Back to Journals » OncoTargets and Therapy » Volume 13
Rat Bone Marrow-Derived Mesenchymal Stem Cells Promote the Migration and Invasion of Colorectal Cancer Stem Cells
Authors Zou W, Zhao J, Li Y, Wang Z ![]() , Yan H, Liu Y, Sun M, Zhuang J, Wang J
, Yan H, Liu Y, Sun M, Zhuang J, Wang J ![]()
Received 12 February 2020
Accepted for publication 7 June 2020
Published 7 July 2020 Volume 2020:13 Pages 6617—6628
DOI https://doi.org/10.2147/OTT.S249353
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Leo Jen-Liang Su
Weiyan Zou,1 Jie Zhao,2 Yi Li,2 Zishu Wang,3 Haiqin Yan,1 Yudong Liu,1 Meiqun Sun,1 Jialu Zhuang,2 Junbin Wang3
1Department of Histology and Embryology, Bengbu Medical College, Bengbu 233004, Anhui Province, People’s Republic of China; 2The Second School of Clinical Medicine, Bengbu Medical College, Bengbu 233004, Anhui Province, People’s Republic of China; 3Department of Oncology, The First Affiliated Hospital of Bengbu Medical College, Bengbu 233004, Anhui Province, People’s Republic of China
Correspondence: Junbin Wang Email [email protected]
Background: Colorectal cancer is one of the most common cancers and the second leading cause of cancer-related deaths worldwide. Targeting cancer stem cells (CSCs) may be a novel strategy for the treatment of colorectal cancer. Previous studies have shown that bone marrow-derived MSCs (BM-MSCs) promote tumor growth and metastasis. However, the role of rat BM-MSCs in the biological behaviors of colorectal CSCs remains unclear until now.
Materials and Methods: BM-MSCs were isolated from rats and characterized. CSCs were enriched from HCT116 cells using the microsphere culture method, and the microspheres incubated for at least 10 passages were termed HCT116-CSCs that were characterized. The effects of rat BM-MSCs on migration and invasion of HCT116-CSCs were examined using transwell migration and invasion assays and xenograft tumor growth assay.
Results: Rat BM-MSCs appeared typical stem cell morphology. Flow cytometry revealed positive CD29 and CD44 expression in rat BM-MSCs at passage 3, and rat BM-MSCs were found to differentiate into osteocytes following incubation in osteogenic induction medium. Microscopy, flow cytometric detection of stem cell surface markers, colony-formation assay and transwell migration and invasion assays characterized the successful preparation of HCT116-CSCs, and subcutaneous injection of HCT116-CSCs produced xenograft tumors in nude mice, while HE staining of the xenograft tumors displayed cancer specimen shapes. Transwell migration and invasion assays showed that rat BM-MSCs promoted the migration and invasion of HCT116-CSCs, and injection of rat BM-MSCs was found to promote the growth of the mouse xenograft tumor derived from HCT116-CSCs.
Conclusion: Rat BM-MSCs promote the migration and invasion of colorectal CSCs, and colorectal CSCs may be a potential target for the therapy against colorectal cancer.
Keywords: bone marrow-derived mesenchymal stem cells, colorectal cancer, cancer stem cells, migration, invasion
Introduction
Colorectal cancer is the fourth most common cancer and the second leading cause of cancer-related deaths worldwide.1 In 2018, there were approximately 1.1 million new cases diagnosed with colorectal cancer and more than 551 thousand people died from this malignancy across the world.2 Currently, the management of colorectal cancer mainly depends on surgery, chemotherapy and radiotherapy.3 Despite great advances in the therapy of colorectal cancer, the outcomes of patients with advanced colorectal cancer remain poor and the 5-year survival rate is unsatisfactory due to tumor recurrence and metastasis and drug resistance.4,6 A search for novel treatments to improve the prognosis of patients with colorectal cancer is therefore given a high priority.7,9
Cancer stem cells (CSCs) are a small proportion of cancer cells that possess self-renewal and pluripotent capabilities and initiate and maintain tumor growth.10 CSCs are thought to be the origin of tumor metastasis, and are responsible for cancer recurrence and chemotherapy resistance.11,13 CSCs were firstly identified in acute myeloid leukemia14 and subsequently in a wide range of solid tumors, including colorectal cancer.15,17 In colorectal cancer, CSCs comprise approximately 1% of the total number of cells within the cancer.18 Currently available evidence shows that CSCs contribute to the development, metastasis, recurrence, chemotherapy resistance, prediction of pathological stages and survival in patients with colorectal cancer.19,21 In addition, targeting CSCs was hypothesized as a novel strategy for the treatment of colorectal cancer.22
Mesenchymal stem cells (MSCs) are a group of plastic-adherent cell populations that are present in multiple tissues, including umbilical cord, bone marrow and fat tissues.23 As multipotent adult stem cells, MSCs can self-renew by dividing and have the potential to differentiate into the osteogenic, chondrogenic, adipogenic, tenogenic, myogenic, or stromal lineages.23 MSC was firstly extracted and are primarily isolated from bone marrow.23,24 In recent decades, bone marrow-derived MSCs (BM-MSCs) have been paid much attention because of their potential use in regenerative medicine and tissue engineering to replace, repair or restore the function of damaged tissues or organs.25,26 Previous studies have demonstrated the success of BM-MSCs for the treatment of hematopoietic pathologies, musculoskeletal diseases, immune disorder diseases, neurodegenerative diseases, cardiovascular disorders, sporty injuries, and other diseases of the gastrointestinal and integumentary systems.27 Under long-term expansion, BM-MSCs may undergo senescence and transformation that contribute to tumor development and metastasis,28 and a recent study reported that BM-MSCs promoted lung cancer growth and metastasis.29 However, the role of rat BM-MSCs in the biological behaviors of colorectal CSCs remains unclear until now. The present study was therefore designed to examine the contribution of rat BM-MSCs to the migration and invasion of colorectal CSCs.
