")
Back to Journals » International Journal of Nanomedicine » Volume 12
Rational design of novel, fluorescent, tagged glutamic acid dendrimers with different terminal groups and in silico analysis of their properties
Authors Martinho N, Silva LC, Florindo HF, Brocchini S, Zloh M , Barata TS
Received 23 February 2017
Accepted for publication 2 June 2017
Published 25 September 2017 Volume 2017:12 Pages 7053—7073
DOI https://doi.org/10.2147/IJN.S135475
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Nuno Martinho,1–3 Liana C Silva,1,4 Helena F Florindo,1 Steve Brocchini,2 Mire Zloh,3 Teresa S Barata2
1Research Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Lisbon, Portugal; 2Department of Pharmaceutics, UCL School of Pharmacy, London, 3School of Life and Medical Sciences, University of Hertfordshire, Hatfield, UK; 4Centro de Química-Física Molecular and IN – Institute of Nanoscience and Nanotechnology, Instituto Superior Técnico, Universidade de Lisboa, Lisboa, Portugal
Abstract: Dendrimers are hyperbranched polymers with a multifunctional architecture that can be tailored for the use in various biomedical applications. Peptide dendrimers are particularly relevant for drug delivery applications due to their versatility and safety profile. The overall lack of knowledge of their three-dimensional structure, conformational behavior and structure–activity relationship has slowed down their development. Fluorophores are often conjugated to dendrimers to study their interaction with biomolecules and provide information about their mechanism of action at the molecular level. However, these probes can change dendrimer surface properties and have a direct impact on their interactions with biomolecules and with lipid membranes. In this study, we have used computer-aided molecular design and molecular dynamics simulations to identify optimal topology of a poly(L-glutamic acid) (PG) backbone dendrimer that allows incorporation of fluorophores in the core with minimal availability for undesired interactions. Extensive all-atom molecular dynamic simulations with the CHARMM force field were carried out for different generations of PG dendrimers with the core modified with a fluorophore (nitrobenzoxadiazole and Oregon Green 488) and various surface groups (glutamic acid, lysine and tryptophan). Analysis of structural and topological features of all designed dendrimers provided information about their size, shape, internal distribution and dynamic behavior. We have found that four generations of a PG dendrimer are needed to ensure minimal exposure of a core-conjugated fluorophore to external environment and absence of undesired interactions regardless of the surface terminal groups. Our findings suggest that NBD-PG-G4 can provide a suitable scaffold to be used for biophysical studies of surface-modified dendrimers to provide a deeper understanding of their intermolecular interactions, mechanisms of action and trafficking in a biological system.
Keywords: dendrimers, peptide dendrimers, molecular dynamics, fluorescence, CHARMM, structure-activity, surface properties
Introduction
Dendrimers belong to a class of hyperbranched polymers with well-defined radial architecture that has been extensively studied for biomedical applications.1 The topology of dendrimers can be described by their core, internal branches (or spacers) and terminal groups. By using various monomers to build different topological architectures, it is possible to design their size, their shape, the surface groups available for interaction and overall physicochemical properties.2–4 Similar to other drug delivery systems, one of the key requirements is to have an adequate safety profile.5 In this regard, peptide dendrimers consisting of natural amino acids are attractive candidates for use in biological systems due to their potential biodegradability and biocompatibility.6 Peptide dendrimers can be synthesized either by linear combinations of amino acids assembled at a branching point7,8 or by using amino acids as focal branching points themselves (eg, glutamic acid [GLU],9 lysine [LYS]10,11 and aspartic acid12). Owing to side chain size and the combination of amino acids, these peptide dendrimers might have an asymmetric arrangement that will create different local microenvironments within the dendrimer and be suitable to different surface modifications.13–15
Although extensively studied, the mechanism of action of bioactive dendrimers is still difficult to probe experimentally.16 One way to track dendrimers during their interaction with biomolecules and cells is to conjugate a fluorescent probe at the surface of these carriers. However, there are two main drawbacks of using this approach. First, fluorescent probes may randomly be distributed at the surface. This would result in a heterogeneous population of dendrimers, ranging from non-labelled dendrimers to dendrimers with multiple fluorophores, where the proximity between the probes might lead to possible quenching effects.17 Second, the fluorophore probes can themselves interact with cells and interfere or mask the real interaction of dendrimers with these biological systems.18,19 Furthermore, the targeted drug delivery achieved by conjugation of different monomers to surface groups13,14 may be affected by the introduction of a fluorophore on the surface.
In order to avoid this kind of interferences, phosphorus dendriplexes with a fluorophore attached to the core have been synthesized to study their interaction with cells.20 Thus, designing dendrimers that have a defined number of probes that are hindered from the external environment is of uppermost importance. However, there is still an overall lack of knowledge of the behavior of these molecules at the atomistic level,8 and consequently, computational methods can provide a suitable tool to explore the behavior of these macromolecules.
Prediction and rationalization of the 3D structure of dendrimers can be a difficult task due to their structural complexity and due to the lack of a dedicated force field (FF). The latter is particularly difficult to develop due to the diversity and high degree of chemical space that dendrimers encompass. Several degrees of complexity from coarse grained to full-atom simulations21,22 have been utilized to understand how dendrimers interact with drugs and other biomolecules at the molecular interface. In this regard, molecular dynamics (MD) simulations have been shown to be a suitable method to predict the behavior of dendrimers and structure15,22 and therefore are useful for the rational design of dendrimers.
The aim of this work was to study the potential of a poly(L-glutamic acid) (PG) dendrimer backbone for its ability to incorporate a fluorophore (of different size) into its core. The goal of this approach was to determine the generation at which the fluorophore can be protected from the external environment, thus ensuring that the probe does not interfere with the interaction of PG dendrimers with other biomolecules. Furthermore, different surface modifications were studied to determine their overall effect on the structural properties of the modified core dendrimers. We have used all-atom MD simulations using the CHARMM FF as an FF suitable to study structural properties of dendrimers.23 These simulations were used to describe the structure and conformational behavior of PG dendrimers, as well as the effects of modifying the dendrimer core by attaching different size fluorophores, namely, nitrobenzoxadiazole (NBD) or Oregon Green 488 (ORG). NBD was chosen due to its small size and optical properties that are suitable to monitor polarity changes in the surrounding environment, whereas the ORG was selected due to its bigger size and suitable spectroscopic properties to be used in monitoring cell events. We further evaluated the effects of diverse terminal groups with different charges on the structure of the dendrimers by using GLU, LYS or tryptophan (TRP; Figure 1). This study demonstrates the use of MD simulation to provide an insight into the versatility of the PG dendrimers that can be further explored for different biomedical applications.
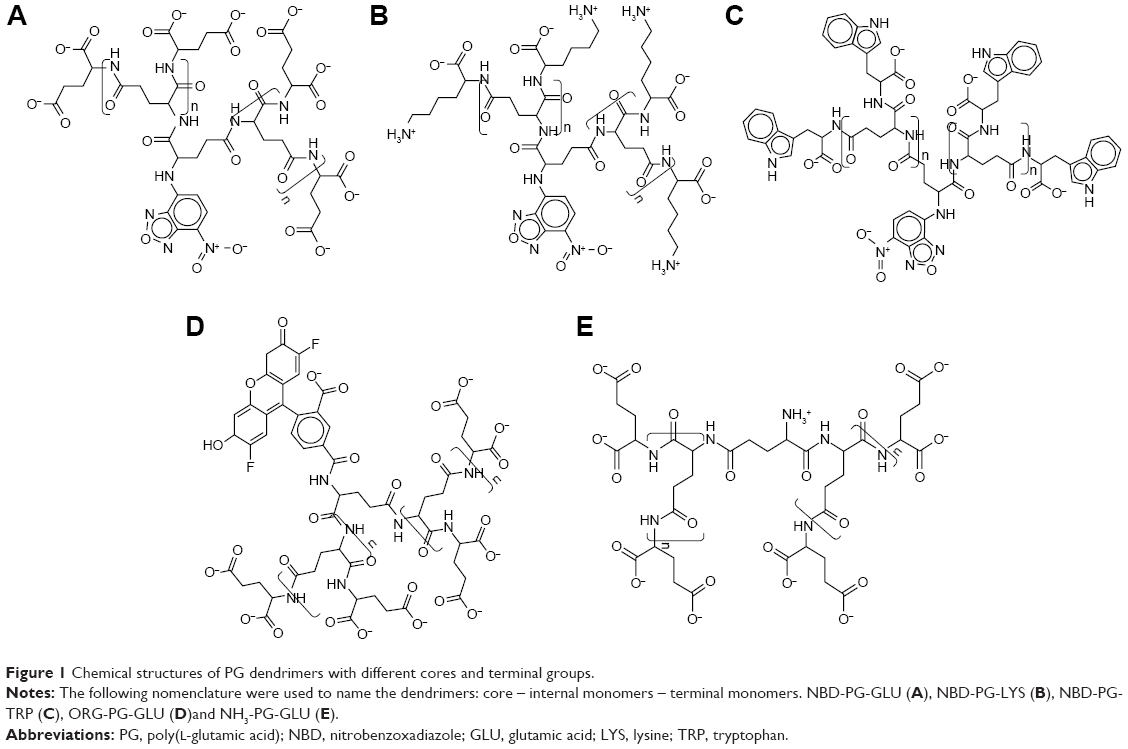 | Figure 1 Chemical structures of PG dendrimers with different cores and terminal groups. |
Methods
Monomer parameterization
Sets of parameters for GLU, LYS and TRP residues were prepared to be compatible with CHARMM 36a FF. These parameters, already available in the XPLOR format as CHARMM 22 FF, were updated to match the syntax of the CHARMM 36a. Moreover, additional parameters were added for a peptide bond between the side chain of the GLU and a new residue using a procedure similar to one previously reported in the literature.24 The topology blocks and parameters for NBD and ORG were obtained using ParamChem,25,26 assuming the transferability of the CGenFF FF and its compatibility with CHARMM 36a FF.25,26 The patches for new bonds between both fluorophores and GLU residues were assigned using ParamChem, ensuring that the overall charge was maintained.
