Back to Journals » OncoTargets and Therapy » Volume 13
Rapamycin Reduces Cervical Cancer Cells Viability in Hypoxic Condition: Investigation of the Role of Autophagy and Apoptosis
Authors Rezazadeh D, Norooznezhad AH ![]() , Mansouri K, Jahani M, Mostafaie A, Mohammadi MH, Modarressi MH
, Mansouri K, Jahani M, Mostafaie A, Mohammadi MH, Modarressi MH
Received 15 February 2020
Accepted for publication 28 April 2020
Published 18 May 2020 Volume 2020:13 Pages 4239—4247
DOI https://doi.org/10.2147/OTT.S249985
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Yao Dai
Davood Rezazadeh,1,2 Amir Hossein Norooznezhad,2 Kamran Mansouri,2 Mozhgan Jahani,2 Ali Mostafaie,2 Mohammad Hossein Mohammadi,3 Mohammad Hossein Modarressi1
1Department of Molecular Medicine, School of Advanced Technologies in Medicine, Tehran University of Medical Sciences, Tehran, Iran; 2Medical Biology Research Center, Health Technology Institute, Kermanshah University of Medical Sciences, Kermanshah, Iran; 3HSCT Research Center, Laboratory Hematology and Blood Banking Department, School of Allied Medical Sciences, Shahid Beheshti University of Medical Science, Tehran, Iran
Correspondence: Mohammad Hossein Modarressi Email [email protected]
Background: Rapamycin has been known as an anti-cancer agent that affects different malignancies such as glioblastoma and prostate cancer. However, there are few studies concerning rapamycin effects on the cervical cancer cells. In this study, it was aimed to investigate the possible effect of rapamycin on a cervical cancer cell line and explored the possible mechanism(s) and pathway(s) for this agent.
Materials and Methods: To do so, HeLa cells as cervical cancer cell line were used and treated with different concentrations of rapamycin under both normoxic and hypoxic conditions. Then, cell viability assays, Western blot, quantitative real-time polymerase chain reaction (QR-PCR), acridine orange and acridine orange/propidium iodide staining were performed to evaluate rapamycin effect on the mentioned cell line.
Results: The results showed that autophagy and apoptosis-related genes increased significantly in rapamycin-treated HeLa cells compared to controls. Moreover, cervical cancer cell death by rapamycin-induced autophagy in hypoxia was greater than normoxia compared with controls. In this study, it was showed that autophagy induction by rapamycin can mediate programmed cell death of cervical cancer cells, especially in hypoxic condition.
Conclusion: These findings provide a new evidence that rapamycin may inhibit hypoxic HeLa cell proliferation through the trigger of programmed cell death, facilitating the development of novel anti-cancer therapy.
Keywords: rapamycin, cervical cancer, autophagy, apoptosis, programed cell death
Introduction
Rapamycin is an antibiotic, anti-cancer, and immunosuppressant compound that can induce cancer cell death by inhibiting the activity of the mammalian target of rapamycin (mTOR). Cancer cells often use the mTOR pathway as a mechanism to enhance their proliferation and growth. Also, mTOR signaling plays a key role in the autophagy process in tumor cells. Thus, the inhibitory effect of rapamycin on the mTOR activity may enhance the autophagy flux in cells and decrease tumor growth. Furthermore, it has been reported that rapamycin not only promotes the formation of new autophagosomes but also induces autophagosome-lysosome fusion.1–4 Evolutionarily, autophagy has been known as a conserved catabolic manner related to the sequestration and transport of organelles and proteins to the lysosomes for degradation. Autophagy is initiated by way of formation of the phagophore or isolated membrane (nucleation), which expands (elongation) and fuses to form a double-membrane structure termed autophagosome. Then after, autophagosomes fuse with lysosomes in order to degrade their content. The autophagy requires numerous conserved proteins, most of them known as autophagy-related proteins (Atgs), functioning at different stages of this process.5–8
Apoptosis and autophagy are activated in response to hypoxia, starvation, and endoplasmic reticulum (ER) stress-inducing chemical substances. Thus, they can lead to removing damaged organelles, protein aggregates, and invading pathogens. In addition, autophagy may initially be caused to defend the cells by using sequestering and degrading the damaged organelles. Therefore, as soon as a certain level of intracellular harm is reached, apoptosis and autophagy may serve to eliminate the damaged cells from most cancer tissues by cell death induction.
So, because of rapamycin effect on mTOR activity as one of the key factors involved in cancer cell autophagy and growth,9–11 we aimed to investigate this drug effect on cervical cancer in both hypoxia and normoxia condition.
