Back to Journals » Clinical Interventions in Aging » Volume 18
Rapamycin Responds to Alzheimer’s Disease: A Potential Translational Therapy
Authors Hou SJ, Zhang SX, Li Y ![]() , Xu SY
, Xu SY ![]()
Received 7 July 2023
Accepted for publication 25 September 2023
Published 2 October 2023 Volume 2023:18 Pages 1629—1639
DOI https://doi.org/10.2147/CIA.S429440
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Nandu Goswami
Si-Jia Hou,1 Sheng-Xiao Zhang,2 Yang Li,1 Sui-Yi Xu1
1Department of Neurology, Headache Center, The First Hospital of Shanxi Medical University, Taiyuan, Shanxi Province, 030001, People’s Republic of China; 2Department of Rheumatology, The Second Hospital of Shanxi Medical University, Taiyuan, Shanxi Province, 030009, People’s Republic of China
Correspondence: Yang Li; Sui-Yi Xu, Department of Neurology, Headache Center, The First Hospital of Shanxi Medical University, Jiefangnan Road 85, Taiyuan, Shanxi Province, 030001, People’s Republic of China, Tel +86 15035182003 ; +86 15103513579, Email [email protected]; [email protected]
Abstract: Alzheimer’s disease (AD) is a sporadic or familial neurodegenerative disease of insidious onset with progressive cognitive decline. Although numerous studies have been conducted or are underway on AD, there are still no effective drugs to reverse the pathological features and clinical manifestations of AD. Rapamycin is a macrolide antibiotic produced by Streptomyces hygroscopicus. As a classical mechanistic target of rapamycin (mTOR) inhibitor, rapamycin has been shown to be beneficial in a variety of AD mouse and cells models, both before the onset of disease symptoms and the early stage of disease. Although many basic studies have demonstrated the therapeutic effects of rapamycin in AD, many questions and controversies remain. This may be due to the variability of experimental models, different modes of administration, dose, timing, frequency, and the availability of drug-targeting vehicles. Rapamycin may delay the development of AD by reducing β-amyloid (Aβ) deposition, inhibiting tau protein hyperphosphorylation, maintaining brain function in APOE ϵ4 gene carriers, clearing chronic inflammation, and improving cognitive dysfunction. It is thus expected to be one of the candidates for the treatment of Alzheimer’s disease.
Keywords: β-amyloid, chronic inflammation, therapeutic effect, macrolide antibiotic
Introduction
Alzheimer’s disease (AD) is a sporadic or familial neurodegenerative disease of insidious onset with progressive cognitive decline, accounting for 50–60% of all dementia patients.1 As the global population ages, the prevalence of AD is increasing year by year. Currently, approximately 50 million people have dementia worldwide, and are expected to reach 152 million by 2050, which will put enormous pressure on society and the economy.2 The main pathological features of AD are the presence of dense senile plaques (SPs) and neurofibrillary tangles (NFTs) in the brain, resulting in massive neuronal and synaptic loss and brain atrophy. Current hypotheses on the pathogenesis of AD include β-amyloid (Aβ) deposition, microtubule-associated protein tau tangles, defective autophagy, insulin resistance, cerebrovascular dysfunction, inflammatory response, mitochondrial dysfunction, and oxidative stress. Although numerous studies have been done or are underway on AD, there have been no effective drugs to reverse the pathological features and clinical manifestations of AD.
Recent studies suggest rapamycin might be a new drug for AD.3 Rapamycin is a macrolide antibiotic produced by Streptomyces hygroscopicus.4 As a classical mechanistic target of rapamycin (mTOR) inhibitor, rapamycin has multiple functions in regulating cell metabolism, cytoskeleton formation, cell life span, and autophagy.5 In addition, rapamycin prolonged the life span of mice,6 and subsequent studies gradually confirmed the anti-aging effects of rapamycin.7,8 In addition, increasing age is the greatest risk factor for the development of AD, and delaying aging may retard AD. Rapamycin has also been shown to be beneficial in a variety of AD mouse and cell models, both before the onset of disease symptoms and the early stage of disease, with improvements observed in AD mice and cells following rapamycin treatment.9–12 Therefore, it is presumed that rapamycin may be a potential translational drug for AD.13
Rapamycin Reduces Aβ Deposition and Promotes Aβ Clearance
Soluble oligomeric Aβ and insoluble β-amyloid are highly neurotoxic and can induce neuronal degeneration. The brain quickly clears Aβ under normal circumstances. However, in the brain of a patient with AD, there is an imbalance between Aβ generation and clearance. The abnormally activated mTOR affects Aβ metabolism by inhibiting autophagy and increasing insulin resistance, thus hindering Aβ clearance.14,15 mTOR activity was closely related to Aβ. AD cells and animal models found higher mTOR signaling pathway activity in cells and animal models with Aβ accumulation.16 The mechanism may be due to the over-activation of the mTOR pathway promoted by the aggregated Aβ. Although whether the mTOR pathway has a direct effect on Aβ production by β and γ secretases remains unelucidated, it has been shown that increasing sirtuin 1 (SIRT1) expression by inhibiting mTOR can inhibit Aβ production.17 SIRT1 is a key regulator of α-secretase and therefore of the Aβ production process.18
Rapamycin enhances autophagy by inhibiting mTOR signaling, regulates cellular metabolism, improves Aβ clearance, and reduces Aβ levels (Figure 1). For example, the treatment of an 8-month-old 3xTg-AD mice with rapamycin revealed a significant reduction in Aβ immunoreactivity of neurons in the CA1 region of the hippocampus.19 In the PDAPP transgenic mouse model, Aβ plaque deposition was reduced in mice by feeding rapamycin.9 Moreover, the addition of rapamycin to PC12 cells activated intracellular autophagy and upregulated Aβ-induced Beclin-1 expression, thereby increasing cell viability, suggesting that rapamycin protects neuronal cells by reducing Aβ levels through enhanced autophagy.20 In addition, rapamycin increases SIRT1 expression by inhibiting mTOR, and SIRT1 can also negatively regulate mTOR.21 The interaction between mTOR and SIRT1 continuously amplifies SIRT1 activity through a positive feedback loop, inhibits mTOR signaling, regulates Aβ production, and clears Aβ by increasing autophagy. Furthermore, rapamycin activates autophagy and inhibits Aβ production by regulating signaling pathways, such as Wnt/GSK3β/β-catenin,12 insulin/insulin-like growth factor-1 (IGF-1)22,23 and Calcium/calmodulin-dependent protein kinase kinase β/ AMP-activated protein kinase (CaMKKβ/AMPK)24 to clear and reduce Aβ deposition.
