Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
Real-world analysis of the Celgene Global Drug Safety database: early discontinuation of lenalidomide in patients with myelodysplastic syndromes due to non-serious rash
Authors Weiss L, Gary D, Swern A, Freeman J, Sugrue M
Received 11 April 2015
Accepted for publication 19 June 2015
Published 4 September 2015 Volume 2015:11 Pages 1355—1360
DOI https://doi.org/10.2147/TCRM.S86449
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Lilia Weiss,1 Dianna Gary,1 Arlene S Swern,2 John Freeman,1 Mary M Sugrue3
1Global Drug Safety, Celgene Corporation, Summit, 2Biometrics, Celgene Corporation, Berkeley Heights, 3Medical Affairs, Celgene Corporation, Summit, NJ, USA
Background: Lenalidomide is approved for treating transfusion-dependent anemia due to lower-risk del(5q) myelodysplastic syndromes (MDS). In clinical trials, rash was common, although severe rash was infrequent. To examine rash in patients with MDS treated with lenalidomide in the real world, the Celgene Global Drug Safety database was analyzed and compared with clinical trials.
Materials and methods: Adverse event reports in the post-marketing setting and in the MDS-003/004 clinical trials were analyzed by action taken with lenalidomide, seriousness/grade, time to onset, and treatment duration.
Results: Globally, 16,942 reports representing 36,793 adverse events from the post-marketing setting were submitted to the Global Drug Safety database between December 27, 2005 and June 13, 2013. Most rash adverse events were non-serious (Global Drug Safety database, 91%) or grade 1/2 (MDS-003/004 trials, 87%–93%). Unexpectedly, rash, occurring at a median of 9 days after treatment initiation, was the leading cause of permanent discontinuation of lenalidomide. Seventy-two percent of non-serious rash adverse events led to early permanent discontinuation within two cycles, while in the MDS-003/004 pivotal clinical trials, only 2%–3% of rash adverse events led to permanent discontinuation.
Conclusion: Non-serious rash was the most common reason for permanent discontinuation of lenalidomide in real-world settings. Managing lenalidomide-related rash using published recommendations might improve treatment duration and optimize patient outcomes.
Keywords: adverse events, safety, post-marketing setting
Introduction
Lenalidomide was approved in the USA in December 2005 for the treatment of transfusion-dependent anemia due to International Prognostic Scoring System lower-risk (low-risk or intermediate 1-risk) myelodysplastic syndromes (MDS) associated with del(5q), with or without additional cytogenetic abnormalities.1 In June 2013, lenalidomide was approved in Europe for a similar indication in patients with transfusion-dependent anemia due to lower-risk MDS associated with isolated del(5q).2
Lenalidomide has been demonstrated to be highly effective in patients with del(5q) MDS, with manageable adverse events (AEs).3 The efficacy and safety of lenalidomide in patients with transfusion-dependent, lower-risk, del(5q) MDS was studied in two pivotal clinical trials, ie, the single-arm Phase II MDS-003 study (n=148)4 and the double-blind, placebo-controlled Phase III MDS-004 study (n=205).5 In these studies, neutropenia and thrombocytopenia were the most common AEs and most frequently led to dose modifications.4,5 The most common grade 3/4 AEs reported in the MDS-003 study were neutropenia (55%), thrombocytopenia (44%), anemia (7%), leukopenia (6%), and rash (6%),4 and in the MDS-004 study for lenalidomide 5 mg/10 mg were neutropenia (74%/75% of patients), thrombocytopenia (33%/41%), leukopenia (13%/9%), anemia (6%/3%), and deep vein thrombosis (1%/6%).5 Discontinuation due to any AEs was reported in 20% of patients in MDS-003 and 8.7%/17.4% of patients receiving lenalidomide 5 mg/10 mg in MDS-004.4,5
A recent meta-analysis examined the risk of rash in patients with hematologic malignancies, including non-del(5q) MDS, treated with lenalidomide in clinical trials.6 The analysis demonstrated that treatment with lenalidomide was significantly associated with an increased risk of rash compared with placebo (relative risk 1.7%; P<0.001). An incidence of all-grade rash of 27.2% and an incidence of high-grade (grade ≥3) rash of 3.6% were found in the trials examined. The mechanism of rash associated with lenalidomide is not completely understood, and it remains unclear whether it is attributable to the direct antitumor effects and/or indirect immunomodulatory effects of lenalidomide.7 Studies have implicated ERK and phosphatidylinositide 3-kinase/Akt in the mechanism of rash associated with other agents,8,9 and it has been hypothesized that lenalidomide may promote rash through similar mechanisms in the epidermis via alteration of keratinocyte growth and survival.6 Generally, studies have not reported any pharmacological or clinical implications of rash associated with lenalidomide, although investigators in one study postulated that rash may be an early predictor of tumor response.10 Of note, in patients with MDS, onset of erythroid response (red blood cell transfusion independence) can take three or more cycles of lenalidomide treatment.1,4,5 Therefore, to achieve the best outcomes, early recognition and appropriate management of toxicities, including rash, are important to maintain patient quality of life and to optimize the dose and duration of lenalidomide treatment.