Materials and Methods
Chemicals and Reagents
Primary Antibodies were rabbit anti-HIF-1α (Santa Cruz, USA). Acridine orange and rapamycin were obtained from Sigma and Invivogen, respectively. Quantitative RT-PCR using Takara Kit was purchased Takara (Japan). All other reagents were obtained from Sigma-Aldrich (Taufkirchen, Germany)
Cell Culture
The HeLa cell line was obtained from the Pasteur Institute of Iran. The cells were cultured in DMEM-F12 medium with 10% fetal bovine serum, 100 μg/mL streptomycin and 100 U/mL penicillin. Cells were maintained at 37°C in a humidified CO2 incubator (5% CO2). Twelve hours before treatment, cells were seeded into a 24-well plate at a density of 1–2 × 105 cells per well. The HeLa cell culture dishes were placed into a hypoxia chamber inside the cell culture incubator and exposed to hypoxia (1% oxygen). Hypoxia was obtained by flushing, low oxygen gas (by injecting nitrogen to displace oxygen). Another set of cell culture dishes were cultured under normoxic condition.
Cell Viability Assay
HeLa cells were seeded at 5 × 103 cells per well in a 96-well culture plate with 10% fetal bovine serum (FBS). After 24 h, cells were treated with 100, 200 and 400 nM of Rapamycin in both hypoxic and normoxic conditions. After 48 and 72 htreatment, MTT assay was done as discussed below. The cells were pulsed with 3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide (MTT, Sigma, USA) 20 μL/well [5 mg/mL in PBS] to measure cell viability. The purple-blue MTT formazan precipitate was dissolved in 200 μL of DMSO and swirled for 30 min. Thereafter, absorbance was measured at 570 nm, with background subtraction of 630 nm using an enzyme-linked immunosorbent assay (ELISA) plate reader. The experiments were repeated six times.
QRT-PCR
Total RNA samples were extracted from the cells using Trizol reagent (Invitrogen), by the supplier protocol. Forward and reverse primers of target genes were designed by an acceptable primer design software. qRT-PCR was performed on RNA samples using Takara Kit by Rotor Gene 6000 system (Corbett Research, Australia) in a 96-well plate (15 min at 95°C, 40 cycles of 15 s at 95°C, 15 s at 58°C and 20 s at 72°C). Finally, melting was done at 60–95°C (at an increment of 0.5°Cper step and a holding time of 5 s for each step). Relative quantification analysis was performed using the Relative expression software tool (REST). This analysis used the sample’s crossing point, the efficiency of the reactions, the number of cycles completed and other values to compare the samples and create the ratios, and results were reported as a normalized ratio. The following primers were used for real-time RT-PCR listed in Table 1.
 |
Table 1 Primers Used for Real-Time RT-PCR |
Western Blot
Cell lysates were incubated in 0.075 M Tris buffer (pH 7.6) and 9 M urea. Bradford’s method was used to assess protein concentration of cell lysates, and finely 40 μg of total proteins was separated by 8–10% gel SDS-PAGE under reducing conditions. Then, sample proteins were transferred to a polyvinylidene fluoride membrane (PVDF). The membrane was incubated with a blocking buffer (PBS containing 5% non-fat milk) for 2 h at 25°C. Thereafter, they were immunoblotted with respective primary antibodies and then incubated with specific secondary antibodies. After washing, HIF-1α was detected using a horseradish peroxidase-conjugated reaction. Protein levels were normalized relative to β-actin protein levels. The results were analyzed with TotalLab2 software (Wales, UK).
Acidic Vesicle Detection
Detection of acidic vesicle formation during autophagy was performed by acridine orange staining protocol followed by fluorescence analysis in normoxic and hypoxic conditions with or without rapamycin. In brief, HeLa cells were washed with PBS, fixed in 4% paraformaldehyde for 15 min at room temperature, and then stained with 1 μg/μL of acridine orange for 30 min in the dark. Acridine orange can accumulate in acidic compartments, emitting bright red fluorescence by fluorescence microscopy (Nikon, Japan, TS100), the intensity of which is proportional to the degree of acidity and volume of the compartment and showed autophagy amount.
Acridine Orange-Propidium Iodide (PI) Staining
eLa cells were seeded in 24-well tissue culture plates (5×105 cells/well), treated with or without Rapamycin (50, 100 and 150 nM) for 48 h under normoxia or hypoxia and the cells were washed twice with PBS. Then, the cells were stained with a mixture of acridine orange (3 μg/mL) and propidium iodide (10 μg/mL) and observed immediately using a fluorescence microscope (Nikon-TS100, Japan).
Statistical Analysis
Continuous variables were presented as means ± SD, and statistical comparisons were made by the independent samples t-test, one-way ANOVA followed by the Tukey’s test. Statistical analyses were performed by GraphPad Prism software v6.0 (GraphPad Software, San Diego, California, USA). Any P-value <0.05 was considered to be statistically significant. Each point or column represents the mean ± SD (n = 4–6). *P < 0.05, ** P < 0.01, ***P < 0.001.
Results
HIF-1α Expression
Western blot analysis was used for HIF-1α protein level evaluation in the HeLa cells under hypoxia (1% O2) and normoxia (20% O2). Results showed that HIF1-α amount significantly increased after 48 h of incubation in hypoxia condition in comparison with the cells in normoxia (Figure 1).