 |
Figure 1 The mechanism of mammalian target of rapamycin (mTOR) and signaling pathway of mTORC1. In the nervous system, brain-derived neurotrophic factor (BDNF), various neuropeptides, insulin, and insulin-like growth factor 1 (IGF-1) activate mTORC1 in neurons. BDNF first activates mTORC1 receptor tyrosine kinase, which further activates the phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) pathway and the RAS extracellular signal-regulated protein kinase (ERK) pathway. Activated Akt and ERK activate Ras homolog enriched in brain protein (Rheb) to activate mTORC1 by inhibiting the mTORC1 inhibitor tuberous sclerosis protein-complex (TSC). AKT also acts as a signaling node between mTORC1 and mTORC2, which indirectly activates mTORC1 by inhibiting TSC through Akt phosphorylation. Elevated mTORC1 activity phosphorylates Unc-51-like kinase 1 (ULK1) and transcription factor EB (TFEB) to suppress autophagy. Furthermore, it also activates 4EBP1 and P70S6K, forming a transcription initiation complex to improve translation efficiency and accelerate protein synthesis. Rapamycin acts on the over-activated mTORC1 in the AD brain, restoring its normal activity and slowing down AD progression, but it also acts mTORC2, which may have some side effects. |
Rapamycin Inhibits the Hyperphosphorylation of Tau Protein
Hyperphosphorylated tau protein aggregates form paired helical filaments in NFTs as one of the key hallmarks of AD.25 Tau protein hyperphosphorylation directly promotes the formation of NFTs, which affect axoplasmic transport function by disintegrating the microtubule system, whereas NFTs do not require alteration of microtubule integrity to have a significant impact on fast axonal transport, leading to neurodegeneration and reduced memory and cognitive function. Overactivation of mTOR signaling may be an important factor in the formation of tau protein hyperphosphorylation.26 Aberrantly activated mTOR drives excessive tau mRNA translation through activation of downstream targets p70 ribosomal S6 protein kinase (p70S6K) and eIF4E-binding protein 1 (4EBP1) (Figure 1). Autopsy of AD brain tissue reveals phosphorylated p70S6K associated with increased tau protein.27,28 Phospho-eukaryotic translation initiation factor 4E (p-eIF4E), p-mTOR, and p-4EBP1 were significantly increased in AD brains and showed a significant positive correlation with total tau and phospho-tau (p-tau).29,30 Over-activated mTOR signaling can also indirectly up-regulate tau protein phosphorylation by inhibiting protein phosphatase 2A (PP2A), which can regulate tau protein phosphorylation from multiple sites.31 In addition, elevated mTOR activity inhibits autophagy and affects the degradation of abnormally phosphorylated tau proteins.32
Since the mTOR signaling pathway is involved in the translation, phosphorylation, and degradation of tau protein, rapamycin may improve cognitive dysfunction by inhibiting the mTOR signaling pathway, suppressing tau mRNA translation, enhancing autophagy, and reducing tau protein hyperphosphorylation. It was shown that rapamycin could reduce hyperphosphorylated tau protein in the 3xTg-AD mouse model, and the mechanism may be to enhance the clearance of hyperphosphorylated tau protein by enhancing autophagy and delaying the neurotoxic effect of NFTs.19 In addition, rapamycin controls glycogen synthase kinase 3β (GSK3β) function by affecting signaling pathways such as PI3K/Akt/mTOR and Wnt/GSK3β/β-catenin, thereby regulating tau protein hyperphosphorylation.12,33 In human neuroblastoma SH-SY5Y cells, rapamycin reduced the phosphorylation of tau protein ser214 by regulating cyclic adenosine monophosphate (cAMP) dependent protein kinase,34 suggesting that rapamycin not only promotes the clearance of phosphorylated tau protein by enhancing autophagy but also inhibits the hyperphosphorylation of tau protein.
Rapamycin Maintains Brain Function in APOE ε4 Gene Carriers
The epsilon4 allele of the apolipoprotein E gene (APOE ε4) is the strongest genetic risk factor for late-onset AD and plays an important role in the AD pathogenesis.35 Several studies have shown that cerebral blood flow (CBF) decreases over time in asymptomatic APOE ε4 carriers prior to dementia onset. Inadequate cerebral perfusion leads to insufficient neuronal nutrition as well as oxidative stress, disruption of blood-brain barrier (BBB) integrity, and reduced cerebrovascular reactivity (CVR).36,37 A magnetic resonance diffusion tensor imaging study finds that white matter (WM) integrity is compromised in healthy APOE ε4 carriers.38 These abnormalities can impair cognitive function in APOE ε4 carriers.
