example, on oral carcinoma,137 where the authors also discuss problems associated with pre-processing, filtering and data normalization for downstream analysis. No consensus pre-processing guidelines currently exist for some quantitative platforms (such as Illumina HM450K in this example), with prediction heavily reliant on machine-learning methods, and only partial information typically available for screening or to signpost clinical outcomes.116,127,130,133,137 Successful application of machine learning and data-mining methods for complex genomic data inevitably relies on exploiting information on the inherent data structure and different data types, as well as attention to practical implementation and interpretation.
Widening the scope: new directions
Ageing, as a major risk factor in many diseases, has stimulated considerable epigenetic research efforts over recent years. The role of stochastic epigenetic variation as a driving force in evolving health and development of disease has been considered,25 while quantitative aspects of human ageing rates have been investigated through genome-wide methylation profiles.34 In an epigenome-wide study, the authors discuss both cross-sectional and longitudinal DNA methylation changes and have identified more than 60 novel age-associated CpG sites, endorsing increased susceptibility to disease.138 The dynamics of DNA methylation in ageing have also been explored through integrative data analyses,139 while an investigation of epigenetic regulation of ageing has looked at the relationship between environmental inputs and genomic stability.36
Epigenetic imprinting, regulation, modulation and inheritance questions have also been investigated for metabolic diseases, such as diabetes and obesity,11,27 heart disease,31,32 respiratory impairment140 and others. Important evidence in recent years has also linked neuropsychiatric disorders with epigenetic marks as biomarkers of disease mechanisms and progression and of lifestyle exposure. In a recent paper, for example, the authors consider reconciliation of diverse data and discuss the efficacy of cross-tissue analysis, particularly combined with blood-based studies, for assessment of effectiveness of longitudinal courses of treatment.30 Epigenetic investigation in brain function and behavioral studies is at a relatively early stage, but interest is growing rapidly, for example in pediatric psychology29,141 and more generally.39,142 The impact of early life experience on the epigenetics of neural development can have persistent effects into adulthood143 and is being examined in the context of childcare, family structure and parenting practices. Computational models are also being developed for behavior and neural activity associated with anxiety traits and mental illness, and a recent review discusses the interplay of environmental factors with epigenetic regulation and plasticity in order to explore development of psychiatric disorders.144
New model paradigms are still being explored to describe fundamental epigenetic mechanisms and processes involved in phenotypic plasticity. One such proposed145 advocates the use of insect-based models to represent environmental or lifestyle insults affecting epigenetic regulation, since insects have the ability to produce distinct phenotypic variants from the same genotype through transcriptional reprogramming. The authors argue that not only does this imply relative cost-effectiveness in realizing experimental results, but also enables epigenetic trans-generational effects of environmental factors to be investigated in relation to cancers, neurodegeneration, ageing and infectious diseases.
Epigenetic inheritance and its role in evolutionary biology continue to pose many unanswered questions. It has been suggested, for example, that epigenetic drift has distinct evolutionary advantages,35 while investigation of epigenetic modulators and their implications for gene expression and therapeutics, in particular, is a major target for future research.33,37
Conclusion
Many discussions on computational epigenetics have focused on generation of data and the ever-increasing wealth and diversity of large-scale databases and tools to mine them. However, this is still only part of the story. Managing and mapping for “-omics” studies are important steps, but the real challenge now is interpretation of these data in order to quantify risk and drive therapeutic development and disease management. System complexity means that questions posed are already challenging basic analysis, and it is clear that more sophisticated model frameworks and novel bioinformatic approaches will be demanded in order to draw meaningful statistical inferences for disease groups and individual profiles. There are indications in recent work that this overarching challenge is now being targeted. Newly emerging theories and model paradigms, efforts at integrative analyses involving multiple data types and the emergence of epigenetic biomarkers offer potential to address disease in novel ways, developing new directions for therapeutic strategies and preventive medicine. The role of computational epigenetics in developing the theories, models and methods required to make sense of complex biological and medical data cannot be overestimated.
Acknowledgment
The authors thank Dr IA Balaur for providing Figure 1, Neo4j schematic, and for useful discussions on graph databases.