A recent study found that reporting of AEs in oncology publications from randomized trials was suboptimal and characterized by substantial selectivity and heterogeneity, and thus may leave physicians with insufficient information for clinical decision-making.11 Furthermore, the safety profile of a drug cannot be entirely known from limited clinical trial data alone and should be supplemented by real-world post-marketing clinical experience. After being approved for commercial distribution, drugs continue to be monitored by manufacturers for any issues affecting patient welfare and to ensure that the necessary measures are taken to reduce the risk and increase the benefits of the drugs.
A recent routine monitoring of the safety of lenalidomide using the internal Celgene Global Drug Safety database revealed that in patients with MDS treated in a post-marketing setting, non-serious rash was the leading cause of permanent discontinuation of lenalidomide. This unexpected observation and the dearth of information provided on non-serious rash and its management in published lenalidomide clinical trials highlighted lenalidomide-associated rash management as an unmet need and an important step toward optimal patient outcomes. Here, we present the most common AEs associated with lenalidomide in patients with MDS as reported to the Celgene Global Drug Safety database. Rash AEs from both the Global Drug Safety database and the MDS-003/004 pivotal clinical trials were examined in detail.
Materials and methods
Data sources
A retrospective analysis of the Celgene Global Drug Safety database was conducted for AE reports submitted between December 27, 2005 (date of approval in the USA) and June 13, 2013 for patients with MDS exposed to lenalidomide in the post-marketing setting. The Global Drug Safety database consists of all AEs reported for Celgene products. This analysis was specific to spontaneous reports by health care professionals (physicians, pharmacists, nurses, and others) and consumers (patients, family members, lawyers, and others) from post-marketing settings.
Reporting of AEs by health care professionals and consumers to the product manufacturers and regulatory authority (US Food and Drug Administration) is voluntary in the USA. In contrast, European Union competent authorities may impose specific obligations on health care professionals to report suspected AEs to product manufacturers or to the authority itself.
The AE data analysis included reports from lenalidomide-treated patients with MDS, time to onset of the AE, severity and outcome of the AE, and action taken with lenalidomide due to the AE. Complete information on the selected endpoints was not always reported. Possible actions taken with lenalidomide included permanent discontinuation, dose modification (defined as dose reduction or interruption), or no dose change. Patients may have been represented in the database by more than one AE report.
For comparison of the real-world and clinical trial safety profile of lenalidomide in patients with MDS, relevant AE data from two multicenter clinical trials investigating treatment with lenalidomide in patients with transfusion-dependent anemia and low-risk or intermediate 1-risk MDS with del(5q) abnormalities were included. MDS-003 was a multicenter, open-label, non-controlled, single-arm Phase II study of lenalidomide 10 mg daily for 21 or 28 days of 28-day treatment cycles (ClinicalTrials.gov NCT00065156).4 MDS-004 was a multicenter, double-blind, placebo-controlled, three-arm Phase III study of two doses of lenalidomide versus placebo. In the double-blind phase, patients received lenalidomide 10 mg daily for 21 of 28 days, lenalidomide 5 mg daily for 28 of 28 days, or placebo in 28-day cycles (ClinicalTrials.gov NCT00179621).5 Full inclusion and exclusion criteria and dose modifications due to treatment-emergent AEs have been described previously.4,5 The following dose modifications were recommended for rash AEs in the MDS-003 and MDS-004 clinical trials: lenalidomide was to be interrupted and restarted at the next lower dose level for grade 3 non-desquamating rash and discontinued for desquamating (blistering) rash or grade 4 non-desquamating rash.
