Back to Journals » Neuropsychiatric Disease and Treatment » Volume 12
Randomized controlled trials – a matter of design
Authors Spieth PM, Kubasch AS, Penzlin AI, Illigens BM, Barlinn K, Siepmann T
Received 6 December 2015
Accepted for publication 18 March 2016
Published 10 June 2016 Volume 2016:12 Pages 1341—1349
DOI https://doi.org/10.2147/NDT.S101938
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Peter Markus Spieth,1,2 Anne Sophie Kubasch,3 Ana Isabel Penzlin,4 Ben Min-Woo Illigens,2,5 Kristian Barlinn,6 Timo Siepmann2,6,7
1Department of Anesthesiology and Critical Care Medicine, University Hospital Carl Gustav Carus, Technische Universität Dresden, 2Center for Clinical Research and Management Education, Division of Health Care Sciences, Dresden International University, 3Pediatric Rheumatology and Immunology, Children’s Hospital, University Hospital Carl Gustav Carus, Technische Universität Dresden, 4Institute of Clinical Pharmacology, University Hospital Carl Gustav Carus, Technische Universität Dresden, Dresden, Saxony, Germany; 5Department of Neurology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, USA; 6Department of Neurology, University Hospital Carl Gustav Carus, Technische Universität Dresden, Dresden, Saxony, Germany; 7Radcliffe Department of Medicine, John Radcliffe Hospital, University of Oxford, Oxford, Oxfordshire, UK
Abstract: Randomized controlled trials (RCTs) are the hallmark of evidence-based medicine and form the basis for translating research data into clinical practice. This review summarizes commonly applied designs and quality indicators of RCTs to provide guidance in interpreting and critically evaluating clinical research data. It further reflects on the principle of equipoise and its practical applicability to clinical science with an emphasis on critical care and neurological research. We performed a review of educational material, review articles, methodological studies, and published clinical trials using the databases MEDLINE, PubMed, and ClinicalTrials.gov. The most relevant recommendations regarding design, conduction, and reporting of RCTs may include the following: 1) clinically relevant end points should be defined a priori, and an unbiased analysis and report of the study results should be warranted, 2) both significant and nonsignificant results should be objectively reported and published, 3) structured study design and performance as indicated in the Consolidated Standards of Reporting Trials statement should be employed as well as registration in a public trial database, 4) potential conflicts of interest and funding sources should be disclaimed in study report or publication, and 5) in the comparison of experimental treatment with standard care, preplanned interim analyses during an ongoing RCT can aid in maintaining clinical equipoise by assessing benefit, harm, or futility, thus allowing decision on continuation or termination of the trial.
Keywords: randomized clinical trials, RCT, validity, study design, CONSORT
Introduction
With respect to study design, randomized controlled trials (RCTs) as well as analysis of quantitatively synthesized RCT data are considered the gold standard for evaluating efficacy in clinical research and constitute evidence for medical treatment. Thus, RCT data are guiding physicians toward evidence-based therapy. However, interpretability of RCT data can be jeopardized by systematic error (bias), random error, or limited generalizability; problems that are usually rooted in shortcomings in study design. Choosing the appropriate RCT design is pivotal to produce data that can be translated into clinical practice.1,2 This review summarizes relevant aspects of design and interpretation of RCTs with the aim of providing the clinician with relevant background information when translating current research findings into clinical practice. Moreover, it reflects on the principle of equipoise, an ethical concept that is increasingly important when large multicentric studies are dominating the impact of medical science on clinical practice.
Design of clinical trials
Types and phases of studies
Clinical studies can be separated into nonexperimental or observational and experimental or RCTs. Nonexperimental research include case reports, case series, cross-sectional, and prospective observational studies, such as case–control and cohort studies. These types of research studies often generate important insights but cannot provide causal inferential value. RCTs may result in high-quality data, enabling the description of causal relationships, and thus forms the basis of evidence-based medicine.3,4
From the methodological point of view, observational studies are investigating both, the exposure and the outcome, whereas experimental studies are observing the outcome of an assigned exposure. The major advantage of RCTs is the straightforward investigation of cause–effect relationships with minimal bias and confounding factors.
In RCTs, a predefined study sample is built out of the target population (eg, patients with the respective diagnosis) and randomly assigned to different groups (eg, standard treatment or placebo vs new treatment). The observed effects of investigational treatments at defined time points constitute predefined end points.
Clinical trials are commonly classified into phases. Each phase is characterized by its design and sample size. Phase I trials usually test the interventions in healthy volunteers and aim to address safety issues as well as pharmacokinetics and dose–response characteristics. Phase II trials are designed to determine the evidence of activity or optimal dosage. Phase III trials are usually pivotal studies designed to provide data for approval by authorities testing new interventions either against placebo or against standard treatment for superiority or noninferiority, respectively. Phase IV studies assess long-term safety data and are often conducted to receive approval for expanded indications after initial approval of the intervention. Although there is a considerable variability in timing and number of patients enrolled in the different study phases, a rule of thumb is that Phase I studies enroll up to 100 healthy volunteers over a period of up to 2 years, Phase II usually up to 300 patients up to 3 years, and Phase III >1,000 patients for 3–4 years.5,6 For drug development studies, Phase III trials are often classified as Phase IIIa (before submission for approval authorities) and IIIb (after approval).