Disclosure
The authors report no conflicts of interest in this work.
References
Bentley DR. Decoding the human genome sequence. Hum Mol Genet. 2000;16:2353–2358. | ||
Lander ES, Linton LM, Birren B, et al; International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. | ||
Fusco G, Minelli A. Phenotypic plasticity in development and evolution: facts and concepts. Philos Trans R Soc Lond B Biol Sci. 2010;365(1540):547–556. | ||
Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature. 2010;465(7299):721–727. | ||
Barrett T, Troup DB, Wilhite SE, et al. NCBI GEO: archive for functional genomics data sets—10 years on. Nucleic Acids Res. 2011;39(Database issue):D1005–D1010. | ||
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. | ||
Herrero J, Muffato M, Beal K, et al. Ensembl comparative genomics resources. Database (Oxford). 2016;2016:bav096. | ||
MacArthur J, Bowler E, Cerezo M, et al. The new NHGRI-EBI catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017;45(Database issue):D896–D901. | ||
NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2017;45(Database issue):D12–D17. | ||
Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell. 2013;152(6):1237–1251. | ||
Seki Y, Williams L, Vuguin PM, Charron MJ. Minireview: epigenetic programming of diabetes and obesity: animal models. Endocrinology. 2012;153(3):1031–1038. | ||
Suvà ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339(6127):1567–1570. | ||
Hu Z, Tee WW. Enhancers and chromatin structures: regulatory hubs in gene expression and diseases. BioSci Rep. 2017;37(2):BSR20160183. | ||
Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23(7):781–783. | ||
Tammen SA, Friso S, Choi SW. Epigenetics: the link between nature and nurture. Mol Aspects Med. 2013;34(4):753–764. | ||
Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153:38–55. | ||
Basso M, Sleiman S, Ratan RR. Looking above but not beyond the genome for therapeutics in neurology and psychiatry: epigenetic proteins and RNAs find new focus. Neurotherapeutics. 2013;10(4):551–555. | ||
Alelú-Paz R, Ashour N, González-Corpas A, Ropero S. DNA methylation, histone modifications, and signal transduction pathways: a close relationship in malignant gliomas pathophysiology. J Signal Transduct. 2012;2012:956–958. | ||
Le Roy G, Dimaggio PA, Chan EY, et al. A quantitative atlas of histone modification signatures from human cancer cells. Epigenetics Chromatin. 2013;6(1):20. | ||
Du J, Johnson LM, Jacobsen SE, Patel DJ. DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol. 2015;16(9):519–532. | ||
Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10(5):295–304. | ||
Ioudinkova ES, Barat A, Pichugin A, et al. Distinct distribution of ectopically expressed histone variants H2A.Bbd and macroH2A in open and closed chromatin domains. PLoS One. 2012;7(10):1101–1113. | ||
Falls JG, Pulford DJ, Wylie AA, Jirtle RL. Genomic imprinting: implications for human disease. Am J Pathol. 1999;154:635–647. | ||
Bjornsson HT, Brown LJ, Fallin MD, et al. Epigenetic specificity of loss of imprinting of the IGF2 gene in Wilms tumors. J Natl Cancer Inst. 2007;99(16):1270–1273. | ||
Feinberg AP, Irizarry RA. Evolution in health and medicine Sackler colloquium: stochastic epigenetic variation as a driving force of development, evolutionary adaptation, and disease. Proc Natl Acad Sci U S A. 2010;107 Suppl 1:1757–1764. | ||
Burggren W. Epigenetic inheritance and its role in evolutionary biology: re-evaluation and new perspectives. Biology (Basel). 2016;5(2):24–46. | ||
Van Dijk SJ, Tellam RL, Morrison JL, Muhlhausler BS, Molloy PL. Recent developments on the role of epigenetics in obesity and metabolic disease. Clin Epigenetics. 2015;7:66. | ||