Dendrimer assembly and equilibration
Initial dendrimer structures were generated by their in silico assembly, resembling their divergent synthesis (from core to terminal groups). The dendrimers were written as a sequence of monomers, and the patches for their assembly were prepared using a previously published procedure27 implemented in scripts for automation.28 The first monomer of GLU served as a trifunctional selective core, in which two focal points (carboxyl groups) were used for branching, while the third one (amine group) was used to connect a selected fluorophore. Files to generate the 3D structures of dendrimers containing the sequences of monomers (eg, NBD GLU GLU GLU …) and the connectivities established between the monomer residues through the description of patches were prepared as input files for XPLOR. These were then used in the XPLOR v3.429 to generate the 3D structure of the dendrimer through a series of simulated annealing protocols in a vacuum. The peptide bonds were kept in a trans conformation by introducing restraints since this is the most likely conformation to occur in hyperbranched dendrimers due to their peptidic nature.
Dendrimers were then immersed in a TIP3P water box model, with a distance between 15 and 20 Å in all directions, using the solvate module in VMD v.1.9.1, followed by the addition of ions, enough to neutralize the charge of the system and obtain a NaCl concentration of 150 mM, using autoionize module. The solvated system was then submitted to minimization and simulated annealing protocol in NAMD v.2.930 using periodic boundary conditions. The system was heated up to 500 K to remove a possible misfolding of the dendrimer by increasing the temperature 5 K each time and simulating for 1,000 fs at 1 fs/step for each temperature increase. The system was then cooled down in a similar way to 310 K. This also allowed rectifying steric clashes due to inappropriate contacts of water with the dendrimer.2
MD settings
Production runs were carried out in NAMD v.2.9.30 Dendrimers were first minimized for 2,500 steps through a steepest-descendent minimization. Each dendrimer was then submitted to MD simulation run for 45 ns at a constant pressure of 1 bar. Each run was performed with periodic boundary conditions and the integration step of 2 fs/step. The temperature was kept constant at 310 K using Langevin thermostat with a coupling constant of 5 ps−1, and the integration time step was 2 fs. The SHAKE algorithm was used to keep all covalent bonds at their equilibrium value. Electrostatics were evaluated using smooth particle mesh Ewald (PME) method with the van der Waals (vdW) cutoff defined as 12 Å, with a pair list of 10 Å and a real space cutoff of 14 Å.
Analysis
The obtained trajectories were analyzed using VMD, XPLOR, PyMOL, Bio3D,31,32 ProDy, MDTraj and in-house tools. The end of equilibration of the simulations for each system was assessed using a radius of gyration and root-mean-square deviation (RMSD). The trajectories of production runs were analyzed by calculating a range of molecular properties, including solvent surface accessible area, monomer density distribution, hydrogen interactions and shape properties.
RMSD and root-mean-square fluctuation (RMSF)
The RMSD was calculated using all dendrimer atoms to quantify the dissimilarity of the two structures once superimposed. The similarity between conformers was calculated after translational and rotational superimposition of structures.33 The RMSD values of superimposed structures were calculated using the following equation:
|
|
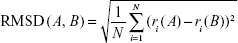
where ri(A) and ri(B) are the Cartesian coordinates of the ith atom in the conformations A and B and N is the total number of atoms. Contrary to symmetric peptide dendrimers,33 no special overlap rules have to be introduced during RMSD measurement. In fact, PG dendrimers are asymmetric as they contain the fluorophore, and thus, the superimposition always occurs between the correct monomers.
Principal component analysis (PCA)
PCA was performed to reduce the dimensionality of the data and identify the conformational space that a molecule occupies during the simulation. The translational and rotational motions of all alpha carbon atoms (since they are common in all the amino acids used) were eliminated by superimposing the structures, and the 3N × 3N covariance matrix was created with the Cartesian coordinates (Equation 2). The sets of eigenvectors and eigenvalues that represent the motion were generated by the diagonalization of the covariance matrix.34,35 PCA was performed on the last 10 ns of each production simulation. The covariance matrix was defined as:
|
|
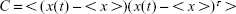
where <> denotes the average ensemble and x(t) is the structure at time t.
Radius of gyration (Rg)
The radius of gyration was calculated from the gyration tensor, a size descriptor based on the distribution of atoms from the center of mass (cm; Figure 2):
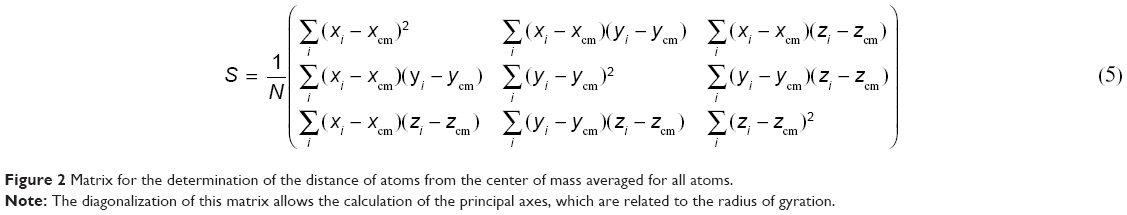 | Figure 2 Matrix for the determination of the distance of atoms from the center of mass averaged for all atoms. |
The diagonalization of the matrix renders the principal axis, λ1, λ2, λ3 (Equation 4), where
|
(3) |
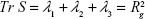
These eigenvalues correspond to the variances of the coordinates along the principal axes and were used to calculate other related shape properties.
Alternatively, the size of the dendrimer (when it reaches a spherical entity) can be measured by the embedding sphere radius (R) or span (Equation 4). This was measured by calculating the farthest atoms from the center of mass, ie, the radius that encompasses completely all atoms of the molecule:36
|
(4) |
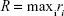
where ri is the distance of the ith atom from the center of mass.
Hydration radius (RH)
Hydration radius (RH) is a representation of an effective radius for molecules, and it can be correlated to experimental data, such as viscosity or pulsed field gradient spin-echo NMR.11 The values of RH for both symmetric and asymmetric branching dendrimers were calculated from 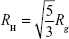 , using Rg.11,37 Additionally, RH was evaluated by measuring the distribution of water molecules inside the dendrimer and the distance from the center of mass, where the water density reaches 95% of the bulk value.11 These values were calculated from the radial pair distribution function, determined from the center of mass of the dendrimer, and relate to the different layers of hydration in these dendrimers.
, using Rg.11,37 Additionally, RH was evaluated by measuring the distribution of water molecules inside the dendrimer and the distance from the center of mass, where the water density reaches 95% of the bulk value.11 These values were calculated from the radial pair distribution function, determined from the center of mass of the dendrimer, and relate to the different layers of hydration in these dendrimers.
Solvent accessible surface area (SASA)
SASA was obtained by following the rolling sphere method using a radius of either 1.4 Å for the whole dendrimer or 3 Å for the fluorophore using the SASA module available in VMD.
Hydrogen bonding
The total number of hydrogen bonds was determined by monitoring the hydrogen bond formation for each frame. Hydrogen bonds were defined using a geometrical criterion with a maximum donor–acceptor distance of 3.5 Å and a hydrogen–donor–acceptor angle inferior to 30°.
Radial distribution of atoms
The distribution of mass was measured through the radial pair distribution function from either the fluorophore or the center of mass of the whole dendrimer. From these two points, several mass distribution functions were calculated:38
- Distribution of all atoms belonging to the dendrimers
- Distribution of the terminal groups
- Distribution of water
- Distribution of ions
The probability distribution of finding an atom in a spherical volume of radius r from a center of mass, a selected atom or a selected group is given in Equation 6. This probability was measured by
|
|
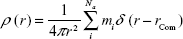
where Na is the number of atoms, mi is the ith atom mass and rcom is the distance from the dendrimer center of mass or alternatively from the fluorophore.
