Back to Journals » Clinical Ophthalmology » Volume 14
Randomized, Double-Masked, Placebo-Controlled Dose Escalation Study of TAK-639 Topical Ophthalmic Solution in Subjects with Ocular Hypertension or Primary Open-Angle Glaucoma
Authors Martin P ![]() , Cohen A, Uddin S
, Cohen A, Uddin S ![]() , Epelbaum L, Josiah S
, Epelbaum L, Josiah S
Received 19 December 2019
Accepted for publication 2 March 2020
Published 20 March 2020 Volume 2020:14 Pages 885—896
DOI https://doi.org/10.2147/OPTH.S242932
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Patrick Martin, Alisa Cohen, Sharif Uddin, Laura Epelbaum, Serene Josiah
Shire, A Takeda Company, Lexington, MA, USA
Correspondence: Patrick Martin Email [email protected]
Purpose: TAK-639 is a topical, nine-amino acid, synthetic, C-type natriuretic peptide analog in Phase 1 development for the treatment of ocular hypertension (OHT) and primary open-angle glaucoma (POAG). TAK-639 is postulated to lower intraocular pressure (IOP) through a novel mechanism of action (MOA) that increases trabecular meshwork outflow. We investigated the safety and tolerability of TAK-639 in subjects with OHT or POAG.
Methods: This was a phase 1, multicenter, randomized, double-masked, placebo-controlled, single- and multiple-dose escalation study. Subjects (aged 18– 90 years) with OHT or POAG were randomized 5:2 to TAK-639 or placebo. Three dose levels were planned (0.1%, 0.3%, 0.6% TAK-639), each with four dosing regimens (QD, BID, TID, QID). Safety measures included treatment-emergent adverse events (TEAEs) and ophthalmologic examinations. Pharmacokinetics and pharmacodynamics (reduction of IOP) were also evaluated.
Results: In total, 63 subjects were randomized and received 0.1%, 0.3% and 0.6% TAK-639, as single dose, QD, or BID, and 0.1% and 0.3% TID. The study was terminated before 0.6% TID or QID dosing cohorts were studied; instead, 0.6% BID was repeated in a new cohort. TEAEs were instillation related and of mild-to-moderate intensity. There were no TEAEs leading to premature discontinuation, and no serious TEAEs. The most common treatment-related TEAEs were instillation site pain and transient corneal staining with fluorescein. There were no clinically significant concerns across dose groups for all other safety measures, including drop comfort, best corrected visual acuity, slit-lamp biomicroscopy, and corneal epithelial integrity. Little or no systemic exposure was observed. There was a marginal reduction in IOP in one cohort at the highest dose (0.6%) and regimen (BID) tested, suggesting biological plausibility of targeting the trabecular meshwork through this mechanism.
Conclusion: TAK-639 was generally well tolerated up to 0.6% BID. Further non-clinical studies will improve understanding of the MOA and the penetration of TAK-639 to the anterior chamber.
Keywords: glaucoma, intraocular pressure, ocular hypertension, SHP-639, TAK-639
Introduction
Glaucoma is a leading cause of irreversible vision loss, and its incidence is expected to rise over time as populations age.1 Primary open-angle glaucoma (POAG) is the most common form of glaucoma and is associated with elevated intraocular pressure (IOP) and damage to the optic nerve.2 Ocular hypertension (OHT) is a condition of elevated IOP without optic nerve damage, which can develop into POAG.2 Conventional glaucoma treatments focus on lowering IOP, which has been effective in reducing the risk of developing POAG in multiple clinical trials, as well as slowing the progression of the disease itself.2
IOP elevation occurs when aqueous humor outflow is impaired, either through the trabecular meshwork (conventional pathway) or the uveoscleral outflow (unconventional) pathway. The trabecular meshwork has an essential role in the maintenance of IOP homeostasis, through the regulation of resistance to aqueous humor outflow. Trabecular impairment, often a result of trabecular cell damage (oxidative or vascular damage) or remodeling of the surrounding extracellular matrix, can result in increased outflow resistance leading to sustained IOP elevation; a primary risk factor for glaucoma.3–5 Alternatively, IOP elevation can be the result of increased aqueous humor production relative to outflow.6 The main classes of topical ocular hypertensive agents target two of these mechanisms, by decreasing the aqueous humor production rate (eg, carbonic anhydrase inhibitors, β-blockers, and α-adrenergic agonists) or increasing aqueous humor drainage through the uveoscleral outflow pathway (eg, prostaglandin analogs, α-adrenergic agonists, and cholinergic agonists). A relatively new approach is to increase aqueous humor outflow facility through the trabecular meshwork,7 as is achieved with rho kinase inhibitors via a reduction of contractility of the trabecular meshwork.8 Another ophthalmic drug that can target the trabecular meshwork is latanoprostene bunod. This drug was recently approved in the United States, and lowers IOP by increasing outflow of aqueous humor through both the trabecular meshwork and uveoscleral pathway.9
The use of glaucoma treatments are often associated with local and systemic side effects, including changes to the ocular surface. The most commonly reported effects with prostaglandin analogs are periorbitopathy, skin pigmentation and hypertrichosis, eyelash growth, and meibomian gland dysfunction, while inflammatory changes of the cornea, corneo-scleral limbus, conjunctival stroma, and conjunctiva-associated lymphoid tissue are also frequently observed.10,11 Further, inflammation of the conjunctiva can adversely impact the success rate of filtration surgery.12 In some patients, the disease can become refractory to specific topical ocular hypotensive drugs,13 with a proportion of patients failing to respond adequately to available medical or surgical therapies,14 necessitating the combination of different drug classes to achieve adequate IOP lowering.2,13,15 Therefore, there is a need for additional treatments with novel mechanisms of action and the potential to be used alone or in combination with existing topical treatments.
The atrial natriuretic peptides play a role in the regulation of aqueous humor dynamics, with C-type natriuretic peptide (CNP) appearing to be the most efficacious for lowering IOP in animal models. CNP selectively binds and activates the type B natriuretic peptide receptor (NPR-B).16 In rabbits, intracameral or intravitreal injection of CNP increased cyclic guanosine monophosphate concentrations in the aqueous humor, resulting in a lowering of IOP.17,18
CNP is a 22-amino acid cyclic peptide and, as with other atrial natriuretic peptides,19 cannot penetrate the cornea to lower IOP when administered topically. To address this issue, a synthetic, cornea-permeable, nine-amino acid analog of CNP has been developed for the treatment of OHT and POAG.20,21 This compound is postulated to relax the trabecular meshwork via NPR-B activation, thus increasing trabecular meshwork outflow resulting in a lowering of IOP. TAK-639 has demonstrated potential as an ocular hypotensive agent in preclinical studies. One study showed that TAK-639 caused a dose–response reduction in IOP in mice, and in the same study, in vivo experiments demonstrated that TAK-639 increased aqueous humor outflow facility, reduced aqueous humor formation, and reduced episcleral venous pressure.21 In a second study conducted in normotensive rabbits, dogs, and monkeys, a single topical dose of TAK-639 had a significant IOP-lowering effect that correlated well with increases in TAK-639 exposure in aqueous humor.20
In the present study, we evaluated the safety and tolerability of single and multiple ascending doses of TAK-639 ophthalmic solution in subjects with OHT or POAG. We also evaluated the reduction of IOP following different TAK-639 dosing regimens.