Rapamycin has been shown to improve impaired brain function in APOE ε4 transgenic mice, restore brain glucose uptake and BBB integrity, and may serve as an intervention to prevent vascular abnormalities and the progression of early cognitive dysfunction in APOE ε4 carriers.39,40 Cognitively normal APOE ε4 carriers developed Aβ plaques, and cerebrovascular metabolic and structural abnormalities decades before the onset of cognitive impairment.41 Is rapamycin able to maintain normal brain function and prevent Aβ deposition in APOE ε4 carriers before Aβ deposition occurs? Ai-Ling et al used the E4FAD mouse model, which expresses the human APOE ε4 gene as well as overexpresses Aβ.42 It was found that rapamycin restored CBF and vascular function, enhanced Aβ clearance by the BBB, reduced free fatty acid levels, maintained white matter structural integrity, restored brain function in young adult mice carrying the APOE ε4 gene, and reduced the risk of AD development in E4FAD mice.43
Rapamycin Clears Chronic Inflammation in AD
Chronic inflammation is a major factor in the development and progression of AD.44 Neuroinflammation may be present for many years before the clinical onset of AD, and central inflammation is closely related to peripheral inflammation. Infections with various pathogens (viruses, bacteria, parasites, and many other microorganisms),45 surgery, critical illness, and dysbiosis of the gut microbiota can elicit a systemic immune response, which in turn triggers a central immune response.46 Pathogenic microorganisms, immune cells and inflammatory mediators directly or indirectly damage neuronal cells, inducing Aβ deposition and tau protein hyperphosphorylation. Rapamycin has multiple anti-inflammatory mechanisms, including affecting inflammatory mediators, and immune cells, and restoring the BBB. Nuclear factor-κB (NF-κB) is the key mediator of the anti-inflammatory effect of rapamycin.47 Rapamycin exerts anti-inflammatory effects by down-regulating p65, interleukin-1β (IL-1β), tumor necrosis factor-alpha (TNF-α), and other inflammatory factors of NF-κB in AD mice.48 Rapamycin selectively enhances the activity of the inflammatory factor interleukin-6 (IL-6) signaling pathway.49 IL-6 has a complex role in the central nervous system in AD, stimulating the synthesis of β-amyloid precursor protein,50 but also exerts a neuroprotective effect by activating the phagocytic activity of microglia to degrade Aβ.51 Therefore, how rapamycin affects AD via IL-6 needs to be further explored.
In addition, rapamycin increases the expression of regulatory T cells (Treg).52,53 Treg cells, as important immunomodulatory cells, play an important role in regulating tissue inflammation, avoiding over-activation of the immune system and maintaining lymphocyte homeostasis. In AD, impaired immune phenotype and function of Treg accelerates the development of inflammation and thus the development of AD.54 Rapamycin can exert anti-inflammatory effects by enhancing Treg expression, thereby improving the development of AD. Rapamycin also inhibits mTOR activity and prevents BBB breakdown, limiting the entry of pro-inflammatory and neurotoxic factors from the blood into the brain parenchyma.55 Towner et al found that rapamycin may play an important therapeutic role in inhibiting neuroinflammation by restoring cerebrovascular function and the blood-brain barrier in a rat model of lipopolysaccharide-induced brain inflammation.56
Rapamycin Improves Cognitive Dysfunction in AD
Extensive experimental evidence that rapamycin improves AD-induced cognitive dysfunction.57–59 Amyloid precursor protein/ Presenilin 1 (APP/PS1) mice treated with rapamycin showed significant improvement in memory.58 Rapamycin enhances synaptic plasticity and synaptic protein expression by increasing mitochondrial autophagy, preventing cytochrome C-mediated apoptosis, and reducing oxygen species.60 Synaptic plasticity and its associated proteins play an important role in the development and maturation of the nervous system, learning and memory, and influence cognitive function.61
Rapamycin breaks the negative feedback mechanism of mTOR on insulin signaling by modulating the mTOR signaling pathway (Figure 1), promotes glucose utilization by neurons, improves energy metabolism, enhances local synaptic protein synthesis, maintains neuronal survival and increases synaptic plasticity, and improves learning and memory functions.62 Although the PI3K/Akt/GSK3β/mTOR axis is a key mechanism for regulating synaptic plasticity, such as long term depression (LTD) and long term potentiation (LTP),63 sustained over-activation of mTOR in AD leads to abnormal translation of synaptic proteins, which may impair higher mental functions severely, such as fragile X disorder and may also lead to over-translation of APP.64 Upregulation of mTOR signaling leading to abnormal LTP in the CA1 region of the hippocampus was observed in an animal model of tuberous sclerosis, and short-term treatment with rapamycin rescued synaptic plasticity, memory, and behavioral deficits.65 In addition to directly improving hippocampal synaptic plasticity, rapamycin improves synaptic function indirectly by reducing Aβ deposition and tau protein hyperphosphorylation, thereby delaying the development of AD.57,66 Rapamycin improved learning ability and spatial memory in PDAPP transgenic mice, and LC3 immunoreactive spots in the hippocampal CA1 region were increased and Aβ42 levels were reduced in the brains of transgenic mice after rapamycin administration.9 Rapamycin increases neuronal autophagic activity through inhibition of mTOR may contribute to reduced Aβ and improved cognitive function in the brain of AD mice.
Controversy Over the Effect of Rapamycin on AD
Although many basic studies have demonstrated the therapeutic effects of rapamycin in AD, many questions and controversies remain, which may be due to the variability of experimental models, different modes of administration, dose, timing, frequency, and the availability of drug-targeting vehicles.
We searched the PubMed database for the last 5 years by using the search formula “rapamycin Alzheimer’s disease [Title/Abstract] not review”, suggesting that the main models are hippocampal injection of Aβ-induced AD mice, intracerebroventricular injection of zinc sulfate-induced AD mice, hAPP (J20) mice, APP/PS1 mice, 3×Tg-AD mice, SH‑SY5Y cells, HEK293T cells, and neuroblastoma N2a cells (Table 1). Although the models were constructed based on the Aβ deposition hypothesis and the microtubule-associated protein tau entanglement hypothesis, the whole-brain spatial distribution of Aβ plaques67 and the extent of Aβ-induced tau protein phosphorylation differed between models.68 In vivo AD models at different ages and in vitro AD models at different backgrounds represent different stages of AD pathophysiology, and rapamycin may then exhibit different effects. Subsequently, in the in vivo models, there are various modes of administration, such as intraperitoneal and tail vein injections, and gavage and feed administration; the dose ranges from 0.24–10 mg/kg/2d; the duration of administration can be 7 days or 8 months; the frequency can be continuous administration into the diet, once daily or one day apart. Other studies have combined rapamycin with transferrin decorated-nanostructured lipid carriers69 and reactive oxygen species (ROS) responsive targeted micelles70 to effectively deliver rapamycin to lesions in the central nervous system for targeted therapeutic effects. In in vitro experiments, factors such as cell source, cell type, gene transfection, incubation conditions, concentration and time of rapamycin treatment are all experimental variables, and in vitro experiments are more susceptible to human factors than in vivo experiments, resulting in differences in experimental results.