Shape of dendrimers
The relative shape anisotropy (κ2 in Equation 7),39 or asphericity, was calculated from the eigenvalues of the gyration tensor.
|
|
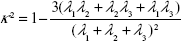
This parameter is limited to the values between 0 and 1. For highly symmetric spherical structures, the value is close to 0, and it reaches 1 for ideal chains (when all points lie on a line). Alternatively, asphericity parameter b can also be calculated (Equation 8),39 which measures the deviation from a spherical symmetry:
|
|
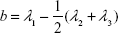
Analogously, the sphericity index can also describe the deviation from spherical symmetry (Equation 9):
|
|
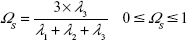
The values are limited between 0 for flat molecules and 1 to describe perfect spheres.
Results and discussion
Generation of PG dendrimers
We have developed a scaffold composed of a PG skeleton by linking fluorophores of different sizes to the core via free amine and adding variable terminal groups to allow functionalization of dendrimers for different applications (Figure 1).
The initial structures of dendrimers were relaxed using a simulated annealing protocol in a vacuum, which resulted in a significant increase in RMSD for all dendrimers within the first few nanoseconds during equilibration. The RMSD change increased in higher generations of dendrimers (Figure S1). This may be attributed to the removal of initial steric clashes between monomers and unfavorable interactions of PG dendrimers with solvent molecules. Depending on the dendrimer generation, simulations took between 5 and 25 ns to equilibrate, which is similar to other dendrimer simulations.8 Therefore, the last 20 ns of each simulation was used to ensure that the equilibrated systems were used during the evaluation of the properties of PG dendrimers. Despite the possibility of dendrimers being highly flexible and that the RMSD might not by itself indicate that equilibration was truly reached, this sampling should be sufficient to account for the dynamic behavior of PG dendrimers.
Effect of the presence of a small fluorophore in the dendrimer core
MD simulations were carried out on all PG dendrimers with a free –NH3 group on the core GLU and with a small fluorophore (NBD) attached to the core GLU to reveal the lowest generation of the dendrimer in which the fluorophore was fully hidden from the solvent.
The radius of gyration and hydration radius, the ratio between the gyration tensors, the relative shape anisotropy and asphericity were calculated to evaluate the differences in the overall size and shape of the dendrimers following the incorporation of the NBD to the PG core. The size of the dendrimers can be represented by its radius of gyration (Rg). These values showed to be constant for each dendrimer throughout the trajectories, suggesting that there is no significant variation of the size of dendrimers due to the change in their conformations. Moreover, no significant differences in size between the NH3- and NBD-PG series were observed as exemplified by their maximum radius (Rmax; Table 1). The distribution of Rg follows a normal distribution with a narrow variation in size indicating no major shift in the conformation throughout the simulation time (Table 1 and Figure 3, top).
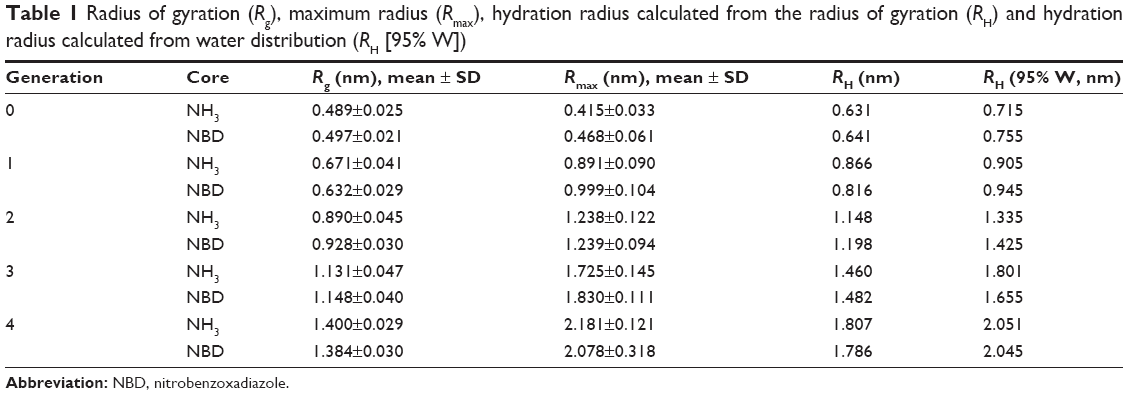 | Table 1 Radius of gyration (Rg), maximum radius (Rmax), hydration radius calculated from the radius of gyration (RH) and hydration radius calculated from water distribution (RH [95% W]) |
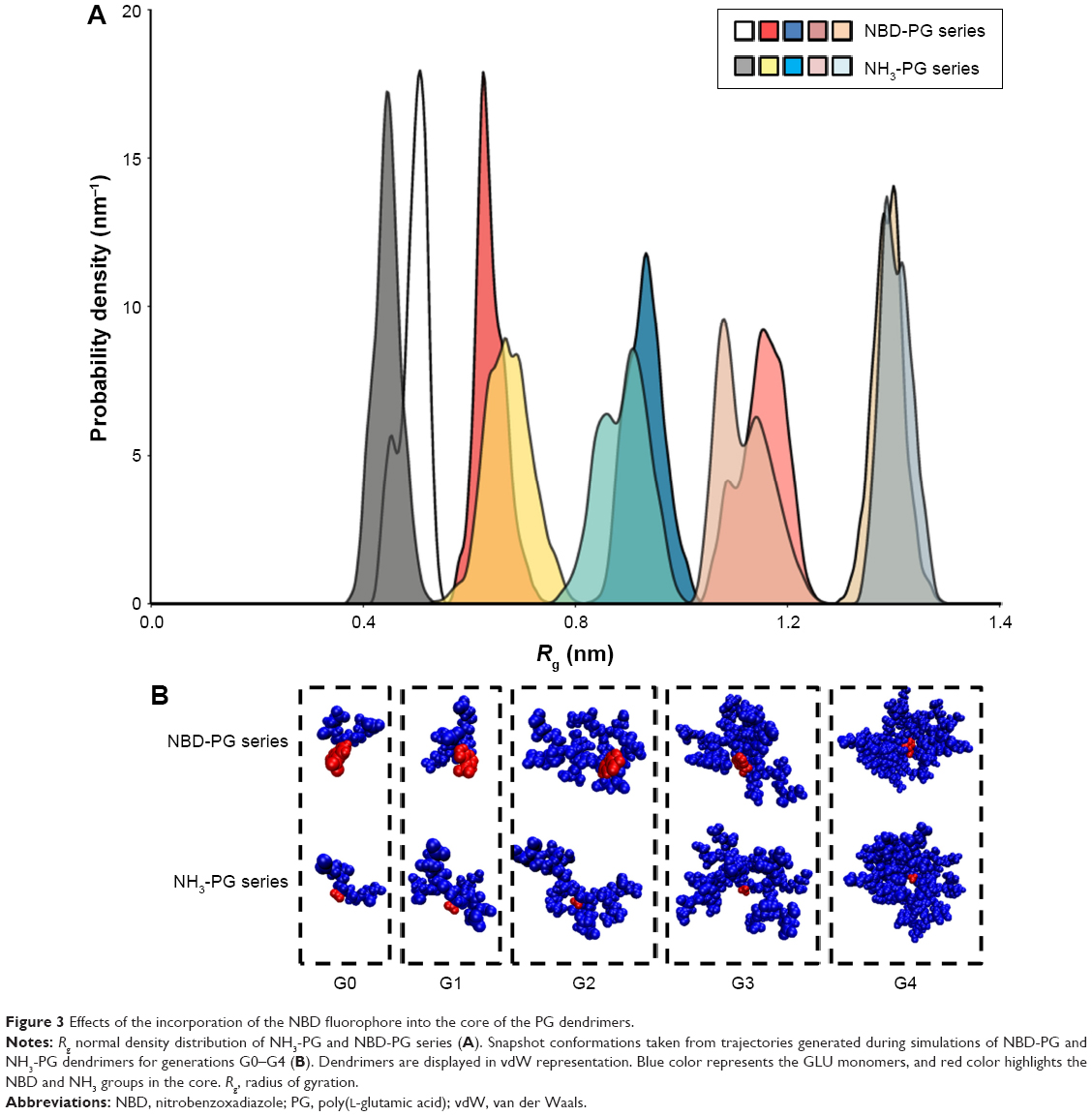 | Figure 3 Effects of the incorporation of the NBD fluorophore into the core of the PG dendrimers. |
The Rg for both series increased exponentially with the number of generations and molecular weight (MW). This is a common observation in dendrimers and results from the increase in MW being much faster than the molecular volume. This also indicated that no restrictions in an incremental increase of generations due to a higher number of terminal groups were observed.
These similarities are also reflected in the snapshots of the vdW representation of these PG dendrimers (Figure 3, bottom). Similar to the previously described peptide poly-L-lysine (PLL) dendrimers,11 PG dendrimers become spherical at higher generations and have a porous structure. This type of architecture is useful as there is potential to incorporate drug molecules within the dendrimer40 and can adopt different conformations. Even at G4, NBD-PG dendrimers showed different conformations, ranging from a more tubular form with the extension of a single branch, exposing the interior of the dendrimer to a more spherical and compact structure (Figures S2 and S3). Since most of the charged groups are located on the surface, the different conformations can lead to distinctive local charge distributions that may affect their interactions with biomacromolecules.