Methods
Study Design
This was a multicenter, randomized, double-masked, placebo-controlled, single- and multiple-dose escalation study conducted at four sites in the United States. We planned to study three dose levels (0.1%, 0.3%, and 0.6% TAK-639), each consisting of four dosing regimen cohorts: A (once daily [QD]), B (twice daily [BID]), C (three times daily [TID]), and D (four times daily [QID]). Eligible subjects were randomized in a 5:2 ratio to receive either TAK-639 or placebo, respectively. A study committee evaluated raw safety data (including the first 14 days of data from subjects in the ongoing cohort) at regular time points, to make decisions on progressing to the next dose level or dosing regimen. The study committee consisted of the medical monitor, coordinating principal ophthalmologist, and ad hoc study team members as required from biostatistics, pharmacokinetics (PK), drug safety, and clinical operations. In addition, all study investigators were invited to (though not required to attend) all dosing review meetings and had the opportunity to comment or ask questions. Any cohort could have been repeated to confirm findings with regard to safety or IOP response. The study was conducted in accordance with the principles of the Declaration of Helsinki and all legal and regulatory requirements.
Cohorts A1, A2, and A3 started with a single dose treatment period (SDTP) in which subjects were randomized and received a single dose of TAK-639 or placebo in the designated eye. After completion of the assessments on day 2 of the SDTP, subjects entered a washout period of 3‒14 days (Table 1). Following a decision to commence the multiple dose treatment period (MDTP), the same dose of TAK-639 or placebo was self-administered QD in both eyes for 28 days (MDTP QD). Cohorts B, C, and D were multiple-dosing regimen cohorts, whereby subjects were randomized to receive TAK-639 (BID, TID, or QID consecutively) or placebo in both eyes for 28 days (Table 2). The 0.6% TID or the QID (0.1%, 0.3%, 0.6%) dosing cohorts were planned but not conducted, as the study was terminated before dosing at these levels. Instead, the 0.6% BID cohort was repeated in order to confirm the tolerability of TAK-639 at this dose strength and regimen. In all cohorts, subjects returned to the site to undergo antidrug antibody sampling approximately 60 days after the last dose of study medication (day 88).
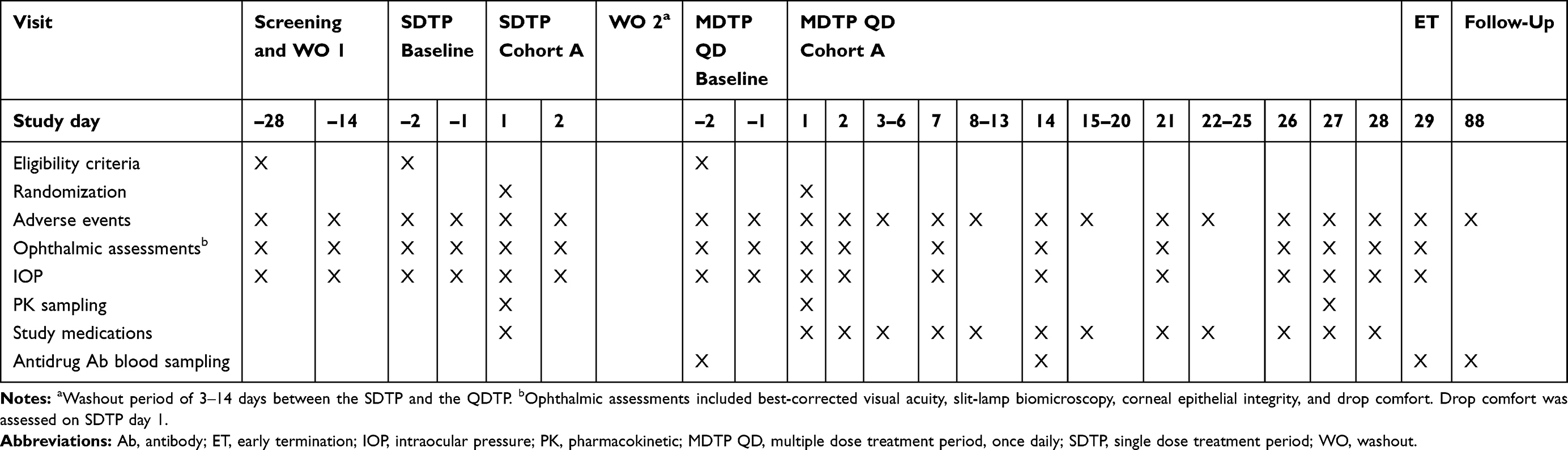 |
Table 1 Schedule of Key Events: Patients Who Received Single Dose and Once Daily Multiple Dosing (Cohort A) |
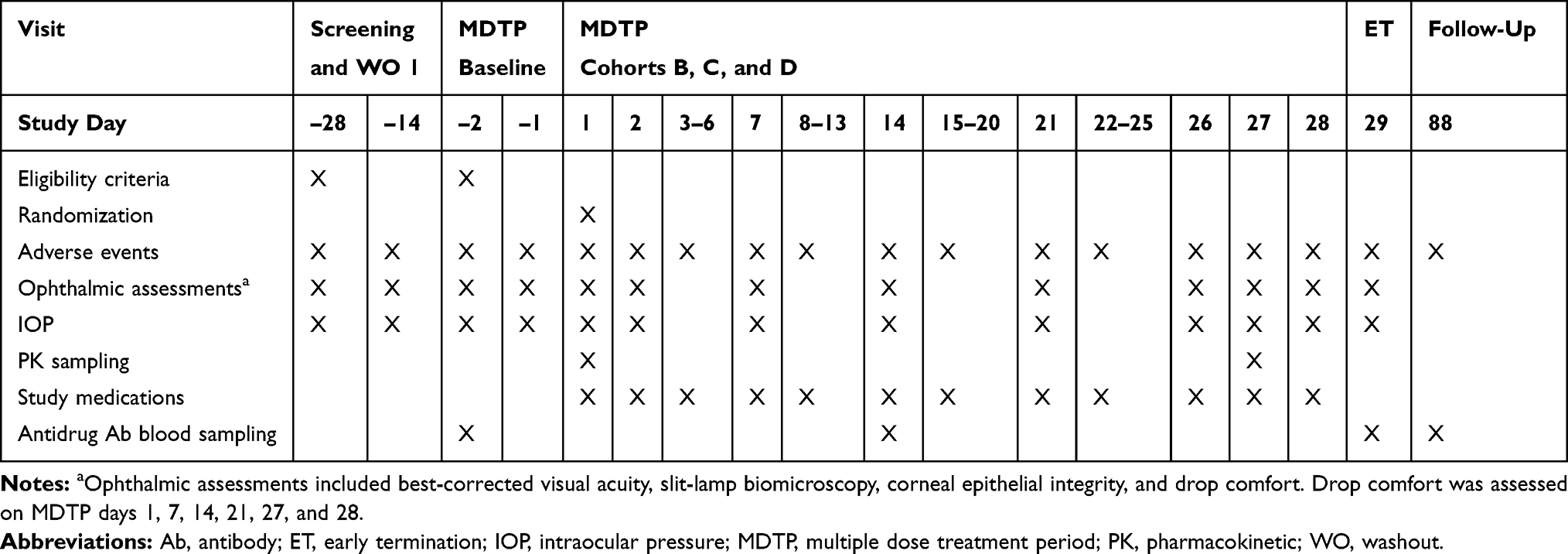 |
Table 2 Schedule of Key Events: Patients Who Received BID or TID Dosing (Cohorts, B, C, D) |
Designated Study Eye
For both the SDTP and MDTP treatments, the designated study eye was the eye with the highest mean baseline (mean on days ‒2 and ‒1) IOP at the pre-dose time point. If both eyes had equal mean baseline IOPs, the eye with the highest mean IOP at the +2 hr time point was the designated study eye. If both eyes had equal mean IOPs at the +2 hr time point, the eye with the highest mean IOP at the +4 hr time point was the designated study eye. If both eyes had equal mean IOPs at the +4 hr time point, the eye with the highest mean IOP at the +8 hr time point was the designated study eye. If both eyes had equal mean IOPs at the +8 hr time point, the right eye was the designated study eye.