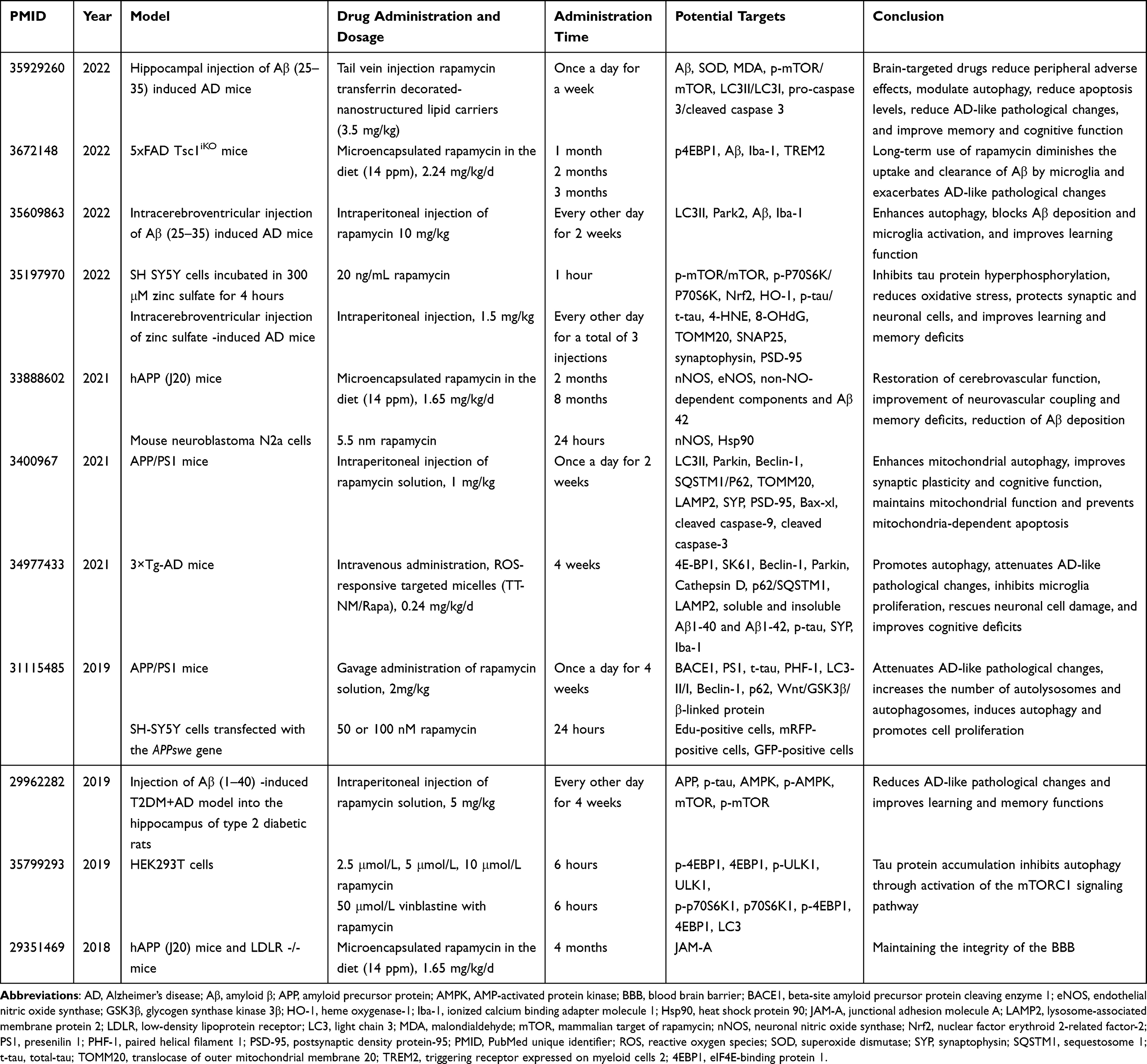 |
Table 1 Therapeutic Targets and Effects of Rapamycin on Different AD Models |
Thus, the above numerous factors influencing the experiment have led to controversy about the effect of rapamycin on AD. However, mTOR signaling has complex effects on the AD brain. The different stage of AD models, the effect of rapamycin on microglia in the brain, and the side effects of rapamycin remain unexplored. mTOR signaling pathway is not only involved in a variety of cellular functions in the brain,71 but is also present in many types of brain cells, such as mature neurons, microglia,72 astrocytes,73 and neural stem cells.74 Rapamycin lacks specific targeted therapeutic effects and potential side effects may occur with non-selective autophagy induction in vivo.75 To address this issue, a rapamycin formulation carried by transferrin decorated-nanostructured lipid carriers (RAPA-Tf-NLCS) was utilized to target neurons in AD lesions. Both mTOR activity and autophagy were regulated to levels similar to control, and moderate mTOR activity was found to achieve the expectant therapeutic effects.69 mTOR pathway is not necessarily overly upregulated in AD because previous studies have shown that mTOR signaling is unchanged or even downregulated in AD models and that activation of mTOR has a neuroprotective effect.76 The Tg2576 model revealed that inhibited mTOR signaling was associated with impaired hippocampal synaptic plasticity, which was associated with reduced expression of the downstream targets p-p70S6K and p-4E-BP1, and that upregulation of mTOR signaling prevented Aβ-induced synaptic damage.