In previous studies, the correlation between the MW and Rg of dendrimers was found to be described by a power law of Rg ~ MWX, where X was found to be equal to 0.34 for spherical particles with an average density of monomers (ie, the similar distribution of monomers throughout the dendrimer).11,37 The correlation between MW and Rg for both NH3- and NBD-PG dendrimer series was found to have a correlation factor of 0.35 and 0.39, respectively. As a result, the NH3-PG series followed a similar trend to previously studied peptide dendrimers,37 whereas the NBD-PG series had a slightly higher value of 0.39. This minor difference in the correlation is attributed to the higher density in the core from the NBD without significant changes in the Rg.
The RH values, additional indicators of effective radii, measured by the water distribution showed 5%–20% higher than those calculated directly from the Rg (Table 2). These higher values can be attributed to the porous and extended structure of PG backbone allowing water to penetrate deeply into dendrimer interior and interact strongly with the terminal groups as well. On the other hand, the RH values calculated from Rg did not consider the properties of terminal groups and their interactions with the water, thus correlating only to the size of the molecules. Overall, both calculations were higher than Rg, which is attributed to the presence of higher number of interactions of these dendrimers with water, where the hydration layer at G4 was ~0.7 nm for both dendrimers, with and without NBD. Notably, no significant differences in the radii of gyration, maximum radii and hydration radii were observed at G4.
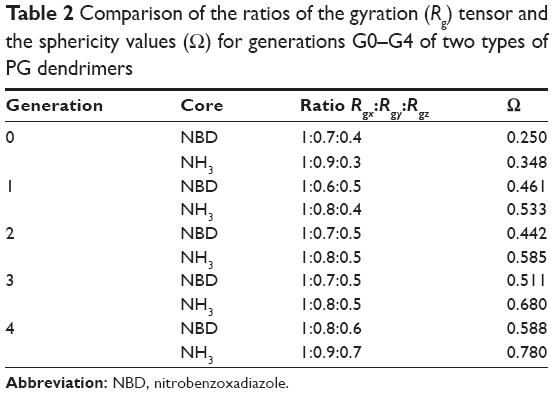 | Table 2 Comparison of the ratios of the gyration (Rg) tensor and the sphericity values (O) for generations G0–G4 of two types of PG dendrimers |
To further explore the possible differences caused by the introduction of NBD, the shape descriptors of the dendrimers were calculated. Since the shape of the dendrimers can ultimately contribute to their interaction with the biological target, the decomposition of the radius of gyration tensors is a good approach to explore the distribution of residues in the three dimensions and thus their shape. The ratios of Rgy/Rgx and Rgz/Rgx can be used to evaluate the shapes of molecules with values closer to 1, indicating spherical shape for dendrimers at higher generations (Table 2). These ratios (Rgy/Rgx and Rgz/Rgx) were similar for NH3- and NBD-PG dendrimers at generations G3 and G4, while differences were observed for lower generations (Table 2). This is a result of the significant influence of NBD on the center of mass and the gyration tensors at lower generations. The increase in generations also resulted in the increase in the spherical shape of the dendrimers, albeit these molecules are not forming a perfect sphere as observed by the differences in the Rgz/Rgx (Table 2).
The increase in spherical nature of both series of dendrimers was also pinpointed by measuring the asphericity (κ2) of these dendrimers, where at higher generations, a value close to zero was observed (Figure 4). Except for generation 0, no significant differences were observed between the NBD- and NH3-PG series, reiterating that NBD does not significantly affect the shape at higher generations. Similar values of κ2 were reported for PLL dendrimers,11 and it was found to be a feature present in both symmetric (eg, poly(amidoamine) [PAMAM]) and asymmetric dendrimers.11 Overall, these results further support the previous observations that the incorporation of NBD did not influence the shape of the dendrimers at higher generations and that the PG dendrimers are not perfect spheres.
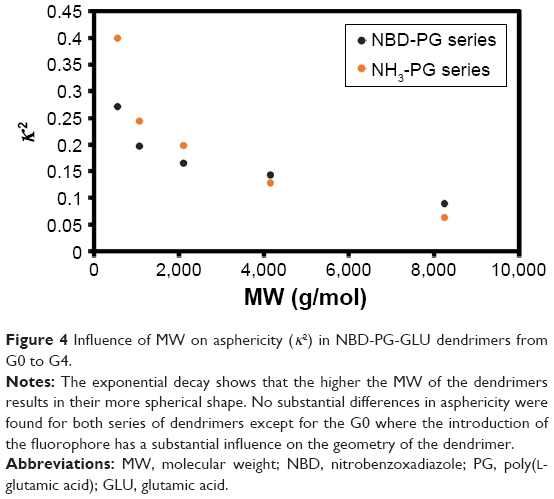 | Figure 4 Influence of MW on asphericity (κ2) in NBD-PG-GLU dendrimers from G0 to G4. |
Other than just the overall size and shape, the internal structure and motion of the dendrimers also take an important role in their behavior while interacting with biological systems. Dendrimers exhibit both local and collective motions (essential movements) that happen at the same time, and their visualization throughout the MD simulation is difficult. They appear as random movements; however, the motions follow defined patterns. PCA was used to identify the overall dominant conformational patterns and overall patterns of motion in the trajectories of dendrimers (Figure S4). Except for NH3-PG-G0, the first principal component (PC) accounted for 30%–50% of the overall motion. Furthermore, three to five PCs were sufficient to explain 70%–80% of the total variance in motion. In the case of NH3-PG-G0, the first two PCs described the overall behavior of the dendrimer due to the lower complexity of the structure. For both series at lower generations, the overall space occupied was similar, whereas at higher generations, the space explored had differences for PC2 and PC3 of dendrimers with different cores. This could be due to the fact that at higher generations, one type of motion might be occurring on different branches for various dendrimers possibly due to their asymmetry. Nevertheless, in visual inspection of all PG dendrimer motions, one to five PCs showed to be related to twisting, stretching and bending of the terminal branches (example of three major PCs is shown for a small G1 NBD-PG dendrimer in Figure 5).
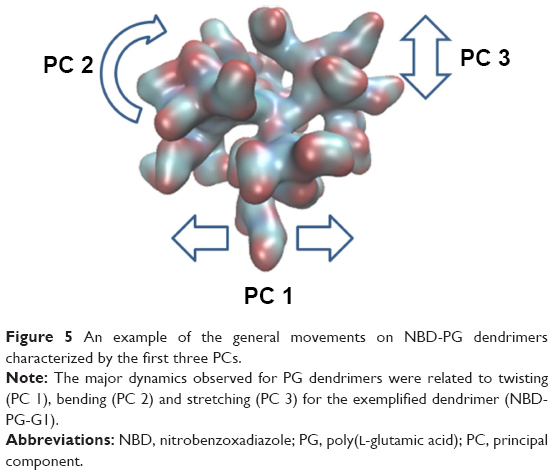 | Figure 5 An example of the general movements on NBD-PG dendrimers characterized by the first three PCs. |
For higher generations, the projection of the two major PCs reveals a U-shape form. This type of shape has been attributed to random diffusion of motion in proteins, allowing only to inform on more accessible degrees of freedom.41 However, for dendrimers, due to their specific connections between monomers, this may be the only degrees of freedom allowed since they cannot be denatured. As a result, both types of dendrimers, with and without NBD, revealed similar motions and dynamics. Furthermore, the data showed that the terminal groups contributed more to the dynamic behavior of the dendrimers and that the introduction of the NBD did not significantly impair the natural motion of PG dendrimers.
Dendrimer mass distribution was measured through the radial mass density from the center of mass (Figures 6 and S5). For both NH3 and NBD series, the density was higher in the core and thus in agreement with the core-dense model of dendrimers found for PAMAM or PLL dendrimers. Both series of PG dendrimers (NH3-PG and NBD-PG) showed similar distributions with only minor differences at lower generations again due to the deviation of the center of mass upon NBD incorporation. Therefore, at G3 and G4, the NBD no longer had a major influence on the mass distribution, and therefore, no significant differences were observed for both series.