Research question and hypothesis
Designing an RCT starts with the development of a clinically relevant research question. Depending on the research question, the underlying hypothesis will commonly either aim at superiority (comparative trial) or at noninferiority (equivalence trial) of one intervention vs another.7,8 The intention behind a superiority trial is that Intervention A is superior to Intervention B, whereas noninferiority trials are designed to prove that the new treatment is at least as good as the standard therapy in terms of efficacy but offers other advantages such as lower costs, lower toxicity, improved side effect profile, or improved forms of administration compared to the standard of care. Although straightforward on the first approach, the validity of noninferiority trials is sometimes jeopardized by the lack of efficacy of the standard treatment as well as the appropriate choice of noninferiority margins. The noninferiority margin represents a prespecified acceptable inferiority, which represents the least clinically relevant difference among groups, and preserves superiority when compared to placebo treatment. The noninferiority margin has to be defined a priori and determines the sample size of the trials as well as the objective of the trial.
Common study designs
Parallel and crossover designs are the two standard designs for RCTs.3,9 Following randomization, subjects will be assigned either to receive Intervention A or B (or C, D, E, etc) throughout the entire study period (parallel design), or subjects are first treated with Intervention A followed by Intervention B and vice versa (crossover design). Crossover trials can be powerful, since every individual serves as their own control, thus variability due to interindividual differences is excluded.9,10
Whereas randomization is a powerful tool to ensure validity in parallel-designed studies, special precautions have to be considered in crossover studies to avoid or at least account for possible carryover effects. Carryover effects are defined as effects that “carry over” from one condition, eg, exposure or treatment, to another. Besides randomization of the treatment sequence, wash-in and wash-out periods of appropriate length are commonly used in crossover studies to avoid carryover effects. If more than two interventions are compared in a crossover study, the latin square design can be used. A latin square design describes a specific (n × n) matrix filled with one symbol, each representing one intervention, in each row and column, vice versa. Latin square sequences are generated to ensure that every intervention is followed or preceded just once by any other intervention tested. The assumption behind the latin square design is that if there are carryover effects, they are better controlled than by simple randomization because of the fixed sequence generated.
To test for treatment effects of combined interventions, factorial study designs have been proposed where individuals are randomly assigned to receive two or more interventions.10,11 The factorial study design increases the study efficiency because it allows for assessment of multiple interventions within the same trial. Factorial designs allow for testing the effects of each factor on the response variable as well as the effects of the interacting factors on the response variable.
Validity of clinical trials
There have been quality indicators defined for RCTs, and these indicators should be considered in both design and reporting of RCTs.5,8,12 These quality indicators include internal and external validity.
Internal validity
High internal validity means that the differences observed between groups are related to the intervention tested in the trial. This means, for example, that the reduction in mortality in the study population is really attributed to the intervention and not to other factors, such as age, sex, or comedications. The internal validity of a clinical trial is directly related to appropriate design, conduction, and reporting of the study. The two main threats to internal validity are bias and random error. Bias hereby refers to a systematic error that leads to a systematic deviation of the results from the truth due to flaws in the design, conduction, or reporting of the trial.5,8,13 Typical sources of bias are flaws in collection, statistical analysis, or interpretation of study data. Consequently, the true difference between study groups may be either under- or overestimated.