Although many studies have shown that inhibition of the mTOR signaling pathway can significantly reduce AD-like neuropathological features and improve cognitive function in mouse models, these mouse studies have administered rapamycin before or early in the development of pathological features. Different states of the lysosomal system occur in mice of different ages and stages of AD disease. Few studies have been performed on aged AD mice aged ≥18 months, and most studies have used mice aged ≤15 months. Moreover, the use of rapamycin restored abnormally enlarged lysosomes in the cortex of AD mice to the size of controls and increased the number of lysosomes, enhanced lysosomal activity, promoted autophagosome-lysosome fusion, and cleared Aβ and p-tau.70 Rapamycin was found to increase the number of autophagosomes and autophagic lysosomes in SH-SY5Y cells stably transfected with the APPswe gene. Rapamycin has an autophagy-promoting effect on Aβ-depositing cells and attenuates further cellular damage by Aβ, which may be one of the mechanisms by which rapamycin can be used to treat patients who have already developed pathologic features.12 However, the lysosomal system in the brain is less expressed and slower in the later stages of AD and with aging.77 Rapamycin inhibits mTOR and activates autophagy to clear Aβ. Decreased lysosomal clearance leads to autophagosomes accumulation, causing autophagic stress and promoting the amyloid plaques formation.78 Thus rapamycin has a complex, even harmful effect on late AD. Clinical trials on the efficacy of rapamycin should be conducted in MCI patients at risk of progression to AD and in patients with a recent diagnosis of AD,3 or that clinical trials should focus on exploring the efficacy of rapamycin in middle-aged populations with early amyloid plaque and accumulation of microtubule-associated protein tau (MAPT) tangles.77 Therefore, rapamycin may have a more significant effect on preventing AD and delaying the progression of early AD. Currently, there is a Phase I clinical trial conducted in 2020 (NCT04200911) on the feasibility and safety of rapamycin in the treatment of MCI patients that have just been carried out (www.clinicaltrials.gov). However, whether individualized treatment for patients with mid- to late-stage AD79 can enter the therapeutic time window for rapamycin remains unclear.80
Furthermore, rapamycin inhibits microglia activation in the brain and reduces microglia proliferation, yet microglia activation may play a dual role in the AD pathogenesis.81 However, acute microglia activation reduces Aβ deposition by increasing phagocytosis or clearance, but it was found that the use of rapamycin for 2–3 months inhibited mTOR activity and reduced TREM2 expression in microglia of AD mice, weakening the uptake and clearance of Aβ by microglia and aggravating AD-like pathological changes in the brains of AD mice.72 Therefore, detailed longitudinal characterization of microglia activation in relation to AD pathogenesis should be explored immediately, and whether the inhibitory effects of rapamycin on various phenotypes of microglia in different dosage forms, as well as routes, frequency and doses of administration, are therapeutic for AD. In addition, the different side effects of rapamycin may have hindered its introduction as an anti-neurodegenerative agent. Although genetic and pharmacological inhibition of mTORC1 can slow the AD progression and other neurodegenerative diseases, side effects, such as glucose intolerance, diabetes, and immunosuppression resulting from long-term rapamycin inhibition of mTORC2 are of concern; thus, exploring additional rapamycin-targeted agents in the future may be an effective solution to reduce adverse effects.
Conclusion
Rapamycin may delay the development of AD by reducing Aβ deposition, inhibiting tau protein hyperphosphorylation, maintaining brain function in APOE ε4 gene carriers, clearing chronic inflammation, and improving cognitive dysfunction, and is expected to be one of the candidates for treating Alzheimer’s disease.
Funding
This study was supported by grants from Shanxi Provincial Health and Family Planning Commission (No. 2017033); Doctoral Fund of the First Hospital of Shanxi Medical University (No. YB161706, No. BS03201631, No. SD2215).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hogh P. [Alzheimer’s disease]. Ugeskr Laeger. 2017;179(12):V09160686. Danish.
2. Collaborators GBDDF. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health. 2022;7(2):e105–e125. doi:10.1016/S2468-2667(21)00249-8
3. Kaeberlein M, Galvan V. Rapamycin and Alzheimer’s disease: time for a clinical trial? Sci Transl Med. 2019;11(476):eaar4289. doi:10.1126/scitranslmed.aar4289
4. Maiese K. Stem cell guidance through the mechanistic target of rapamycin. World J Stem Cells. 2015;7(7):999–1009. doi:10.4252/wjsc.v7.i7.999
5. Chang YF, Hu WM. [Roles of mammalian target of rapamycin signaling and autophagy pathway in Alzheimer’s disease]. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2019;41(2):248–255. Chinese. doi:10.3881/j.issn.1000-503X.10802