Materials and Methods
Cell Line and Culture
Human colorectal cancer HCT116 cell line was purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai, China), and incubated in McCoy’s 5A medium (Invitrogen; Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS; GIBCO, Rockville, MD, USA), 100 IU/mL penicillin (GIBCO; Rockville, MD, USA) and 100 μg/mL streptomycin (GIBCO; Rockville, MD, USA).
Animals
Four- to six-week-old rats of the SD strain, each weighing 80 to 100 g, were purchased from the Laboratory Animal Center of Anhui Medical University (Hefei, China), and BALB/c nude mice at ages of 4 to 6 weeks, each weighing 18 to 20 g, were obtained from Changzhou Cavens Laboratory Animal Co. Ltd. (Changzhou, China). All animals were housed in a specific-pathogen-free (SPF) facility at the Laboratory Animal Center of Bengbu Medical College (Bengbu, China), and were given free access to food and water.
Preparation and Characterization of Rat BM-MSCs
SD rats were sacrificed by CO2 inhalation, and the bilateral femurs and tibias were collected. Following removal of muscles, both ends of the femur and tibia were cut down, and the marrow cavity was flushed with low-glucose Dulbecco’s Modified Eagle’s Medium (L-DMEM; GIBCO, Rockville, MD, USA) using a 5 mL syringe. The cell suspensions collected from the marrow cavity were centrifuged at 4°C, 1000 r/min for 5 min. Then, the supernatant was discarded, and the sediment was re-suspended in 100 mL L-DMEM supplemented with 10% FBS. Cells were then seeded onto T25 culture flasks (Corning; Lowell, MA, USA) at a density of 1 × 106 to 1 × 108 cells/mL at a saturated humidity atmosphere containing 5% CO2 at 37°C. A half amount of the medium was changed 48 hours after plating and once every 3 to 4 days thereafter. The cell morphology and growth were observed under an Axiovert 200 inverted microscope (Carl Zeiss Meditec AG; Jena, Germany), when cells were defined as passage 0. Following culture for 10 to 12 days when cell confluence reached 80% to 90%, each flask was added with 1 mL pancreatin containing 0.25% EDTA (GIBCO; Rockville, MD, USA) for incubation of 30 s, and then the pancreatin was removed. Then, cells were digested with pancreatin again at 37°C for 1 min, immediately added with 2 to 3 mL L-DMEM supplemented with 10% FBS, and gently pipetted. The digested cells were passaged at a ratio of 1:2 or 1:3 (numbering: 1.25 to 1.5 × 105 cells/mL), when cells were defined as passage 1, and all non-digested cells were discarded. Subsequently, cells were passaged once every 3 to 7 days, which were defined as passages 2, 3, etc., and cells at passages 5 to 8 were harvested for the subsequent experiments.
Log-phase rat BM-MSCs at passage 3 were seeded onto 6-well culture plates (Corning; Lowell, MA, USA) at a density of 3 × 104 cells/well. After the cell confluence reached 80% to 90%, the medium was changed to osteogenic induction medium (GIBCO; Rockville, MD, USA), and the cell culture was then changed once every 3 to 4 days. Following 21-day induction, the plates were collected, washed twice in PBS, fixed in 95% ethanol at 4°C for 10 min, rinsed twice with distilled water, stained with 0.1% Alizarin Red S in 1% Tris-HCl (pH 8.3) at 37°C for 30 min, and rinsed twice in distilled water. Finally, the differentiation of rat BM-MSCs was observed under a microscope.
To detect the surface markers of rat BM-MSCs, log-phase BM-MSCs at passage 3 were digested with 2.5 g/L pancreatin containing 0.25% EDTA. Then, cells were harvested, centrifuged, and the supernatant was discarded. After the density was adjusted to 5 × 105 cells/mL, cells were then added with 300 μL fluorescence-activated cell sorting (FACS) buffer, gently pipetted to prepare single-cell suspensions, and transferred to 5 FACS tubes with 50 μL in each tube. Then, cells were incubated with fluorescein isothiocyanate (FITC)-conjugated rat anti-mouse CD29 (CD29-FITC) antibody (eBioscience, Inc.; San Diego, CA, USA), phycoerythrin (PE)-conjugated rat anti-mouse CD44 (CD44-PE) antibody (eBioscience, Inc.; San Diego, CA, USA), FITC-conjugated rat anti-mouse CD34 (CD34-FITC) antibody (eBioscience, Inc.; San Diego, CA, USA) and PE-conjugated rat anti-mouse CD45 (CD45-PE) antibody (eBioscience, Inc.; San Diego, CA, USA), while an isotype control was assigned for each assay. Cells were incubated on ice for 30 min, washed with 1 mL PBS, centrifuged at 1500 r/min for 5 min, washed twice in PBS, and added with the fixation solution to a total volume of 250 μL in each tube. Subsequently, single-cell suspensions were formulated and analyzed on a DXP Athena flow cytometer (Cytek; Fremont, CA, USA).
Preparation of CSCs Microsphere
CSCs were enriched from HCT116 cells using the microsphere culture method.30 Briefly, HCT116 cells were digested with pancreatin containing 0.25% EDTA, which was terminated with serum-containing medium. Then, cells were centrifuged, and the supernatant was discarded. The sediment was rinsed twice in PBS, and re-suspended in complete stem cell medium (DMEM/F12 medium supplemented with 10 ng/mL B27, 20 ng/mL EGF, 10 ng/mL bFGF and 10 ng/mL LIF), and the cell number was counted. Then, cells were seeded onto ultra-low adhesive petri dishes at a density of 1 × 104 cells/mL, and incubated at a saturated humidity atmosphere containing 5% CO2 at 37°C. The microsphere formation was observed under an inverted microscope. Semi-quantitative medium changes were done once every 2 to 3 days, and cell passage was completed once every 5 to 8 days. The microspheres were incubated for at least 10 passages to enrich CSCs from HCT116 cells, which were termed HCT116-CSCs.
