Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 14
Randomized dose-finding study of batefenterol via dry powder inhaler in patients with COPD
Authors Crim C ![]() , Watkins ML, Bateman ED, Feldman GJ, Schenkenberger I, Kerwin EM
, Watkins ML, Bateman ED, Feldman GJ, Schenkenberger I, Kerwin EM ![]() , Crawford C
, Crawford C ![]() , Pudi K, Ho S
, Pudi K, Ho S ![]() , Baidoo C, Castro-Santamaria R
, Baidoo C, Castro-Santamaria R ![]()
Received 12 October 2018
Accepted for publication 23 January 2019
Published 8 March 2019 Volume 2019:14 Pages 615—629
DOI https://doi.org/10.2147/COPD.S190603
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Courtney Crim,1 Michael L Watkins,1 Eric D Bateman,2 Gregory J Feldman,3 Isabelle Schenkenberger,4 Edward M Kerwin,5 Catriona Crawford,6 Krishna Pudi,7 Shuyen Ho,8 Charlotte Baidoo,9 Ramiro Castro-Santamaria10
1GSK, Research and Development, Research Triangle Park, NC, USA; 2Division of Pulmonology, Department of Medicine, University of Cape Town, Cape Town, South Africa; 3S. Carolina Pharmaceutical Research, Spartanburg, SC, USA; 4Klinische Forschung Berlin GbR, Berlin, Germany; 5Clinical Trials Division, Crisor LLC, Clinical Research Institute, Medford OR, USA; 6GSK, Global Medical, Stockley Park, Uxbridge, Middlesex, UK; 7Upstate Pharmaceutical Research, Greenville, SC, USA; 8PAREXEL International, Durham, NC, USA; 9GSK, Clinical Statistics, Stockley Park, Uxbridge, Middlesex, UK; 10GSK, Research and Development, Collegeville, PA, USA
Background: Batefenterol is a novel bifunctional muscarinic antagonist β2-agonist in development for COPD. The primary objective of this randomized, double-blind, placebo-controlled, active comparator, Phase IIb study was to model the dose–response of batefenterol and select a dose for Phase III development.
Patients and methods: Patients aged ≥40 years with COPD and FEV1 ≥30% and ≤70% predicted normal were randomized equally to batefenterol 37.5, 75, 150, 300, or 600 µg, placebo, or umeclidinium/vilanterol (UMEC/VI) 62.5/25 µg once daily. The primary and secondary endpoints were weighted-mean FEV1 over 0–6 hours post-dose and trough FEV1, analyzed by Bayesian and maximum likelihood estimation Emax of dose–response modeling, respectively, on day 42.
Results: In the intent-to-treat population (N=323), all batefenterol doses demonstrated statistically and clinically significant improvements from baseline vs placebo in the primary and secondary endpoints (191.1–292.8 and 182.2–244.8 mL, respectively), with a relatively flat dose–response. In the subgroup reversible to salbutamol, there were greater differences between batefenterol doses. Lung function improvements with batefenterol ≥150 µg were comparable with those with UMEC/VI. Batefenterol was well tolerated and no new safety signals were observed.
Conclusion: Batefenterol 300 µg may represent the optimal dose for Phase III studies.
Keywords: bifunctional, bronchodilator, dual-pharmacophore, dose–response, muscarinic antagonist β2-agonist
Introduction
The pharmacological management of COPD aims primarily to improve symptoms and quality of life, optimize lung function, reduce exacerbations, and improve exercise tolerance.1 Inhaled bronchodilators, including long-acting β2-adrenergic agonists (LABAs), long-acting muscarinic antagonists (LAMAs), and inhaled corticosteroids (ICS) are the mainstays of therapy for patients with COPD.1 In addition, combining an inhaled LAMA with a LABA improves lung function more effectively than the individual components.2–4 This approach is recommended in global COPD strategy documents if symptoms do not improve with a single bronchodilator.1 Products combining inhaled LABA and LAMA within a single device include umeclidinium (UMEC) with vilanterol trifenatate (VI) and indacaterol with glycopyrronium.
A new approach to dual bronchodilation is offered by batefenterol, a novel bifunctional muscarinic antagonist β2-agonist (MABA). Batefenterol combines muscarinic antagonism (M2 and M3 receptors) and β2-agonism in a single molecule and is currently in development for the treatment of COPD.5 The MABA approach provides several advantages over combination therapy, including delivery of a fixed ratio of LABA and LAMA to the lung, with a uniform ratio of respective activities at the cellular level.6 Triple therapy with LABA, LAMA, and ICS delivered via a single inhaler has recently become available;7,8 however, the dual-pharmacophore nature of batefenterol may offer a simplified manufacturing process by reducing the number of agents to be co-formulated for triple therapy and also enable the potential development of quad therapy delivered in a single inhaler.
The Phase IIb study assessed the dose–response, efficacy, and safety of five doses of once-daily batefenterol (37.5–600 μg) administered via dry powder inhaler (ELLIPTA-DPI [owned by or licensed to the GlaxoSmithKline (GSK) group of companies]) in patients with COPD. The primary objective was to model the dose–response of batefenterol and select an appropriate dose for Phase III development.
Materials and methods
A randomized, placebo-controlled, double-blind, parallel-group, dose-ranging study was conducted at 26 centers in South Africa, the USA, and Germany between November 2015 and July 2016. The study protocol and informed consent were reviewed and approved by local ethics committees/institutional review boards (Table S1) in accordance with the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use Good Clinical Practice.
Study population
Eligible patients were males or females aged ≥40 years with established COPD according to the American Thoracic Society/European Respiratory Society (ATS/ERS) definition;9 a post-salbutamol FEV1/FVC ratio ≤0.70; FEV1 ≥30%, and ≤70% of predicted normal at visit 1 (using ERS reference equations10); current/former smokers with a ≥10 pack-year history. Patients took the COPD Assessment Test at randomization and at day 42.
Exclusion criteria included a current diagnosis of asthma; poorly controlled COPD (defined as acute worsening of COPD that required treatment in the 6 weeks prior to screening or hospitalization in the 12 weeks prior to screening); a history of >1 moderate or severe COPD exacerbation in the 12 months prior to screening; disease preventing the use of anticholinergic agents; any significant disease other than COPD. Patients were required to discontinue ICS, ICS/LABA, LABA/LAMA (2 weeks), as well as LABA (~10 days) or LAMA (7 days) prior to screening. Other exclusion criteria, including prohibited medications, are shown in the Supplementary materials and Table S2. A minimum peak inspiratory flow was neither an inclusion nor exclusion criterion. Patients were withdrawn from the study if they experienced a COPD exacerbation, pneumonia, pregnancy, or abnormalities in liver chemistry (Supplementary materials), 12-lead electrocardiogram (ECG) (Supplementary materials), or laboratory parameters.
Study design
Patients attended a pre-screening visit (visit 0) to obtain informed consent before changes could be made to their medication. If no changes were required, the pre-screening and screening visits (visit 1) were conducted on the same day (Figure 1). Patients entered a 2-week, single-blind, placebo run-in period to assess baseline salbutamol use and were randomized equally at day 1 (visit 2) to one of seven treatment groups: batefenterol 37.5, 75, 150, 300, or 600 μg, UMEC/VI 62.5/25 μg, or placebo once daily (Figure 1). Doses were selected based on results from a previous dose-finding study of batefenterol administered via DISKUS (MAB115032; owned by or licensed to the GSK group of companies)11 to provide an appropriate range for the determination of the minimal, optimally effective, and safe batefenterol dose. The placebo arm was included to measure the absolute effect of each dose to allow a robust determination of the dose–response of batefenterol. The UMEC/VI active control was included to aid evaluation of the batefenterol response as a bifunctional molecule.