The four main sources of bias in clinical trials are selection bias, performance bias, detection bias, and attrition bias.14,15 Selection bias is usually controlled by randomization. The main goal of randomized trials is therefore to assure that each individual has an equal probability to be assigned to one or the other treatment. Randomization also allows to balance known and unknown confounders in order to make control and treatment groups as balanced as possible. Practically, this can be realized by random allocation (eg, by using opaque envelopes, allocation tables, or computer-based random number generators).8,16 Allocation concealment is another key factor for successful randomization, this concept means that all people who are involved in the recruitment process cannot predict the next group assignment.17,18 Allocation concealment prevents specific patients from being assigned to one or the other group by the investigator, possibly resulting in dysbalance or systematic exaggeration of study results. Allocation concealment can be realized by separating the person who is generating random allocation and the person who is recruiting patients. In addition, performance bias occurs if there is insufficient adherence to the study protocol either by the participant or the investigator. Although data on adherence are important for the interpretation of RCTs, they are usually underreported.7
Detection bias refers to systematically different outcome assessments among study groups. Detection bias may occur if the outcome assessment is affected by the group membership of the assessed individual. The risk of detection bias may increase with close subject or assessor interaction or increased margin of discretion by the assessor. For both performance and detection bias, the major source is the individual perception of the investigator about the advantages or disadvantages of the different investigational treatments. The most important measure to reduce these sources of bias is the blinding of the investigators including the study physician. While balancing for possible confounders is easy in large RCTs, design of smaller trials should employ techniques such as block randomization or a priori stratification to reduce the risk of dysbalanced groups due to confounders. However, stratification techniques must be defined a priori and are associated with special requirements regarding the statistical analysis. Typical examples of confounders in RCTs are sex, age, and severity of disease. Another way to address confounding is to employ multivariable analysis methods to adjust for the effects of confounders.1,19 It is worth noting that adjusting for confounders needs to be done before randomization since postrandomization adjustment for covariates jeopardizes the effects of randomization. Performance bias is the consequence of systematic intergroup differences in the investigational intervention or of exposure factors that are not related to the intervention.20 This form of bias can be avoided through blinding. Blinding can be single-blind (if just the patient or the assessor do not know the group assignment), double-blind (if both, the assessor and the patient do not know the group assignment), and triple-blind (if additionally, the person who performs the statistical analysis is not aware of the group assignment). Blinding techniques are frequently limited when investigational new medical devices or procedures are tested, as these can usually be easily distinguished from control conditions. In placebo-controlled pharmacological trials, effort should be put into producing a placebo that is as similar as possible to the study drug. For safety reasons, rapid unblinding should be possible in the case of adverse events or emergencies.
Attrition bias occurs if there are systematic differences in the number of participants dropping out of the study among the study groups.3 There are reasons for drop outs such as 1) participants may withdraw informed consent, 2) participants may become uncontactable, and 3) participants or investigators violate the study protocol or refuse to continue treatment for whatever reason. To control for attrition bias, most studies are nowadays analyzed according to the intention-to-treat (ITT) approach, which means all participants that have been randomized will be included in the final analysis, regardless whether they completed the study or dropped out.10 In addition to the ITT principle, the handling of missing data should also be reported. There are different types of missing data. Data missing completely at random means that the lost data are independent from observable variables and unobservable parameters of interest; therefore, the remaining subjects with full data sets are still fully randomized from the sample population, and the further analysis will not be biased by the missing data. Missing at random describes a situation where the missing data are not random but cannot be related to a mechanism depending on unobserved data. Missing not at random means that data are neither missing completely at random nor missing at random, for example, when the missing data are related to the reason why this data are missing. There are different ways to handle missing data in clinical trials such as the last observation carried forward method, multiple imputation techniques, or mixed methods in case of missing values within repeated measures.10
Although bias can be voluntarily reduced by appropriate study design and good clinical practice, the chance of random error remains due to the intrinsic variability of the measured data by pure chance. The most feasible way to improve the estimate of random error is to recruit a sufficient number of subjects21 according to a sample size calculation or power analysis.5 Random error can be divided into two types of errors with respect of hypothesis testing. Type I error refers to the probability of drawing a false-positive conclusion by incorrect rejection of a true null hypothesis, whereas Type II error refers to the probability of a false-negative conclusion by rejecting a false null hypothesis.5 In a simplified way, Type I error describes the phenomenon of detecting a difference that is not really present, whereas a Type II error leads to neglecting an effect that is really present. Given these facts, it become obvious that sample size calculation needs to be performed a priori.5,13
External validity
External validity describes the extent to which the results of an RCT can be generalized into clinical practice and the general population.14 This means, for example, if an RCT showed that the antihypertensive Drug A is able to reduce the mortality in the study sample of hypertonic patients as compared to placebo, Drug A can also reduce the mortality in all hypertonic patients. It is worth noting that internal validity of a study is the prerequisite of its external validity since incorrect data due to missing internal validity can, per se, not be applied to the general population.8 Even if internal validity is assumed, insufficient external validity may reduce the clinical relevance of an RCT.17 The study sample of an RCT is defined as the patients enrolled in the trial out of the population of theoretically accessible patients (target population). Practically, the target population represents all patients with the disease that should be studied, whereas the accessible population includes all patients who could be screened. Finally, the study sample is constituted by all patients who have been successfully enrolled into the trial. By carefully setting inclusion and exclusion criteria, specific study samples can be selected, but it also bears the risk of jeopardizing generalizability. Van Spall et al19 reviewed the eligibility criteria of RCTs published in high-impact general medical journals and showed that exclusion criteria are often not well reported and that women, children, and elderly patients are commonly excluded from RCTs.