Characterization of HCT116-CSCs
To detect the expression of HCT116-CSCs surface markers CD133 and CD44, log-phase HCT116 cells and HCT116-CSCs were prepared into single-cell suspensions and adjusted to a density of 5 × 106 cells/mL. Cells were added with 300 μL FACS buffer, gently pipetted to prepare single-cell suspensions, and transferred to 5 FACS tubes with 50 μL in each tube. Then, cells were incubated with FITC-conjugated rat anti-human CD44 (CD44-FITC) antibody (Invitrogen; Carlsbad, CA, USA) and PE-conjugated mouse anti-human CD133 (CD133-PE) antibody (Invitrogen; Carlsbad, CA, USA) on ice in darkness for 30 min, while an isotype control was assigned for each assay. Then, cells were washed with 1 mL PBS, centrifuged at 1500 r/min for 5 min, washed twice in PBS, and added with fixation solution to a total volume of 250 μL in each tube. Subsequently, single-cell suspensions were formulated and subjected to flow cytometric analysis.
To observe cell growth, a colony-formation assay was performed. Briefly, log-phase HCT116 cells and HCT116-CSCs were prepared into single-cell suspensions and seeded onto 6-well plates at a density of 200 cells per well, with three duplicate wells assigned for each type of cells. Cells were incubated in McCoy’s 5A medium supplemented with 10% FBS, and the medium was changed once every 3 to 4 days. Following 2-week incubation, the 6-well plate was removed and the medium was discarded. Cells were washed twice in PBS, fixed in neutral methanol at room temperature for 20 min, stained with crystal violet for 20 min and washed twice in PBS. Cell colony with more than 30 cells was visualized, counted under a microscope, and the colony-formation rate was calculated using the following formula: colony-formation rate (%) = number of cell colonies/number of cells seeded × 100%.
To observe cell migration and invasion, transwell migration and invasion assays were performed. Briefly, log-phase HCT116 cells and HCT116-CSCs were prepared into single-cell suspensions and transferred to Transwell chambers at a density of 4 × 104 cells per chamber. Cells were re-suspended in 200 μL serum-free McCoy’s 5A medium, and 800 μL McCoy’s 5A medium supplemented with 10% FBS was transferred to the lower chamber, with three duplicate chambers assigned for each type of cells. Cells were incubated at 37°C containing 5% CO2 for 12 to 24 hours. Then, the chamber was collected, and the medium in the upper chamber was removed. Following removal of non-migrated cells, the chamber was washed twice or thrice in PBS, fixed in neutral methanol for 20 min, washed twice or thrice in PBS, stained with crystal violet for 20 min and washed twice or thrice in PBS to remove the residual crystal violet. Cell was observed and counted under a microscope. For the transwell invasion assay, the chamber bottom was coated with a mixture of Matrigel and serum-free McCoy’s 5A medium at a ratio of 1:8, and other experimental procedures were the same with the migration assay.
To observe the tumor xenograft growth, a tumor-bearing mouse xenograft model was established. Briefly, 1 × 105 cells log-phase HCT116 cells (left groin) and HCT116-CSCs (right groin) were subcutaneously injected into the left and right groins of a nude mouse, and all 5 mice were sacrificed 4 weeks post-injection. The tumor xenograft growth was observed and its histology was visualized using HE staining.
Effect of Rat BM-MSCs on Migration and Invasion of HCT116-CSCs
To assess the effect of rat BM-MSCs on the migration and invasion of HCT116-CSCs, transwell migration and invasion assays were performed. Four groups were assigned in the lower chamber, including the control group (DMEM/F12 medium), BM-MSCs group (rat BM-MSCs incubated in DMEM/F12 medium), 10% FBS group (DMEM/F12 medium + 10% FBS) and rat BM-MSCs + 10% FBS group (rat BM-MSCs incubated in DMEM/F12 medium supplemented with 10% FBS), with three duplicate wells in each group. Log-phase rat BM-MSCs at the 7th passage were seeded onto 24-well plates at a density of 5 × 104 cells/well and plated for 24 hours. Then, cells were washed twice in PBS and added with 800 μL DMEM/F12 medium. Log-phase HCT116-CSCs were prepared into single-cell suspensions, transferred to the lower chamber (3 × 104 cells) in each group, and re-suspended in 200 μL serum-free DMEM/F12 medium. Cells were incubated at 37°C containing 5% CO2 for 24 to 48 hours. Then, the chamber was collected, and the medium in the upper chamber was removed. Following removal of non-migrated cells, the chamber was washed twice or thrice in PBS, fixed in neutral methanol for 20 min, washed twice or thrice in PBS, stained with crystal violet for 20 min and washed twice or thrice in PBS to remove the residual crystal violet. Cell was observed and counted under a microscope. For transwell invasion assay, the chamber bottom was coated with Matrigel, and other experimental procedures were the same with the migration assay.
To observe the effect of rat BM-MSCs on tumor xenograft growth, a tumor-bearing mouse xenograft model was established. Briefly, rat BM-MSCs and HCT116-CSCs were prepared into single-cell suspension by digestion with pancreatin containing 0.25% EDTA, washed twice in PBS and counted. BALB/c nude mice were randomly assigned into 5 groups, of 5 mice in each group. Mice in the S + injection group were subcutaneously injected with 1 × 105 HCT116-CSCs, mice in the S + S co-injection group 1 were co-injected with 1 × 105 HCT116-CSCs and 1 × 105 BM-CSCs subcutaneously, mice in the S + S co-injection group 2 were co-injected with 1 × 105 HCT116-CSCs and 2 × 105 rat BM-CSCs subcutaneously, mice in the S + V co-injection group 1 were subcutaneously injected with 1 × 105 HCT116-CSCs and intravenously injected with 1 × 105 rat BM-CSCs via the tail vein, and mice in the S + V co-injection group 2 were subcutaneously injected with 1 × 105 HCT116-CSCs and intravenously injected with 2 × 105 rat BM-CSCs via the tail vein (Figure 1A). Since the presence of the xenograft tumor, the longest diameter and the shortest diameter of the tumor were measured once every 3 days for successive 6 weeks. Then, all mice were sacrificed, and xenograft tumors were collected. The tumor volume was measured and intrapulmonary metastasis was observed. The histological changes of the xenograft tumors and intrapulmonary metastasis were observed.