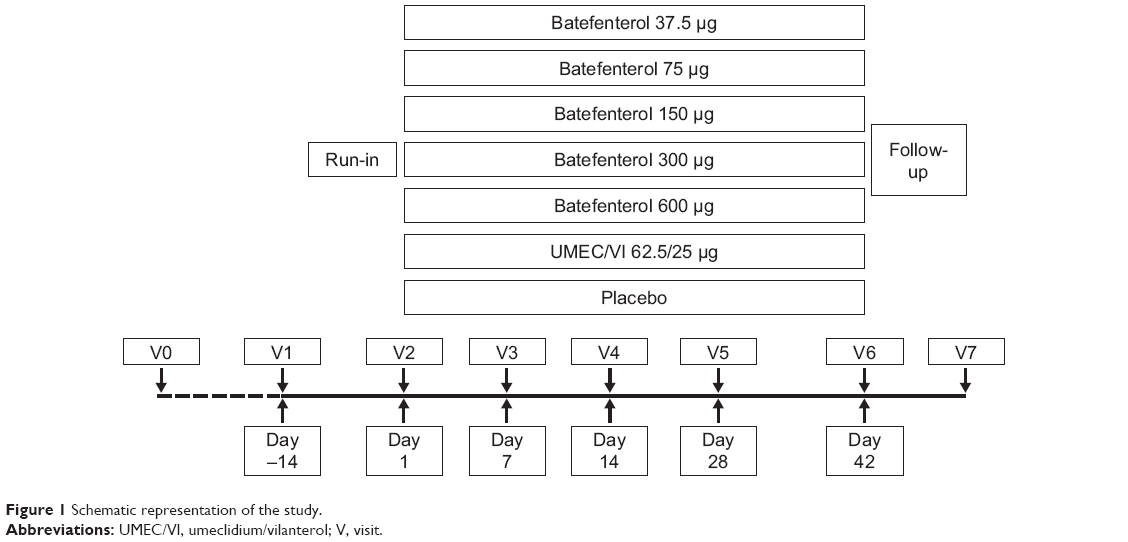 | Figure 1 Schematic representation of the study. |
A telephone-based interactive voice response system was used to randomize and register patients and to provide medication assignment information.
Randomization was stratified by reversibility to salbutamol, defined as an increase in FEV1 ≥12% and ≥200 mL following administration of four puffs of salbutamol via a metered dose inhaler.
Once-daily morning treatments were administered via ELLIPTA-DPI for 42 days and both patients and physicians were blinded to treatment allocation. Adherence was assessed by the DPI dose counter. Inhaled salbutamol was provided for symptomatic relief during the run-in and treatment periods, except in the 4-hour period prior to spirometry. Patients recorded the number of times rescue salbutamol was used over the last 24 hours, as well as any medical problems and medications used, in a diary. Patients visited the clinic on days 1 (randomization), 7, 14, 28, and 42 (Figure 1). Spirometry was performed at every visit. Trough spirometry was obtained 23 and 24 hours after the previous day’s dose of blinded study medication at visits 2–6; trough FEV1 was defined as the mean of the two assessments. Six-hour post-dose serial spirometry was conducted on days 1 and 42 (visits 2 and 6).
Efficacy and safety assessments
The primary efficacy endpoint was weighted-mean (WM) FEV1 over 0–6 hours post-dose at day 42. The secondary endpoint was trough FEV1 at day 42.
Safety assessments included the incidence of adverse events (AEs), 12-lead ECG, vital signs, laboratory assessments, and COPD exacerbations.
The primary efficacy analysis was based on the intent-to-treat (ITT) population, which included all randomized patients who received at least one dose of study medication. Additional efficacy analyses were conducted in the reversible population, which consisted of all patients in the ITT population who showed reversibility to salbutamol as previously described. Safety was analyzed in the ITT population.
Statistical analysis
The primary objective was to model the dose–response of batefenterol and select an appropriate dose for Phase III development. The primary efficacy endpoint was analyzed using a three-parameter Bayesian Emax model of the dose–response curve with data from the ITT population. The secondary efficacy endpoint was analyzed using the maximum likelihood estimation (MLE) method of Emax dose–response modeling in the ITT population.
Supportive analyses of the primary and secondary endpoints were undertaken in the ITT population using a mixed models repeated measures (MMRM) model with an unstructured variance–covariance matrix.
Dose–response modeling was repeated in the reversible population to evaluate consistency of the dose–response with the ITT population. Further details of the statistical analysis, including description of the Bayesian Emax and MMRM models, are provided in Supplemental materials.
Ethics approval and informed consent
Each participant provided written informed consent; the study protocol and informed consent were reviewed and approved by local ethics committees/institutional review boards in accordance with the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use Good Clinical Practice. Details of the ethics committees/institutional review boards are provided in Table S1.
Results
Study population
A total of 585 patients were screened, of which 324 were randomized; the ITT population comprised 323 patients (one patient was randomized in error) (Figure 2). Overall, 93% of the population completed the study, with similar proportions completing within each treatment group (Figure 2). Five patients withdrew from the study due to lack of efficacy, that is, a COPD exacerbation (5/323, 2%); four in the batefenterol 300 μg group (4/47, 9%).
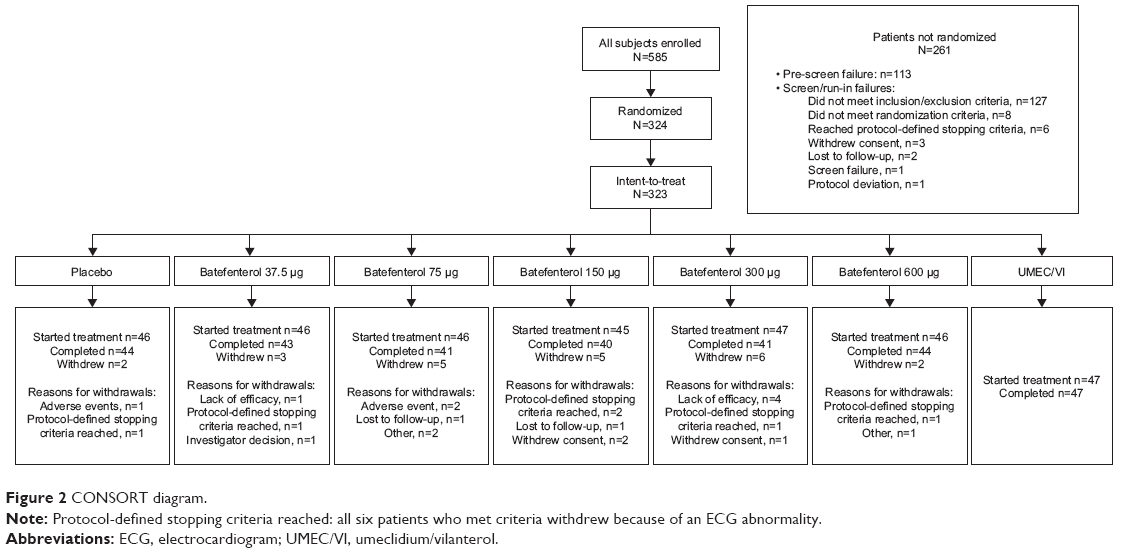 | Figure 2 CONSORT diagram. |
Demographics were similar across the groups (Table 1). Mean age was 62.6 years (SD: 7.78) and 56% of patients were males. The batefenterol 75 μg group had higher proportion of men (72%) and a greater FEV1, while the batefenterol 150 μg group had a lower proportion of men (42%).