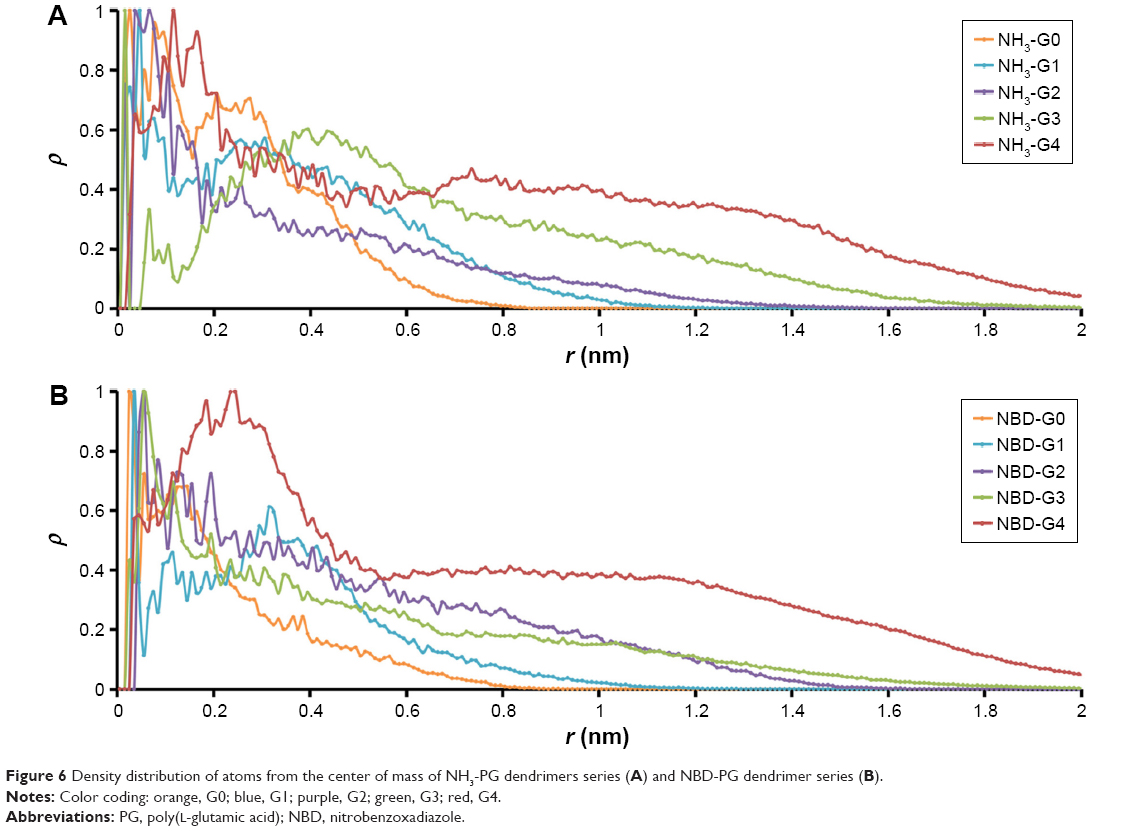 | Figure 6 Density distribution of atoms from the center of mass of NH3-PG dendrimers series (A) and NBD-PG dendrimer series (B). |
Regarding the distribution of ions around dendrimers, it was observed that negatively charged ions (Cl−) had a lower distribution around the dendrimer than positively charged ions (Na+), as it would be accepted for a dendrimer with a negatively charged terminal group. These ions accumulated near the dendrimer surface with higher generation dendrimers for both series, showing the maximum peak distribution at the same distance as the terminal groups (Figure S5). This suggests that ions are complexed with charged terminal groups in the same manner. Consequently, it is expected that cationic charged molecules will be attracted to the dendrimer, whereas anionic molecules will be repelled. This will result in different sets of molecules that could be conjugated to promote specific interactions with biomacromolecules.
Overall, the introduction of the NBD fluorophore did not influence the most relevant properties of size, shape, internal structure and motion of dendrimers from two series. Therefore, generations G0–G4 of the NBD dendrimers were evaluated to determine at which generation the fluorophore could be incorporated inside the scaffold with minimal interaction with the external environment. SASA measurements were carried out for the fluorophore (NBD) residue. The classic rolling probe of 1.4 Å showed not to be adequate for this measurement, as the probe would enter inside the dendrimer into the void space and would not give adequate representation of their exposure to the external environment. As a result, a 3 Å probe was found to be an adequate size to perform these measurements.
SASA values for the NBD showed that there was a significant decrease in NBD SASA values at G4 (Figure 7). This is also observed by visual inspection throughout the trajectory, in which it is apparent that the NBD is well embedded inside the dendrimer at this generation. All analyses from the obtained trajectories indicate that generation 4 is required to fully cover the fluorophore and that at this generation, the incorporation of this small fluorophore has minimal to none interference with the overall surface properties. Thus, G4 was selected to study further modifications on the dendrimer to understand how the overall structure can be tuned for different applications.
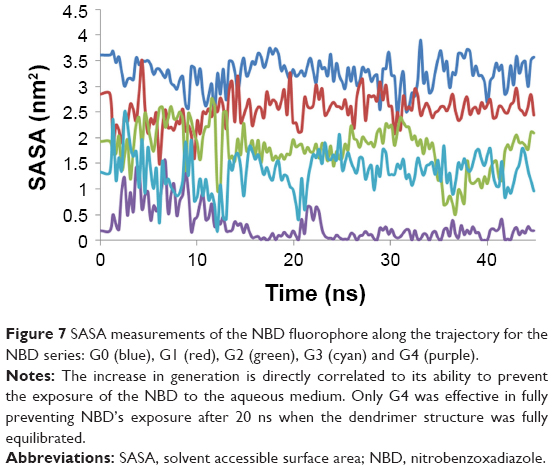 | Figure 7 SASA measurements of the NBD fluorophore along the trajectory for the NBD series: G0 (blue), G1 (red), G2 (green), G3 (cyan) and G4 (purple). |
Effect of using a bigger fluorophore
Since different probes with distinct spectra might be required to study multiple phenomena experimentally, ORG was linked to the core at G4 to evaluate the effect of the introduction of a bigger fluorophore (Figure 8).
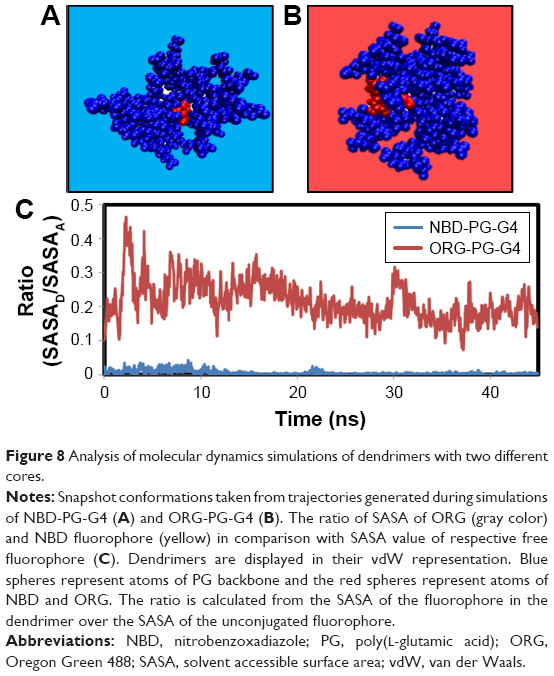 | Figure 8 Analysis of molecular dynamics simulations of dendrimers with two different cores. |
As observed in Figure 8, no major structural differences could be observed between NBD-PG-G4 and ORG-PG-G4. However, the gyration tensor and sphericity showed that ORG-PG-G4 dendrimer is more spherical than NBD-PG-G4 as the ratios of the gyration tensor and the sphericity were found to be 1:0.9:0.7 and 0.795, respectively. This can be attributed to the incorporation of a bulkier monomer that introduces the bias on the gyration tensor calculations. However, the bulkier nature of ORG prevented a complete coverage of the fluorophore by the dendrimer, even at the G4 (Figure 8). This is because ORG is big enough to extend to the terminal groups resulting in ~20% of the fluorophore still exposed to the solvent, as shown by SASA measurements. In order to fully encompass ORG, a higher generation of PG dendrimers might be necessary. However, producing dendrimers with generations higher than G4 increases the difficulty of their synthesis and potentially could lead to increased toxicity. Nevertheless, ORG-PG-G4 could still potentially be useful as a probe since the introduction of the larger fluorophore did not cause any major structural modifications, and 80% of ORG’s volume was embedded within the interior of the dendrimer.
Surface modification by changing the terminal groups
The interaction of dendrimers with biomolecules is often driven by their terminal groups. For this reason, we further modified the NBD-PG dendrimers to display different surface groups and thus potentially modulate their biological interaction. Two modifications were pursued by having a G4 dendrimer with LYS or TRP at the surface. These modifications allow the NBD-PG dendrimer to display a zwitterionic behavior (LYS terminal groups) or have a hydrophobic surface while being water-soluble type of dendrimer (TRP terminal groups). These modifications may also allow the conjugation of different types of therapeutics, thus ensuring their delivery using these dendrimers.
The LYS modification had no effect on the overall Rg of PG dendrimers (Table 3). However, the modification with TRP significantly increased the Rg (1.424 nm, Student’s t-test, p<0.05) compared to any other G4 so far described. This can be attributed to the bulkier side chain of TRP and the fact that the ring decreases the flexibility of the side chain and forms pi–pi stacking interactions, thus locking the dendrimer terminal groups to prevent their folding into the interior. This can also be observed by the number of particles at the surface and thus to the extended gyration tensor.
 | Table 3 Radius of gyration (Rg), maximum radius (Rmax), radius of hydration (RH) calculated using two different approaches and sphericity (O) of NBD-PG-LYS-G4 and NBD-PG-TRP-G4 |
Both dendrimers were found to be in more compact conformations throughout the simulated time. The Rmax for dendrimers with LYS and TRP surface modifications branches (1.576 and 1.596 nm, respectively) was smaller in comparison with the GLU terminal residues (2.078 nm; Table 3). This is due to the electrostatic interactions established between the carboxyl and amine groups for the LYS substitution and the pi–pi stacking in the case of TRP. In fact, some snapshots showed pi–pi interactions between the rings of the NBD and the TRP, and evidenced the backfolding of some of the terminal groups toward the interior of the dendrimer (Figure 9). This also results in a more oblate shape (gyration tensor ratio of 1:0.8:0.6), even though only minor differences in asphericity were found for LYS and TRP surface modifications, 0.0488 and 0.0813 respectively.