 |
Figure 1 Xenograft tumor growth assay reveals the effect of BM-MSCs on the migration and invasion of HCT116-CSCs. (A) Mice are inoculated with BM-MSCs and HCT116-CSCs; (B) Nude mice xenograft tumors; (C) Volumes of nude mice xenograft tumors at various time points; (D) Weight of nude mice xenograft tumors at various time points; (E) Lungs of nude mice harboring xenograft tumors; (F) Intrapulmonary metastasis 6 weeks post-inoculation of BM-MSCs and HCT116-CSCs; (G) HE staining of xenograft tumors (× 400); (H) HE staining of lung specimens of nude mice harboring xenograft tumors (× 400). *Indicates metastatic foci from tumors (× 100). Ho indicates the amplification of the sites marked by * (× 400). **P < 0.01; ***P < 0.001. |
Ethical Statement
This study was approved by the Ethics Review Committee of Bengbu Medical College (permission number: BBMC-2016-JC00201), and all efforts were made to minimize animal suffering and to reduce the number of animals used in the experiments. All experimental procedures were performed in accordance with the international Guidelines for the Use and Care of Laboratory Animals, Regulation of the People’s Republic of China on the Administration of Human Genetic Resources, and the National Regulations for the Management of Laboratory Animals in China.
Data Analysis
All data were entered into Microsoft Excel 2017 (Microsoft; Redmond, WA, USA), and all statistical analyses were done using the statistical software SPSS version 21.0 (SPSS, Inc.; Chicago, IL, USA). Multi-group comparisons were performed with one-way analysis of variance (ANOVA), followed by the SNK test, and differences of proportions were tested for statistical significance with chi-square test. A P value of < 0.05 was considered statistically significant.
Results
Characterization of Rat BM-MSCs
The rat BM-MSCs at day 7 of passage 0 (Figure 2A1) and at passage 3 (Figure 2A2) presented a fibroblast-like and spindle-shaped morphology. Flow cytometry detected positive CD29 and CD44 expression and negative CD45 and CD34 expression in rat BM-MSCs at passage 3 (Figure 2B). In addition, the rat BM-MSCs at passage 3 had the potential for differentiation along the osteogenic lineages, as determined by Alizarin Red S staining. Following incubation in osteogenic induction medium for 21 days, rat BM-MSCs were found to differentiate into osteocytes (Figure 2C).
 |
Figure 2 Characterization of rat bone marrow-mesenchymal stem cells (BM-MSCs). (A) Microscopic observation shows that the isolated BM-MSCs have a fibroblast-like and spindle-shaped morphology (× 100). 1, BM-MSCs on day 7 of passage 0; 2, BM-MSCs at the third passage; (B) Flow cytometry detects positive CD29 and CD44 expression and negative CD34 and CD45 expression in passage 3 BM-MSCs; (C) Following incubation in osteogenic induction medium for 21 days, BM-MSCs were stained with Alizarin Red S. Osteocytes differentiation is evidenced by calcium deposits stained with Alizarin Red S (C2), while untreated BM-MSCs show no calcium deposits (C1) (× 100). |
Biological Features of HCT116-CSCs
Following incubation complete stem cell medium for 24 hours, most HCT116 cells showed a single-cell suspension growth pattern with small sizes and circular, transparent and bright shapes; however, there are a small proportion of cells adherent to the dish wall. On day 3 after incubation, cells formed clusters and the cell size was enlarged, with cells appearing transparent, bright and circular shapes. Then, the size of cell clusters gradually increased. Typical microsphere morphology was formed on day 5 after incubation, and the cell number s increased, which appeared a suspension growth pattern (Figure 3A1). With the microsphere passage, there was an increase in the formation of microspheres, and less and less cells were adherent to the wall. There was almost no cells adherent to the wall until 6 passages (Figure 3A2). Flow cytometry detected a higher percentage of CD133+CD44+ cells in parental HCT116 cells (79.5%) than in HCT116-CSCs (94.2%, Figure 3B). Colony-formation assay revealed more colonies of HCT116-CSCs than that of parental HCT116 cells (Figure 3C). Transwell migration and invasion assays revealed a stronger migrating and invasive ability of HCT116-CSCs than that of parental HCT116 cells (Figure 4A). In addition, injection of parental HCT116 cells did not form tumors, while subcutaneous injection of HCT116-CSCs produced xenograft tumors in nude mice, and HE staining of the xenograft tumors showed cancer specimen shapes (Figure 4B).