Subjects
Eligible subjects were women (non-pregnant and non-lactating) and men aged 18–90 years who had bilateral OHT or stable early POAG, with acceptable Humphrey visual fields and a best-corrected visual acuity (BCVA) in both eyes of ≥65 letters on the Early Treatment Diabetic Retinopathy Study chart. On day ‒1, all subjects had mean IOP ≥24 mmHg at 08:00 and ≥22 mmHg at 10:00 in at least one eye, with an IOP difference of <4 mmHg between eyes at both of these time points. If only one eye met this criterion, then it was the designated study eye for pharmacodynamic (PD) analysis; this eye was also used for dosing in cohort A SDTP.
Subjects were excluded if they had an ocular pathology or any other relevant condition that could affect visual function or interfere with study data collection procedures. Other exclusion criteria were a history of angle closure, ocular surgery, microinvasive glaucoma surgery device insertion, laser surgery, a history of significant ocular trauma or ocular disease, or evidence of ocular infection, inflammation, degeneration, or dystrophy. Prohibited medications included topical ocular hypotensive drugs, corticosteroids, belladonna alkaloids, cannabinoids, opioids, B-type natriuretic peptides, amantadine, and any new prescription drug medication for chronic disease, including those medications that affect IOP. The use of all preserved artificial tears and any ocular gels and other lubricants was prohibited at all times during the study, and the use of nonpreserved artificial tears was allowed only during washout periods.
Safety Assessments
Adverse events (AEs) were coded using the Medical Dictionary for Regulatory Activities (MedDRA, version 20). Treatment-emergent AEs (TEAEs) were defined as AEs that occurred after the first dose of study medication. Ophthalmologic examinations of both eyes were performed at the visits specified in Tables 1 and 2, and included BCVA and slit-lamp biomicroscopy. In addition, graded assessments of the anterior segment were conducted, including corneal epithelial integrity by fluorescein staining (three corneal zones [inferior, central, and superior], and were graded on a 4-point scale from 0 = none to 4 = severe, giving a maximum score of 12). Drop comfort was assessed weekly from day 1 through day 27 immediately post-instillation, and at 1, 2, 3, 5, 10, and 15 mins post-instillation. An 11-point scale was used, in which 0 = very comfortable and 10 = very uncomfortable.
PK and PD Analyses
Blood samples were centrifuged to obtain plasma, and concentrations of plasma TAK-639 were determined at the visits specified in Tables 1 and 2 using a fully validated bioanalytical method based on liquid chromatography with mass spectrometry. Where appropriate, PK parameters for TAK-639 were calculated based on plasma concentration–time data using a non-compartmental approach (Phoenix® WinNonlin® version 6.4 or higher, Certara USA, Inc., Princeton, NJ, USA). They included maximum observed concentration (Cmax); time to reach Cmax (tmax); area under the concentration–time curve from time zero to the time of the last measurable concentration (AUC0–t); AUC over the dosing interval (AUC0–τ); and AUC from time zero extrapolated to infinity (AUC0–∞). The presence of antidrug antibodies in serum was determined qualitatively by antigen capture enzyme-linked immunosorbent assay. Reduction in IOP was used as a PD biomarker. IOP (in mmHg) was measured using Goldmann applanation tonometry.
Statistical Analyses
The safety set consisted of all subjects who were randomized and who received at least one dose of study medication. The PK set consisted of all subjects in the safety set for whom the primary PK data were considered sufficient and interpretable. The PD set consisted of all subjects in the safety set for whom the primary PD data were evaluable.
The number of subjects in this study was not based on statistical power considerations because the statistical analyses were primarily descriptive in nature. However, if at least seven subjects completed each cohort, then there was to be a ≥65–85% probability of observing at least one occurrence of any AE, with a true incidence rate of ≥15–25% for any given dose group.
All plasma PK analyses were performed using the PK set. For accumulation ratios, the 95% confidence interval for the arithmetic mean of the difference on the logarithmic scale (natural base) was calculated and the results were back-transformed to give the ratio of geometric means, together with the corresponding 95% confidence interval. Before statistical comparisons, dose-dependent parameters (Cmax, AUC0–t, AUC0–τ, and AUC0–∞) were normalized to a common dose, assuming linearity. Additionally, the linear dose proportionality was evaluated for Cmax, AUC0–τ, AUC0–t, and AUC0–∞ using the power function model (eg, Cmax = α·Doseβ or equivalently log[Cmax] = log [α] + β·log[Dose], where Cmax was considered to increase linearly proportional to dose if β is not significantly different from 1.0). All PD analyses were performed using the PD set.