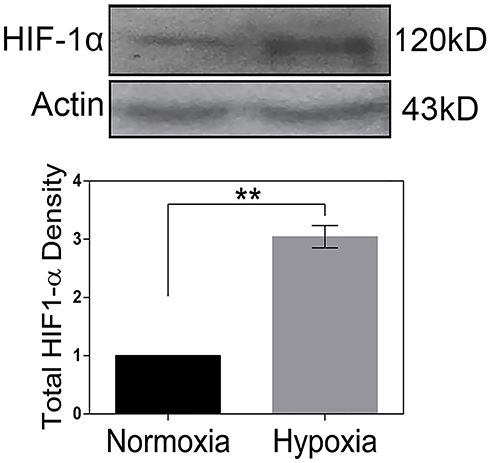 |
Figure 1 Analysis of HIF-1α protein level in Hela cells. Quantification of the protein bands in Western blot analysis carried out using densitometric analysis (TotalLab software, Wales, UK). Protein amounts were normalized against beta-actin and compared with the control. Each data point was presented as mean ± SD from 3 independent experiments. **P < 0.01. |
Rapamycin Increases Autophagy in HeLa Cells Under Hypoxia Rather Than Normoxia
To detect autophagy amount, HeLa cells were treated with 100 nM and 200 nM of rapamycin for 48 h under normoxic and hypoxic conditions. Then, autophagy related-genes (Atgs) such as Beclin 1, Bnip3, Bnip3L, LC3A, LC3B and Atg5 mRNA levels were assessed by qRT-PCR in the presence or absence of rapamycin. Related results showed increased expressions of Beclin 1, Atg5, LC3A and LC3B in rapamycin-treated HeLa cells compared with untreated cells in normoxia and hypoxia. LC3A and LC3B mRNA expression levels were much higher in rapamycin-treated HeLa cells compared with untreated ones in the normoxia (Figure 2). Basal Atgs expression increased under normoxia and hypoxia in the presence of rapamycin. mRNA levels of Bnip3 and Bnip3L were elevated in rapamycin-treated cells in hypoxia but not in normoxia. Therefore, the expression of two mentioned genes was significantly increased in rapamycin-treated cells in hypoxia compared with untreated cells and the same results obtained by comparison of rapamycin-treated in normoxia with treated cells in hypoxia. It seems it was due to the different effects of rapamycin on Bnip3 and Bnip3L under hypoxia compared with normoxia. These data indicate the main role for rapamycin as a positive inducer of autophagy during hypoxia rather than normoxia. Rapamycin led to modest but significant up-regulation of Atg levels in HeLa cells in normoxia while, Atg levels were much higher in treating cells under hypoxia in two rapamycin concentrations (Figure 2A, B, D and E). Furthermore, a comparison of the rapamycin-treated cells in normoxia with treated-cells in hypoxia showed an increased mRNA in autophagy-related genes in both 100 nm and 200 nM rapamycin concentrations (Figure 2C and F).
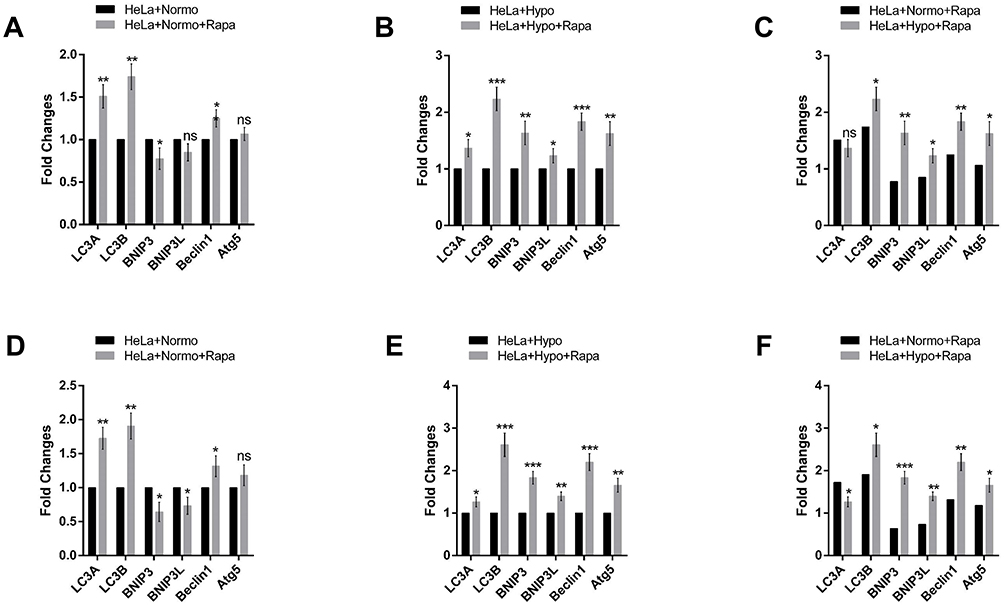 |
Figure 2 Real-time PCR Analysis of autophagy-related genes. (A–C) Real-time PCR Analysis of autophagy-genes such as Beclin 1, ATG-5, Bnip3, Bnip3L, LC3A and LC3B, in HeLa cells under normoxia and hypoxia for 48 h with or without 100 nM Rapamycin treatment. (D–F) Real-time PCR Analysis of Autophagy-Related Genes such as Beclin 1, ATG-5, Bnip3, Bnip3L, LC3A and LC3B, in HeLa cells under normoxia and hypoxia for 48 h with or without 200 nM Rapamycin treatment each data point was presented as mean ± SD from 3–4 independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001. |
Furthermore, acridine orange analysis showed an increased autolysosome amount in HeLa cells under rapamycin treatment. The cytoplasmic orange compartment (autolysosome) was much higher in rapamycin-treated HeLa cells incubated in hypoxia rather than normoxia (Figure 3).