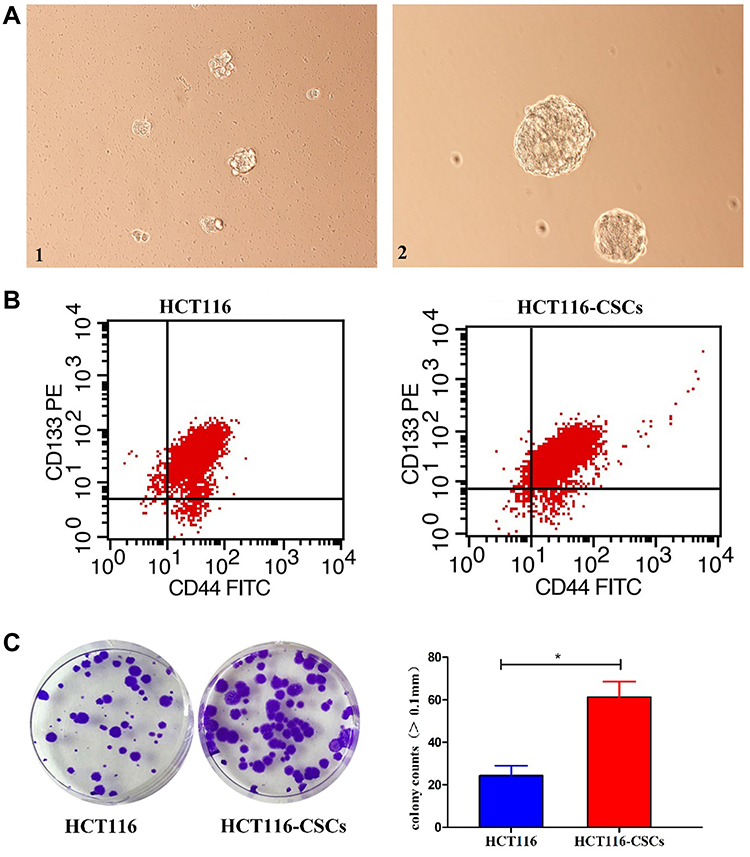 |
Figure 3 Characterization and growth of HCT116-CSCs. (A) HCT116-CSCs culture (× 200). 1, HCT116-CSCs on day 5 after incubation; A2, HCT116-CSCs on day 5 after successive 6 passages of incubation; (B) Flow cytometry detects the percentages of CD133+CD44+ cells in parental HCT116 cells and HCT116-CSCs; (C) Colony-formation assay measures the colony formation of parental HCT116 cells and HCT116-CSCs. *P < 0.05. |
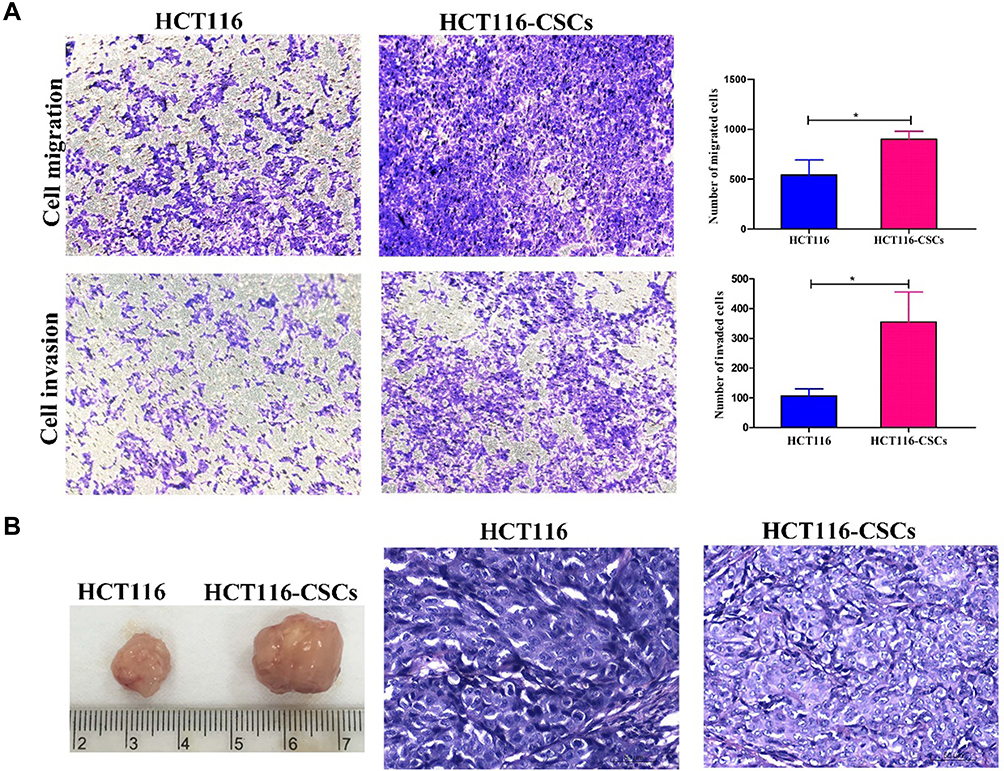 |
Figure 4 In vitro and in vivo migration and invasion of HCT116 cells and HCT116-CSCs. (A) Transwell migration and invasion assays detects the migration and invasion of HCT116 cells and HCT116-CSCs (× 200); (B) HCT116-CSCs-derived xenograft tumors (left) and HE staining of the tumors (right, × 400). *P < 0.05. |
Rat BM-MSCs Promote Migration and Invasion of HCT116-CSCs
Transwell migration and invasion assays revealed BM-MSCs promoted the migration and invasion of HCT116-CSCs in the absence or presence of FBS in the lower chamber, and more cells migrated and invaded through the membrane in the rat BM-MSCs + 10% FBS group than in the 10% FBS group (Figure 5A–C). Our data demonstrate that rat BM-MSCs promote the migration and invasion of HCT116-CSCs regardless of the medium.
