")
Back to Journals » Clinical Interventions in Aging » Volume 14
RAB10: an Alzheimer’s disease resilience locus and potential drug target
Authors Tavana JP, Rosene M , Jensen NO , Ridge PG, Kauwe JSK, Karch CM
Received 27 October 2018
Accepted for publication 6 December 2018
Published 28 December 2018 Volume 2019:14 Pages 73—79
DOI https://doi.org/10.2147/CIA.S159148
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Walker
Justina P Tavana,1,* Matthew Rosene,2,* Nick O Jensen,2 Perry G Ridge,1 John SK Kauwe,1,3 Celeste M Karch2,4
1Department of Biology, Brigham Young University, Provo, UT 84602, USA; 2Department of Psychiatry, Washington University in St Louis, St Louis, MO, USA; 3Department of Neuroscience, Brigham Young University, Provo, UT 84602, USA; 4Hope Center for Neurological Disorders, Washington University in St Louis, St Louis, MO, USA
*These authors contributed equally to this work
Abstract: Alzheimer’s disease (AD) is mainly a late-onset neurodegenerative disorder. Substantial efforts have been made to solve the complex genetic architecture of AD as a means to identify therapeutic targets. Unfortunately, to date, no disease-altering therapeutics have been developed. As therapeutics are likely to be most effective in the early stages of disease (ie, before the onset of symptoms), a recent focus of AD research has been the identification of protective factors that prevent disease. One example is the discovery of a rare variant in the 3'-UTR of RAB10 that is protective for AD. Here, we review the possible genetic, molecular, and functional role of RAB10 in AD and potential therapeutic approaches to target RAB10.
Keywords: Alzheimer’s disease, RAB10, retromer, APP, resilience, GTPase
Alzheimer’s disease: overview
Alzheimer’s disease (AD) is mainly a late-onset neurodegenerative disease defined clinically by progressive memory decline.1 AD currently affects nearly 50 million people worldwide, and this number is expected to rapidly increase as the population ages (http://www.alz.co.uk/). Neuropathologically, AD is characterized by neuronal loss and the accumulation of amyloid plaques and neurofibrillary tangles in the brain, much of which occurs many years before the onset of clinical symptoms.2 While clinical diagnosis of AD is challenging because it is dependent primarily on observations of family members and caregivers, changes in protein analytes in plasma and cerebrospinal fluid (CSF) and brain imaging, although not widely used in clinical practice, may facilitate AD diagnosis.2 AD can broadly be defined either as early-onset, occurring prior to age 65, or the much more common late-onset, occurring after age 65 (>96.5% of total cases).
Genetic architecture of AD
The genetics of early-onset AD (EOAD) are relatively well understood. Autosomal dominant mutations occurring in APP, PSEN1, or PSEN2 give rise to a subset of EOAD cases. More than 200 pathogenic mutations have been reported worldwide (http://www.molgen.ua.ac.be/admutations/).3 Clinically, EOAD mutation carriers exhibit disease onset prior to 65 years of age.4 Neuropathologically, EOAD and late-onset AD (LOAD) are indistinguishable.5 Unfortunately, the genetics of LOAD is more complex.
Among individuals with LOAD, there is strong evidence for genetic heritability of disease.6–10 The single largest genetic risk factor for LOAD is APOE.11 APOE plays critical roles in cholesterol transport, neuroplasticity, and inflammation.12 APOE regulates Aβ metabolism directly through binding and clearance of Aβ13 and indirectly via the LRP1 receptor.14 Moreover, APOE influences tau-mediated neurodegeneration.15 Two different APOE alleles, ε4 and ε2, affect risk for AD. Risk for developing AD is 3-fold higher in individuals carrying one copy of the ε4 allele and 12-fold higher in individuals carrying 2 copies of APOE ε4.16 Conversely, APOE ε2 confers resilience to AD, while the ε3 allele is considered neutral risk.16
Genetic variants that influence risk for AD can be broadly classified into two groups: common and rare. The majority of common variants (minor allele frequency [MAF] >5%) were identified using genome-wide association studies (GWAS).17 With the exception of variants in APOE, all other common variants have small effect sizes on AD risk (ORs =1.08–1.22, 0.73–0.94).17 While GWAS variants provide insights into disease processes, none of the common risk variants for AD that have been identified by GWAS have clear functional effects and efforts to identify functional variants in the regions of GWAS variants have largely been unsuccessful.
The advent of next-generation sequencing has enabled the sequencing of whole exomes and genomes, resulting in progressively larger AD datasets and providing insights into the contribution of low-frequency (MAF 1%–5%) and rare variants (MAF <1%) to the genetic architecture of AD. Using a variety of study designs, multiple rare variants have been identified that affect risk for AD.18–22 Rare variants typically have much larger effect sizes on disease risk than common variants identified by GWAS. Rare variants are also more likely to occur in coding or regulatory regions and, thus, are more likely to be functional. Thus, rare variants represent effective therapeutic targets.