6. Harrison DE, Strong R, Sharp ZD, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460(7253):392–395. doi:10.1038/nature08221
7. Selvarani R, Mohammed S, Richardson A. Effect of rapamycin on aging and age-related diseases-past and future. Geroscience. 2021;43(3):1135–1158. doi:10.1007/s11357-020-00274-1
8. Kaeberlein M. Translational geroscience: a new paradigm for 21(st) century medicine. Transl Med Aging. 2017;1:1–4. doi:10.1016/j.tma.2017.09.004
9. Spilman P, Podlutskaya N, Hart MJ, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One. 2010;5(4):e9979. doi:10.1371/journal.pone.0009979
10. Lin AL, Zheng W, Halloran JJ, et al. Chronic rapamycin restores brain vascular integrity and function through NO synthase activation and improves memory in symptomatic mice modeling Alzheimer’s disease. J Cereb Blood Flow Metab. 2013;33(9):1412–1421. doi:10.1038/jcbfm.2013.82
11. Pierce A, Podlutskaya N, Halloran JJ, et al. Over-expression of heat shock factor 1 phenocopies the effect of chronic inhibition of TOR by rapamycin and is sufficient to ameliorate Alzheimer’s-like deficits in mice modeling the disease. J Neurochem. 2013;124(6):880–893. doi:10.1111/jnc.12080
12. Chen J, Long Z, Li Y, Luo M, Luo S, He G. Alteration of the Wnt/GSK3beta/beta-catenin signalling pathway by rapamycin ameliorates pathology in an Alzheimer’s disease model. Int J Mol Med. 2019;44(1):313–323. doi:10.3892/ijmm.2019.4198
13. Richardson A, Galvan V, Lin AL, Oddo S. How longevity research can lead to therapies for Alzheimer’s disease: the rapamycin story. Exp Gerontol. 2015;68:51–58. doi:10.1016/j.exger.2014.12.002
14. Li L, Zhang S, Zhang X, et al. Autophagy enhancer carbamazepine alleviates memory deficits and cerebral amyloid-beta pathology in a mouse model of Alzheimer’s disease. Curr Alzheimer Res. 2013;10(4):433–441. doi:10.2174/1567205011310040008
15. Son SM, Song H, Byun J, et al. Altered APP processing in insulin-resistant conditions is mediated by autophagosome accumulation via the inhibition of mammalian target of rapamycin pathway. Diabetes. 2012;61(12):3126–3138. doi:10.2337/db11-1735
16. Caccamo A, Maldonado MA, Majumder S, et al. Naturally secreted amyloid-beta increases mammalian target of rapamycin (mTOR) activity via a PRAS40-mediated mechanism. J Biol Chem. 2011;286(11):8924–8932. doi:10.1074/jbc.M110.180638
17. Medvedik O, Lamming DW, Kim KD, Sinclair DA. MSN2 and MSN4 link calorie restriction and TOR to sirtuin-mediated lifespan extension in Saccharomyces cerevisiae. PLoS Biol. 2007;5(10):e261. doi:10.1371/journal.pbio.0050261
18. Bonda DJ, Lee HG, Camins A, et al. The sirtuin pathway in ageing and Alzheimer disease: mechanistic and therapeutic considerations. Lancet Neurol. 2011;10(3):275–279. doi:10.1016/S1474-4422(11)70013-8
19. Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285(17):13107–13120. doi:10.1074/jbc.M110.100420
20. Xue Z, Zhang S, Huang L, He Y, Fang R, Fang Y. Increased expression of Beclin-1-dependent autophagy protects against beta-amyloid-induced cell injury in PC12 cells [corrected]. J Mol Neurosci. 2013;51(1):180–186. doi:10.1007/s12031-013-9974-y
21. Ghosh HS, McBurney M, Robbins PD. SIRT1 negatively regulates the mammalian target of rapamycin. PLoS One. 2010;5(2):e9199. doi:10.1371/journal.pone.0009199
22. El-Ami T, Moll L, Carvalhal Marques F, Volovik Y, Reuveni H, Cohen E. A novel inhibitor of the insulin/IGF signaling pathway protects from age-onset, neurodegeneration-linked proteotoxicity. Aging Cell. 2014;13(1):165–174. doi:10.1111/acel.12171
23. Moll L, Ben-Gedalya T, Reuveni H, Cohen E. The inhibition of IGF-1 signaling promotes proteostasis by enhancing protein aggregation and deposition. FASEB J. 2016;30(4):1656–1669. doi:10.1096/fj.15-281675
24. Vingtdeux V, Chandakkar P, Zhao H, d’Abramo C, Davies P, Marambaud P. Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-beta peptide degradation. FASEB J. 2011;25(1):219–231. doi:10.1096/fj.10-167361
25. Tang Z, Ioja E, Bereczki E, et al. mTor mediates tau localization and secretion: implication for Alzheimer’s disease. Biochim Biophys Acta. 2015;1853(7):1646–1657. doi:10.1016/j.bbamcr.2015.03.003
26. Shen W, Lu K, Wang J, Wu A, Yue Y. Activation of mTOR signaling leads to orthopedic surgery-induced cognitive decline in mice through beta-amyloid accumulation and tau phosphorylation. Mol Med Rep. 2016;14(4):3925–3934. doi:10.3892/mmr.2016.5700
27. An WL, Cowburn RF, Li L, et al. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. Am J Pathol. 2003;163(2):591–607. doi:10.1016/S0002-9440(10)63687-5
28. Pei JJ, Hugon J. mTOR-dependent signalling in Alzheimer’s disease. J Cell Mol Med. 2008;12(6B):2525–2532. doi:10.1111/j.1582-4934.2008.00509.x
29. Li X, Alafuzoff I, Soininen H, Winblad B, Pei JJ. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J. 2005;272(16):4211–4220. doi:10.1111/j.1742-4658.2005.04833.x
30. Li X, An WL, Alafuzoff I, Soininen H, Winblad B, Pei JJ. Phosphorylated eukaryotic translation factor 4E is elevated in Alzheimer brain. Neuroreport. 2004;15(14):2237–2240. doi:10.1097/00001756-200410050-00019