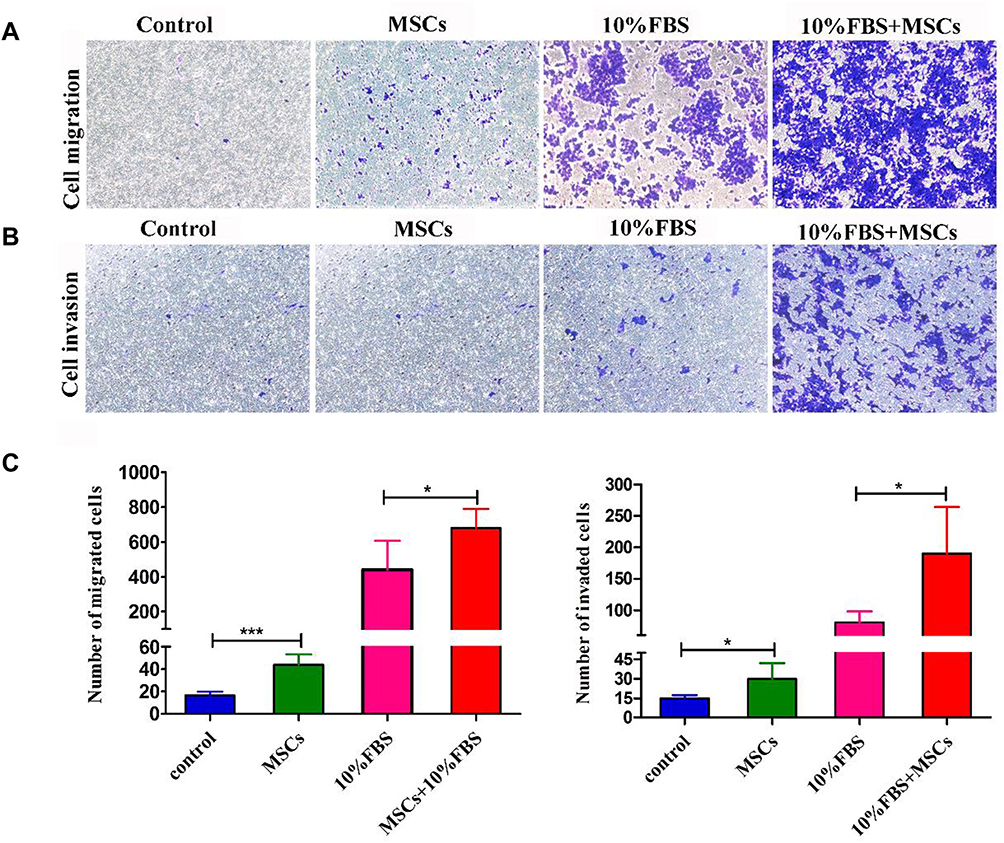 |
Figure 5 Effects of BM-MSCs on the migration and invasion of HCT116-CSCs. (A and B) Transwell migration and invasion assays detect cells in the BM-MSCs group, the 10% FBS group and the BM-MSCs + 10% FBS group crossing the membrane; however, more cells in the BM-MSCs + 10% FBS group are found to penetrate through the membrane (× 200); (C) Comparison of the number of cells penetrating through the membrane. *P < 0.05; ***P < 0.001. |
Xenograft Tumor Growth Assay
Xenograft tumors were produced in the S + injection group, S + S co-injection group 1 and S + S co-injection group 2; however, the tumor volume and weight were significantly greater in the S + S co-injection group 1 and S + S co-injection group 2 than in the S + injection group (P < 0.05), and the tumor volume and weight were significantly greater in the S + S co-injection group 2 than in the S + S co-injection group 1 (P < 0.05) (Figure 1B–D). No metastatic foci were found in the lung of 5 nude mice in the S + injection group, while there were 3 and 5 mice with intrapulmonary metastases in the S + S co-injection group 1 and S + S co-injection group 2, respectively (Figure 1E). The xenograft tumor volume and weight was greater in the S + V co-injection group 1 and S + V co-injection group 2 than in the S + injection group (P < 0.05), and the tumor volume and weight was greater in the S + V co-injection group 2 than in the S + V co-injection group 1 (P > 0.05); however, the tumor volume and weight was lower in the S + V co-injection group 1 and S + V co-injection group 2 than in the S + S co-injection group 1 and S + S co-injection group 2 (Figure 1C and D). In addition, there were 2 and 5 mice with intrapulmonary metastases in the S + V co-injection group 1 and S + V co-injection group 2, respectively (Figure 1F–H).
Discussion
Although great advances have been recently achieved in the management of colorectal cancer, the long-term outcomes of this common malignancy remain unsatisfactory,3 which is mainly because of the tumor occurrence and chemotherapy resistance.31,32 The success of MSC-based therapy for colorectal cancer has been documented in both experimental and clinical models; however, there are still a number of questions that remain to be investigated.33,35 Our data showed, for the first time, that rat BM-MSCs promoted the migration and invasion of colorectal CSCs, which is of great value for development of novel treatments for colorectal cancer.
Currently, the role of BM-MSCs in colorectal cancer remains in dispute.28 Human BM-MSCs were found to stimulate invasion, survival and tumorigenesis of colorectal cancer through the release of soluble neuregulin 1 (NRG1), activating the HER2/HER3-dependent PI3K/AKT signalling cascade in colorectal cancer cells, and high transmembrane NRG1 expression was found to correlate with poor prognosis in colorectal cancer,36 and in vitro and in vivo assays showed that human BM-MSCs-secreted IL-8 promoted the angiogenesis and growth of colorectal cancer.37 In addition, human BM-MSCs were reported to promote colorectal cancer progression via CCR5, and serum levels of CCL3 and CCL4 could be predictive biomarkers for the prognosis of patients with colorectal cancer.38 However, there are studies reporting that BM-MSCs ameliorate the tumorigenesis of inflammatory bowel disease in mice,39,40 and BM-MSCs-derived exosomes overexpressing miR-16-5p were found to suppress proliferation, migration, and invasion and promote apoptosis of colorectal cancer cells by downregulating ITGA2.41 However, the contribution of BM-MSCs to colorectal CSCs remains unknown until now.
Although colorectal CSCs comprise a very small proportion of total cells found in colorectal cancer, they have shown a crucial role not only in the establishment of the primary colorectal cancer but also in the metastasis of the disease, owing to their self-renewal capacity,18,19 and CSCs have been identified as a promising therapeutic target for colorectal cancer.42 Hereby, we aimed to examine the effect of rat BM-MSCs on the progression of colorectal CSCs, so as to provide insights into the identification of novel therapeutic targets for colorectal cancer.