The MDS-003 and MDS-004 studies conformed to the Declaration of Helsinki and were approved by the institutional review boards or independent ethics committees of participating institutions. All patients provided written informed consent.
Data analysis
AEs reported to the Global Drug Safety database were coded using the Medical Dictionary for Regulatory Activities (MedDRA). For this analysis, AEs were further grouped by medical concept. For example, thrombocytopenia included the MedDRA preferred terms “thrombocytopenia” and “platelet count decreased”, and rash was defined to encompass the MedDRA high-level terms “rashes, eruptions, and exanthems” and “erythemas”. AEs were also analyzed by their severity. Serious AEs were defined as those requiring hospitalization or intervention or that were disabling, an important medical event, life-threatening, or fatal. Non-serious AEs included all other AE reports. AEs were analyzed by their respective outcomes: recovered, including recovering with sequelae; not recovered, including AEs that did not resolve before death from another cause; and death. AE reports missing the action taken with lenalidomide were excluded from the analysis.
AEs in the MDS-003 and MDS-004 studies were coded using MedDRA, and severity was captured by seriousness and graded using the National Cancer Institute Common Terminology Criteria for Adverse Events version 2.0 (MDS-003)4 and version 3.0 (MDS-004).5
Results
Overall, there were 16,942 reports representing 36,793 AEs in patients with MDS exposed to lenalidomide submitted to the Global Drug Safety database during the reporting period between December 2005 and June 2013. The majority (73%) of AE reports included the action taken with lenalidomide. Globally, approximately 42,132 patients received lenalidomide for the treatment of MDS in the commercial setting during the reporting period.
AEs reported and actions taken with lenalidomide due to AEs
Consistent with the known safety profile of lenalidomide,4,5 the ten most common AEs reported were neutropenia, thrombocytopenia, infections, rash, anemia, fatigue, diarrhea, pancytopenia, pruritus, and nausea and vomiting (Table 1). Of all AEs, 22% led to dose modifications and 18% led to permanent discontinuation of lenalidomide (Table 1). Neutropenia and thrombocytopenia were the most common AEs leading to dose modification. Unexpectedly, rash was the most common AE leading to permanent discontinuation of lenalidomide. In clinical trials of lenalidomide for the treatment of patients with MDS, rash, although common, rarely led to discontinuation of lenalidomide. The leading cause of permanent discontinuation of lenalidomide was thrombocytopenia in MDS-003 and neutropenia in MDS-004.
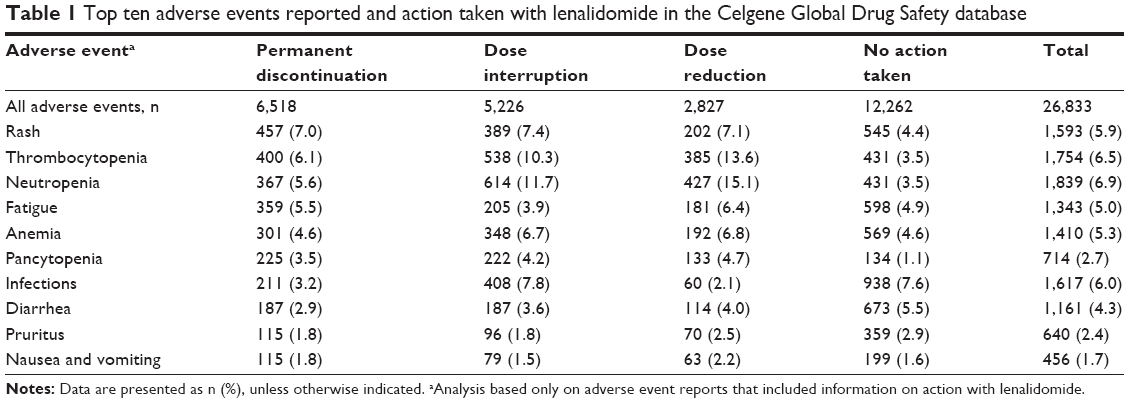 | Table 1 Top ten adverse events reported and action taken with lenalidomide in the Celgene Global Drug Safety database |
Experience with rash in the real world versus clinical trials
Most of the rash AEs from the post-marketing setting were reported as non-serious (91% non-serious versus 9% serious). Of those, 29% (52% serious and 27% non-serious) were reported by patients or physicians to be related to lenalidomide treatment. In clinical trials, 87% and 93% of all rash AEs were reported as grade 1/2 (assumed non-serious) and 13% and 7% were reported as grade 3/4 (assumed serious) for MDS-003 and MDS-004, respectively.