Reporting of RCT data
In 1996, the Consolidated Standards of Reporting Trials (CONSORT) were introduced to improve the reporting of RCT data and enhance the quality of newly designed clinical trials.22 The CONSORT statements have been revised and updated twice since then to adapt for new methodological evidence and for the implementation of additional experiences.2,12,13 The CONSORT 2010 statement provides a template for the flow diagram of the study process (Figure 1) and a 25-item checklist (Table 1) of information that should be reported in both groups in parallel randomized trials.4,13,23
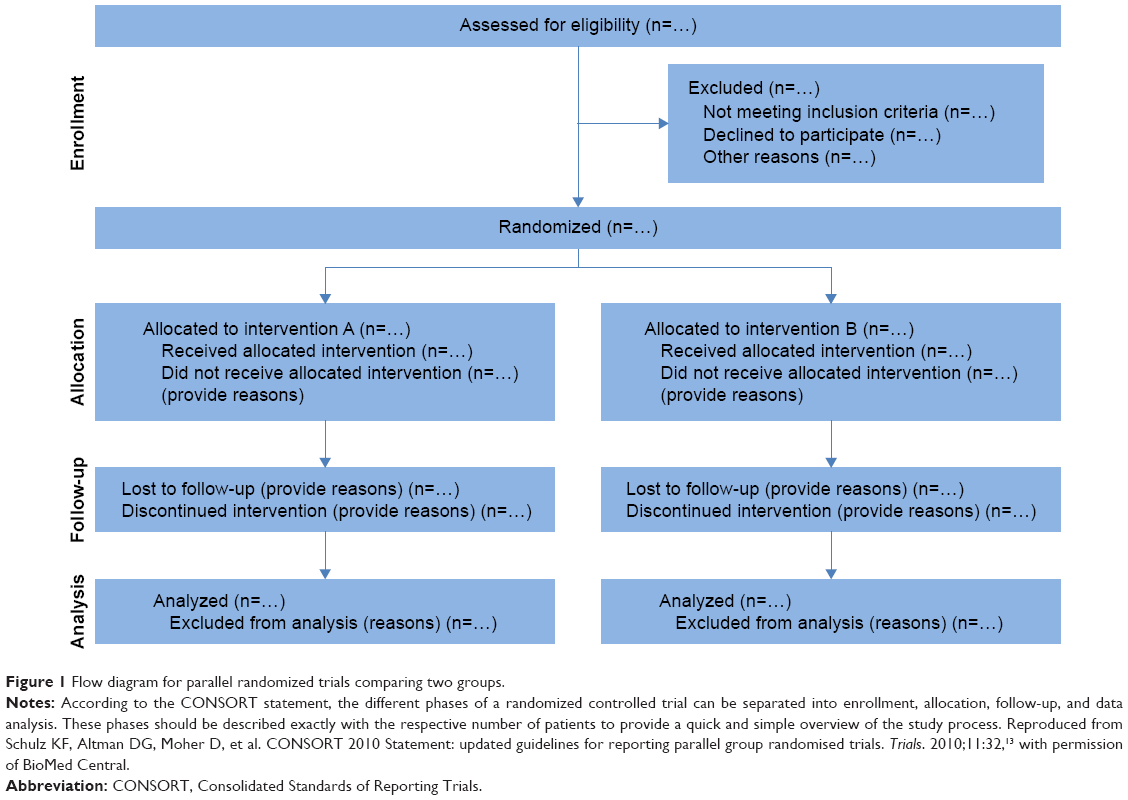 | Figure 1 Flow diagram for parallel randomized trials comparing two groups. |
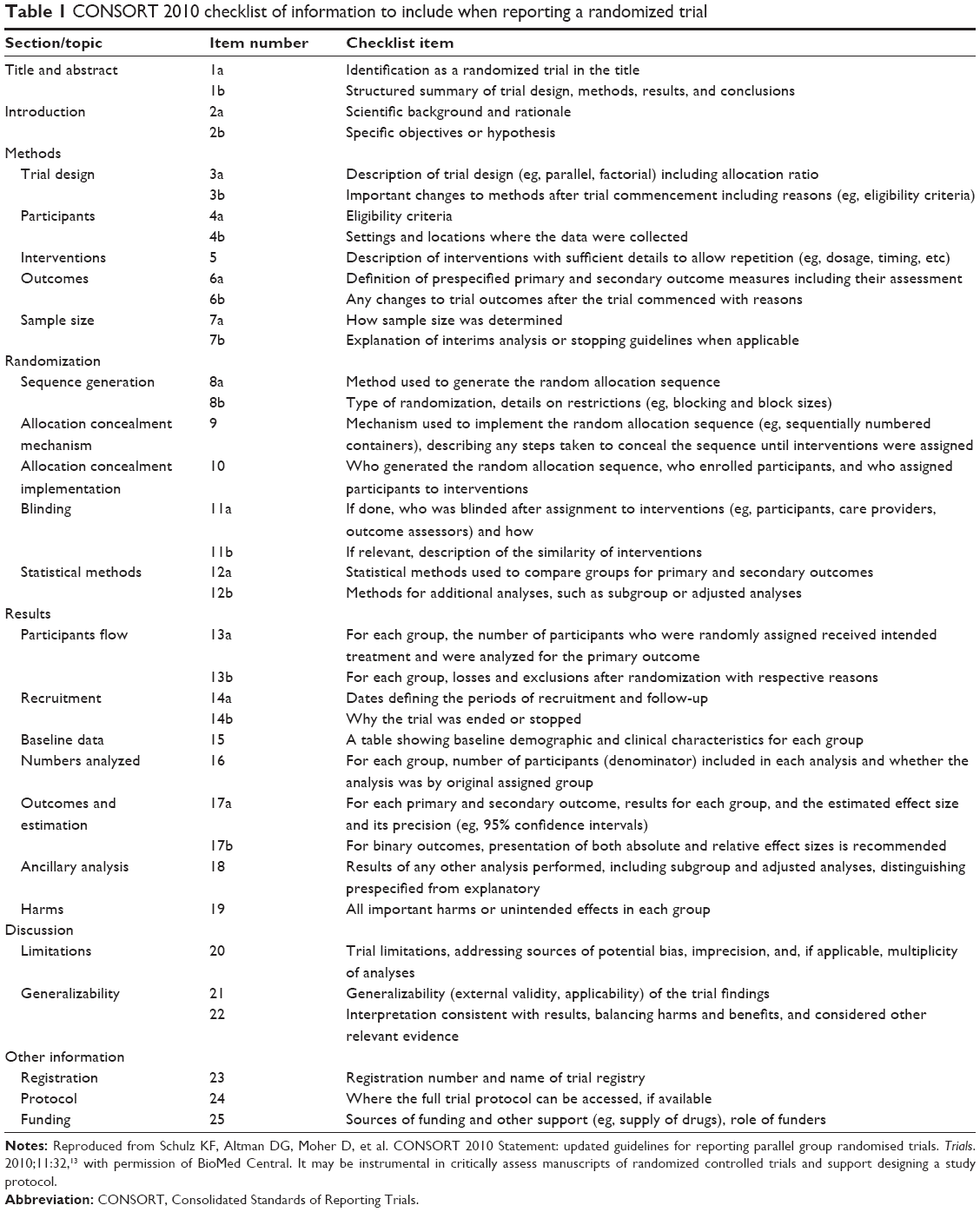 | Table 1 CONSORT 2010 checklist of information to include when reporting a randomized trial |
Additional extensions and modifications have been published for cluster-randomized trials,8,24 noninferiority and equivalence trials,25–27 nonpharmacological treatments,9,28 herbal interventions,9,29 and pragmatic trials.11,30 Nowadays, the majority of medical journals state in their authors instructions that reporting of clinical trial data must be in accordance with the CONSORT statements and that illustration of the sequence of study procedures must comply with the CONSORT flow chart design (Figure 1) to be considered for publication. Thus, both the CONSORT statements and the CONSORT flow chart have become a widely used standard in scientific medical publishing.
To improve transparency of clinical trials, databases such as ClinicalTrials.gov have been founded to track changes between the planned and the published study and to keep researchers updated about ongoing clinical trials. In these databases, studies have to be registered before the enrollment of the first patient. The majority of medical journals ask for the registration in a study database as a prerequisite for publication.11,31 However, there are still numerous clinical trials without complying to these standards published, especially since many of the journals, editors, and reviewers are not reinforcing the strict compliance with these quality assurance measures.