In this study, we enriched colorectal CSCs from HCT116 cells using a microsphere culture system, and HCT116-CSCs were characterized by microscopy, flow cytometric detection of stem cell surface markers, colony-formation assay and transwell migration and invasion assays. In addition, a mouse xenograft tumor growth assay revealed that subcutaneous injection of HCT116-CSCs produced xenograft tumors in nude mice, and HE staining of the xenograft tumors showed cancer specimen shapes. These findings indicated the success of isolation of HCT116-CSCs. To observe the effect of rat BM-MSCs on HCT116-CSCs progression, we isolated BM-MSCs from rats, and the isolated cells appeared typical stem cell morphology. Furthermore, flow cytometry revealed positive CD29 and CD44 expression in rat BM-MSCs at passage 3, and rat BM-MSCs were found to differentiate into osteocytes following incubation in osteogenic induction medium. These data confirmed the successful isolation of BM-MSCs from rats. Then, transwell migration and invasion assays revealed that rat BM-MSCs promoted the migration and invasion of HCT116-CSCs regardless of the medium, and the mouse xenograft tumor growth assay showed that injection of rat BM-MSCs promoted the growth of the xenograft tumor derived from HCT116-CSCs. Previous studies have demonstrated that human BM-MSCs promote the progression of colorectal cancer,36,38 which is in agreement with the findings from the present study. Nevertheless, this is the first report to demonstrate that rat-derived BM-MSCs promote the progression of colorectal CSCs. It is therefore hypothesized that the inhibition of the interaction between rat BM-MSCs and colorectal CSCs may provide the possibility of a novel therapeutic strategy for colorectal cancer.
Conclusions
In summary, the results of the present study demonstrate that rat BM-MSCs promote the migration and invasion of colorectal CSCs. Our data further confirm colorectal CSCs as a target for the therapy against colorectal cancer, and provide new insights into the treatment of colorectal cancer. However, further studies to investigate the contribution of human-derived BM-MSCs to colorectal cancer progression and the mechanisms underlying the mediation of rat BM-MSCs in the migration and invasion of colorectal CSCs seem justified.
Funding
This study was supported by the grants from the Key Project of the Natural Science Funding in Anhui Provincial Colleges and Universities (grant nos. KJ2018A0245 and KJ2019A0295) and the Natural Science Foundation of Anhui Province (grant no. 1808085MC54) and Bengbu Medical College Science Fund for Innovative Research Team (grant no. BYKC201908).
Disclosure
The authors declare no conflicts of interests in this work.
References
1. Dekker E, Tanis PJ, Vleugels JLA, Kasi PM, Wallace MB. Colorectal cancer. Lancet. 2019;394(10207):1467–1480. doi:10.1016/S0140-6736(19)32319-0
2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
3. Brown KGM, Solomon MJ, Mahon K, O’Shannassy S. Management of colorectal cancer. BMJ. 2019;366:l4561. doi:10.1136/bmj.l4561
4. Kuipers EJ, Grady WM, Lieberman D, et al. Colorectal cancer. Nat Rev Dis Primers. 2015;1:15065. doi:10.1038/nrdp.2015.65
5. Van der Jeught K, Xu HC, Li YJ, Lu XB, Ji G. Drug resistance and new therapies in colorectal cancer. World J Gastroenterol. 2018;24:3834–3848. doi:10.3748/wjg.v24.i34.3834
6. Mahar AL, Compton C, Halabi S, Hess KR, Weiser MR, Groome PA. Personalizing prognosis in colorectal cancer: a systematic review of the quality and nature of clinical prognostic tools for survival outcomes. J Surg Oncol. 2017;116:969–982. doi:10.1002/jso.24774
7. Dzunic M, Petkovic I, Cvetanovic A, Vrbic S, Pejcic I. Current and future targets and therapies in metastatic colorectal cancer. J BUON. 2019;24:1785–1792.
8. Lee CL, Veeramani S, Molouki A, et al. Virotherapy: current trends and future prospects for treatment of colon and rectal malignancies. Cancer Invest. 2019;37:393–414. doi:10.1080/07357907.2019.1660887
9. Sur DG, Colceriu M, Sur G, et al. MiRNAs roles in the diagnosis, prognosis and treatment of colorectal cancer. Expert Rev Proteomics. 2019;16:851–856. doi:10.1080/14789450.2019.1659732
10. Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006;355(12):1253–1261. doi:10.1056/NEJMra061808
11. Croker AK, Allan AL. Cancer stem cells: implications for the progression and treatment of metastatic disease. J Cell Mol Med. 2008;12(2):374–390. doi:10.1111/j.1582-4934.2007.00211.x
12. Atashzar MR, Baharlou R, Karami J, et al. Cancer stem cells: a review from origin to therapeutic implications. J Cell Physiol. 2020;235(2):790–803. doi:10.1002/jcp.29044
13. Yadav AK, Desai NS. Cancer stem cells: acquisition, characteristics, therapeutic implications, targeting strategies and future prospects. Stem Cell Rev Rep. 2019;15(3):331–355. doi:10.1007/s12015-019-09887-2
14. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730–737. doi:10.1038/nm0797-730
15. Eun K, Ham SW, Kim H. Cancer stem cell heterogeneity: origin and new perspectives on CSC targeting. BMB Rep. 2017;50:117–125. doi:10.5483/BMBRep.2017.50.3.222
16. Ajani JA, Song S, Hochster HS, Steinberg IB. Cancer stem cells: the promise and the potential. Semin Oncol. 2015;42:S3–S17. doi:10.1053/j.seminoncol.2015.01.001
17. Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. 2007;58(1):267–284. doi:10.1146/annurev.med.58.062105.204854
18. Munro MJ, Wickremesekera SK, Peng L, Tan ST, Itinteang T. Cancer stem cells in colorectal cancer: a review. J Clin Pathol. 2018;71(2):110–116. doi:10.1136/jclinpath-2017-204739
19. Wahab SMR, Islam F, Gopalan V, Lam AK. The identifications and clinical implications of cancer stem cells in colorectal cancer. Clin Colorectal Cancer. 2017;16:93–102. doi:10.1016/j.clcc.2017.01.011
20. Anderson EC, Hessman C, Levin TG, Monroe MM, Wong MH. The role of colorectal cancer stem cells in metastatic disease and therapeutic response. Cancers (Basel). 2011;3(1):319–339. doi:10.3390/cancers3010319
21. Fanali C, Lucchetti D, Farina M, et al. Cancer stem cells in colorectal cancer from pathogenesis to therapy: controversies and perspectives. World J Gastroenterol. 2014;20:923–942. doi:10.3748/wjg.v20.i4.923
22. Szaryńska M, Olejniczak A, Kobiela J, Spychalski P, Kmieć Z. Therapeutic strategies against cancer stem cells in human colorectal cancer. Oncol Lett. 2017;14:7653–7668. doi:10.3892/ol.2017.7261
23. Bianco P. “Mesenchymal” stem cells. Annu Rev Cell Dev Biol. 2014;30(1):677–704. doi:10.1146/annurev-cellbio-100913-013132
24. Friedenstein AJ, Latzinik NW, Grosheva AG, Gorskaya UF. Marrow microenvironment transfer by heterotopic transplantation of freshly isolated and cultured cells in porous sponges. Exp Hematol. 1982;10:217–227.