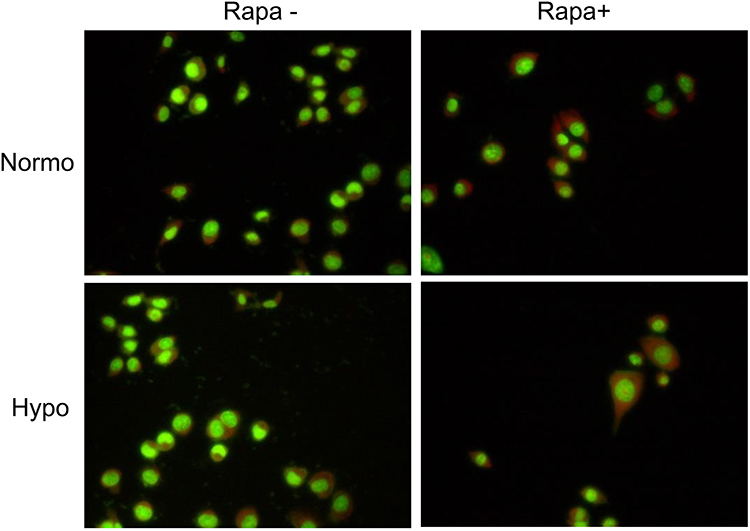 |
Figure 3 Autophagosome formation in HeLa cells. Acridine Orange analysis of HeLa cells with or without 200 nM Rapamycin in both normoxic and hypoxic conditions. Abbreviations: H+H, HeLa-Hypoxia; H+N, HeLa-Normoxia; Rapa, Rapamycin. |
Rapamycin Induced Apoptosis in HeLa Cells Under Hypoxia Rather Than Normoxia
In order to determine rapamycin effect on chromatin condensation and fragmentation, as morphological features of apoptosis, HeLa cells were treated with 100 nM and 200 nM of rapamycin for 48 h under normoxia and hypoxia. As shown in control (no drug treatment) HeLa cells were stained with uniform green fluorescence and no apoptotic features were observed using acridine orange-PI staining. Following rapamycin treatment of cells for 48 h, obvious morphological changes and green apoptotic HeLa cells containing apoptotic characteristics such as cell blebbing and chromatin condensation were observed (Figure 4A). The results suggest that rapamycin induced HeLa cell apoptosis. Furthermore, rapamycin effect on apoptosis-related genes including Bcl-2 and Bax expression was assessed by real-time PCR. This finding showed an increased Bax-Bcl-2 ratio in rapamycin-treated cells in comparison with control (Figure 4B). Since autophagy and apoptosis have a close cross-talk together, it seems that apoptosis induction is partly due to autophagy activation in rapamycin-treated HeLa cells.
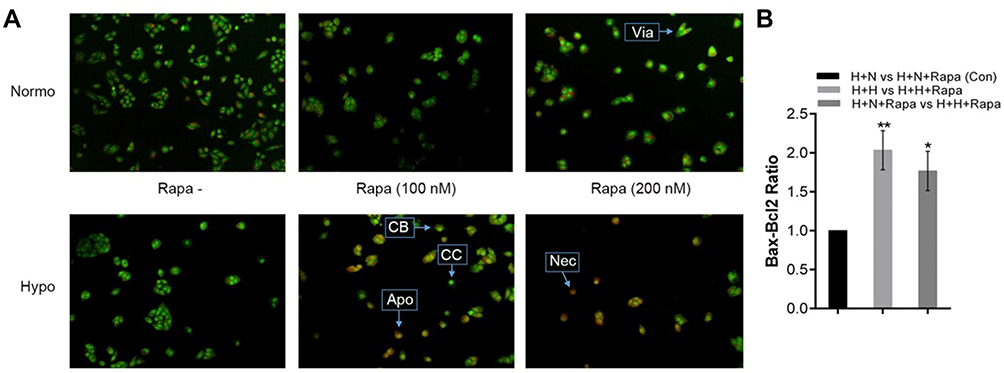 |
Figure 4 Rapamycin induces cell death in HeLa cells. (A) Acridine orange-PI staining in Hela cells incubated for 48 h under hypoxia and normoxia with or without rapamycin (100 and 200 nM). Increased orange staining is indicative of cell death induction. (B) Real time-PCR Analysis of apoptosis-related genes such as Bax and Bcl-2 in Rapamycin-treated cells compared with untreated cells was incubated under normoxic and hypoxic conditions for 48 h. *P < 0.05 and **P < 0.01. Abbreviations: Via, Viable cell; Apo, Apoptotic cell; Nec, Necrotic cell; CB, Cell blebbing; CC, Chromatin condensation; H+H, HeLa-Hypoxia; H+N, HeLa-Normoxia; Rapa, Rapamycin. |
Rapamycin Reduces Cell Viability in HeLa Cells Under Hypoxia in Comparison with Normoxia
The cell viability of HeLa cells was tested using MTT assays. At the normoxia, changes in cell proliferation or survival of rapamycin-treated cells did not significant. The viability of HeLa cells treated with 100 nM/l of rapamycin in normoxia was 85% greater than untreated cells. In 200 and 400 nM of rapamycin, the viability of treated cells under normoxia was lower than control (80% and 70%, respectively) which indicates that rapamycin treatment in normoxia was moderately cytotoxic to HeLa cells (Figure 5A).