Archer T, Oscar-Berman M, Blum K, Gold MS. Epigenetic modulation of mood disorders. J Genet Syndr Gene Ther. 2013;4(120):1000120. | ||
Roth TL, Sweatt JD. Annual research review: epigenetic mechanisms and environmental shaping of the brain during sensitive periods of development. J Child Psychol Psychiatry. 2010;52(4):398–408. | ||
Bakulski KM, Halladay A, Hu VW, Mill J, Fallin MD. Epigenetic research in neuropsychiatric disorders: the “Tissue Issue”. Curr Behav Neurosci Rep. 2016;3(3):264–274. | ||
Cao Y, Lu L, Liu M, et al. Impact of epigenetics in the management of cardiovascular disease: a review. Eur Rev Med Pharmacol Sci. 2014; 18(20):3097–3104. | ||
Berezin A. Epigenetics in heart failure phenotypes. BBA Clin. 2016;6:31–37. | ||
Feinberg AP, Koldobskiy MA, Göndör A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet. 2016;17(5):284–299. | ||
Hannum G, Guinney J, Zhao L, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49(2):359–367. | ||
Teschendorff AE, West J, Beck S. Age-associated epigenetic drift: implications, and a case of epigenetic thrift? Hum Mol Genet. 2013;22(R1):R7–R15. | ||
Benayoun BA, Pollina EA, Brunet A. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat Rev Mol Cell Biol. 2015;16(10):593–610. | ||
Swaminathan V, Reddy BA, Ruthrotha Selvi B, Sukanya MS, Kundu TK. Small molecule modulators in epigenetics: implications in gene expression and therapeutics. Subcell Biochem. 2007;41:397–428. | ||
Balaur I, Saqi M, Barat A, et al. EpiGeNet: a graph database of interdependencies between genetic and epigenetic events in colorectal cancer. J Comput Biol. 2016;23:1–12. | ||
Romanoski CE, Glass CK, Stunnenberg HG, Wilson L, Almouzni G. Epigenomics: roadmap for regulation. Nature. 2015;518(7539):314–316. | ||
Halsberger A, Varger F, Karlic H. A model for epigenetic mechanisms in cancer development. Med Hypotheses. 2006;67(6):1448–1454. | ||
Liu Y, Siegmund KD, Laird PW, Berman BP. Bis-SNP: combined DNA methylation and SNP calling for Bisulfite-seq data. Genome Biol. 2012;13(7):R61. | ||
Anink-Groenen LC, Maarlerveld TR, Verschure PJ, Bruggeman FJ. Mechanistic stochastic model of histone modification pattern formation. Epigenetics Chromatin. 2014;7(1):30. | ||
Capra JA, Kostka D. Modeling DNA methylation dynamics with approaches from Phylogenetics. Bioinformatics. 2014;30(17):i408–i414. | ||
Erdel F, Greene EC. Generalized nucleation and looping model for epigenetic memory of histone modifications. Proc Natl Acad Sci U S A. 2016;113(29):E4180–E4189. | ||
Raghavan K, Ruskin HJ, Perrin D, Goasmat F, Burns J. Computational micromodel for epigenetic mechanisms. PLoS One. 2010;5(11):e14031. | ||
Sneppen K, Dodd IB. A simple histone code opens many paths to epigenetics. PLoS Comput Biol. 2012;8(8):e1002643. | ||
Ruskin HJ, Perrin D. Computational methods in epigenetic research. In: Wells RD, Bond JS, Klinman J, Siler Masters BS, Bell E, editors. Molecular Life Sciences: An Encyclopedic Reference. New York: Springer; 2014;1–8. | ||
Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381–395. | ||
Rose NR, Klose RJ. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim Biophys Acta. 2014;1839(12):1362–1372. | ||
Ushijima T, Sasako M. Focus on gastric cancer. Cancer Cell. 2004;5(2):121–125. | ||
Perrin D, Ruskin HJ, Niwa T. Cell type-dependent, infection-induced, aberrant DNA methylation in gastric cancer. J Theor Biol. 2010;264(2):570–577. | ||
Harrison LG. The Shaping of Life. The Generation of Biological Pattern. Cambridge: Cambridge University Press; 2011. | ||
Raghavan K, Ruskin HJ. Modelling DNA methylation dynamics. In: Tatarinova T, editor. DNA Methylation - From Genomics to Technology. InTech; 2012. | ||