Finally, alternative study designs can provide additional insights into the genetics of AD. For example, the use of quantitative endophenotypes (eg, CSF levels of protein analytes such as Aβ and tau;23–25 mitochondrial copy number;26 metabolic efficiency;27,28 measurements of progressive brain atrophy;29 among others) and the analysis of mitochondrial genomic variation30 have expanded our understanding of the genetic architecture underlying disease onset and disease course.
Using the genetics of AD to identify therapeutic targets
Genes and pathways that regulate AD pathogenesis may represent viable targets for treating disease. For example, therapies targeting the removal of Aβ or inhibition of Aβ production are currently in clinical trials. These strategies are based on our understanding of the mechanisms by which rare mutations in APP, PSEN1, and PSEN2 drive AD pathogenesis in EOAD and focus on inhibition of the deposition of Aβ in the brain. Similarly, targeting APOE function has been investigated for its therapeutic capacity.31–33 AD risk genes implicated in immune function, endocytosis, and lipid biology represent novel avenues of therapeutic development. Unfortunately, to date, more than 90% of all clinical trials have failed. Given the expected increase in disease incidence, developing effective therapeutics is imperative. For the remainder of this review, we discuss the potential role of RAB10 as a therapeutic target.
Protective factors in AD: RAB10
Common and rare variants have been identified that reduce the risk for AD.3 Those with the strongest protective effect include APOE ε2 and APP-A673T.34,35 Thus, genes and pathways involved in increasing AD risk may also confer resilience to disease. To identify additional genetic variants that confer resilience to AD, there is a need to develop novel study designs that focus on those individuals carrying protective factors for AD. In one such case, family pedigrees were identified with a statistical excess of AD mortality (ie, families with a higher number of AD deaths than expected relative to similarly sized pedigrees; termed high-risk pedigrees). Within these high-risk pedigrees, elderly individuals with genetic risk for AD (ie, APOE ε4 carriers) but who had escaped AD were evaluated using whole-genome sequencing. Ridge et al identified a rare variant (rs142787485) in the 3′-UTR of RAB10 that confers resilience to AD.10 In a cell model of APP metabolism, silencing of Rab10 expression leads to a significant decrease in Aβ42 and the Aβ42/40 ratio, consistent with prior reports.10,36 Interestingly, independent of the RAB10 SNP, individuals with a neuropathologic diagnosis of AD exhibited significantly higher levels of RAB10 compared to neuropathology-free controls.10 Thus, genetic and molecular evidences support a role for RAB10 in AD pathogenesis.
RAB10 in AD pathology
RAB10 is expressed in all cell types in the brain.37 RAB10 is activated by phosphorylation at Thr73, which is mediated by LRRK2, a protein kinase associated with Parkinson’s disease.38,39 This phosphorylation event may represent a pathologic feature in the brains of AD patients.38 pRAB10-Thr73 was observed to colocalize with neurofibrillary tangles in AD brains as well as with granulovascular degeneration, neuropil threads, and dystrophic neurons.38 Double immunofluorescence staining of these structures further revealed positive staining for phosphorylated tau. However, this colocalization of pRAB10-Thr73 and phosphorylated tau was not complete, with some instances showing neuropil threads or dystrophic neurons containing only phosphorylated tau or only pRAB10-Thr73. Interestingly, dystrophic neurons near amyloid plaques stained positive for pRAB10-Thr73, while the amyloid plaques themselves were negative for pRAB10.38 Total RAB10 staining, however, did not differ between AD and control brains. Thus, while pRAB10-Thr73 is associated with AD pathology, it remains unclear whether this represents excess activation or aberrant function of RAB10.
RAB10: function in development
RAB10 is a member of the RAB family of small GTPases, which are key regulators of vesicular trafficking. RAB proteins are cyclically controlled, requiring a GDP–GTP exchange that is facilitated by RAB guanine nucleotide exchange factors.40 However, unlike most of the other RAB proteins, which have relatively specific roles within intracellular transport, RAB10 is relatively unique because it carries out a wide variety of functions and has multiple subcellular localizations.41
Rab10 plays essential functional roles in development: mice are embryonic lethal when both alleles are deleted.42 In addition, RAB10 plays a role in maintenance and regulation of endoplasmic reticulum (ER) morphology.43–45 When RAB10 is mutated or constitutively inactive (eg, GDP-locked), the ER contains more cisternae, suggesting that RAB10 plays a critical role in maintaining and/or generating tubule extension and fusion in the ER.43 RAB10 function has also been linked to neuronal morphology and polarization, playing a critical role in axonal development and dendrite arborization.46,47 During neuronal development, RAB10 associates with plasmalemmal precursor vesicles, which are linked to kinesin 1 via c-Jun N-terminal protein kinase-interacting protein 1.48 Together this complex mediates anterograde transport of RAB10-positive vesicles to axonal tips, promoting axonal growth.48 Thus, RAB10 function is critical for proper neuronal function.