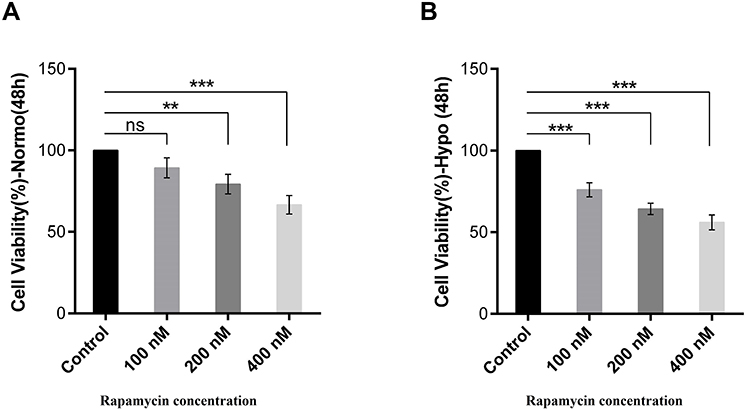 |
Figure 5 Rapamycin reduced cell viability in HeLa cells. (A) Cell viability amount analyzed by MTT assay in rapamycin-treated cells compared with control after 48-h incubation with 100, 200, 400 nM rapamycin in normoxia. (B) Cell viability amount analyzed by MTT assay in rapamycin-treated cells compared with control after 48-h incubation with 100, 200, 400 nM in hypoxia. n = 3, mean ± SD, **P < 0.01, ***P < 0.001. |
The viability of treated cells with 100, 200 and 400 nM rapamycin under hypoxia was lower than 75%, 65% and 60%, respectively, in comparison with control, indicating that rapamycin treatment in hypoxia was more cytotoxic to HeLa cells (Figure 5B). We observed decreased cell viability in HeLa cells with rapamycin in hypoxia compared with control rather than rapamycin-treated cells within normoxia in two rapamycin concentrations. We have found that cells that incubated for 48 h and treatment by 100 and 200 nM/l rapamycin decreased the proliferation amount under hypoxia rather than normoxia. Comparison of the rapamycin-treated cells in hypoxia with treated-cells in normoxia showed a lower survival rate indicating that rapamycin induced cell death by hypoxia is higher than normoxia. Since autophagy induction can lead to apoptosis promotion via pro-apoptotic molecules such as Bnip3, Bnip3L and Atg5 as a cell dead inducer molecule,12,13 it seems that rapamycin has the main role in hypoxia-induced cell death through autophagy induction.
Discussion
Rapamycin as an anti-cancer drug can effect cell viability and growth by inhibition of mTOR activity and signaling. mTOR is one of the important factors in autophagy regulation which its inactivation can result in cancer cell death. Autophagy, a protein and organelle degradation mechanism is a critical process in normal development and responds to environmental stress.14 However, there are opposite reports claiming autophagy can induce cell survival or death.13,15,16 So, in the present study, we investigated the mechanisms by which rapamycin promotes autophagy by mTOR inactivation and the role of autophagy in cancer cell death under normoxic and hypoxic conditions.