McGovern AP, Powell BE, Chevassut TJT. A dynamic multi-compartmental model of DNA methylation with demonstrative predictive value in hematological malignancies. J Theor Biol. 2012;310:14–20. | ||
Karmaus W, Ziyab AH, Everson T, Holloway JW. Epigenetic models and mechanisms in the origins of asthma. Curr Opin Allergy Clin Immunol. 2013;13(1):63–69. | ||
Bush WS, Moore JH. Genome-wide association studies. PLoS Comput Biol. 2012;8(12):e1002822. | ||
Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet. 2011;12(8):529–541. | ||
Flanagan JM. Epigenome-wide association studies (EWAS): past, present, and future. Methods Mol Biol. 2015;1238:51–63. | ||
Roznovat IA, Ruskin HJ. Theoretical cross-comparative analysis on dynamics of small intestine and colon crypts during cancer initiation. IET Syst Biol. 2015;9(6):259–267. | ||
Ku WL, Girvan M, Yuan GC, Sorrentino F, Ott E. Modeling the dynamics of bivalent histone modifications. PLoS One. 2013;8(11): e77944. | ||
Rohlf T, Steiner L, Przybilla J, Prohaska S, Binder H, Galle J. Modeling the dynamic epigenome: from histone modifications towards self-organizing chromatin. Epigenomics. 2012;4(2):205–219. | ||
Bronner C, Chataigneau T, Schini-Kerth VB, Landry Y. The “Epigenetic Code Machinery”, ECREM: a promising drugable target of the epigenetic cell memory. Curr Med Chem. 2007;14(25):2629–2641. | ||
Peschansky VJ, Wahlestedt C. Non-coding RNAs as direct and indirect modulators of epigenetic regulation. Epigenetics. 2014;9(1):3–12. | ||
Hansen KD, Timp W, Bravo HC, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43(8):768–775. | ||
Dinalankara W, Bravo HC. Gene expression signatures based on variability can robustly predict tumor progression and prognosis. Cancer Inform. 2015;14:71–81. | ||
Kanz C, Aldebert P, Althorpe N, et al. The EMBL nucleotide sequence database. Nucleic Acids Res. 2005;33(Database issue):D29–D33. | ||
Liu L, Li Y, Li S, et al. Comparison of next-generation sequencing systems. J Biomed Biotechnol. 2012;2012:251364. | ||
Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17(6):333–351. | ||
Lowe R, Shirley N, Bleackley M, Dolan S, Shafee T. Transcriptomics technologies. PLoS Comput Biol. 2017;13(5):e1005457. | ||
Bock C, Lengauer T. Computational epigenetics. Bioinformatics. 2008;24(1):1–10. | ||
Lim SJ, Tan TW, Tong JC. Computational epigenetics: the new scientific paradigm. Bioinformation. 2010;4(7):331–337. | ||
Epigenie. Epigenetic databases, tools and resources. Available from: http://epigenie.com/epigenetic-tools-and-databases/. Accessed June 20, 2017. | ||
Fingerman IM, Zhang X, Ratzat W, Husain N, Cohen RF, Schuler DG. NCBI epigenomics: whats new for 2013. Nucleic Acids Res. 2013;41:D221–D225. | ||
Medvedeva YA, Lennartsson A, Ehsani R, et al. EpiFactors: a comprehensive database of human epigenetic factors and complexes. Database (Oxford). 2015;2015:bav067. | ||
Barat A, Ruskin HJ. A manually curated novel management system for genetic and epigenetic molecular determinants of colon cancer. Open Colorectal Cancer J. 2010;3:36–46. | ||
He X, Chang S, Zhang J, et al. MethyCancer: the database of human DNA methylation and cancer. Nucleic Acids Res. 2008;36(Database issue):D836–D841. | ||
Forbes SA, Beare D, Gunasekaran P, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(Database issue):D805–D811. | ||
Ongenaert M, Van Neste L, De Meyer T, Menschaert G, Bekaert S, Van Criekinge W. PubMeth: a cancer methylation database combining text mining and expert annotation. Nucleic Acids Res. 2008;36(Database issue):D842–D846. | ||