Rates of lenalidomide discontinuation or dose modification in the real world and in clinical trials were examined by seriousness or severity grade of rash, respectively (Table 2). Of 1,593 rash AEs in the Global Drug Safety database with information on action taken with lenalidomide, 29% led to permanent discontinuation of lenalidomide, whereas dose modification was reported in 37% and no change in dose was reported in 34%. Of non-serious rash events, 26% reported permanent discontinuation of lenalidomide. In the MDS-003 and MDS-004 trials, only 3% (three grade 3/4 rash AEs) and 2% (one grade 1/2 rash AE) of rash AEs, respectively, resulted in permanent discontinuation of lenalidomide, whereas 11% and 5% of rash AEs led to dose modifications; the majority (86% and 93%, respectively) had no change in lenalidomide dosing. Reported time to onset of rash and duration of treatment with lenalidomide in patients with rash were also examined in the real-world setting (Global Drug Safety database reports) and in clinical trials (Table 3). In the real-world setting, median time to rash onset was shortest in patients who ultimately discontinued treatment (9 days), with a median duration of treatment of only 28 days. Upon closer examination, it was found that non-serious rash was the leading cause of early (within the first two treatment cycles) permanent discontinuation. Dates of lenalidomide treatment were provided in 56% of AE reports, and of those reporting on rash, 72% led to early permanent discontinuation.
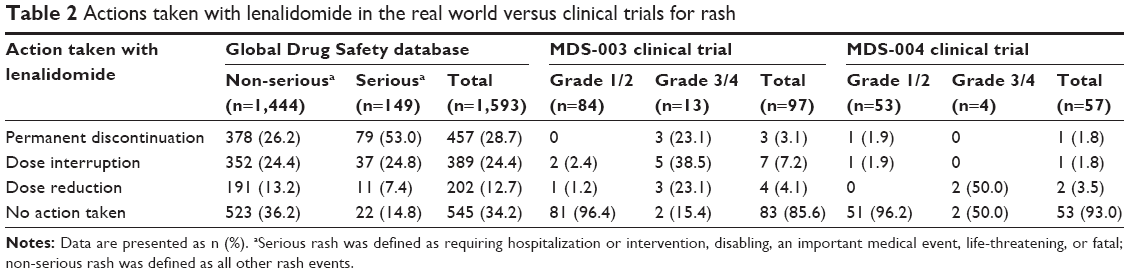 | Table 2 Actions taken with lenalidomide in the real world versus clinical trials for rash |
 | Table 3 Time to onset and duration of treatment by action taken with lenalidomide due to rash |
In the MDS-003 and MDS-004 trials, only four rash AEs resulted in permanent discontinuation. In general, there was no clear trend toward early median onset of rash leading to permanent discontinuation with lenalidomide in the MDS-003 and MDS-004 trials.
Data on outcomes of rash events were also available for 63% of reports (1,005 of 1,593) in the Global Drug Safety database that included information on action taken with lenalidomide (Table 4). Rates of rash recovery did not vary greatly regardless of lenalidomide dose interruption (79.5%), dose reduction (85.6%), or permanent discontinuation (87.9%). Rash did not result in death in any instances reported.