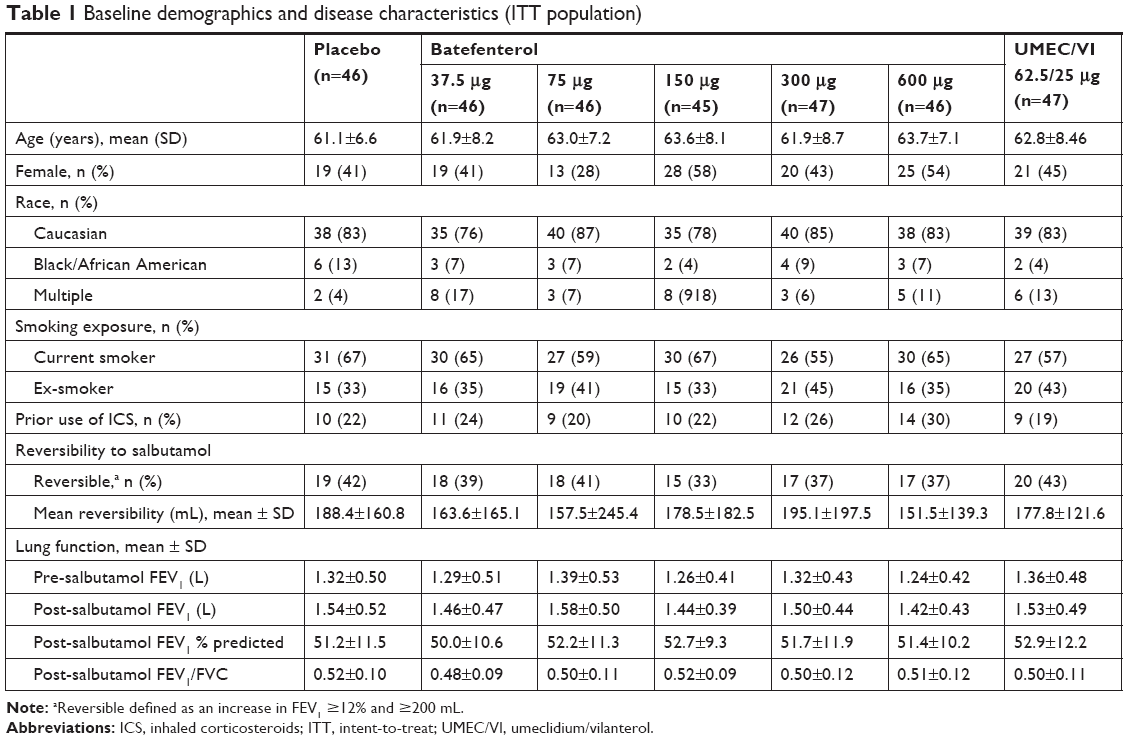 | Table 1 Baseline demographics and disease characteristics (ITT population) |
Efficacy results
The Bayesian Emax model for the change from baseline in the WM FEV1 over 0–6 hours post-dose on day 42 is shown in Figure 3. All batefenterol doses demonstrated statistically and clinically significant improvements in the primary endpoint vs placebo (191.1–292.8 mL) in the ITT population (Table 2, Figure 4A). Incremental improvements were observed with escalating doses of batefenterol, with the differences becoming progressively smaller with each dose increase (Table 2, Figure 4A). Pairwise comparisons between UMEC/VI and batefenterol 300 and 600 μg for the primary endpoint were numerically similar, with credible intervals around zero and posterior Bayesian probabilities of ~50% for the change associated with UMEC/VI were found to be greater than the change with batefenterol (Figure 4B), indicating that the two treatments had comparable effects.