Fang YC, Lai PT, Dai HJ, Hsu WL. MethInfoText 2.0: gene methylation and cancer relation extraction from biomedical literature. BMC Bioinformatics. 2011;12:471. | ||
Grunau C, Renault E, Rosenthal A, Roizes G. MethDB—a public database for DNA methylation data. Nucleic Acids Res. 2001;29(1):270–274. | ||
Zou D, Sun S, Li R, Liu J, Zhang J, Zhang Z. MethBank: a database integrating next-generation sequencing single-base resolution DNA methylation programming data. Nucleic Acids Res. 2015;43(Database issue):D54–D58. | ||
Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18(11):1427–1431. | ||
Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE—a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2015;43(Database issue):D298–D299. | ||
Mariño-Ramírez L, Levine KM, Morales M, et al. The Histone Database: an integrated resource for histones and histone fold-containing proteins. Database (Oxford). 2011;2011:bar048. | ||
Khare SP, Habib F, Sharma R, Gadewal N, Gupta S, Galande S. HIstome—a relational knowledgebase of human histone proteins and histone modifying enzymes. Nucleic Acids Res. 2012;40(Database issue):D337–D342. | ||
Gendler K, Paulsen T, Napoli C. CHROMDB: the chromatin database. Nucleic Acids Res. 2008;36(Database issue):D298–D302. | ||
Shipra A, Chetan K, Rao MR. CREMOFAC—a database of chromatin remodelling factors. Bioinformatics. 2006;22(23):2940–2944. | ||
Wang Q, Huang J, Sun H, et al. CR Cistrome: a ChIP-Seq database for chromatin regulators and histone modification linkages in human and mouse. Nucleic Acids Res. 2014;42(Database issue):D450–D458. | ||
GigaOM. Twitter open-sources the home of its social graph. Available from: http://gigaom.com/2010/04/12/twitter-open-sources-the-home-of-its-social-graph. Accessed February 8, 2017. | ||
AllegroGraph. AllegroGraph RDFStore Web 30s database. Available from: http://franz.com/agraph/allegrograph. Accessed February 4, 2017. | ||
Neo4j. Neo4j world’s leading graph database. Available from: http://neo4j.com. Accessed January 20, 2017. | ||
Johnson D, Connor AJ, McKeever S, et al. Semantically linking in silico cancer models. Cancer Inform. 2014;13 Suppl 1:133–143. | ||
Henkel R, Wolkenhauer O, Waltemath D. Combining computational models, semantic annotations and simulation experiments in a graph database. Database(Oxford). 2015;2015:bau130. | ||
Chelliah V, Juty N, Ajmera I, et al. BioModels: ten-year anniversary. Nucleic Acids Res. 2015;43(D1):D542–D548. | ||
Low Y, Gonzalez J, Kyrola A, Bickson D, Guestrin C, Hellerstein JM. Distributed Graphlab: a framework for machine learning and data mining in the cloud. Proc VLDB Endowment. 2012;5(8):716–727. | ||
Corbellini A, Mateos C, Godoy D, Zunino A, Schiaffino S. An architecture and platform for developing distributed recommendation algorithms on large-scale social networks. J Inf Sci. 2015;41(5):686–704. | ||
Michels KB, Binder AM, Dedeurwaerder S, et al. Recommendations for the design and analysis of epigenome-wide association studies. Nat Methods. 2013;10(10):949–955. | ||
Birney E, Smith GD, Greally JM. Epigenome-wide association studies and the interpretation of disease-omics. PLoS Genet. 2016;12(6):e1006105. | ||
Scacheri PC, Crawford GE, Davis S. Statistics for ChIP-chip and DNase hypersensitivity experiments on NimbleGen arrays. Methods Enzymol. 2006;411:270–282. | ||
Krueger F, Kreck B, Franke A, Andrews SR. DNA methylome analysis using short bisulfite sequencing data. Nat Methods. 2012;9(2):145–151. | ||
Pedersen BS, Eyring K, De S, Yang IV, Schwartz DA. Fast and accurate alignment of long bisulfite-seq reads. Bioinformatics. 2014:1–2. | ||
Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27(11):1571–1572. | ||