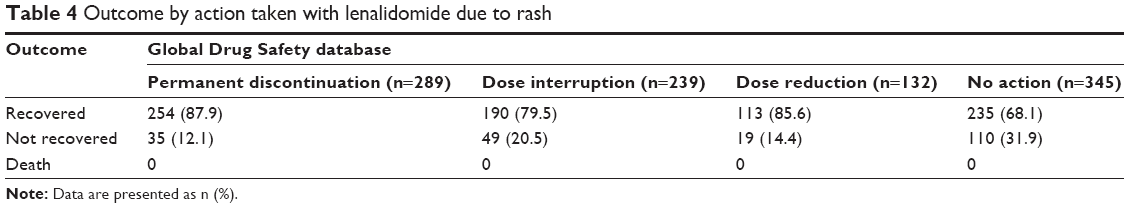 | Table 4 Outcome by action taken with lenalidomide due to rash |
Discussion
Clinical trial publications may not adequately equip health care professionals with a comprehensive view of the safety profile of a prescribed medication. Safety information may be incomplete and generally focused on grade 3/4 AEs.11 Discussion of common but less severe AEs is often neglected.
Consistent with published clinical trial data, this analysis of the Celgene Global Drug Safety database showed that neutropenia and thrombocytopenia were the most commonly reported AEs leading to dose modification of lenalidomide in MDS. Unexpectedly, it was found that non-serious rash, occurring early in treatment (within two cycles), was the most common reason for permanent discontinuation of lenalidomide, suggesting differences in management of rash in the real world versus clinical trials. Although thrombocytopenia and neutropenia appear to be managed in line with clinical trial data4,5 and label recommendations,1,2 our data suggest that non-serious rash is often managed with permanent discontinuation of lenalidomide. This is inconsistent with clinical trial data from MDS-003 and MDS-004, in which only 3% of patients with rash had permanent discontinuation of lenalidomide, whereas the majority of patients with rash did not require any dosing modification or treatment discontinuation.
This database relies on spontaneous reporting of AEs, and AE reporting by health care professionals and consumers is generally not compulsory. Requests for follow-up information on incomplete reports require the permission of the reporter, which is not always granted. Privacy laws vary by country. Some countries do not allow the manufacturer to contact the reporter for additional information. Accordingly, underreporting is a well recognized limitation of spontaneous reporting systems.12 Additional limitations include variability in the quality of reports and the inability to confidently determine whether reported events were a result of the drug and/or the disease state.13 Therefore, it should be noted that while this database allows for an exploration of relative rates of AEs, actions taken with lenalidomide, and outcomes, numbers represent only an approximation of the real-world experience rather than an incidence/prevalence. More accurate estimation of the “true” incidence/prevalence of rash and how it is managed in a “real-world” setting would require a prospective registry or a widespread retrospective study/chart review. Nonetheless, post-marketing studies using spontaneous reporting remain an important tool for assessing drug safety.
Guidelines for managing lenalidomide-associated rash are outlined in the lenalidomide package insert.1,2 Interruption or discontinuation of lenalidomide should be considered for grade 2/3 rash. Permanent discontinuation is recommended if exfoliative or bullous rash, Stevens–Johnson syndrome, or toxic epidermal necrolysis is suspected, and for grade 4 rash. The package insert guidelines are complemented by additional published recommendations for the management of rash associated with lenalidomide treatment.6,14 Giagounidis et al14 observed that in patients with MDS, rash and other non-hematologic AEs were generally self-limiting and did not often require treatment. Rash generally resolves in 2–3 weeks without interruption of lenalidomide. Mild to moderate grade 1/2 maculopapular or morbilliform rash may be treated with topical corticosteroids and antihistamines until rash resolves.6,14 For severe or persistent rash, including intolerable grade 2 rash, interruption of lenalidomide treatment for 7–14 days is recommended until resolution of symptoms.6,14 Antihistamines, topical steroids, or short courses of oral steroids may also be used as needed. Generally, the authors find that lenalidomide can be restarted without recurrence of rash, and permanent discontinuation is not recommended.6,14
Considering that in patients with MDS, onset of red blood cell transfusion independence can take three or more cycles of treatment with lenalidomide,1,4,5 early recognition and adequate management of rash are important to maintain patient quality of life and to optimize treatment outcomes. Although health care professionals may be familiar with management of hematologic AEs associated with lenalidomide treatment, which is frequently discussed in the literature, they may be less familiar with management recommendations for non-serious rash AEs. There is a need for further education of health care professionals and their patients with MDS on the lenalidomide safety profile and management of expected AEs. Non-serious rash should be managed by following published practical recommendations, with dose modification/interruption as appropriate, and not permanent discontinuation, to optimize the duration and dose of lenalidomide treatment to achieve best outcomes.