Results
Study Population
The study was conducted from May 10, 2017, to May 30, 2018. A total of 111 subjects were enrolled, 63 patients were randomized to treatment, and 62 subjects completed the study (Figure 1). All subjects across all regimens were included in the safety population. All subjects in the QD and TID dosing regimens and the repeat 0.6% BID cohort completed the study. One subject in the BID dosing regimen (TAK-639 0.1% BID) discontinued the study (per sponsor request) due to a distal fibular fracture of the right ankle that required hospitalization. Overall, subject demographics and baseline disease characteristics were well balanced across treatment groups, dosing regimens, and dosing levels (Tables 3 and 4).
 |
Table 3 Baseline Characteristics in Subjects Who Received Single Dose and Once-Daily Dosing (Randomized Set) |
 |
Table 4 Baseline Characteristics in Subjects Who Received BID or TID Dosing (Randomized Set) |
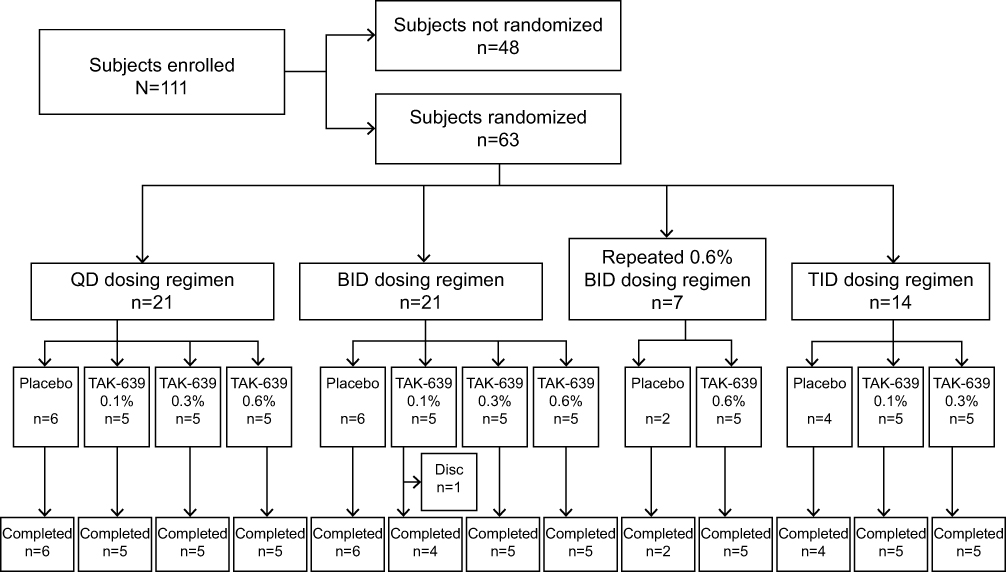 |
Figure 1 Subject disposition. Abbreviations: BID, twice daily; disc, discontinued; TID, three times daily; QD, once daily. |
Nearly all subjects across dose groups and regimens had an ocular medical history. The ocular histories of the study and non-study eyes were similar. In the QD dosing group, the ocular medical history of the study eye was mostly open-angle glaucoma (TAK-639, 60.0%; placebo, 83.3%), OHT (TAK-639, 53.3%; placebo, 16.7%), dry eye (TAK-639, 53.3%; placebo, 50.0%), and cataract (TAK-639, 46.7%; placebo, 33.3%). In the BID dosing group, the ocular medical history of the study eye was mostly cataract (TAK-639, 80.0%; placebo, 100.0%), open-angle glaucoma (TAK-639, 73.3%; placebo, 83.3%), OHT (TAK-639, 40.0%; placebo, 83.3%), and dry eye (TAK-639, 40.0%; placebo, 33.3%). In the repeat 0.6% BID cohort, the ocular medical history of the study eye was mostly cataract (TAK-639, 60.0%; placebo, 0) and OHT (TAK-639, 60.0%; placebo, 50.0%). In the TID dose regimen, the ocular medical history of the study eye was mostly cataract (TAK-639, 60.0%; placebo, 50.0%), OHT (TAK-639, 60.0%; placebo, 75.0%), and open-angle glaucoma (TAK-639, 40.0%; placebo, 25.0%).
Safety and Tolerability
Overall, no deaths or TEAEs leading to premature discontinuation were reported in this study. The most common TEAEs associated with the use of TAK-639 were instillation site pain and transient corneal staining with fluorescein (Tables 5 and 6).
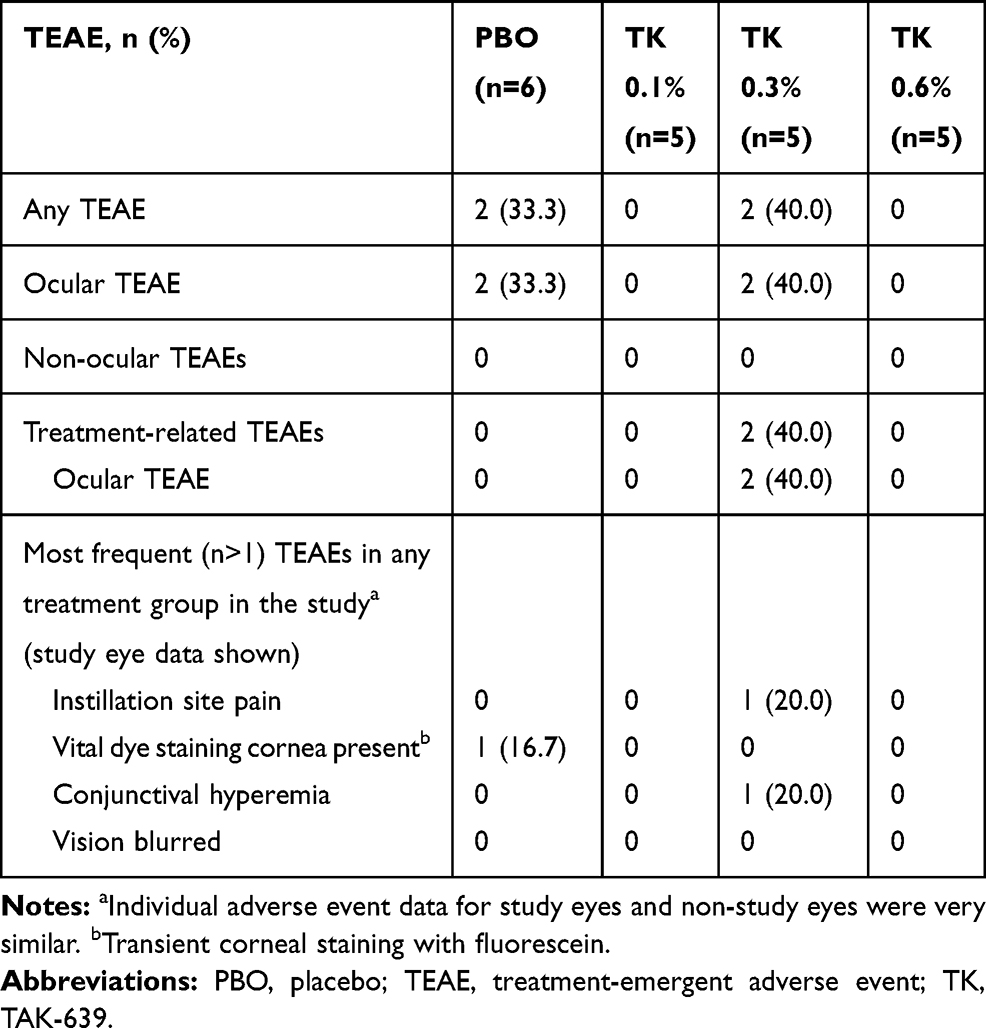 |
Table 5 Summary of TEAEs in Subjects Who Received Once Daily Dosing (Safety Set) |
During the SDTP and washout period, one of five subjects receiving TAK-639 0.3% QD had ocular TEAEs of conjunctival hyperemia, instillation site pain, and vital dye staining cornea present (transient corneal staining with fluorescein) in the study eye; these were all considered related to the study medication and mild in intensity. No other TEAEs occurred during the SDTP.