Sun D, Xi Y, Rodriguez B, et al. MOABS: model based analysis of bisulfite sequencing data. Genome Biol. 2014;15:R38. | ||
Dolzhenko E, Smith AD. Using beta-binomial regression for high-precision differential methylation analysis in multifactor whole-genome bisulfite sequencing experiments. BMC Bioinformatics. 2014;15:215. | ||
Lea AJ, Tung J, Zhou X. A flexible, efficient binomial mixed model for identifying differential dna methylation in bisulfite sequencing data. PLoS Genet. 2015;11(11):e1005650. | ||
Gao S, Zou D, Mao L, et al. BS-SNPer: SNP calling in bisulfite-seq data. Bioinformatics. 2015;31(24):4006–4008. | ||
Simpson JT, Workman RE, Zuzarte PC, David M, Dursi LJ, Timp W. Detecting DNA cytosine methylation using nanopore sequencing. Nat Methods. 2017;14(4):407–410. | ||
Rand AC, Jain M, Eizenga JM, et al. Mapping DNA methylation with high-throughput nanopore sequencing. Nat Methods. 2017;14(4):411–413. | ||
Houseman EA, Christensen BC, Yeh RF, et al. Model-based clustering of DNA methylation array data: a recursive-partitioning algorithm for high-dimensional data arising as a mixture of beta distributions. BMC Bioinformatics. 2008;9:365. | ||
Wang B, Mezlini AM, Demir F, et al. Similarity network fusion for aggregating data types on a genomic scale. Nat Methods. 2014;11(3):333–337. | ||
Sánchez A, Fernández-Real J, Vegas E, et al. Multivariate methods for the integration and visualization of omics data. In: Freitas AT, Navarro A, editors. Bioinformatics for Personalized Medicine. Lecture Notes in Computer Science. Berlin, Heidelberg: Springer; 2012: 6620. | ||
Zhang L, Lin X. Multivariate statistical methods in bioinformatics research. In: Jiang R, Zhang X, Zhang M, editors. Basics of Bioinformatics. Berlin, Heidelberg: Springer; 2013:63–231. | ||
Teschendorff AE, Zheng SC. Cell-type deconvolution in epigenome-wide association studies: a review and recommendations. Epigenomics. 2017;9(5):757–768. | ||
Zheng SC, Beck S, Jaffe AE, et al. Correcting for cell-type heterogeneity in epigenome-wide association studies: revisiting previous analyses. Nat Methods. 2017;14(3):216–217. | ||
Jones MJ, Islam SA, Edgar RD, Kobor MS. Adjusting for cell type composition in DNA methylation data using a regression-based approach. Methods Mol Biol. 2017;1589:99–106. | ||
König IR, Auerbach J, Gola D, et al. Machine Learning and data mining in complex genomic data – a review on the lessons learned in Genetic Analysis Workshop 19. BMC Genet. 2016;17 Suppl 2:1. | ||
Dutkowski J, Kramer M, Surma MA, et al. A gene ontology inferred from molecular networks. Nat Biotechnol. 2013;31(1):38–45. | ||
Raja K, Patrick M, Gao Y, Madu D, Yang Y, Tsoil LC. A review of recent advancement in integrating omics data with literature mining towards biomedical discoveries. Int J Genomics. 2017;2017:6213474. | ||
Leslie R, O’Donnell CJ, Johnson AD. GRASP: analysis of genotype–phenotype results from 1390 genome-wide association studies and corresponding open access database. Bioinformatics. 2014;30(12):i185–i194. | ||
Banerji CRS, Miranda-Saavedra D, Severini S, et al. Cellular network entropy as the energy potential in Waddington’s differentiation landscape. Sci Rep. 2013;3:3039. | ||
Teschendorff AE, Banerji CRS, Severini S, Kuehn R, Sollich P. Increased signaling entropy in cancer requires the scale-free property of protein interaction networks. Sci Rep. 2015;5:9646. | ||
Eason K, Sandanandam A. Molecular or metabolic reprogramming: what triggers tumor subtypes? Cancer Res. 2016;76(18):5195–5200. | ||
Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006;7(1):21–33. | ||
Teschendorff AE, Jones A, Fiegl H, et al. Epigenetic variability in cells of normal cytology is associated with the risk of future morphological transformation. Genome Med. 2012;4(3):24. | ||