Conclusion
Contrary to data from the pivotal MDS-003 and MDS-004 clinical trials, non-serious rash was the most common reason for permanent discontinuation of lenalidomide in a real-world setting. Following published practical recommendations for the management of rash, including non-serious rash, may improve patient quality of life and lead to optimal treatment outcomes.
Acknowledgments
We would like to acknowledge the statistical support received from Jack Shiansong Li and Celgene Corporation, and the medical writing assistance of Jennifer Leslie and Stacey Rose of MediTech Media. The MDS-003 and MDS-004 trials were sponsored by Celgene Corporation.
Disclosure
LW, DG, ASS, JF, and MMS are all employees of and have ownership in Celgene Corporation. The authors report no other conflicts of interest in this work.
References
Revlimid (lenalidomide) [package insert]. Summit, NJ, USA: Celgene Corporation; 2015. | ||
Revlimid (lenalidomide) [summary of product characteristics]. Uxbridge, UK: Celgene Europe Ltd; 2015. | ||
Palumbo A, Freeman J, Weiss L, Fenaux P. The clinical safety of lenalidomide in multiple myeloma and myelodysplastic syndromes. Expert Opin Drug Saf. 2012;11(1):107–120. | ||
List A, Dewald G, Bennett J, et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med. 2006;355(14):1456–1465. | ||
Fenaux P, Giagounidis A, Selleslag D, et al. A randomized phase 3 study of lenalidomide versus placebo in RBC transfusion-dependent patients with low-/intermediate-1-risk myelodysplastic syndromes with del5q. Blood. 2011;118(14):3765–3776. | ||
Nardone B, Wu S, Garden BC, West DP, Reich LM, Lacouture ME. Risk of rash associated with lenalidomide in cancer patients: a systematic review of the literature and meta-analysis. Clin Lymphoma Myeloma Leuk. 2013;13(4):424–429. | ||
Komrokji RS, List AF. Role of lenalidomide in the treatment of myelodysplastic syndromes. Semin Oncol. 2011;38(5):648–657. | ||
Lacouture ME. Mechanisms of cutaneous toxicities to EGFR inhibitors. Nat Rev Cancer. 2006;6(10):803–812. | ||
Nardone B, Nicholson K, Newman M, et al. Histopathologic and immunohistochemical characterization of rash to human epidermal growth factor receptor 1 (HER1) and HER1/2 inhibitors in cancer patients. Clin Cancer Res. 2010;16(17):4452–4460. | ||
Dueck G, Chua N, Prasad A, et al. Interim report of a phase 2 clinical trial of lenalidomide for T-cell non-hodgkin lymphoma. Cancer. 2010;116(19):4541–4548. | ||
Sivendran S, Latif A, McBride RB, et al. Adverse event reporting in cancer clinical trial publications. J Clin Oncol. 2014;32(2):83–89. | ||
Hazell L, Shakir SA. Under-reporting of adverse drug reactions: a systematic review. Drug Saf. 2006;29(5):385–396. | ||
Fontanarosa PB, Rennie D, DeAngelis CD. Postmarketing surveillance – lack of vigilance, lack of trust. JAMA. 2004;292(21):2647–2650. | ||
Giagounidis A, Fenaux P, Mufti GJ, et al. Practical recommendations on the use of lenalidomide in the management of myelodysplastic syndromes. Ann Hematol. 2008;87(5):345–352. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.