Autophagy is a multistep process that each of them is controlled by autophagy-related genes (Atgs) such as Beclin 1, Bnip3, BNIP3L, Bcl-2, Atg5, LC3A and LC3B. Furthermore, it has been indicated that most of these genes are controlled by hypoxia and HIF-1 activity. Hypoxia effect on autophagy has been indicated by its impact on some anti-apoptotic factors including Bcl-2.17
The BNIP3L gene, belonging to the Bcl-2 (BH3-Only) family, can induce autophagy cell death.18 This gene’s product is involved in the pathobiology of various diseases like cancer. The modulated expression of BINP3L during tumor hypoxia can directly influence tumor growth.19 Expression of BNIP3 and BNIP3L is required for an optimum autophagy induction in hypoxic conditions needed their BH3 domains assists in competitively separating Beclin1 from Beclin-Bcl-2 and Beclin-BCL-xL complexes.20–22 BNIP3 and BNIP3L are proteins with pro-apoptotic effects. Studies have shown that silencing BNIP3 gene is directly associated with tumor growth and development under hypoxic condition. Tumors with severe decrease or the lack of BNIP3 protein are highly metastatic to the lymph nodes and have a higher mitotic index. BNIP and BNIP3L proteins interfere with the anti-apoptotic effects of Bcl-2 and BCL-xL and induce apoptosis. Up-regulation of pro-apoptotic molecules such as the aforementioned molecules can induce both autophagy and apoptosis. Moreover, BNIP3 induces hypoxia autophagic cell death in apoptosis-competent cells such as HeLa cells.11,23,24
The LC3A/B presents in both outer and inner membrane of autophagosome, so the expression level of this protein is in balance with autophagosome formation level in mammalian cells25 The ATG5 induces autophagy-dependent cell death and LC3-I to LC3-II conversion. ATG5, regard as an autophagy-dependent cell death inducer, also plays a significant role in apoptosis by acting at the downstream of caspases.26 Other studies have proved that increased ATG5 activates apoptosis through calpain activation. Therefore, the studies have defined two autophagy-mediated pathways for cancer cell death in hypoxia, which are dependent on ATG5, interplayed with endoplasmic reticulum and mitochondria, and tightly regulated by hypoxic condition. In addition, both pathways can also require ATG5-mediated LC3-I to LC3-II conversion.11,27,28 In the present study up-regulation of autophagy-related genes (Atgs) such as Beclin 1, Bnip3, Bnip3L, Atg5, LC3A, LC3B were shown in rapamycin-treated HeLa cells compared with untreated cells. These findings demonstrated Atgs over-induction in rapamycin-treated cells, especially in hypoxia.
Furthermore, we provide evidence that autophagy induction results in an increased amount of apoptotic and non-apoptotic death in response to rapamycin treatment. Mechanistically, rapamycin regulates autophagy by both inducing LC3s expression and inactivating mTOR, which might be indirect.29 Previously, mTOR inhibitors were demonstrated to both increase and decrease the transcriptional expression of certain genes. The mTOR inhibitors can promote cell-cycle arrest by increasing the expression of the cell-cycle inhibitor genes. In the case of LC3s, we observed a transcription-dependent increase in LC3B levels induced by rapamycin.30–32 We have detected that rapamycin-treated HeLa cells decreased the cell viability under hypoxia rather than normoxia using MTT assay. It has been found that the lower cell viability in treating cells under hypoxia compared with the same under normoxia is partly due to autophagy induction. Acridine orange analysis showed an increased autolysosome in HeLa cells under rapamycin treatment, especially in hypoxia. Due data showed an increased Bax-Bcl-2 ratio in rapamycin-treated cells in comparison with control, and apoptosis amount in rapamycin-treated cells was elevated in comparison with untreated cells using acridine orange-PI analysis.
There are few studies concerning the therapeutic efficacy of rapamycin in cervical cancer so, in the present study, we examined this drug effect on HeLa cells. It was shown that rapamycin induces cervical cancer cells death by modulating autophagy and regulating the expression of Atgs and Bcl-2 family proteins, especially in tumor hypoxia (Figure 6). However, HIF1-α is regulated by mTOR and is considered as a substantial molecule for adaptation of cells to stress like hypoxia by regulating hundreds of genes. Essential processes which include autophagy, apoptosis, glucose delivery and glycolysis are regulated in large via HIF1-α. Consequently, HIF1-α is implicated to be of relevance in several medical settings, consisting of tumor growth and development. In hypoxia, mTOR is inactivated, which is assumed to be a part of cells program to keep energy homeostasis. Therefore, it seems, maybe rapamycin affects tumor cell death in mTOR-independent pathways.33–36
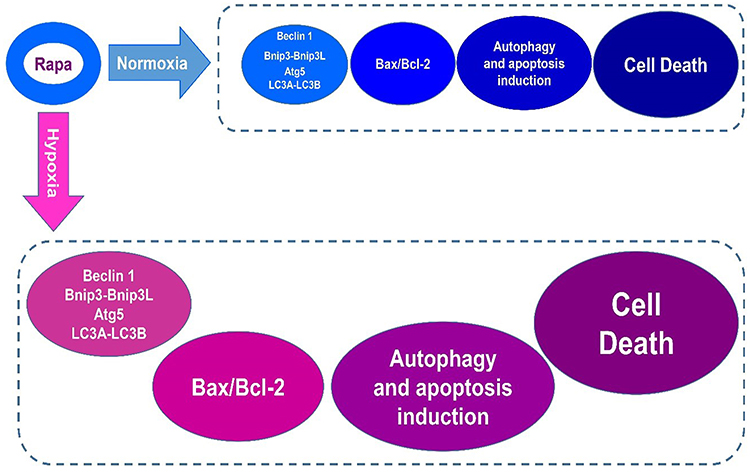 |
Figure 6 Rapamycin induces cervical cancer cell death in hypoxia more than normoxia. |
Conclusion
Our results indicated the main role for rapamycin in autophagy and apoptosis induction in normoxia and hypoxia. These findings showed that rapamycin may be a promising strategy for the treatment of cervical cancer. The critical reaction to rapamycin can be extensively influenced via mobile intrinsic (genotype and cell cycle) and extrinsic factors (dosage, stimulation time, oxygen and nutrient availability). The findings provide a new evidence that autophagy may inhibit the HeLa cell viability under hypoxia through the trigger of PCD, facilitating the development of novel anti-cancer therapy. It can be a main strategy for the cervical cancer treatment by modulating the expression of Atgs and Bcl-2 family proteins, especially in hypoxia-tumor cells. However, studying other cervical cancer cell lines, as well as in-vivo studies, is necessary.