During the MDTP, TAK-639 was generally well tolerated at the 0.1% and 0.3% dose strengths using a single dose, QD, BID, or TID dosing regimen. In the QD dosing group, one of five (20.0%) subjects in the TAK-639 0.3% dose group had a TEAE of conjunctival hyperemia and instillation site pain at the study eye. One of six (16.7%) subjects in the placebo group had a TEAE of transient corneal staining with fluorescein in the study eye. There were no ocular TEAEs reported for the five subjects who received 0.6% QD dosing (Table 5). In the TID dosing group, no subjects had ocular TEAEs at the study eye (Table 6).
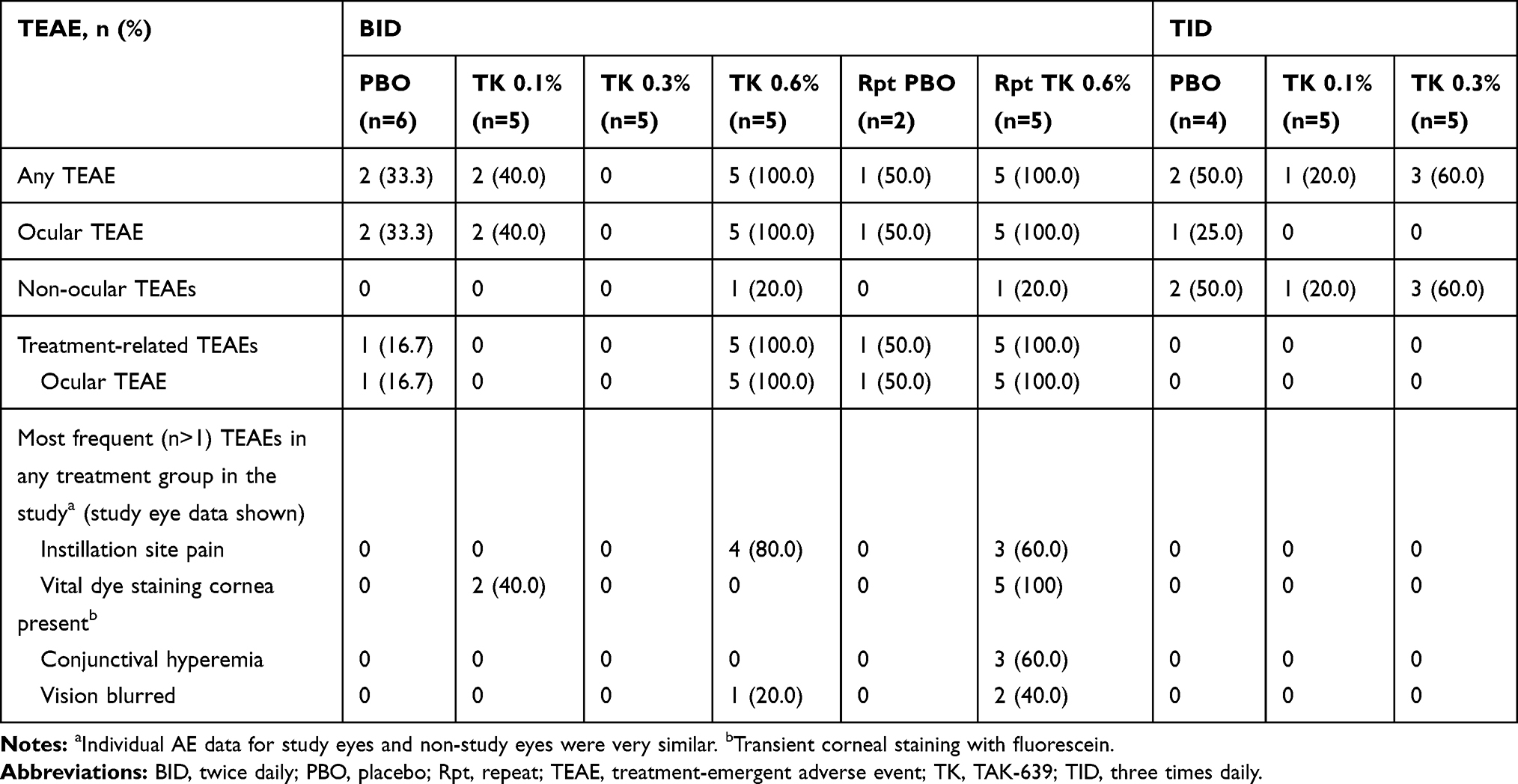 |
Table 6 Summary of TEAEs in Subjects Who Received BID or TID Dosing (Safety Set) |
In the SDTP and MDTP (QD, BID repeat, and TID) dosing regimens, all ocular and non-ocular TEAEs at the study eye were mild in intensity. However, in the 0.6% BID dosing regimen, all five subjects receiving TAK-639 reported moderate instillation site pain or discomfort. This cohort was also notable for the presence of corneal staining at baseline in two of five subjects who received 0.6% BID dosing and one of two subjects who received placebo. In addition, all five subjects had at least one mild TEAE under the system organ class of eye disorders. Given the favorable safety experience of the previous cohorts, the safety results following 0.6% BID dosing were unexpected. To better understand the tolerability of 0.6% BID TAK-639, this dose was repeated in a new cohort. Five subjects in this repeat 0.6% BID dosing cohort reported with ocular TEAEs, of which three had mild instillation site pain and four had mild eye disorders; all five had corneal fluorescein staining (Table 6). In this cohort, conjunctival hyperemia was reported in 3 subjects in the study eye, and one subject receiving placebo in the non-study eye.
No non-ocular TEAEs were reported in the SDTP or MDTP QD dose regimens, and those reported in the other dosing cohorts were not considered related to the study medication. In the BID dose regimen, one of five (20.0%) subjects in the TAK-639 0.6% dose group had a TEAE of epistaxis. In the repeat 0.6% BID cohort, one of five (20.0%) subjects in the TAK-639 group had a TEAE of upper respiratory tract infection. In the TID dose regimen, four of 10 subjects (40.0%) in the TAK-639 group and two of four subjects (50.0%) in the placebo group had non-ocular TEAEs. One of five (20.0%) subjects each in the TAK-639 0.3% TID dose group had a TEAE of upper respiratory tract infection, urinary tract infection, and myalgia. One of five (20%) subjects in the TAK-639 0.1% TID dose group had a TEAE of cellulitis. One of five (25%) subjects each in the placebo TID group had a TEAE of upper respiratory tract infection, herpes zoster, and tooth fracture.