Furthermore, the manuscript should be written in a clear and concise way, there should be a strict separation between results and interpretation, and authors should not overestimate their results, especially in the absence of statistical significance. Potential conflict of interest of each author should be clearly disclaimed, and funding sources should be identified.6,32
Interpretation of RCT data
After assessing internal and external validity by checking adequate randomization, complete reporting of data and interventions, blinding if applicable, and selection of meaningful end points, one should assess the validity of the primary statistical analysis. This means first to assess, if an ITT analysis was performed and how missing data were handled.
If an efficacy analysis is performed after excluding certain subjects, the exclusion criteria should have been defined a priori. It is worth noting that an efficacy analysis should be considered hypothesis generating, and their results should be confirmed in subsequent RCTs. In case of a subgroup analysis, it is necessary to assess whether the subgroups have been defined a priori and whether they are defined by baseline characteristics to consider this analysis valid as confirmatory analysis. If one of these two prerequisites is not fulfilled, the subgroup analysis should be viewed solely as a hypothesis-generating technique warranting further confirmation in a subsequent RCT.
Harhay et al33 reviewed 146 RCTs in the field of intensive care that have been published between 2007 and 2013 in 16 high-impact or critical care journals.8,12 The authors concluded that most RCTs in intensive care have “negative” results and are powered according to unrealistic treatment effects, particularly if mortality was used as primary end point.