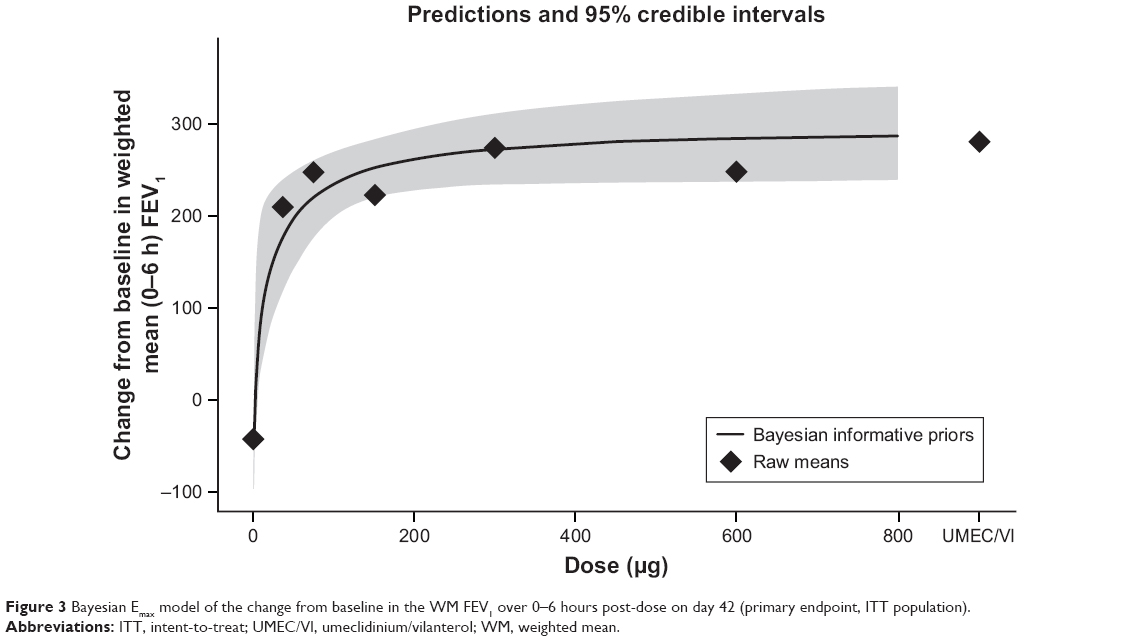 | Figure 3 Bayesian Emax model of the change from baseline in the WM FEV1 over 0–6 hours post-dose on day 42 (primary endpoint, ITT population). |
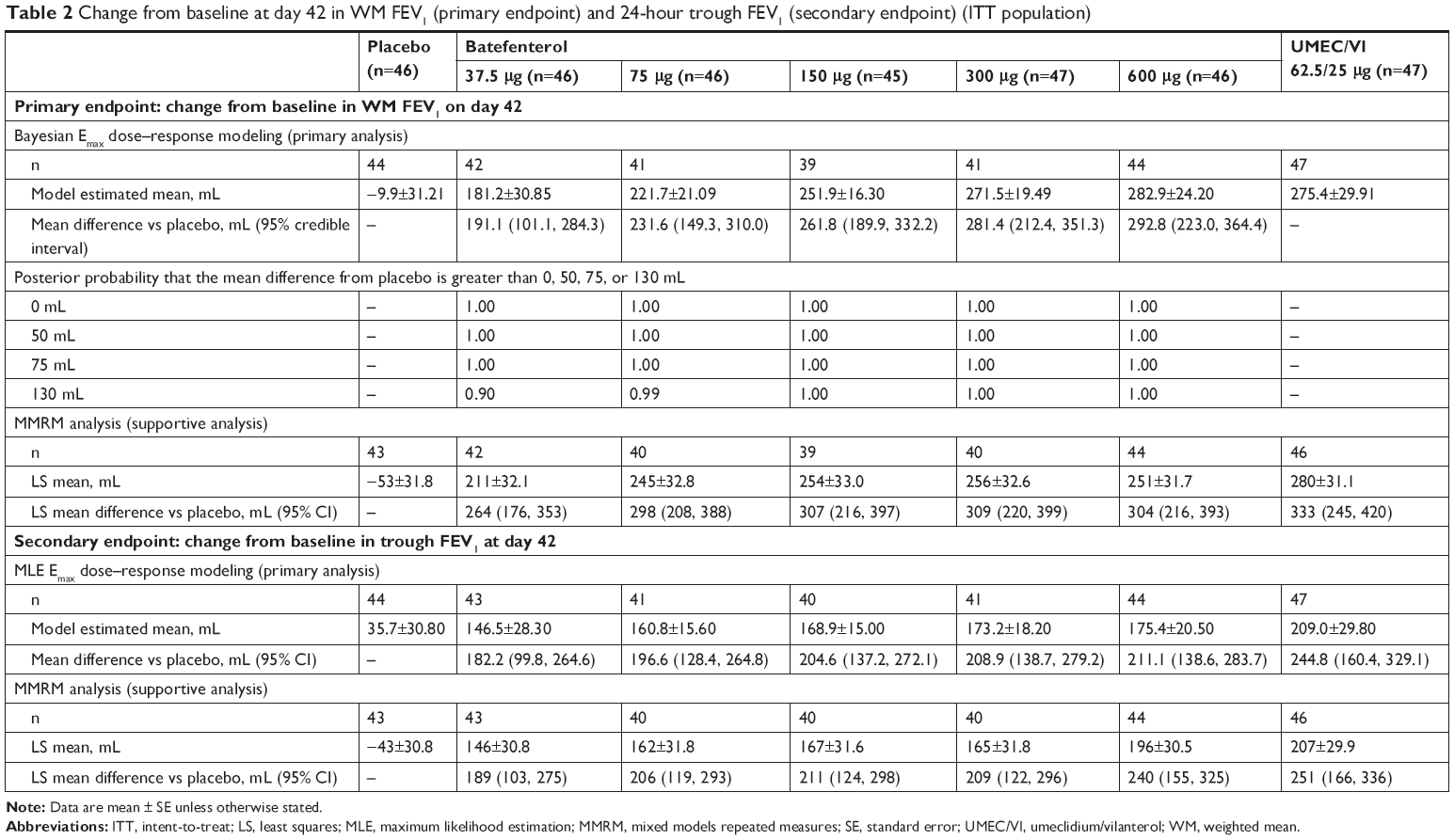 | Table 2 Change from baseline at day 42 in WM FEV1 (primary endpoint) and 24-hour trough FEV1 (secondary endpoint) (ITT population) |
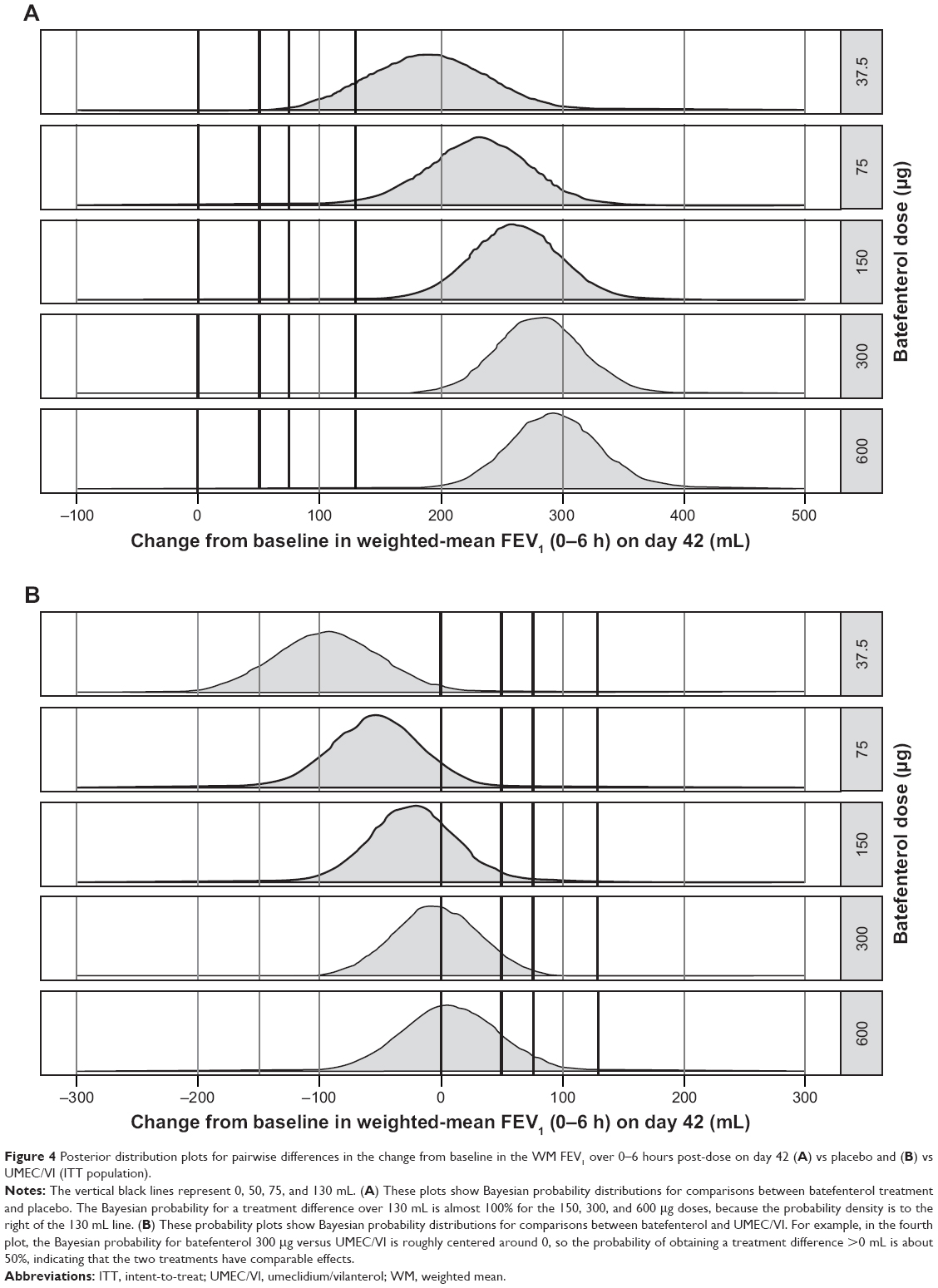 | Figure 4 Posterior distribution plots for pairwise differences in the change from baseline in the WM FEV1 over 0–6 hours post-dose on day 42 (A) vs placebo and (B) vs UMEC/VI (ITT population). |
MMRM analysis in the ITT population supported the results of the Bayesian analysis: all batefenterol doses achieved statistically greater improvements in the primary endpoint than placebo (Table 2; Figure 5).