Ophthalmic Assessments
There were no clinically significant concerns across dose groups for all other safety measures, including drop comfort, BCVA, slit-lamp biomicroscopy, and corneal epithelial integrity.
The highest staining scores for corneal epithelial integrity occurred in the 0.6% BID group followed by the repeat 0.6% BID group, although staining scores in the original 0.6% BID dose group were not considered clinically significant by the investigator. Similarly, the largest increases from baseline were seen with 0.6% BID followed by repeat 0.6% BID dosing. Mean corneal staining scores in the 0.6% BID dose groups increased after each instillation of TAK-639, with the highest staining scores peaking at 2 or 4 hrs after instillation. By 8 or 12 hrs, post-instillation staining score in each of the three zones for each subject was either none (0 points) or trace (1 point). The peak mean staining score for each day (in the study eye) ranged from 8.2 to 10.6 for the original 0.6% BID group and from 2.4 to 4.6 for the repeat 0.6% BID group (maximum possible score was 12.0).
The highest (ie, most uncomfortable) drop comfort scores were observed in the 0.6% BID group followed by the repeat 0.6% BID group. In general, the highest drop comfort scores were observed on day 1 or day 7, suggesting that subjects’ tolerability improved over time.
Most post-baseline changes in BCVA across the SDTP and MDTP dose groups were decreases of five or fewer letters (both study eye and non-study eye). The few exceptions are described below. In the QD dose regimen, the largest post-baseline change was a six- to nine-letter decrease at day 27 and a ≥10-letter decrease at day 21 for one subject (16.7%) in the placebo group (non-study eye). In the BID dose regimen, the worst post-baseline change was a six- to nine-letter decrease for one subject (20.0%) in the original 0.6% BID group (study eye) and one subject (16.7%) in the placebo group (non-study eye).
No slit-lamp biomicroscopy findings for any placebo or TAK-639 dose groups were considered clinically significant at any time during the study.
Antidrug Antibody Testing
Serum samples were collected for antidrug antibody testing throughout the study (Tables 1 and 2). All samples for all subjects were negative for antidrug antibody.
PK Results
Overall, the plasma concentrations of TAK-639 in all treatment groups either in the SDTP or the MDTP treatment period showed low or negligible levels of systemic exposure. For those samples containing quantifiable drug levels, concentrations ranged from below the lower limit of quantitation to 2.12 ng/mL in the 0.3% TAK-639 (QD, BID, or TID) dosing groups, and from the lower limit of quantitation to 10.6 ng/mL for the 0.6% TAK-639 (QD or BID) groups. In a few subjects having sufficient quantifiable data for half-life determination, the elimination half-life was approximately 0.6 hrs. There were insufficient quantifiable data for calculation of AUC, clearance, and volume of distribution.
Systemic exposure with SDTP 0.1% and 0.3% TAK-639 and MDTP 0.1% QD, BID, and TID was negligible (<0.0800 ng/mL limit of quantitation). In the SDTP 0.6% QD cohort, the mean Cmax was 0.264 ng/mL (n=4). In the MDTP 0.3% dosing cohort, there was an apparent increase in systemic exposure as the number of doses per day increased (QD [n=1], mean Cmax day 1 = 0.233 ng/mL; BID [n=1], mean Cmax day 1 = 0.0980 ng/mL; TID [n=3], mean Cmax day 1 = 0.440 ng/mL). A similar trend was observed with MDTP 0.6% dosing (QD [n=5], mean Cmax day 1 = 0.605; BID repeated [n=5], mean Cmax day 1 = 2.72). The study was terminated before the 0.6% TID regimen or any QID dosing were studied.
PD Results
Some marginal reduction in IOP from baseline values was observed, but only at the highest dose group (0.6%) and at the most frequent regimen studied at this dose (BID) (Figure 2). Of note, in the 0.6% BID cohort there was a high degree of variability in IOP change from baseline in the placebo arm.
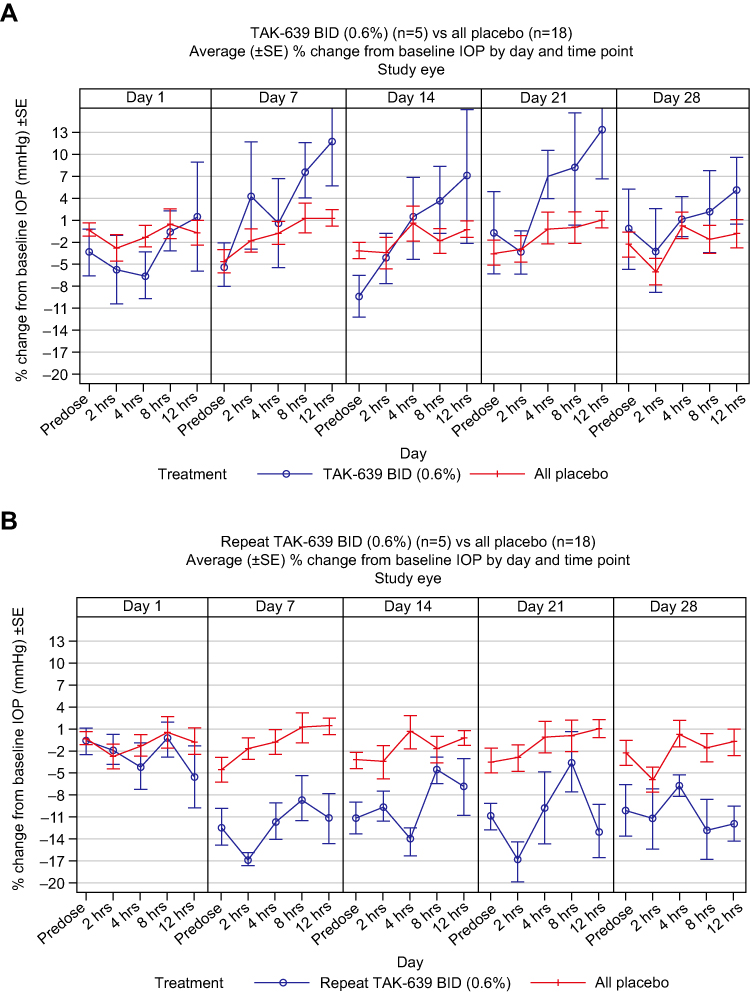 |
Figure 2 Average (±SE) percent change from baseline in IOP by day and time point with (A) TAK-639 0.6% BID and (B) repeat TAK-639 0.6% BID. Abbreviations: BID, twice daily; IOP, intraocular pressure; SE, standard error. |
Discussion
TAK-639 is a topical, nine-amino acid, synthetic, CNP analog in phase 1 development for the treatment of patients with OHT and POAG. The present study investigated the safety and tolerability of TAK-639 ophthalmic solution at single and multiple ascending doses in subjects with bilateral OHT or stable early POAG. TAK-639 ophthalmic solution was generally well tolerated up to the highest dose and regimen tested (0.6% BID). Across the dose groups, TEAEs were instillation related. There were no TEAEs leading to premature study discontinuation and no serious or severe TEAEs. The most common treatment-related TEAEs were instillation site pain and transient corneal staining with fluorescein. No non-ocular TEAEs were reported in the SDTP or MDTP QD dose regimens, and those reported in the other dosing cohorts were not considered treatment related.