Even if RCTs reporting a statistically significant difference, estimates of the magnitude of these effects should be provided to guide the reader if these differences are clinically meaningful. Among the numerous ways how this could be performed, absolute risk reduction (ARR) and relative risk reduction (RRR) as well as the calculation of the number needed to treat (NNT) are commonly used. ARR is calculated by subtracting the risk of one treatment by the risk for the other treatment. For example, in the Acute Respiratory Distress Syndrome Network (ARDSnet) trial comparing lower and traditional tidal volumes in ARDS patients,8,34 the risk of death before discharge was 31% in the low tidal volume group and 39.8% in the group receiving higher tidal volumes. Therefore, the ARR was 8.8% favoring low tidal volume ventilation. The RRR is calculated as the ratio of the relative risk of the treatment group (RR, eg, ratio of both mortality rates) divided by 1 (the relative risk of the control group is by definition 100%). In the example of the ARDSnet study, the RRR can be calculated as (31/39.8) −1=−0.22, representing a RRR of 22% for the low tidal volume group compared to traditional tidal volumes. The RRR frequently leads to overestimation of the treatment effects, so usually the ARR should be reported whenever possible.15,35 The NNT is derived directly from the ARR and is calculated by dividing 1 by the ARR. In the example of the ARDSnet trial, the NNT would be 1/0.088=11.4, which means that 11.4 patients needed to be ventilated with low tidal volumes to experience one patient with an additional response, in this case survival.
Although RRR, ARR, and NNT can provide information about the magnitude of the treatment effect, they are strongly related to the variability of the measured parameter and the sample size. Therefore, confidence intervals should be reported to provide information about the accuracy of the findings.16,36 In the ARDSnet study, the 95% confidence intervals for the difference in mortality between groups were 2.4% and 15.3%.18,34 This means that with a certainty of 95%, the difference in mortality (ARR) between the lower and higher tidal volume groups was between 2.4% and 15.3%.
Principle of equipoise
Depending on the predefined clinical research question and statistical considerations, RCTs are frequently designed to determine superiority, noninferiority, or equivalence of a new (experimental) intervention relative to an established standard-of-care treatment.7,8 Before randomly assigning patients to one or more of the competing study arms, investigators involved in both design and conduction of clinical trials need to be free of any treatment preferences, which means there is genuine uncertainty about the best treatment regimen for the disease of interest.37,38 This so-called principal of equipoise constitutes an ethical prerequisite for conducting an RCT. However, clinical investigators commonly face the moral dilemma where emerging data (eg, arising from preceding Phase II trials) provide a strong signal of efficacy for an experimental treatment, and the existing standard-of-care treatment, although being considered efficacious, is in need of improvement due to only minor impacts on an otherwise unfavorable course of a disease.
For instance, basilar artery occlusion is a devastating disease leading to death in up to 90% of untreated acute ischemic stroke patients, and there is still uncertainty about its preferred treatment regimen.39 Intravenous thrombolysis with tissue plasminogen activator constitutes the only approved therapy for acute ischemic stroke that prevents patients from permanent disability and death when initiated within 4.5 hours from symptom onset.40 Recanalization rates, however, are low, particularly in patients suffering from major occlusions, and majority of patients with basilar artery occlusion do not clinically benefit from intravenous thrombolysis.39,41 On the other hand, endovascular treatment of acute ischemic stroke results in remarkably higher recanalization rates than intravenous thrombolysis, and recently published RCTs have shown strong treatment effects on clinical outcomes in patients treated interventionally for anterior circulation stroke (ie, excluding basilar arty occlusion).42 The current body of scientific knowledge on treatment of basilar artery occlusion (ie, mostly based on meta-analyses of observational studies and registries), however, does not favor one treatment regimen over the other – while intravenous thrombolysis can be initiated in any acute stroke-ready hospital in a timely manner (which is crucial for favorable outcomes), higher recanalization rates achieved with endovascular therapy may be counterbalanced by procedural delays, resulting in longer times to reperfusion.43–45 Consequently, current lack of scientific evidence for acute treatment of basilar artery occlusion justifies null hypothesis testing in this context and meets ethical requirements for clinical equipoise, and meanwhile, a large RCT has been initiated to properly address this question.46,47 Nevertheless, from a clinician investigator’s standpoint and with knowledge of the aforementioned RCT results on endovascular treatment for anterior circulation stroke, personal equipoise may become compromised when withholding endovascular therapy for basilar artery occlusion and will lead to significant moral objections. Once there is no longer clinical or personal equipoise, continuation of and contribution to an RCT should be reconsidered, otherwise serious biases may be introduced (eg, selection bias – less severely affected stroke patients with basilar artery occlusion are more likely cleared for randomization than more severely affected patients).38
Preplanned interims analyses at certain time points or recruited sample sizes during an ongoing RCT aid in maintaining clinical equipoise.48 Trial data are analyzed for benefit, harm, or futility, and decisions on continuation or termination of the trial will be made by an independent data safety monitoring board according to clinical equipoise (eg, large effect size suggests superiority of one treatment over the other and clinical equipoise no longer exists), among others. However, it is worth noting that when repeated significance testing on accumulating data is performed, adjustment of the hypothesis testing procedure is necessary to maintain the overall significance level.