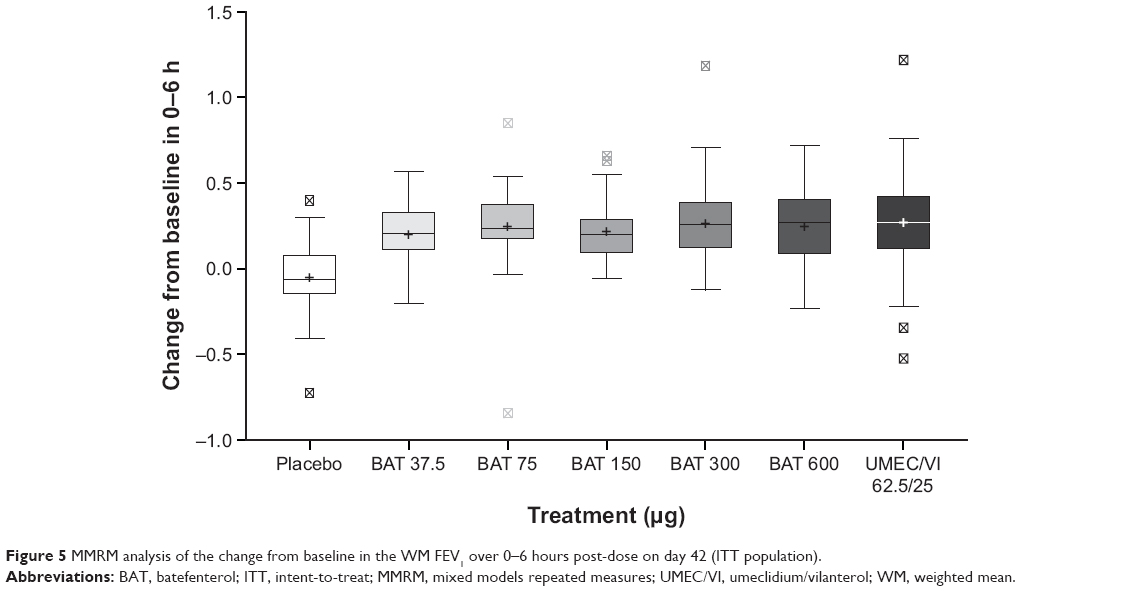 | Figure 5 MMRM analysis of the change from baseline in the WM FEV1 over 0–6 hours post-dose on day 42 (ITT population). |
As in the ITT population, Bayesian analysis of the reversible population demonstrated statistically and clinically significant improvements in the primary endpoint with batefenterol compared to placebo; the mean (95% credible interval) difference vs placebo was 149.2 mL (71.4, 264.5) for 37.5 μg, 208.4 mL (115.4, 318.3) for 75 μg, 263.5 mL (165.0, 364.7) for 150 μg, 305.1 mL (206.4, 403.2) for 300 μg, and 334.1 mL (231.9, 432.7) for 600 μg. Pairwise comparisons between UMEC/VI and batefenterol 300 and 600 μg were numerically similar, with credible intervals around zero and posterior Bayesian probabilities of 40% and 60% for the change associated with UMEC/VI were found to be greater than the change with batefenterol 300 and 600 μg, respectively. MMRM analysis in the reversible population also demonstrated statistically significant differences vs placebo; the least squares mean difference vs placebo was 243 mL (95% CI 84, 402) for 37.5 μg (P=0.003), 241 mL (95% CI 86, 397) for 75 μg (P<0.003), 315 mL (95% CI 146, 485) for 150 μg (P<0.001), 362 mL (95% CI 204, 519) for 300 μg (P<0.001), and 315 mL (95% CI 158, 472) for 600 μg (P<0.001).
In the batefenterol 300 μg group, peak bronchodilation was observed 1-hour post-dose on days 1 and 42: the least squares mean difference in FEV1 vs placebo was 280 mL (95% CI 220, 340) on day 1 and 380 mL (95% CI 280, 480) on day 42, indicating that the peak effect increased over time.
Secondary endpoint: trough FEV1 on day 42
Compared to placebo, all batefenterol doses demonstrated statistically significant improvements in change from baseline trough FEV1 (182–211 mL) at day 42 based on MLE dose–response modeling (Table 2). Incremental improvements in trough FEV1 were observed with escalating doses of batefenterol. Supportive MMRM analysis showed statistically significant differences in all batefenterol and UMEC/VI treatment groups compared to placebo (P<0.001 for all batefenterol doses in the ITT population; Table 2).
Safety
AEs were reported in 22%, 33%, 27%, 30%, and 61% of patients in the batefenterol 37.5, 75, 150, 300, and 600 μg groups, 34% in the UMEC/VI group, and 24% in the placebo group (Table 3). Cough, nasopharyngitis, and dysgeusia were the most commonly reported AEs in the batefenterol groups; there were 2, 1, and 6 cases of dysgeusia in the batefenterol 150, 300, and 600 μg groups, respectively. There were no AE reports of dysphagia. Serious AEs (SAEs) were reported in two patients in the batefenterol 75 μg group (COPD exacerbation, and scrotal abscess), one in the 300 μg group (COPD exacerbation), one in the 600 μg group (pulmonary embolism), and one in the UMEC/VI group (tachycardia). In addition, one patient reported SAEs (hypertensive crisis and panic attack) 4 days after the last dose of batefenterol 600 μg. None of the SAEs were considered drug-related and all were resolved; one SAE (COPD exacerbation in the batefenterol 75 μg group) led to withdrawal from the study. There were no deaths and no clinically relevant effects on laboratory parameters, vital signs, or ECGs. Overall, two patients in the placebo group and five in the batefenterol groups (two from 75 μg and one each from 150, 300, and 600 μg groups) withdrew from the study due to an AE. One patient (from the batefenterol 75 μg group) withdrew from the study due to nonserious AEs of rash and pharyngeal edema, which were considered drug-related by the study investigators.
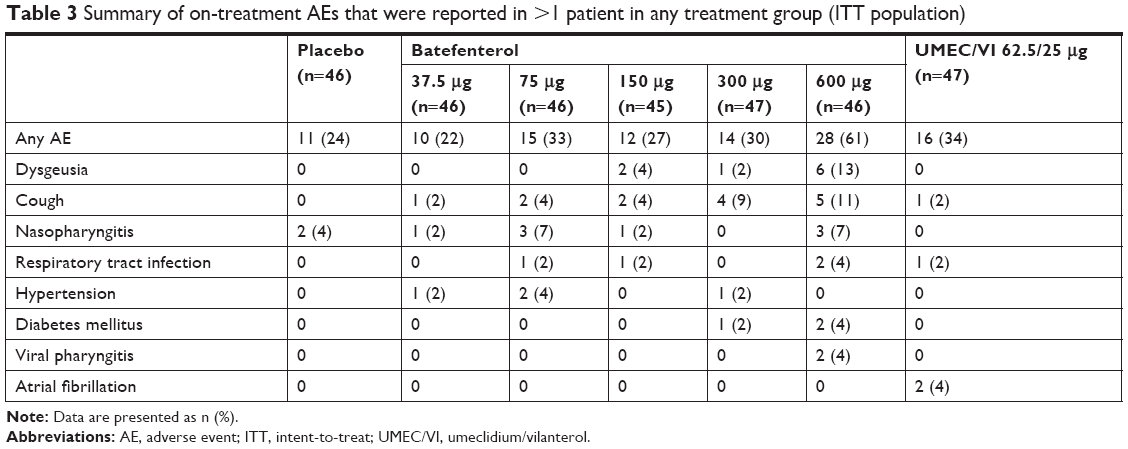 | Table 3 Summary of on-treatment AEs that were reported in >1 patient in any treatment group (ITT population) |
Discussion
This study aimed to determine the minimal, optimally effective dose of batefenterol, with an acceptable safety profile, in patients with stable COPD to take forward to Phase III development. In the ITT population, all batefenterol doses produced statistically significant and clinically meaningful improvements in lung function, measured by WM FEV1 over 0–6 hours post-dose (primary endpoint) and trough FEV1 (secondary endpoint). Incremental improvements in lung function vs placebo were seen with escalating doses of batefenterol, with the difference between doses becoming smaller as the dose increased. Overall, a relatively flat dose–response profile was observed. The previous dose-finding study found an increasing dose–response profile for batefenterol, albeit over a broader range of doses.11
Approximately 40% of patients were reversible to salbutamol, a slightly higher proportion than in the previous dose-finding study (~33%).11 Overall, treatment responses were greater in the reversible subgroup compared with the ITT population, as expected. The reversible subgroup demonstrated a slightly different dose–response pattern compared with the ITT population, with a greater difference between batefenterol doses and a clear improvement in lung function at doses above 75 μg. Data from both the Bayesian and MMRM analyses of the WM FEV1 over 0–6 hours post-dose suggest that the inflection point at the upper plateau of the dose–response curve corresponds approximately with the batefenterol 150 μg dose. This indicates that batefenterol 300 μg may be the optimal once-daily dose. This is similar to the conclusion of a previous dose-finding study, which identified batefenterol 400 μg as the optimal daily dose.11 Identification of a clearer dose–response in the reversible subgroup is not surprising, as it is conceivable that such a subgroup would demonstrate greater response to a bronchodilator and would be more likely to discern dose–response trends.12
Treatment guidelines recommend that patients with COPD receive a single bronchodilator initially, followed by the addition of a second bronchodilator with a different mechanism of action if symptoms do not improve.1 A number of dual LABA/LAMA bronchodilators are now available, including UMEC/VI and indacaterol/glycopyrronium (QVA149).1 In this study, batefenterol produced improvements in lung function compared with those seen with QVA14913,14 and UMEC/VI.15,16 Likewise, spirometric improvements achieved in this study with batefenterol ≥150 μg were compared with those with UMEC/VI; the UMEC/VI 62.5/25 μg lung function response was toward the upper end of the response reported in previous trials, but within the expected range of data variability.15,16 Lung function improvements with batefenterol in this study were also similar to those observed in a previous dose-finding study.10 Direct comparisons of batefenterol with dual bronchodilators in randomized trials will be required, but dose-finding studies provide an early indication that the efficacy of batefenterol will be similar to existing dual bronchodilators. A dual pharmacophore such as batefenterol could simplify the formulation of triple therapies including dual bronchodilation and ICS, which may provide benefits in patients who are not adequately controlled with a LABA/LAMA combination.6,17
We did not analyze our data based on smoking status as the total number of patients in each treatment group was small (n=45–47), with the percentage of current smokers ranging between 55% and 67%. However, it is noteworthy that as part of the regulatory submission for UMEC/VI 62.5/25 μg, efficacy by smoking status was assessed in a post hoc analysis (unpublished data) of combined data from replicate 12-week comparative studies of UMEC/VI 62.5/25 μg vs fluticasone propionate/salmeterol 250/50 μg.18 In the UMEC/VI 62.5/25 μg treatment arm, on the primary endpoint of 0–24-hour weighted mean FEV1 on day 84, the difference between former and current smokers was 17 mL, which is in favor of former smokers. For the secondary endpoint of 24-hour trough FEV1 on day 85, the difference between former and current smokers was 25 mL, which is again in favor of former smokers. We would therefore anticipate small but similar differences in treatment effects in favor of former smokers with batefenterol.