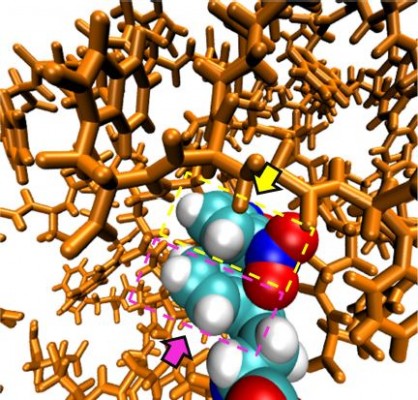
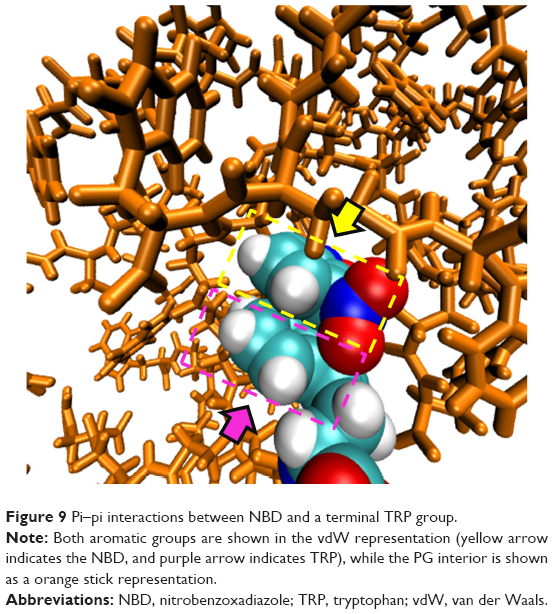 | Figure 9 Pi–pi interactions between NBD and a terminal TRP group. |
This observation was further supported by hydrogen bonding calculations. Dendrimers, and in particular peptide dendrimers, display a complex hydrogen bonding network as the internal groups are near each other. This can translate into increasing compactness of structure and higher stabilization of conformers. NBD-PG-G4, NH3-PG-G4 and ORG-PG-G4 showed a similar average number of hydrogen bonds per residue, ie, 1.86, 2.15 and 1.7, respectively. On the other hand, substituting the terminal groups with TRP and LYS increased hydrogen bonding, 3.1 and 9.69 hydrogen bonds per residue, respectively. The higher number of hydrogen bonds is expected since hydrogen donors were introduced for both residues and, in the case of LYS, a charged protonated amine was also introduced. This may be important as the number of hydrogen bond acceptors and donors, as well as the number of intramolecular hydrogen bonds that can be formed, may influence the number and type of intermolecular interactions established between dendrimers and biomolecules.
The surface charge was also considerably different for dendrimers with distinct surface modifications (Figure 10), which enable substantially distinct intermolecular interactions with biomolecules to occur. Therefore, these dendrimers are potentially suitable for modification to achieve multiple applications. For example, NBD-PG-LYS-G4 could be used to complex with nucleic acid molecules, whereas the TRP substitution can complex with molecules that present both positive and negative charges, as well as hydrophobic surfaces.
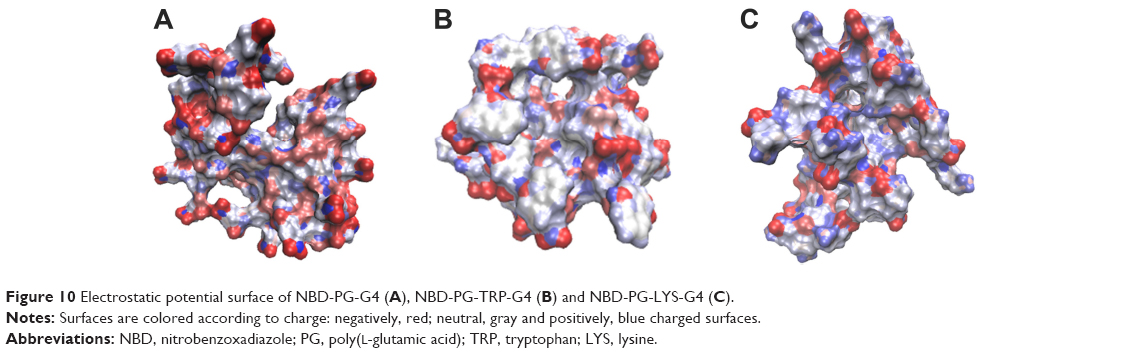 | Figure 10 Electrostatic potential surface of NBD-PG-G4 (A), NBD-PG-TRP-G4 (B) and NBD-PG-LYS-G4 (C). |
Overall, we showed that it is possible to change the terminal groups of the NBD-PG dendrimer to display different charges and hence lipophilicity of their surface while keeping the fluorophore from interacting with the external environment. Other modifications can, therefore, be pursued in order to obtain the desired properties according to intended application. Finally, G4 seems to be the potential candidate generation for drug delivery, presenting a plausible size range for synthesis purposes and affording a range of surface modifications tailored to multiple applications.
Conclusion
Predicting structure of dendrimers to design effective carriers can be a difficult endeavor. Herein, MD simulations were performed to predict the 3D structure of PG dendrimers with different surface modifications to study their capacity to incorporate a fluorophore as an imaging agent. We showed that a G4 PG dendrimer could fully encompass a small fluorophore, without affecting the intermolecular interactions that the dendrimer can form with biomacromolecules. The addition of NBD in the core of G4 PG dendrimers did not introduce significant changes in their size and shape. The incorporation of a bigger fluorophore (ORG) was also inspected. A G4 PG dendrimer did not completely incorporate the fluorophore with 80% of its surface in the dendrimer interior, rendering the use of this fluorophore as an imaging agent possible, albeit not ideal. Finally, dendrimer surface modifications by different amino acids, LYS and TRP, were further explored. These differed not only in size but also in the charges of the surface compared to GLU-terminated dendrimer. The study showed that these different amino acids induce different topological features compared to GLU-terminated PG dendrimers while maintaining the ability to encompass NBD fully. These changes in charge distribution and shape can, therefore, be used to tailor the surface of NBD-PG-G4 for different biological applications via introducing desired interactions with targets. NBD-PG-G4 dendrimers with different surface modifications provide a suitable scaffold to be used to monitor the distribution of dendrimers in a biological system without NBD interfering with interactions with targets or other biomolecular systems.