All ocular and non-ocular TEAEs at the study eye were mild in intensity in the single and multiple dose regimens (QD, BID repeat, and TID). However, in the original 0.6% BID dosing regimen, in which three of seven subjects had corneal fluorescein staining at baseline (two of five in TAK-639 group, one of two in placebo group), unexpected safety results were observed, including moderate instillation site pain or discomfort in all five subjects. Thus, the 0.6% BID TAK-639 dose regimen was repeated in a new cohort, as allowed per protocol, to better understand the tolerability at this (highest) dose strength and regime. In the repeat 0.6% BID cohort, five subjects had ocular TEAEs, of which three had mild instillation site pain and four had mild eye disorders; all five had corneal fluorescein staining with vital dyes.
There were no clinically significant concerns across dose groups for all other safety measures, including drop comfort, BCVA, slit-lamp biomicroscopy, and corneal epithelial integrity. The highest (ie, most uncomfortable) drop comfort scores were observed in the 0.6% BID group followed by the repeat 0.6% BID group. In general, the highest drop comfort scores were observed on day 1 or day 7, suggesting that subjects’ tolerability improved over time. Most post-baseline changes in BCVA across the dose groups were decreases of five or fewer letters. The highest corneal staining scores occurred in the 0.6% BID and repeat 0.6% BID groups.
Ocular abnormalities, as detected by vital staining, are associated with long-term treatment with preservative-containing glaucoma treatments, and also occur with use of non-preserved IOP-lowering medications; patients taking multiple medications to control their ocular symptoms may further increase the risk of ocular surface abnormalities.22 Conjunctival hyperemia is a relatively minor side-effect of glaucoma treatment, but one that can have important implications for treatment adherence.23 Few patients in this study showed evidence of either vital staining or conjunctival hyperemia, although more cases of both were noted with higher doses.
In the present study, PK parameters and reduction of IOP (a PD biomarker) were also evaluated following the different dosing regimens. PK studies showed that there was little or no systemic exposure to study drug at the dose regimen tested. In the few subjects who had sufficient quantifiable data for half-life determination, the elimination half-life was approximately 0.6 hrs. There were insufficient quantifiable data for calculation of other AUC parameters, clearance, or volume of distribution. All subjects were negative for antidrug antibody.
TAK-639 has a novel mechanism of action: it is postulated to relax the trabecular meshwork via NPR-B activation, thus increasing trabecular meshwork outflow and resulting in a lowering of IOP. Such a novel agent has the potential to be used in combination with other topical IOP-lowering drugs, thereby achieving target IOP levels while minimizing the local and systemic side effects associated with current glaucoma treatments. In animal studies, TAK-639 has demonstrated potential as an ocular hypotensive agent.20,21 In the present study of subjects with OHT or POAG, a marginal reduction in IOP was observed in a single cohort at the highest dose (0.6%) and regimen (BID) tested. This suggests biological plausibility of 0.6% TAK-639 BID in targeting the trabecular meshwork, as IOP reduction is an accepted PD endpoint in glaucoma trials.
The present phase 1 study is the first in-human study of TAK-639, and provides promising data on the safety and tolerability of TAK-639 up to 0.6% BID dosing, as well as PK and PD. The findings provide sufficient evidence for further investigation of TAK-639. Additional studies are required, including non-clinical studies to improve understanding of the mechanism of action. In particular, studies should investigate whether changes in the formulation of TAK-639 to improve penetration into the anterior chamber could elicit a greater IOP reduction.
This study has some limitations. We planned to investigate three dose levels (0.1%, 0.3%, and 0.6% TAK-639), each consisting of four dosing regimen cohorts (QD, BID, TID, and QID). However, interim analysis suggested that other formulations and additional dose strengths should be explored to improve the IOP-lowering effect of TAK-639. Consequently, the study was terminated before the 0.6% TID regimen or any QID (0.1%, 0.3%, or 0.6%) dosing cohorts were studied. Another limitation is that the repeat TAK-639 0.6% BID group was the only cohort to show a marginal reduction in IOP compared with placebo. Given the strong PD response in preclinical models, a larger drop in IOP was anticipated. This raises the question as to whether relaxing the trabecular meshwork is ineffective in humans, or if the current formulation of TAK-639 is not reaching the anterior chamber. Future studies to explore the potential of TAK-639 include evaluation of exposure to target tissue in multiple species by adjusting the formulation of TAK-639. In addition, further human studies are needed to assess concentrations of TAK-639 in the aqueous humor and to confirm the reduction in IOP with TAK-639 in patients with OHT or POAG.
Conclusion
TAK-639 at single and multiple ascending doses was generally well tolerated with no clinically significant safety concerns identified up to the highest dose and regimen tested (0.6% BID) in subjects with bilateral OHT or stable early POAG. There was little or no systemic exposure to study drug. A marginal reduction in IOP was observed with 0.6% BID TAK-639, suggesting biological plausibility of targeting the trabecular meshwork. Further studies are required to improve understanding of the mechanism of action and penetration of TAK-639 in the anterior chamber, and to confirm the IOP-reducing effect of TAK-639.
Abbreviations
Ab, antibody; AE, adverse event; AUC0–t, area under the concentration–time curve from time zero to the time of the last measurable concentration; AUC0–τ, area under the concentration–time curve over the dosing interval; AUC0–∞, area under the concentration–time curve from time zero extrapolated to infinity; BCVA, best-corrected visual acuity; BID, twice daily; BMI, body mass index; Cmax, maximum observed concentration; CNP, C-type natriuretic peptide; disc, discontinued; ET, early termination; IOP, intraocular pressure; MDTP, multiple dose treatment period; NPR-B, type B natriuretic peptide receptor; OHT, ocular hypertension; PBO, placebo; PD, pharmacodynamic; PK, pharmacokinetic; POAG, primary open-angle glaucoma; QD, once daily; QID, four times daily; Rpt, repeat; SD, standard deviation; SE, standard error; SDTP, single dose treatment period; TEAE, treatment-emergent adverse event; TID, three times daily; tmax, time to reach maximum observed concentration; TK, TAK-639; WO, washout.
Informed Consent
Informed consent was obtained from each subject prior to performance of any study-related procedures.
Data Sharing Statement
The datasets generated during the current study are not publicly available due to ethical considerations but are available from the corresponding author on reasonable request.