Limitations
Although a systematic review of the literature was performed, this article is influenced by personal opinion and the individual experience of the authors. Owing to the diversity of clinical research, this article offers an overview over commonly used clinical research scenarios but may not cover every aspect of clinical trial design.
Summary of recommendations
In RCTs, the research question should be scientifically relevant and clearly stated. Randomization and stratification techniques should be employed as well as the use of placebo control or blinding whenever possible to reduce the risk of bias. Clinically relevant end points should be defined a priori, and an unbiased analysis and report of the study results should be warranted. Both significant and nonsignificant results should be objectively reported and published. Structured study design and performance as indicated in the CONSORT statement should be employed, as well as the registration of the RCT in a public trial database. Besides careful conduction and interpretation of the study, potential conflict of interests and funding sources should be disclaimed. Finally, the principle of equipoise should be met prior to and during the conduction of an RCT.
Acknowledgments
This work is part of a Master’s thesis (PMS) of the Master’s Program in Clinical Research, Center for Clinical Research and Management Education, Division of Health Care Sciences, Dresden International University, Dresden, Germany. Publication costs were funded by the Open Access Publication Funds of the Technische Universität Dresden.
Disclosure
The authors report no conflicts of interest in this work.
References
Wunsch H, Linde-Zwirble WT, Angus DC. Methods to adjust for bias and confounding in critical care health services research involving observational data. J Crit Care. 2006;21:1–7. | ||
Reith C, Landray M, Devereaux PJ, et al. Randomized clinical trials – removing unnecessary obstacles. N Engl J Med. 2013;369:1061–1065. | ||
Berkman ND, Santaguida PL, Viswanathan M, et al. The Empirical Evidence of Bias in Trials Measuring Treatment Differences. Rockville, MD: Agency for Healthcare Research and Quality (US); 2014. | ||
Collins R, MacMahon S. Reliable assessment of the effects of treatment on mortality and major morbidity, I: clinical trials. Lancet. 2001;357:373–380. | ||
Schulz KF, Grimes DA. Sample size calculations in randomised trials: mandatory and mystical. Lancet. 2005;365:1348–1353. | ||
Lipsky MS, Sharp LK. From idea to market: the drug approval process. J Am Board Fam Pract. 2001;14:362–367. | ||
Zhang Z, Peluso MJ, Gross CP, et al. Adherence reporting in randomized controlled trials. Clin Trials. 2014;11:195–204. | ||
Akobeng AK. Assessing the validity of clinical trials. J Pediatr Gastroenterol Nutr. 2008;47:277–282. | ||
Kabisch M, Ruckes C, Seibert-Grafe M, et al. On the proper use of the crossover design in clinical trials: part 18 of a series on evaluation of scientific publications. Dtsch Arztebl Int. 2012;109:276–281. | ||
Hollis S, Campbell F. What is meant by intention to treat analysis? Survey of published randomised controlled trials. BMJ. 1999;319:670–674. | ||
Whelan DB, Dainty K, Chahal J. Efficient designs: factorial randomized trials. J Bone Joint Surg Am. 2012;94(Suppl 1):34–38. | ||
Moher D, Schulz KF, Altman DG, et al. The CONSORT statement: revised recommendations for improving the quality of reports of parallel-group randomized trials. Lancet. 2001;357:1191–1194. | ||
Schulz KF, Altman DG, Moher D, et al. CONSORT 2010 Statement: updated guidelines for reporting parallel group randomised trials. Trials. 2010;11:32. | ||
Rothwell PM. External validity of randomised controlled trials: “to whom do the results of this trial apply?” Lancet. 2005;365:82–93. | ||
Jüni P, Altman DG, Egger M. Systematic reviews in health care: assessing the quality of controlled clinical trials. BMJ. 2001;323:42–46. | ||
Schulz KF, Grimes DA. Generation of allocation sequences in randomised trials: chance, not choice. Lancet. 2002;359:515–519. | ||
Rothwell PM. Factors that can affect the external validity of randomised controlled trials. PLoS Clin Trials. 2006;1:e9. | ||
Schulz KF, Grimes DA. Allocation concealment in randomised trials: defending against deciphering. Lancet. 2002;359:614–618. | ||
Van Spall HG, Toren A, Kiss A, et al. Eligibility criteria of randomized controlled trials published in high-impact general medical journals: a systematic sampling review. JAMA. 2007;297:1233–1240. | ||
Schulz KF, Grimes DA. Blinding in randomised trials: hiding who got what. Lancet. 2002;359:696–700. | ||
Stephenson JM, Babiker A. Overview of study design in clinical epidemiology. Sex Transm Infect. 2000;76:244–247. | ||