Several authors have suggested that a peak inspiratory flow ≥60 L/min is the minimum requirement for optimal drug delivery with a dry powder inhaler.19,20 However, results have been shown to vary between different inhaler devices (eg, DISKUS, TURBUHALER, and AEROLIZER). DISKUS demonstrated consistency of delivered dose at flows between 30 and 90 L/min, whereas the delivered dose was reduced with flows <90 L/min with the TURBUHALER.21 Similarly, compared with the consistency of delivered fine particle mass with the DISKUS, for both the AEROLIZER and TURBUHALER this fraction was reduced at flows ≤60 L/min.21,22 In this study, all inhaled products were delivered via ELLIPTA, which demonstrates consistency of delivered dose and fine particle mass fraction at flows of 30–90 L/min.23,24 Furthermore, two groups of investigators demonstrated that patients with COPD and an FEV1 <30% predicted normal were able to generate peak inspiratory flows of at least 41.6–52 L/min.25,26 Thus, with correct usage, most patients with COPD should be able to receive an adequate dose with the ELLIPTA device.
No new safety signals were observed after 42 days of treatment with batefenterol. The AE profile of batefenterol 37.5 μg was similar to placebo. The incidence of cough increased with higher doses of batefenterol, occurring in 4% of patients receiving 75 and 150 μg doses, 9% receiving 300 μg, and 11% receiving 600 μg. Other than cough, AE profiles of batefenterol 75–300 μg were similar to placebo and UMEC/VI 62.5/25 μg. In the batefenterol 600 μg group, an increased incidence of dysgeusia (13% of patients) was also observed. No clinically relevant effects on vital signs, ECGs, blood glucose, and potassium levels were observed for any batefenterol dose. Overall, the AE profile of batefenterol was similar to that seen in the previous dose-ranging study.11
In conclusion, treatment with once-daily batefenterol 37.5, 75, 150, 300, or 600 μg for 42 days resulted in statistically and clinically significant improvements in lung function compared to placebo in patients with COPD. No new or unexpected safety signals were observed in this COPD population. These data suggest that batefenterol 300 μg might represent the optimal dose for Phase III studies.
Data availability
Anonymized individual participant data and study documents can be requested for further research from www.clinicalstudydatarequest.com.
Acknowledgments
This study was funded and conducted by GlaxoSmithKline (GSK; study number 201012). Medical writing assistance in the form of developing a draft based on author input and editorial assistance was provided by Matthew Robinson, DPhil, and Clare Slater, PhD CMPP, of Fishawack Indicia Ltd, funded by GSK.
The current affiliation of the author Shuyen Ho is UCB BioSciences Inc., Global Statistics and Innovation, Raleigh, NC, USA. The current affiliation of the author Krishna Pudi is Sierra Clinical Research, Las Vegas, NV, USA.
Author contributions
CB contributed to data analysis and interpretation; C Crim, C Crawford, MLW, RC-S, and SH contributed to study design, data analysis, and interpretation; EDB, EMK, IS, GJF, and KP contributed to data acquisition, analysis, and interpretation. All authors were involved at each stage of manuscript preparation including drafting the article or revising it critically for important intellectual content, approved the final version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare the following real/perceived conflicts of interest: C Crim, MLW, C Crawford, CB, and RC-S are GSK employees and hold shares in GSK; SH holds shares in GSK; EDB has received fees from Novartis, Cipla, Sanofi Regeneron, AstraZeneca, ALK, and Boehringer Ingelheim for advisory board membership, fees from Novartis for participation in speakers’ bureau, fees from Vectura and Actelion for consultancy work, fees from Cipla, Menarini, ALK, AstraZeneca, and Boehringer Ingelheim for lectures, and fees from ICON for participation on a study oversight steering committee, and his institution has received funding from Boehringer Ingelheim, Merck, Takeda, GSK, Hoffman le Roche, Actelion, Chiesi, Sanofi-Aventis, Cephalon, TEVA, Novartis, and AstraZeneca for participation in clinical trials. EMK has participated in advisory boards, speaker panels, or received travel reimbursement from Amphastar, AstraZeneca (Pearl), Forest, Novartis, Sunovion, Teva, and Theravance, has participated in medical advisory boards for Mylan and Oriel, and has performed consulting for Oriel and GSK. GJF’s institution has received funding from GSK for participation in clinical trials; KP and IS report no conflicts of interest in this work.