31. Meske V, Albert F, Ohm TG. Coupling of mammalian target of rapamycin with phosphoinositide 3-kinase signaling pathway regulates protein phosphatase 2A- and glycogen synthase kinase-3 -dependent phosphorylation of Tau. J Biol Chem. 2008;283(1):100–109. doi:10.1074/jbc.M704292200
32. Cai Z, Zhao B, Li K, et al. Mammalian target of rapamycin: a valid therapeutic target through the autophagy pathway for Alzheimer’s disease? J Neurosci Res. 2012;90(6):1105–1118. doi:10.1002/jnr.23011
33. Heras-Sandoval D, Perez-Rojas JM, Hernandez-Damian J, Pedraza-Chaverri J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal. 2014;26(12):2694–2701. doi:10.1016/j.cellsig.2014.08.019
34. Liu Y, Su Y, Wang J, et al. Rapamycin decreases tau phosphorylation at Ser214 through regulation of cAMP-dependent kinase. Neurochem Int. 2013;62(4):458–467. doi:10.1016/j.neuint.2013.01.014
35. Serrano-Pozo A, Das S, Hyman BT. APOE and Alzheimer’s disease: advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021;20(1):68–80. doi:10.1016/S1474-4422(20)30412-9
36. Hays CC, Zlatar ZZ, Meloy MJ, et al. APOE modifies the interaction of entorhinal cerebral blood flow and cortical thickness on memory function in cognitively normal older adults. Neuroimage. 2019;202:116162. doi:10.1016/j.neuroimage.2019.116162
37. Thambisetty M, Beason-Held L, An Y, Kraut MA, Resnick SM. APOE epsilon4 genotype and longitudinal changes in cerebral blood flow in normal aging. Arch Neurol. 2010;67(1):93–98. doi:10.1001/archneurol.2009.913
38. Persson J, Lind J, Larsson A, et al. Altered brain white matter integrity in healthy carriers of the APOE epsilon4 allele: a risk for AD? Neurology. 2006;66(7):1029–1033. doi:10.1212/01.wnl.0000204180.25361.48
39. Lin AL, Butterfield DA, Richardson A. mTOR: alzheimer’s disease prevention for APOE4 carriers. Oncotarget. 2016;7(29):44873–44874. doi:10.18632/oncotarget.10349
40. Lin AL, Jahrling JB, Zhang W, et al. Rapamycin rescues vascular, metabolic and learning deficits in apolipoprotein E4 transgenic mice with pre-symptomatic Alzheimer’s disease. J Cereb Blood Flow Metab. 2017;37(1):217–226. doi:10.1177/0271678X15621575
41. Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019;15(9):501–518. doi:10.1038/s41582-019-0228-7
42. Lin AL, Parikh I, Yanckello LM, et al. APOE genotype-dependent pharmacogenetic responses to rapamycin for preventing Alzheimer’s disease. Neurobiol Dis. 2020;139:104834. doi:10.1016/j.nbd.2020.104834
43. Tai LM, Balu D, Avila-Munoz E, et al. EFAD transgenic mice as a human APOE relevant preclinical model of Alzheimer’s disease. J Lipid Res. 2017;58(9):1733–1755. doi:10.1194/jlr.R076315
44. Hampel H, Caraci F, Cuello AC, et al. A path toward precision medicine for neuroinflammatory mechanisms in Alzheimer’s disease. Front Immunol. 2020;11:456. doi:10.3389/fimmu.2020.00456
45. Piekut T, Hurla M, Banaszek N, et al. Infectious agents and Alzheimer’s disease. J Integr Neurosci. 2022;21(2):73. doi:10.31083/j.jin2102073
46. Bettcher BM, Tansey MG, Dorothee G, Heneka MT. Peripheral and central immune system crosstalk in Alzheimer disease - a research prospectus. Nat Rev Neurol. 2021;17(11):689–701. doi:10.1038/s41582-021-00549-x
47. Zhou M, Xu W, Wang J, et al. Boosting mTOR-dependent autophagy via upstream TLR4-MyD88-MAPK signalling and downstream NF-kappaB pathway quenches intestinal inflammation and oxidative stress injury. EBioMedicine. 2018;35:345–360. doi:10.1016/j.ebiom.2018.08.035
48. Liu YC, Gao XX, Chen L, You XQ. Rapamycin suppresses Abeta25-35- or LPS-induced neuronal inflammation via modulation of NF-kappaB signaling. Neuroscience. 2017;355:188–199. doi:10.1016/j.neuroscience.2017.05.005
49. Zhang Y, He X, Wu X, et al. Rapamycin upregulates glutamate transporter and IL-6 expression in astrocytes in a mouse model of Parkinson’s disease. Cell Death Dis. 2017;8(2):e2611. doi:10.1038/cddis.2016.491
50. Ringheim GE, Szczepanik AM, Petko W, Burgher KL, Zhu SZ, Chao CC. Enhancement of beta-amyloid precursor protein transcription and expression by the soluble interleukin-6 receptor/interleukin-6 complex. Brain Res Mol Brain Res. 1998;55(1):35–44. doi:10.1016/s0169-328x(97)00356-2
51. Erta M, Quintana A, Hidalgo J. Interleukin-6, a major cytokine in the central nervous system. Int J Biol Sci. 2012;8(9):1254–1266. doi:10.7150/ijbs.4679
52. Chapman NM, Chi H. mTOR signaling, Tregs and immune modulation. Immunotherapy. 2014;6(12):1295–1311. doi:10.2217/imt.14.84
53. Mandrioli J, D’Amico R, Zucchi E, et al. Rapamycin treatment for amyotrophic lateral sclerosis: protocol for a Phase II randomized, double-blind, placebo-controlled, multicenter, clinical trial (RAP-ALS trial). Medicine. 2018;97(24):e11119. doi:10.1097/MD.0000000000011119
54. Faridar A, Thome AD, Zhao W, et al. Restoring regulatory T-cell dysfunction in Alzheimer’s disease through ex vivo expansion. Brain Commun. 2020;2(2):fcaa112. doi:10.1093/braincomms/fcaa112
55. Van Skike CE, Jahrling JB, Olson AB, et al. Inhibition of mTOR protects the blood-brain barrier in models of Alzheimer’s disease and vascular cognitive impairment. Am J Physiol Heart Circ Physiol. 2018;314(4):H693–H703. doi:10.1152/ajpheart.00570.2017