Acknowledgments
Funding from the UK Engineering & Physical Sciences Research Council (EPSRC) for the EPSRC Centre for Innovative Manufacturing in Emergent Macromolecular Therapies is gratefully acknowledged. Financial support from the consortium of industrial and governmental users is also acknowledged. NM, LCS and HFF acknowledge Fundação para a Ciência e a Tecnologia – Ministério da Ciência; Tecnologia e Ensino Superior (FCT-MCTES), Portugal (PhD Grant SFRH/BD/87838/2012 (NM); Investigador FCT IF/00437/2014 to LCS and iMed.ULisboa grant UID/DTP/04138/2013). The University of Hertfordshire is acknowledged for providing support to this project.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Svenson S, Tomalia DA. Dendrimers in biomedical applications – reflections on the field. Adv Drug Deliv Rev. 2005;57:2106–2129. | ||
Maingi V, Kumar MVS, Maiti PK. PAMAM dendrimer-drug interactions: effect of pH on the binding and release pattern. J Phys Chem B. 2012;116(14):4370–4376. | ||
Milhem OM, Myles C, McKeown NB, Attwood D, D’Emanuele A. Polyamidoamine Starburst(R) dendrimers as solubility enhancers. Int J Pharm. 2000;197:239–241. | ||
Shi X, Lee I, Chen X, et al. Influence of dendrimer surface charge on the bioactivity of 2-methoxyestradiol complexed with dendrimers. Soft Matter. 2010;6(11):20–27. | ||
De Jong WH, Borm PJ. Drug delivery and nanoparticles: applications and hazards. Int J Nanomedicine. 2008;3(2):133–149. | ||
Sadler K, Tam JP. Peptide dendrimers: applications and synthesis. Rev Mol Biotechnol. 2002;90(3–4):195–229. | ||
Javor S, Delort E, Darbre T, Reymond JL. A peptide dendrimer enzyme model with a single catalytic site at the core. J Am Chem Soc. 2007;129(43):13238–13246. | ||
Filipe LCS, Machuqueiro M, Darbre T, Baptista AM. Unraveling the conformational determinants of peptide dendrimers using molecular dynamics simulations. Macromolecules. 2013;46(23):9427–9436. | ||
Twyman L, Beezer A, Mitchell J. The synthesis of chiral dendritic molecules based on the repeat unit L-glutamic acid. Tetrahedron Lett. 1994;35(25):4423–4424. | ||
Vlasov GP, Korol’kov VI, Pankova GA, et al. Lysine dendrimers and their starburst polymer derivatives: possible application for DNA compaction and in vitro delivery of genetic constructs. Russ J Bioorganic Chem. 2004;30(1):12–20. | ||
Falkovich S, Markelov D, Neelov I, Darinskii A. Are structural properties of dendrimers sensitive to the symmetry of branching? Computer simulation of lysine dendrimers. J Chem Phys. 2013;139(64903):1–8. | ||
Ranganathan D, Kurur S. Synthesis of totally chiral, multiple armed, poly Glu and poly Asp scaffoldings on bifunctional adamantane core. Tetrahedron Lett. 1997;38(7):1265–1268. | ||
Li N, Cai H, Jiang L, et al. Enzyme-sensitive and amphiphilic PEGylated dendrimer-paclitaxel prodrug-based nanoparticles for enhanced stability and anticancer efficacy. ACS Appl Mater Interfaces. 2017;9(8):6865–6877. | ||
Li N, Li N, Yi Q, et al. Amphiphilic peptide dendritic copolymer-doxorubicin nanoscale conjugate self-assembled to enzyme-responsive anti-cancer agent. Biomaterials. 2014;35(35):9529–9545. | ||
Bouvier B. Optimizing the multivalent binding of the bacterial lectin LecA by glycopeptide dendrimers for therapeutic purposes. J Chem Inf Model. 2016;56(6):1193–1204. | ||
Caminade AM, Laurent R, Majoral JP. Characterization of dendrimers. Adv Drug Deliv Rev. 2005;57(15):2130–2146. | ||
Franc G, Mazères S, Turrin CO, et al. Synthesis and properties of dendrimers possessing the same fluorophore(s) located either peripherally or off-center. J Org Chem. 2007;72(23):8707–8715. | ||
Yoo H, Juliano RL. Enhanced delivery of antisense oligonucleotides with fluorophore-conjugated PAMAM dendrimers. Nucleic Acids Res. 2000;28(21):4225–4231. | ||
Kitchens KM, Kolhatkar RB, Swaan PW, Eddington ND, Ghandehari H. Transport of poly(amidoamine) dendrimers across Caco-2 cell monolayers: influence of size, charge and fluorescent labeling. Pharm Res. 2006;23(12):2818–2826. | ||
Kazmierczak-Baranska J, Pietkiewicz A, Janicka M, et al. Synthesis of a fluorescent cationic phosphorus dendrimer and preliminary biological studies of its interaction with DNA. Nucleosides Nucleotides Nucleic Acids. 2010;29(3):155–167. | ||
Bello M, Fragoso-Vázquez J, Correa-Basurto J. Theoretical studies for dendrimer-based drug delivery. Curr Pharm Des. Epub 2017 Feb 28. | ||
Martinho N, Florindo H, Silva L, Brocchini S, Zloh M, Barata T. Molecular modeling to study dendrimers for biomedical applications. Molecules. 2014;19(12):20424–20467. | ||
Kanchi S, Suresh G, Priyakumar UD, Ayappa KG, Maiti PK. Molecular dynamics study of the structure, flexibility, and hydrophilicity of PETIM dendrimers: a comparison with PAMAM dendrimers. J Phys Chem B. 2015;119(41):12990–13001. | ||
Gumbart JC, Beeby M, Jensen GJ, Roux B. Escherichia coli peptidoglycan structure and mechanics as predicted by atomic-scale simulations. PLoS Comput Biol. 2014;10(2):e1003475. | ||
Vanommeslaeghe K, MacKerell AD Jr. Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J Chem Inf Model. 2012;52(12):3144–3154. | ||
Vanommeslaeghe K, MacKerell AD Jr. Automation of the CHARMM general force field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J Chem Inf Model. 2012;52(12):3155–3168. | ||
Barata TS, Brocchini S, Teo I, Shaunak S, Zloh M. From sequence to 3D structure of hyperbranched molecules: application to surface modified PAMAM dendrimers. J Mol Model. 2011;17(11):2741–2749. | ||
Martinho N, Silva LC, Florindo HF, Brocchini S, Barata T, Zloh M. Practical computational toolkits for dendrimer and linear polymer structure design. J Comput Aided Mol Des. Epub 2017. | ||
Schwieters CD, Kuszewski JJ, Tjandra N, Marius Clore G. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160(1):65–73. | ||
Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781–1802. | ||
Grant BJ, Rodrigues APC, ElSawy KM, McCammon JA, Caves LSD. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics. 2006;22(21):2695–2696. | ||
Skjærven L, Yao X, Scarabelli G, Grant BJ. Integrating protein structural dynamics and evolutionary analysis with Bio3D. BMC Bioinformatics. 2014;15:1–11. | ||
Filipe LCS, MacHuqueiro M, Baptista AM. Unfolding the conformational behavior of peptide dendrimers: insights from molecular dynamics simulations. J Am Chem Soc. 2011;133(13):5042–5052. | ||
Hayward S, de Groot BL. Normal modes and essential dynamics. Methods Mol Biol. 2008;443:89–106. | ||
Berendsen HJC, Hayward S. Collective protein dynamics in relation to function. Curr Opin Struct Biol. 2000;10:165–169. | ||
Todeschini R, Consonni V. Descriptors from molecular geometry. Handbook of Chemoinformatics. Wiley-VCH: Weinheim, Germany; 2000:1004–1033. | ||
Neelov I, Falkovich S, Markelov D, Paci E, Darinskii A, Tenhu H. Molecular dynamics of lysine dendrimers. Computer simulation and NMR. Dendrimers in Biomedical Applications. Royal Society Of Chemistry: Cambridge, UK; 2013:204. | ||
Tian W, Ma Y. Effects of valences of salt ions at various concentrations on charged dendrimers. Soft Matter. 2010;6:1308–1316. | ||
Arkin H, Janke W. Gyration tensor based analysis of the shapes of polymer chains in an attractive spherical cage. J Chem Phys. 2013;138(5):1–9. | ||
Al-Jamal KT, Al-Jamal WT, Wang JTW, et al. Cationic poly-L-Lysine dendrimer complexes doxorubicin and delays tumor growth in vitro and in vivo. ACS Nano. 2013;7(3):1905–1917. | ||
Wolf A, Kirschner KN. Principal component and clustering analysis on molecular dynamics data of the ribosomal L11·23S subdomain. J Mol Model. 2013;19(2):539–549. |
Supplementary materials
The following files are available free of charge: document containing information regarding the RMSD and RMSF analysis of the different dendrimer series and further detail on conformational analysis, density profiles and PCA of the different dendrimers under study (PDF).
RMSD analysis of the different series of PG dendrimers
RMSD was used to determine when dendrimers’ structure reached equilibrium. It was observed that the higher the generation, the longer it took to reach equilibrium. For this reason, only the last 20 ns of each simulation was taken and analyzed (Figure S1).
 | Figure S1 RMSD for the different series of PG dendrimers (heavy atoms). |
2D and 3D representation of GLU dendrimers bearing NBD
2D representation of dendrimers is not enough for topological characterization of the dendrimers. Visualization of the 3D structure highlighting the fluorophore showed that G4 is required to fully prevent NBD exposure (Figure S2).
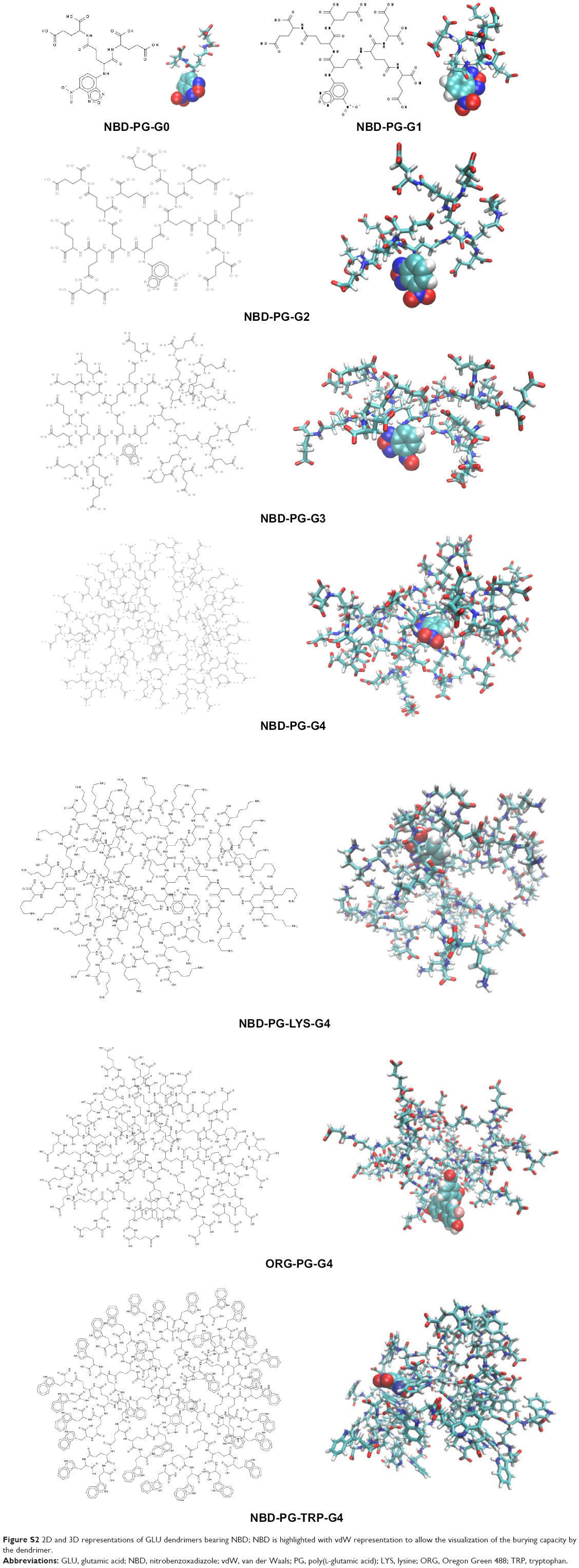 | Figure S2 2D and 3D representations of GLU dendrimers bearing NBD; NBD is highlighted with vdW representation to allow the visualization of the burying capacity by the dendrimer. |
NBD-PG-G4 dendrimer conformations
Once dendrimers’ simulation reached equilibrium, several conformational changes were observed. Most of these corresponded to rotation movements of the terminal monomers where no major changes in the overall structure were observed (Figure S3).