Abbreviations
Rapa, Rapamycin; HIF1-α, hypoxia inducible factor α; Hypo, hypoxia; Normo, normoxia; BNIP3, Bcl-2/Adenovirus E1B 19 kDa Interacting Protein3; PI, Propidium iodide; kDa, kilodalton; DMEM, Dulbecco’s Modified Eagle Medium; Bcl-2, Anti-apoptotic B-cell lymphoma 2; BNIP3L/NIX, Bcl-2/Adenovirus E1B 19 kDa Interacting Protein 3-Like; ATG5, autophagy-related gene 5; LC3A, Microtubule-Associated Protein 1 Light Chain 3 Alpha; LC3B, Microtubule-Associated Protein 1 Light Chain 3 Beta.
Acknowledgments
We thank Dr. Mohammad Hossein Ghahramani and Ms. Zohreh Hosseinkhani for providing critical reagents and excellent technical assistance. Also, it is important to thank the Tehran University of Medical Sciences for grant providing.
Disclosure
The authors declare no actual or potential conflicts of interest in this work.
References
1. Demain AL. Importance of microbial natural products and the need to revitalize their discovery. J Ind Microbiol Biotechnol. 2014;41(2):185–201. doi:10.1007/s10295-013-1325-z
2. Chiarini F, Evangelisti C, McCubrey JA, Martelli AM. Current treatment strategies for inhibiting mTOR in cancer. Trends Pharmacol Sci. 2015;36(2):124–135. doi:10.1016/j.tips.2014.11.004
3. Zhou C, Zhong W, Zhou J, et al. Monitoring autophagic flux by an improved tandem fluorescent-tagged LC3 (mTagRFP-mWasabi-LC3) reveals that high-dose rapamycin impairs autophagic flux in cancer cells. Autophagy. 2012;8(8):1215–1226. doi:10.4161/auto.20284
4. Pratt J, Annabi B. Induction of autophagy biomarker BNIP3 requires a JAK2/STAT3 and MT1-MMP signaling interplay in concanavalin-A-activated U87 glioblastoma cells. Cell Signal. 2014;26(5):917–924. doi:10.1016/j.cellsig.2014.01.012
5. Rogov V, Dötsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell. 2014;53(2):167–178. doi:10.1016/j.molcel.2013.12.014
6. Ma W, Zhang X, Li M, et al. Proapoptotic RYBP interacts with FANK1 and induces tumor cell apoptosis through the AP-1 signaling pathway. Cell Signal. 2016;28(8):779–787. doi:10.1016/j.cellsig.2016.03.012
7. Moon JY, Cho SK. Nobiletin induces protective autophagy accompanied by ER-stress mediated apoptosis in human gastric cancer SNU-16 cells. Molecules. 2016;21(7):E914. doi:10.3390/molecules21070914
8. Lin C-F, Kuo Y-T, Chen T-Y, Chien C-T. Quercetin-rich guava (Psidium guajava) juice in)combination with trehalose reduces autophagy, apoptosis and pyroptosis formation in the kidney and pancreas of type II diabetic rats. Molecules. 2016;21(3):334. doi:10.3390/molecules21030334
9. Pietrocola F, Izzo V, Niso-Santano M, et al. Regulation of autophagy by stress-responsive transcription factors. Semin Cancer Biol. 2013;23(5):310–322. doi:10.1016/j.semcancer.2013.05.008
10. Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833(12):3460–3470. doi:10.1016/j.bbamcr.2013.06.028
11. Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2016;12(1):1–222. doi:10.1080/15548627.2015.1100356
12. Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15(2):81–94. doi:10.1038/nrm3735
13. Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta Mol Cell Res. 2013;1833(12):3448–3459. doi:10.1016/j.bbamcr.2013.06.001
14. White E. The role for autophagy in cancer. J Clin Invest. 2015;125(1):42–46. doi:10.1172/JCI73941
15. Kazemi A, Sadri M, Houshmand M, et al. The anticancer effects of pharmacological inhibition of autophagy in acute erythroid leukemia cells. Anticancer Drugs. 2018;29(10):944–955. doi:10.1097/CAD.0000000000000668
16. Chen K, Shou L-M, Lin F, et al. Artesunate induces G2/M cell cycle arrest through autophagy induction in breast cancer cells. Anticancer Drugs. 2014;25(6):652–662. doi:10.1097/CAD.0000000000000089