References
Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report: gold executive summary. Eur Respir J. 2017;49(3):1700214. | ||
van Noord JA, Aumann JL, Janssens E, et al. Comparison of tiotropium once daily, formoterol twice daily and both combined once daily in patients with COPD. Eur Respir J. 2005;26(2):214–222. | ||
van Noord JA, Aumann JL, Janssens E, et al. Effects of tiotropium with and without formoterol on airflow obstruction and resting hyperinflation in patients with COPD. Chest. 2006;129(3):509–517. | ||
van Noord JA, de Munck DR, Bantje TA, Hop WC, Akveld ML, Bommer AM. Long-term treatment of chronic obstructive pulmonary disease with salmeterol and the additive effect of ipratropium. Eur Respir J. 2000;15(5):878–885. | ||
Hughes AD, Chen Y, Hegde SS, et al. Discovery of (R)-1-(3-((2-chloro-4-(((2-hydroxy-2-(8-hydroxy-2-oxo-1,2-dihydroquinolin-5-yl)ethyl)amino)methyl)-5-methoxyphenyl)amino)-3-oxopropyl)piperidin-4-yl [1,1′-biphenyl]-2-ylcarbamate (TD-5959, GSK961081, batefenterol): first-in-class dual pharmacology multivalent muscarinic antagonist and β2 agonist (MABA) for the treatment of chronic obstructive pulmonary disease (COPD). J Med Chem. 2015;58(6):2609–2622. | ||
Cazzola M, Lopez-Campos JL, Puente-Maestu L. The MABA approach: a new option to improve bronchodilator therapy. Eur Respir J. 2013;42(4):885–887. | ||
Lipson DA, Barnhart F, Brealey N, et al. Once-daily Single-Inhaler triple versus dual therapy in patients with COPD. N Engl J Med. 2018;378(18):1671–1680. | ||
Lipworth B, Kuo CR, Jabbal S. Current appraisal of single inhaler triple therapy in COPD. Int J Chron Obstruct Pulmon Dis. 2018;13:3003–3009. | ||
Celli BR, MacNee W, ATS/ERS Task Force. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23(6):932–946. | ||
Quanjer PH, Stanojevic S, Cole TJ, et al. Multi-ethnic reference values for spirometry for the 3-95-yr age range: the global lung function 2012 equations. Eur Respir J. 2012;40(6):1324–1343. | ||
Wielders PL, Ludwig-Sengpiel A, Locantore N, Baggen S, Chan R, Riley JH. A new class of bronchodilator improves lung function in COPD: a trial with GSK961081. Eur Respir J. 2013;42(4):972–981. | ||
Chowdhury BA, Seymour SM, Michele TM, Durmowicz AG, Liu D, Rosebraugh CJ. The risks and benefits of indacaterol – the FDA’s review. N Engl J Med. 2011;365(24):2247–2249. | ||
Bateman ED, Ferguson GT, Barnes N, et al. Dual bronchodilation with QVA149 versus single bronchodilator therapy: the shine study. Eur Respir J. 2013;42(6):1484–1494. | ||
Mahler DA, Kerwin E, Ayers T, et al. FLIGHT1 and FLIGHT2: efficacy and safety of QVA149 (Indacaterol/Glycopyrrolate) versus its Monocomponents and placebo in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;192(9):1068–1079. | ||
Decramer M, Anzueto A, Kerwin E, et al. Efficacy and safety of umeclidinium plus vilanterol versus tiotropium, vilanterol, or umeclidinium monotherapies over 24 weeks in patients with chronic obstructive pulmonary disease: results from two multicentre, blinded, randomised controlled trials. Lancet Respir Med. 2014;2(6):472–486. | ||
Donohue JF, Maleki-Yazdi MR, Kilbride S, Mehta R, Kalberg C, Church A. Efficacy and safety of once-daily umeclidinium/vilanterol 62.5/25 mcg in COPD. Respir Med. 2013;107(10):1538–1546. | ||
Hughes AD, Jones LH. Dual-pharmacology muscarinic antagonist and β2 agonist molecules for the treatment of chronic obstructive pulmonary disease. Future Med Chem. 2011;3(13):1585–1605. | ||
Donohue JF, Worsley S, Zhu CQ, Hardaker L, Church A. Improvements in lung function with umeclidinium/vilanterol versus fluticasone propionate/salmeterol in patients with moderate-to-severe COPD and infrequent exacerbations. Respir Med. 2015;109(7):870–881. | ||
Janssens W, Vandenbrande P, Hardeman E, et al. Inspiratory flow rates at different levels of resistance in elderly COPD patients. Eur Respir J. 2008;31(1):78–83. | ||
Mahler DA, Waterman LA, Gifford AH. Prevalence and COPD phenotype for a suboptimal peak inspiratory flow rate against the simulated resistance of the Diskus® dry powder inhaler. J Aerosol Med Pulm Drug Deliv. 2013;26(3):174–179. | ||
Prime D, Grant AC, Slater AL, Woodhouse RN. A critical comparison of the dose delivery characteristics of four alternative inhalation devices delivering salbutamol: pressurized metered dose inhaler, Diskus inhaler, Diskhaler inhaler, and Turbuhaler inhaler. J Aerosol Med. 1999;12(2):75–84. | ||
Chew NY, Chan HK. In vitro aerosol performance and dose uniformity between the Foradile Aerolizer and the Oxis Turbuhaler. J Aerosol Med. 2001;14(4):495–501. | ||
Grant AC, Walker R, Hamilton M, Garrill K. The ELLIPTA® dry powder inhaler: design, functionality, in vitro dosing performance and critical task compliance by patients and caregivers. J Aerosol Med Pulm Drug Deliv. 2015;28(6):474–485. | ||
Hamilton M, Leggett R, Pang C, Charles S, Gillett B, Prime D. In vitro dosing performance of the ELLIPTA® dry powder inhaler using asthma and COPD patient inhalation profiles replicated with the electronic lung (eLung™). J Aerosol Med Pulm Drug Deliv. 2015;28(6):498–506. | ||
Altman P, Wehbe L, Dederichs J, et al. Comparison of peak inspiratory flow rate via the Breezhaler®, Ellipta® and HandiHaler® dry powder inhalers in patients with moderate to very severe COPD: a randomized cross-over trial. BMC Pulm Med. 2018;18(1):100. | ||
Prime D, de Backer W, Hamilton M, et al. Effect of disease severity in asthma and chronic obstructive pulmonary disease on Inhaler-Specific inhalation profiles through the ELLIPTA® dry powder inhaler. J Aerosol Med Pulm Drug Deliv. 2015;28(6):486–497. |
Supplementary materials
Exclusion criteria
Patients were not eligible for inclusion in this study if any of the following criteria applied:
- Current diagnosis of asthma
- Respiratory disorders other than COPD, including but not limited to: α–1 antitrypsin deficiency, active tuberculosis, bronchiectasis, sarcoidosis, lung fibrosis, pulmonary hypertension unrelated to COPD, and interstitial lung disease. Allergic rhinitis was not exclusionary
- Other diseases/abnormalities, including uncontrolled hypertension, diabetes, and thyroid disease
- Presence of hepatitis B surface antigen, positive hepatitis C antibody test result at screening (visit 1) or within 3 months prior to first dose of study treatment
- Current or chronic history of liver disease and known hepatic or biliary abnormalities (with the exception of Gilbert’s syndrome or asymptomatic gallstones)
- Current malignancy or previous history of cancer in remission for <5 years prior to visit 1 (localized basal cell or squamous cell carcinoma of the skin that had been resected was not exclusionary); any current or previous history of throat cancer
- Chest X-ray or computed tomography (CT) scan revealing evidence of clinically significant abnormalities not believed to be due to the presence of COPD. A chest X-ray was taken at visit 1 if a chest X-ray or CT scan was not acquired within 6 months prior to visit 1
- History of hypersensitivity or allergy to any β-adrenergic receptor agonist, sympathomimetic, anticholinergic/anti-muscarinic receptor antagonist, or lactose/milk protein
- Diseases preventing use of anticholinergics, for example, narrow-angle glaucoma, prostatic hypertrophy, or bladder neck obstruction
- Poorly controlled COPD, defined as the occurrence of acute worsening of COPD that was managed with corticosteroid and/or antibiotics or that required treatment prescribed by a physician in the 6 weeks prior to screening (visit 1), or subjects who were hospitalized due to acute worsening of COPD within 12 weeks of visit 1
- History of more than one COPD exacerbation (moderate or severe) within 12 months prior to visit 1
- Pneumonia and lower respiratory tract infections requiring the use of antibiotics within 6 weeks prior to visit 1, or pneumonia requiring hospitalization within 12 weeks of visit 1
- Lung volume reduction surgery within 12 months prior to visit 1
- Abnormal and clinically significant 12-lead electrocardiogram
- Clinically significant abnormal findings from clinical chemistry or hematology tests at visit 1
- Medication prior to spirometry: unable to withhold albuterol/salbutamol for the 4-hour period required prior to spirometry testing at each study visit
- Use of any of the excluded medications (Table S2)
- Use of long-term oxygen therapy, described as oxygen therapy prescribed for >12 hours a day. As-needed oxygen use (ie, ≤12 hours/day) was not exclusionary
- Nebulized therapy: regular use (prescribed for use every day, not for as-needed use) of short-acting bronchodilators (eg, albuterol/salbutamol) via nebulized therapy
- Pulmonary rehabilitation: participation in the acute phase of a pulmonary rehabilitation program within 4 weeks prior to visit 1. Subjects who were in the maintenance phase of a pulmonary rehabilitation program were not excluded
- Known or suspected history of alcohol or drug abuse within 2 years prior to visit 1
- Non-adherence with study procedures
- Questionable validity of consent, for example, due to a history of psychiatric disease, intellectual deficiency, poor motivation, and so on
- Affiliation with investigator site
- Inability to read.
 | Table S1 Institutional review boards |
 | Table S2 Excluded medications prior to visit 1 and throughout the study |
Withdrawal/stopping criteria
Liver chemistry withdrawal/stopping criteria
Liver chemistry withdrawal or stopping criteria are schematically represented in Figure S1.