Begg C, Cho M, Eastwood S, et al. Improving the quality of reporting of randomized controlled trials. The CONSORT statement. JAMA. 1996;276:637–639. | ||
Moher D, Hopewell S, Schulz KF, et al. CONSORT 2010 explanation and elaboration: updated guidelines for reporting parallel group randomised trials. BMJ. 2010;340:c869. | ||
Campbell MK, Elbourne DR, Altman DG, et al. CONSORT statement: extension to cluster randomised trials. BMJ. 2004;328:702–708. | ||
Wellek S, Blettner M. Establishing equivalence or non-inferiority in clinical trials: part 20 of a series on evaluation of scientific publications. Dtsch Arztebl Int. 2012;109:674–679. | ||
Schiller P, Burchardi N, Niestroj M, et al. Quality of reporting of clinical non-inferiority and equivalence randomised trials – update and extension. Trials. 2012;13:214. | ||
Piaggio G, Elbourne DR, Altman DG, et al. Reporting of noninferiority and equivalence randomized trials: an extension of the CONSORT statement. JAMA. 2006;295:1152–1160. | ||
Boutron I, Moher D, Altman DG, et al. Extending the CONSORT statement to randomized trials of nonpharmacologic treatment: explanation and elaboration. Ann Intern Med. 2008;148:295–309. | ||
Gagnier JJ, Boon H, Rochon P, et al. Reporting randomized, controlled trials of herbal interventions: an elaborated CONSORT statement. Ann Intern Med. 2006;144:364–367. | ||
Zwarenstein M, Treweek S, Gagnier JJ, et al. Improving the reporting of pragmatic trials: an extension of the CONSORT statement. BMJ. 2008;337:a2390. | ||
De Angelis C, Drazen JM, Frizelle FA, et al. Clinical trial registration: a statement from the International Committee of Medical Journal Editors. N Engl J Med. 2004;351:1250–1251. | ||
Gasparyan AY, Ayvazyan L, Akazhanov NA, et al. Conflicts of interest in biomedical publications: considerations for authors, peer reviewers, and editors. Croat Med J. 2013;54:600–608. | ||
Harhay MO, Wagner J, Ratcliffe SJ, et al. Outcomes and statistical power in adult critical care randomized trials. Am J Respir Crit Care Med. 2014;189:1469–1478. | ||
Network TARDS. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med. 2000;342:1301–1308. | ||
Naylor CD, Chen E, Strauss B. Measured enthusiasm: does the method of reporting trial results alter perceptions of therapeutic effectiveness? Ann Intern Med. 1992;117:916–921. | ||
Gardner MJ, Altman DG. Confidence intervals rather than P values: estimation rather than hypothesis testing. Br Med J (Clin Res Ed). 1986;292:746–750. | ||
Freedman B. Equipoise and the ethics of clinical research. N Engl J Med. 1987;317:141–145. | ||
Fries JF, Krishnan E. Equipoise, design bias, and randomized controlled trials: the elusive ethics of new drug development. Arthritis Care Res. 2004;6:250–255. | ||
Mattle HP, Arnold M, Lindsberg PJ, Schonewille WJ, Schroth G. Basilar artery occlusion. Lancet Neurol. 2011;10:1002–1014. | ||
Emberson J, Lees KR, Lyden P, et al. Effect of treatment delay, age, and stroke severity on the effects of intravenous thrombolysis with alteplase for acute ischaemic stroke: a meta-analysis of individual patient data from randomised trials. Lancet. 2014;384:1929–1935. | ||
Saqqur M, Uchino K, Demchuk AM, et al. Site of arterial occlusion identified by transcranial Doppler predicts the response to intravenous thrombolysis for stroke. Stroke. 2007;38:948–954. | ||
Grotta JC, Hacke W. Stroke neurologist’s perspective on the new endovascular trials. Stroke. 2015;46:1447–1452. | ||
Lindsberg PJ, Mattle HP. Therapy of basilar artery occlusion: a systematic analysis comparing intra-arterial and intravenous thrombolysis. Stroke. 2006;37:922–928. | ||
Kumar G, Shahripour RB, Alexandrov AV. Recanalization of acute basilar artery occlusion improves outcomes: a meta-analysis. J Neurointerv Surg. 2015;7:868–874. | ||
Schonewille WJ, Wijman CA, Michel P, et al; BASICS study group. Treatment and outcomes of acute basilar artery occlusion in the Basilar Artery International Cooperation Study (BASICS): a prospective registry study. Lancet Neurol. 2009;8:724–730. | ||
van der Hoeven EJ, Schonewille WJ, Vos JA, et al; BASICS Study Group. The Basilar Artery International Cooperation Study (BASICS): study protocol for a randomised controlled trial. Trials. 2013;14:200. | ||
Tomsick TA, Khatri P, Jovin T, et al; IMS III Executive Committee. Equipoise among recanalization strategies. Neurology. 2010;74:1069–1076. | ||
Fleming TR, Sharples K, McCall J, Moore A, Rodgers A, Stewart R. Maintaining confidentiality of interim data to enhance trial integrity and credibility. Clin Trials. 2008;5:157–167. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.