25. Barry FP, Murphy JM. Mesenchymal stem cells: clinical applications and biological characterization. Int J Biochem Cell Biol. 2004;36(4):568–584. doi:10.1016/j.biocel.2003.11.001
26. Polymeri A, Giannobile WV, Kaigler D. Bone marrow stromal stem cells in tissue engineering and regenerative medicine. Horm Metab Res. 2016;48(11):700–713. doi:10.1055/s-0042-118458
27. Hoch AI, Leach JK. Concise review: optimizing expansion of bone marrow mesenchymal stem/stromal cells for clinical applications. Stem Cells Transl Med. 2014;3(5):643–652. doi:10.5966/sctm.2013-0196
28. Chu DT, Phuong TNT, Tien NLB, et al. An update on the progress of isolation, culture, storage, and clinical application of human bone marrow mesenchymal stem/stromal cells. Int J Mol Sci. 2020;21:E708. doi:10.3390/ijms21030708
29. Luo D, Hu SY, Liu GX. Multi-channel promotion of lung cancer progress by bone marrow derived mesenchymal stem cells in tumor microenvironment. Chin J Oncol. 2018;40:85–91.
30. Heiskanen A, Hirvonen T, Salo H, et al. Glycomics of bone marrow-derived mesenchymal stem cells can be used to evaluate their cellular differentiation stage. Glycoconj J. 2009;26(3):367–384. doi:10.1007/s10719-008-9217-6
31. Majidinia M, Yousefi B. Long non-coding RNAs in cancer drug resistance development. DNA Repair (Amst). 2016;45:25–33. doi:10.1016/j.dnarep.2016.06.003
32. Majidinia M, Alizadeh E, Yousefi B, Akbarzadeh M, Zarghami N. Downregulation of notch signaling pathway as an effective chemosensitizer for cancer treatment. Drug Res (Stuttg). 2016;66(11):571–579. doi:10.1055/s-0042-111821
33. El-Said MM, Emile SH. Cellular therapy: a promising tool in the future of colorectal surgery. World J Gastroenterol. 2019;25(13):1560–1565. doi:10.3748/wjg.v25.i13.1560
34. Ocansey DKW, Qiu W, Wang J, et al. The achievements and challenges of mesenchymal stem cell-based therapy in inflammatory bowel disease and its associated colorectal cancer. Stem Cells Int. 2020;2020:7819824. doi:10.1155/2020/7819824
35. Majidinia M, Reiter RJ, Shakouri SK, et al. The multiple functions of melatonin in regenerative medicine. Ageing Res Rev. 2018;45:33–52. doi:10.1016/j.arr.2018.04.003
36. De Boeck A, Pauwels P, Hensen K, et al. Bone marrow-derived mesenchymal stem cells promote colorectal cancer progression through paracrine neuregulin 1/HER3 signalling. Gut. 2013;62:550–560. doi:10.1136/gutjnl-2011-301393
37. Wang J, Wang Y, Wang S, et al. Bone marrow-derived mesenchymal stem cell-secreted IL-8 promotes the angiogenesis and growth of colorectal cancer. Oncotarget. 2015;6:42825–42837. doi:10.18632/oncotarget.5739
38. Nishikawa G, Kawada K, Nakagawa J, et al. Bone marrow-derived mesenchymal stem cells promote colorectal cancer progression via CCR5. Cell Death Dis. 2019;10(4):264. doi:10.1038/s41419-019-1508-2
39. Zheng XB, He XW, Zhang LJ, et al. Bone marrow-derived CXCR4-overexpressing MSCs display increased homing to intestine and ameliorate colitis-associated tumorigenesis in mice. Gastroenterol Rep (Oxf). 2019;7:127–138. doi:10.1093/gastro/goy017
40. Chen Z, He X, He X, et al. Bone marrow mesenchymal stem cells ameliorate colitis-associated tumorigenesis in mice. Biochem Biophys Res Commun. 2014;450:1402–1408. doi:10.1016/j.bbrc.2014.07.002
41. Xu Y, Shen L, Li F, Yang J, Wan X, Ouyang M. microRNA-16-5p-containing exosomes derived from bone marrow-derived mesenchymal stem cells inhibit proliferation, migration, and invasion, while promoting apoptosis of colorectal cancer cells by downregulating ITGA2. J Cell Physiol. 2019;234:21380–21394. doi:10.1002/jcp.28747
42. Iyer DN, Sin WY, Ng L. Linking stemness with colorectal cancer initiation, progression, and therapy. World J Stem Cells. 2019;11:519–534. doi:10.4252/wjsc.v11.i8.519
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.