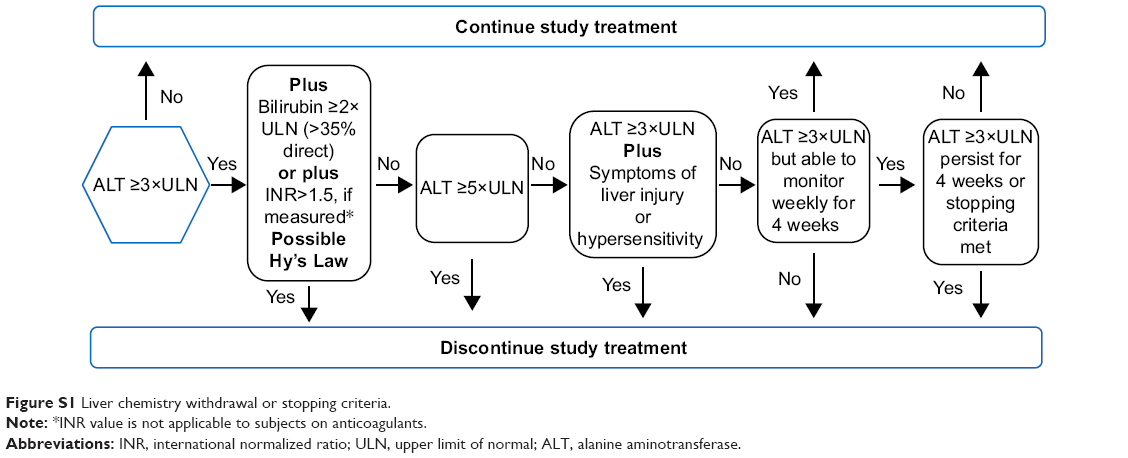 | Figure S1 Liver chemistry withdrawal or stopping criteria. |
12-lead electrocardiogram (ECG) withdrawal criteria
For this study, an abnormal and clinically significant 12-lead ECG that would preclude a subject from entering the trial is defined as a 12-lead tracing that is interpreted as, but not limited to, any of the following:
- Sinus bradycardia <45 bpm
- Sinus tachycardia ≥110 bpm
- Multifocal atrial tachycardia (wandering atrial pacemaker with rate >100 bpm)
- PR interval >240 ms
- Evidence of Mobitz II second-degree or third-degree atrioventricular block
- Pathological Q waves (defined as wide [>0.04 seconds] and deep [>0.4 mV (4 mm with 10 mm/mV setting)] or >25% of the height of the corresponding R wave, providing the R wave was >0.5 mV [5 mm with 10 mm/mV setting]) appearing in at least two contiguous leads
- Evidence of ventricular ectopic couplets, bigeminy, trigeminy, or multifocal premature ventricular complexes
- For subjects without complete right bundle branch block: QTc(F) ≥450 ms or an ECG that is unsuitable for QT measurements (eg, poorly defined termination of the T wave)
- For subjects with complete right bundle branch block: QTc(F) ≥480 ms or an ECG that is unsuitable for QT measurements (eg, poorly defined termination of the T wave)
- ST-T wave abnormalities (excluding nonspecific ST-T wave abnormalities)
- Clinically significant conduction abnormalities (eg, Wolff–Parkinson–White syndrome or bifascicular block defined as complete left bundle branch block or complete right bundle branch block with concomitant left fascicular block)
- Clinically significant arrhythmias (eg, atrial fibrillation with rapid ventricular response and ventricular tachycardia).
○ | Note: Sinus bradycardia should be confirmed by two additional readings at least 5 minutes apart |
○ | Note: Sinus tachycardia should be confirmed by two additional readings at least 5 minutes apart |
○ | Note: prior evidence (ie, ECG obtained at least prior to 12 months) of pathological Q waves that are unchanged are not exclusionary and the investigator will determine if the subject is precluded from entering the study |
○ | Note: All potentially exclusionary QT measurements (corrected or uncorrected) should be confirmed by two additional readings at least 5 minutes apart. The final assessment will be based on averaged QTc value of triplicate ECGs |
○ | Note: Prior evidence (ie, ECG obtained at least 12 months prior) of ST-T abnormalities that are unchanged are not exclusionary and the investigator will determine if the subject is precluded from entering the study |
Statistical analysis
The sample size was determined by simulations and assurance (probability of success) calculations based on Bayesian methods. It was determined that 40 evaluable patients per treatment group would provide a high chance of achieving the study objectives. Statistical assurance was high for hypothesis testing. For example, there was ~90% assurance that the 150-μg dose would provide a 130 mL improvement in FEV1 over placebo. With 40 patients per group, the half-width of the 95% credible interval for the dose that produced an average improvement of 130 mL versus placebo was approximately within twofold of the estimate. For example, if the estimate was 80 μg, the 95% credible interval was expected to range between 40 and 160 μg; in this case, the 160-μg dose would provide assurance that the average treatment effect would be at least 130 mL greater than placebo.
Summary of safety statistics were prepared for the intent-to-treat (ITT) population. SAS version 9.1 or later was used for analysis.
Bayesian and mixed models repeated measures (MMRM) models
The Bayesian Emax model can accommodate various dose–response curves and has three parameters, encapsulated in the following formula:
|
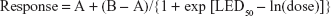
where A is the mean response at dose 0; B is the mean response at dose = ∞; and LED50 = ln(ED50) = natural logarithm of the dose that yields a mean response of (A + B)/2.
This formula was used to fit existing Phase IIb data as starting points for the simulations. Bayesian informative priors on the Emax parameters were then constructed directly from the existing data.
Terms fitted to the Emax model included treatment group and baseline FEV1 value. The Emax dose–response model that best fitted the observed data was used to estimate and predict the change from baseline in weighted-mean FEV1 over 0–6 hours across the dose range investigated for batefenterol. Hypothesis testing was performed after the selection of the best fitting model and assessed whether the selected dose would reject a null hypothesis of a treatment effect on FEV1 of 0, 50, 75, or 130 mL greater than placebo. The secondary efficacy endpoint was analyzed using the maximum likelihood estimation method of Emax dose–response modeling in the ITT population with terms including treatment group and baseline FEV1.
The MMRM model included terms for treatment group, smoking status, country, sex, inhaled corticosteroid usage, reversibility, and visit (except for serial measures when analysis was performed for each visit separately). The MMRM analysis for trough FEV1 and FVC included FEV1 and FVC measurements, respectively, at day 1 (baseline) as covariate, and days 7, 14, 28, and 42 as response. Data were presented as least squares (LS) mean change from baseline with standard error for each treatment group and/or LS mean treatment differences with 95% CIs.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.