Ethics and Consent Statement
The study protocol, protocol amendments, the final approved informed consent document, relevant supporting information, and all types of subject recruitment information were submitted to and approved by the ethics committee/Institutional Review Board (Western IRB [WIRB] Panel 1, Puyallup, WA, USA; Sterling IRB, Atlanta, GA, USA; Quorum, Seattle, WA, USA; Wills Eye Hospital IRB, Philadelphia, PA, USA) and regulatory agency (as appropriate) prior to study initiation. This study was conducted in accordance with Good Clinical Practice as described in Code of Federal Regulations Title 21 Parts 50, 56, and 312 and the International Conference on Harmonisation Good Clinical Practice guidelines. Compliance with these regulations and guidelines also constitutes compliance with the ethical principles described in the Declaration of Helsinki.
Acknowledgments
Under direction of the authors, Nasser Malik and Lisa O’Brien, employees of Excel Medical Affairs, provided writing assistance for this manuscript. Writing and editorial support was funded by Takeda.
Author Contributions
All authors contributed to conception and design of the study, data acquisition, data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This study was funded by Shire, a Takeda company.
Disclosure
Sharif Uddin is an employee of and holds stock/stock options in Shire, a Takeda company. Patrick Martin, Alisa Cohen and Serene Josiah were employees of and held stock/stock options in Shire at the time that this study was conducted. Laura Epelbaum is a consultant to Shire/Takeda and does not hold any stock/stock options in the company. The authors report no other conflicts of interest in this work.
References
1. Tham Y-C, Li X, Wong TY, Quigley HA, Aung T, Cheng C-Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology. 2014;121(11):2081–2090. doi:10.1016/j.ophtha.2014.05.013
2. Prum BE
3. Vranka JA, Kelley MJ, Acott TS, Keller KE. Extracellular matrix in the trabecular meshwork: intraocular pressure regulation and dysregulation in glaucoma. Exp Eye Res. 2015;133:112–125. doi:10.1016/j.exer.2014.07.014
4. Carreon T, van der Merwe E, Fellman RL, Johnstone M, Bhattacharya SK. Aqueous outflow - a continuum from trabecular meshwork to episcleral veins. Prog Retin Eye Res. 2017;57:108–133. doi:10.1016/j.preteyeres.2016.12.004
5. Sacca SC, Pulliero A, Izzotti A. The dysfunction of the trabecular meshwork during glaucoma course. J Cell Physiol. 2015;230(3):510–525. doi:10.1002/jcp.24826
6. Goel M, Picciani RG, Lee RK, Bhattacharya SK. Aqueous humor dynamics: a review. Open Ophthalmol J. 2010;4:52–59. doi:10.2174/1874364101004010052
7. Liebmann JM, Lee JK. Current therapeutic options and treatments in development for the management of primary open-angle glaucoma. Am J Manag Care. 2017;23(15 Suppl):S279–S292.
8. Schehlein EM, Robin AL. Rho-associated kinase inhibitors: evolving strategies in glaucoma treatment. Drugs. 2019;79(10):1031–1036. doi:10.1007/s40265-019-01130-z
9. Fingeret M, Gaddie IB, Bloomenstein M. Latanoprostene bunod ophthalmic solution 0.024%: a new treatment option for open-angle glaucoma and ocular hypertension. Clin and Exp Optom. 2019;102(6):541–550. doi:10.1111/cxo.12853
10. Di Staso S, Agnifili L, Cecannecchia S, Di Gregorio A, Ciancaglini M. In vivo analysis of prostaglandins-induced ocular surface and periocular adnexa modifications in patients with glaucoma. In Vivo. 2018;32(2):211–220. doi:10.21873/invivo.11227
11. Di Staso S, Agnifili L, Ciancaglini M, Murano G, Borrelli E, Mastropasqua L. In vivo scanning laser confocal microscopy of conjunctival goblet cells in medically-controlled glaucoma. In Vivo. 2018;32(2):437–443. doi:10.21873/invivo.11259
12. Mastropasqua R, Fasanella V, Brescia L, et al. In vivo confocal imaging of the conjunctiva as a predictive tool for the glaucoma filtration surgery outcome. Invest Ophthalmol Vis Sci. 2017;58(6):BIO114–BIO120. doi:10.1167/iovs.17-21795
13. Weinreb RN, Aung T, Medeiros FA. The pathophysiology and treatment of glaucoma: a review. JAMA. 2014;311(18):1901–1911. doi:10.1001/jama.2014.3192
14. Zhou Z, Althin R, Sforzolini BS, Dhawan R. Persistency and treatment failure in newly diagnosed open angle glaucoma patients in the United Kingdom. Br J Ophthalmol. 2004;88(11):1391–1394. doi:10.1136/bjo.2003.037713
15. Inoue K. Managing adverse effects of glaucoma medications. Clin Ophthalmol. 2014;8:903–913. doi:10.2147/opth.S44708
16. Koller K, Lowe D, Bennett G, et al. Selective activation of the B natriuretic peptide receptor by C-type natriuretic peptide (CNP). Science. 1991;252(5002):120–123. doi:10.1126/science.1672777
17. Takashima Y, Taniguchi T, Yoshida M, et al. Ocular hypotension induced by intravitreally injected C-type natriuretic peptide. Exp Eye Res. 1998;66(1):89–96. doi:10.1006/exer.1997.0403
18. Fernández-Durango R, Moya FJ, Rípodas A, de Juan JA, Fernández-Cruz A, Bernal R. Type B and type C natriuretic peptide receptors modulate intraocular pressure in the rabbit eye. Eur J Pharmacol. 1999;364(2–3):107–113. doi:10.1016/S0014-2999(98)00828-0
19. Korenfeld MS, Becker B. Atrial natriuretic peptides. Effects on intraocular pressure, cGMP, and aqueous flow. Invest Ophthalmol Vis Sci. 1989;30(11):2385–2392.
20. Savinainen A, Prusakiewicz JJ, Oswald J, Spencer E. Reduction of intraocular pressure by SHP639, a novel C-type natriuretic peptide analog, and its exposure in aqueous humor of normotensive Dutch belted rabbits and beagle dogs. Presented at: Association for Research in Vision and Ophthalmology 2018 Annual Meeting; April 29–May 3; 2018; Honolulu, HI. Poster B0035.
21. Millar JC, Savinainen A, Josiah S, Pang IH. Effects of TAK-639, a novel topical C-type natriuretic peptide analog, on intraocular pressure and aqueous humor dynamics in mice. Exp Eye Res. 2019;188:107763. doi:10.1016/j.exer.2019.107763
22. Pflugfelder SC, Baudouin C. Challenges in the clinical measurement of ocular surface disease in glaucoma patients. Clin Ophthalmol. 2011;5:1575–1583. doi:10.2147/OPTH.S24410
23. Masumoto H, Tabuchi H, Yoneda T, et al. Severity classification of conjunctival hyperaemia by deep neural network ensembles. J Ophthalmol. 2019;2019:7820971. doi:10.1155/2019/7820971
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.