Timp W, Feinberg AP. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer. 2013;13(7):497–510. | ||
Plass C, Pfister SM, Lindroth AM, Bogatyrova O, Claus R, Lichter P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat Rev Genet. 2013;14:765–780. | ||
Barat A, Ruskin HJ. Comparative correlation structure of colon cancer locus specific methylation: characterization of patient profiles and potential markers across 3 array-based datasets. J Cancer. 2015;6(8):795–811. | ||
Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–1356. | ||
Forough F, Iman R, D’agnillo M, Porter L, Rueda L, Ngom A. An integrative approach for identifying network biomarkers of breast cancer subtypes using genome, interactome and transcriptome data. J Comput Biol. 2017;24(8):756–766. | ||
Lanigan F, Brien GL, Fan Y, et al. Delineating transcriptional networks of prognostic gene signatures refines treatment recommendations for lymph node-negative breast cancer patients. FEBS J. 2015;282(18):3455–3473. | ||
Barat A, Ruskin HJ, Byrne AT, Prehn JH. Integrating colon cancer microarray data: associating locus-specific methylation groups to gene expression-based classifications. Microarrays (Basel). 2015;4(4):630–646. | ||
Sadanandam A, Lyssiotis CA, Homicsko K, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19:619–625. | ||
Marisa L, de Reyniès A, Duval A, et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med. 2013;10:e1001453. | ||
Isella C, Terrasi A, Bellomo SE, et al. Stromal contribution to the colorectal cancer transcriptome. Nat Genet. 2015;47(4):312–319. | ||
Munchel S, Hoang Y, Zhao Y, et al. Targeted or whole genome sequencing of formalin fixed tissue samples: potential applications in cancer genomics. Oncotarget. 2015;6(28):25943–25961. | ||
Moran B, Das S, Smeets D, et al. Assessment of concordance between fresh-frozen and formalin-fixed paraffin embedded tumor DNA methylation using a targeted sequencing approach. Oncotarget. 2017;8(29):48126–48137. | ||
Lim AM, Wong NC, Pidsley R, et al. Genome-scale methylation assessment did not identify prognostic biomarkers in oral tongue carcinomas. Clin Epigenetics. 2016;8:74. | ||
Florath I, Butterbach K, Müller H, Bewerunge-Hudler M, Brenner H. Cross-sectional and longitudinal changes in DNA methylation with age: an epigenome-wide analysis revealing over 60 novel age-associated CpG sites. Hum Mol Genet. 2014;23(5):1186–1201. | ||
Yuan T, Jiao Y, de Jong S, Ophoff RA, Beck S, Teschendorff AE. An integrative multi-scale analysis of the dynamic DNA methylation landscape in aging. PLoS Genet. 2015;11(2):e1004996. | ||
Salam MT. Asthma epigenetics. Adv Exp Med Biol. 2014;795:183–199. | ||
Nugent NR, Goldberg A, Uddin M. Topical review: the emerging field of epigenetics: informing models of pediatric trauma and physical health. J Pediatr Psychol. 2016;41(1):55–64. | ||
Roth TL. Epigenetic mechanisms in the development of behavior: advances, challenges, and future promises of a new field. Dev Psychopathol. 2013;25(4 Pt 2):1279–1291. | ||
Champagne FA. Epigenetics of mammalian parenting. In: Narvaez D, Valentino K, Fuentes A, McKenna JJ, Gray P, editors. Ancestral Landscapes in Human Evolution: Culture, Childrearing and Social Wellbeing. Oxford School; 2014. | ||
Raymond JG, Steele JD, Seriès P. Modeling trait anxiety: from computational processes to personality. Front Psychiatry. 2017;8:1. | ||
Mukherjee K, Twyman RM, Vilcinskas A. Insects as models to study the epigenetic basis of disease. Prog Biophys Mol Biol. 2015;118(1–2):69–78. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.