 | Figure S3 Snapshots of the different conformations observed throughout the trajectory of NBD-PG-G4 dendrimer. |
Principal component analysis
PCA was performed to determine the conformational space that a molecule occupies and the general motion of these dendrimers. For most of the simulations, three to five PCs were sufficient to explain most of the motions of dendrimers. Most of these motions can be attributed to movements of the terminal monomers (Figure S4).
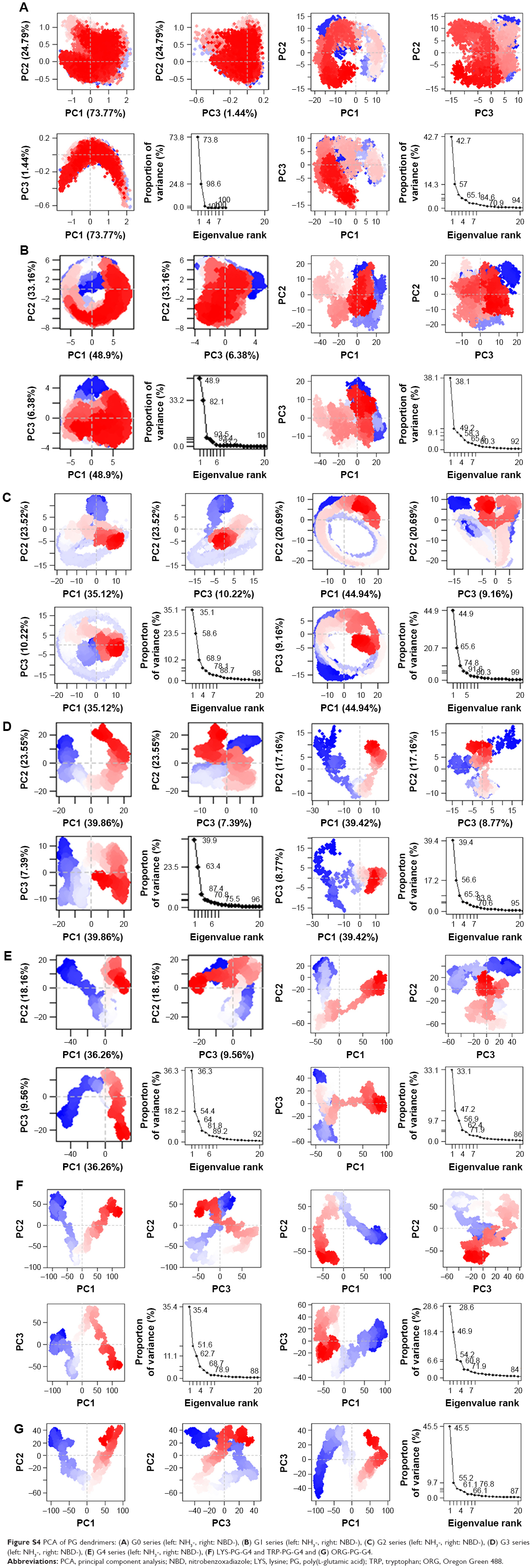 | Figure S4 PCA of PG dendrimers: (A) G0 series (left: NH3-, right: NBD-), (B) G1 series (left: NH3-, right: NBD-), (C) G2 series (left: NH3-, right: NBD-), (D) G3 series (left: NH3-, right: NBD-), (E) G4 series (left: NH3-, right: NBD-), (F) LYS-PG-G4 and TRP-PG-G4 and (G) ORG-PG-G4. |
Density distribution of PG dendrimers
The density of atoms distributed from the center of the molecule was calculated for all series of dendrimers. Modification of the terminal groups with different charge resulted in a different ion distribution. This indicated the formation of ion complexation with the dendrimer. Water molecules were found to be well embedded in the dendrimer and formed a layer around the dendrimer as well (Figure S5).
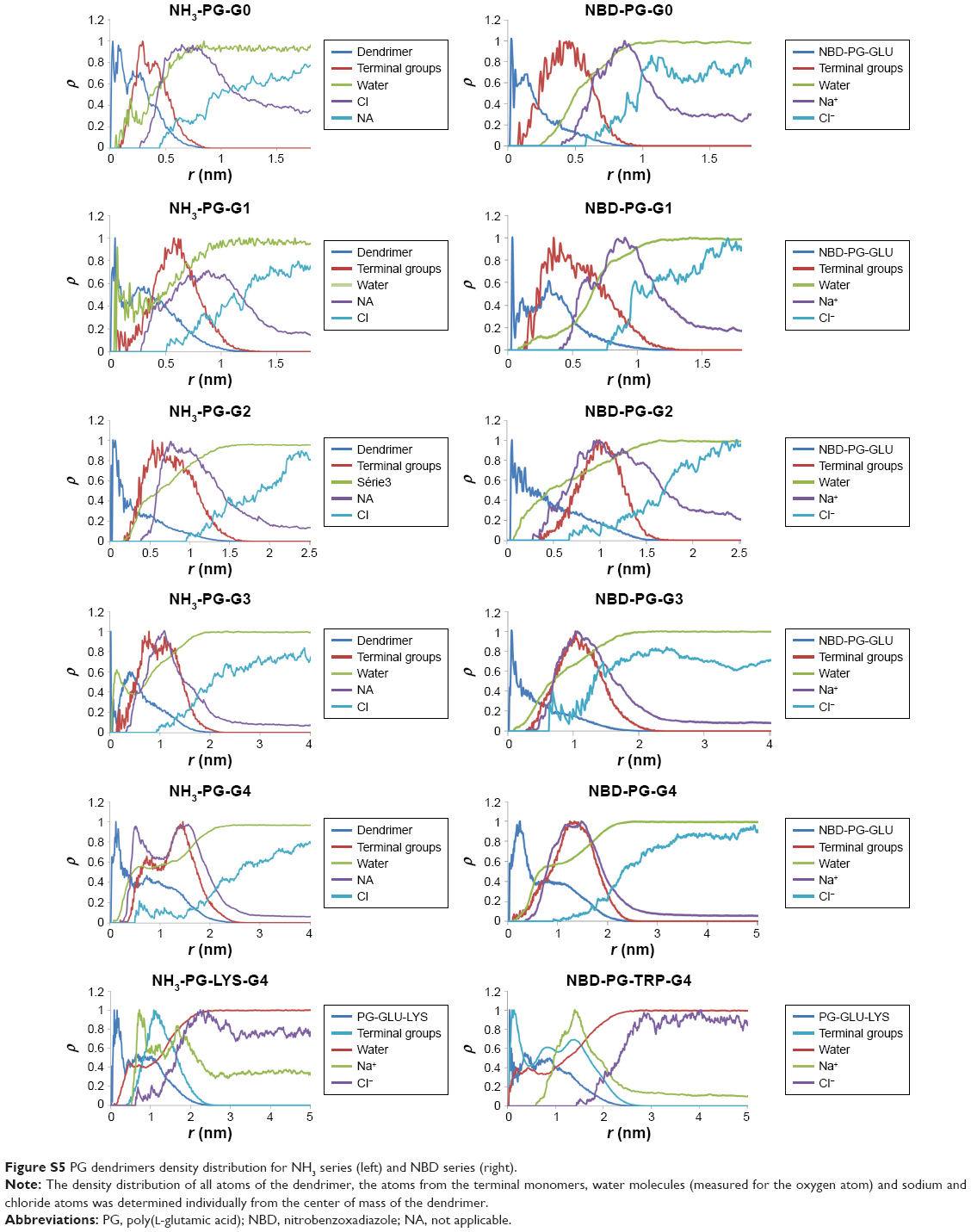 | Figure S5 PG dendrimers density distribution for NH3 series (left) and NBD series (right). |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.