56. Towner RA, Gulej R, Zalles M, et al. Rapamycin restores brain vasculature, metabolism, and blood-brain barrier in an inflammaging model. Geroscience. 2021;43(2):563–578. doi:10.1007/s11357-021-00363-9
57. Van Skike CE, Lin AL, Roberts burbank R, et al. mTOR drives cerebrovascular, synaptic, and cognitive dysfunction in normative aging. Aging Cell. 2020;19(1):e13057. doi:10.1111/acel.13057
58. Wang X, Xia W, Li K, Zhang Y, Ge W, Ma C. Rapamycin regulates cholesterol biosynthesis and cytoplasmic ribosomal proteins in hippocampus and temporal lobe of APP/PS1 mouse. J Neurol Sci. 2019;399:125–139. doi:10.1016/j.jns.2019.02.022
59. Caccamo A, De Pinto V, Messina A, Branca C, Oddo S. Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer’s disease-like cognitive and pathological deficits by restoring hippocampal gene expression signature. J Neurosci. 2014;34(23):7988–7998. doi:10.1523/JNEUROSCI.0777-14.2014
60. Wang H, Fu J, Xu X, Yang Z, Zhang T. Rapamycin activates mitophagy and alleviates cognitive and synaptic plasticity deficits in a mouse model of Alzheimer’s disease. J Gerontol a Biol Sci Med Sci. 2021;76(10):1707–1713. doi:10.1093/gerona/glab142
61. Li S, Selkoe DJ. A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Abeta oligomers from Alzheimer’s brain. J Neurochem. 2020;154(6):583–597. doi:10.1111/jnc.15007
62. O’Neill C, Kiely AP, Coakley MF, Manning S, Long-Smith CM. Insulin and IGF-1 signalling: longevity, protein homoeostasis and Alzheimer’s disease. Biochem Soc Trans. 2012;40(4):721–727. doi:10.1042/BST20120080
63. Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A. 2002;99(1):467–472. doi:10.1073/pnas.012605299
64. Garelick MG, Kennedy BK. TOR on the brain. Exp Gerontol. 2011;46(2–3):155–163. doi:10.1016/j.exger.2010.08.030
65. Ehninger D, Han S, Shilyansky C, et al. Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis. Nat Med. 2008;14(8):843–848. doi:10.1038/nm1788
66. Majumder S, Richardson A, Strong R, Oddo S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One. 2011;6(9):e25416. doi:10.1371/journal.pone.0025416
67. Whitesell JD, Buckley AR, Knox JE, et al. Whole brain imaging reveals distinct spatial patterns of amyloid beta deposition in three mouse models of Alzheimer’s disease. J Comp Neurol. 2019;527(13):2122–2145. doi:10.1002/cne.24555
68. Kam TI, Gwon Y, Jung YK. Amyloid beta receptors responsible for neurotoxicity and cellular defects in Alzheimer’s disease. Cell Mol Life Sci. 2014;71(24):4803–4813. doi:10.1007/s00018-014-1706-0
69. Khonsari F, Heydari M, Sharifzadeh M, Valizadeh H, Dinarvand R, Atyabi F. Transferrin decorated-nanostructured lipid carriers (NLCs) are a promising delivery system for rapamycin in Alzheimer’s disease: an in vivo study. Biomater Adv. 2022;137:212827. doi:10.1016/j.bioadv.2022.212827
70. Xu S, Yang P, Qian K, et al. Modulating autophagic flux via ROS-responsive targeted micelles to restore neuronal proteostasis in Alzheimer’s disease. Bioact Mater. 2022;11:300–316. doi:10.1016/j.bioactmat.2021.09.017
71. Querfurth H, Lee HK. Mammalian/mechanistic target of rapamycin (mTOR) complexes in neurodegeneration. Mol Neurodegener. 2021;16(1):44. doi:10.1186/s13024-021-00428-5
72. Shi Q, Chang C, Saliba A, Bhat MA. Microglial mTOR Activation Upregulates Trem2 and Enhances beta-Amyloid Plaque Clearance in the 5XFAD Alzheimer’s Disease Model. J Neurosci. 2022;42(27):5294–5313. doi:10.1523/JNEUROSCI.2427-21.2022
73. Zhang Z, Yan J, Bowman AB, Bryan MR, Singh R, Aschner M. Dysregulation of TFEB contributes to manganese-induced autophagic failure and mitochondrial dysfunction in astrocytes. Autophagy. 2020;16(8):1506–1523. doi:10.1080/15548627.2019.1688488
74. Zoungrana LI, Krause-Hauch M, Wang H, et al. The interaction of mTOR and Nrf2 in neurogenesis and its implication in neurodegenerative diseases. Cells. 2022;11(13):2048.
75. Tramutola A, Lanzillotta C, Di Domenico F. Targeting mTOR to reduce Alzheimer-related cognitive decline: from current hits to future therapies. Expert Rev Neurother. 2017;17(1):33–45. doi:10.1080/14737175.2017.1244482
76. Ma T, Hoeffer CA, Capetillo-Zarate E, et al. Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer’s disease. PLoS One. 2010;5(9):e12845.
77. Carosi JM, Sargeant TJ. Rapamycin and Alzheimer disease: a double-edged sword? Autophagy. 2019;15(8):1460–1462. doi:10.1080/15548627.2019.1615823
78. Carosi JM, Sargeant TJ. Rapamycin and Alzheimer disease: a hypothesis for the effective use of rapamycin for treatment of neurodegenerative disease. Autophagy. 2023;19(8):2386–2390. doi:10.1080/15548627.2023.2175569
79. Norwitz NG, Querfurth H. mTOR mysteries: nuances and questions about the mechanistic target of rapamycin in neurodegeneration. Front Neurosci. 2020;14:775. doi:10.3389/fnins.2020.00775
80. Arriola Apelo SI, Lamming DW. Rapamycin: an InhibiTOR of aging emerges from the soil of Easter Island. J Gerontol a Biol Sci Med Sci. 2016;71(7):841–849. doi:10.1093/gerona/glw090
81. Sarlus H, Heneka MT. Microglia in Alzheimer’s disease. J Clin Invest. 2017;127(9):3240–3249. doi:10.1172/JCI90606
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.