17. Qiu Y, Li P, Ji C. Cell death conversion under hypoxic condition in tumor development and therapy. Int J Mol Sci. 2015;16(10):25536–25551. doi:10.3390/ijms161025536
18. Kubli DA, Gustafsson ÅB. Mitochondria and mitophagy the yin and yang of cell death control. Circ Res. 2012;111(9):1208–1221. doi:10.1161/CIRCRESAHA.112.265819
19. Chourasia AH, Macleod KF. Tumor suppressor functions of BNIP3 and mitophagy. Autophagy. 2015;11(10):1937–1938. doi:10.1080/15548627.2015.1085136
20. Chien C-T, Shyue S-K, Lai M-K. Bcl-xL augmentation potentially reduces ischemia/reperfusion induced proximal and distal tubular apoptosis and autophagy. Transplantation. 2007;84(9):1183–1190. doi:10.1097/01.tp.0000287334.38933.e3
21. Zhang J, Ney P. NIX induces mitochondrial autophagy in reticulocytes. Autophagy. 2008;4(3):354–356. doi:10.4161/auto.5552
22. Novak I, Kirkin V, McEwan DG, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11(1):45–51. doi:10.1038/embor.2009.256
23. Labi V, Erlacher M, Kiessling S, Villunger A. BH3-only proteins in cell death initiation, malignant disease and anticancer therapy. Cell Death Differ. 2006;13(8):1325–1338. doi:10.1038/sj.cdd.4401940
24. Karpathiou G, Sivridis E, Koukourakis M, et al. Autophagy and Bcl‐2/BNIP3 death regulatory pathway in non‐small cell lung carcinomas. APMIS. 2013;121(7):592–604. doi:10.1111/apm.12026
25. Luo S, Rubinsztein D. Atg5 and Bcl-2 provide novel insights into the interplay between apoptosis and autophagy. Cell Death Differ. 2007;14(7):1247–1249. doi:10.1038/sj.cdd.4402149
26. Ryter SW, Mizumura K, Choi AM. The impact of autophagy on cell death modalities. Int J Cell Biol. 2014;2014:502676. doi:10.1155/2014/502676
27. Pyo J-O, Yoo S-M, Ahn -H-H, et al. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun. 2013;4(1):2300. doi:10.1038/ncomms3300
28. Liu B, Wen X, Cheng Y. Survival or death: disequilibrating the oncogenic and tumor suppressive autophagy in cancer. Cell Death Dis. 2013;4(10):e892. doi:10.1038/cddis.2013.422
29. Gammoh N, Lam D, Puente C, Ganley I, Marks PA, Jiang X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc Natl Acad Sci U S A. 2012;109(17):6561–6565. doi:10.1073/pnas.1204429109
30. Loos C, Syrovets T, Musyanovych A, Mailänder V, Landfester K, Simmet T. Amino-functionalized nanoparticles as inhibitors of mTOR and inducers of cell cycle arrest in leukemia cells. Biomaterials. 2014;35(6):1944–1953. doi:10.1016/j.biomaterials.2013.11.056
31. Majchrzak A, Witkowska M, Smolewski P. Inhibition of the PI3K/Akt/mTOR signaling pathway in diffuse large B-cell lymphoma: current knowledge and clinical significance. Molecules. 2014;19(9):14304–14315. doi:10.3390/molecules190914304
32. Kim J, Moon IS, Goo T-W, Moon -S-S, Seo M. Algae undaria pinnatifida protects hypothalamic neurons against endoplasmic reticulum stress through Akt/mTOR signaling. Molecules. 2015;20(12):20998–21009. doi:10.3390/molecules201219744
33. Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013;493(7432):338–345. doi:10.1038/nature11861
34. Semba H, Takeda N, Isagawa T, et al. HIF-1 [alpha]-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat Commun. 2016;18(7):11635. doi:10.1038/ncomms11635
35. Zou M, Lu N, Hu C, et al. Beclin 1-mediated autophagy in hepatocellular carcinoma cells: implication in anticancer efficiency of oroxylin A via inhibition of mTOR signaling. Cell Signal. 2012;24(8):1722–1732. doi:10.1016/j.cellsig.2012.04.009
36. Mansouri K, Mostafie A, Rezazadeh D, Shahlaei M, Modarressi MH. New function of TSGA10 gene in angiogenesis and tumor metastasis: a response to a challengeable paradox. Hum Mol Gen. 2016;25(2):233–244. doi:10.1093/hmg/ddv461
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.