A role for RAB10 and APP in the retromer pathway
RAB10 has been linked to many roles involved in intracellular transport (Figure 1), including but not limited to a role in endocytic recycling. In Caenorhabditis elegans, RAB10 is required for α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-type glutamate receptor recycling in postsynaptic membranes.49 RAB10 is likely functional upstream of RME-1/EHD, a protein involved in recycling endosome tubulization and endosome function.50–52 RAB10 is expressed at the interface of early endosomes and recycling endosomes. These structures are morphologically defective in rab-10 mutant C. elegans models.50,51 RAB10 is required for endosomal recruitment of CNT-1.53 This likely contributes to biogenesis and/or maintenance of recycling endosome compartments. RAB10 could also play a role in promoting the maturation of early endosomes to recycling endosome. RAB10 is also involved in protein degradation. RAB10 and RAB3A are essential for lysosomal exocytosis and plasma membrane repair.54,55 In addition, RAB10 associates with lipid droplets.56 Complexes of RAB10–EHBP1–EHD2 promote autophagic engulfment and degradation of lipid droplets.57 Thus, these functional roles for RAB10 in recycling and degradation make it an interesting candidate protein in AD pathogenesis.
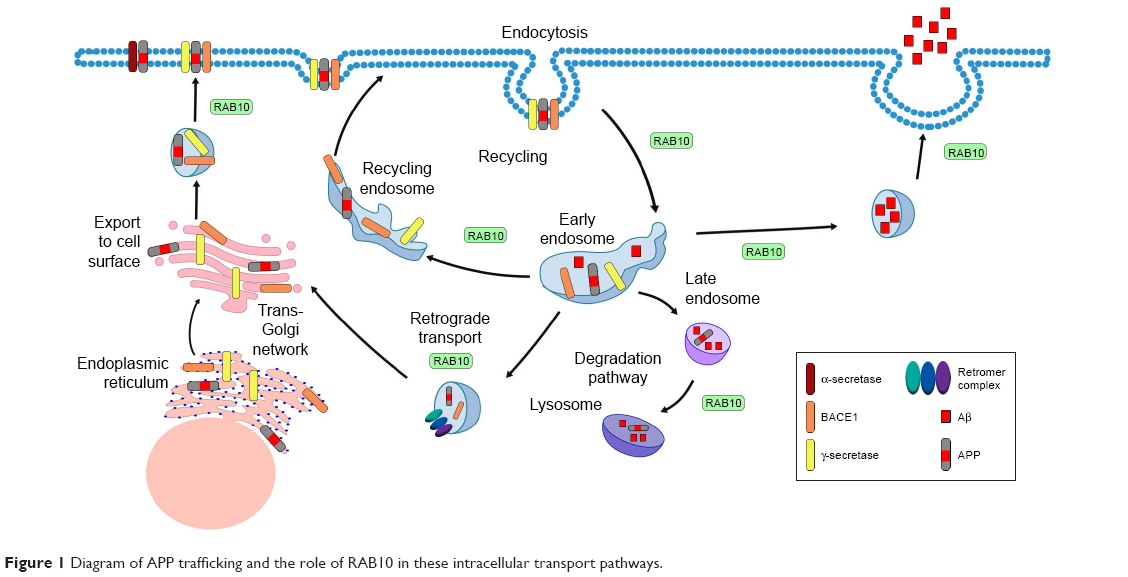 | Figure 1 Diagram of APP trafficking and the role of RAB10 in these intracellular transport pathways. |
RAB10 is involved in transport of proteins from the early endosome to the trans-Golgi network (TGN), where it may also function in the retromer pathway.58 The retromer complex consists of Vps26, Vps35, Vps29, SNX1, SNX2, SNX5, and SNX6.59 The retromer complex regulates transport of protein cargo from endosomes to the TGN by the retrograde pathway or to the cell surface through the recycling pathway.60
To form the amyloid plaques that define a primary component of AD pathology, the transmembrane APP must first be processed into Aβ peptides. APP is thought to be retrieved from the cell surface and trafficked to the early endosomes where it is initially cleaved by β-site APP-cleaving enzyme 1 (BACE1) to generate a C-terminal fragment (β-CTF).61,62 The β-CTF fragments are then trafficked via retromer-dependent transport to the TGN, which facilitates further cleavage by γ-secretase to produce Aβ40.63 The retromer complex and its receptors can transport APP and BACE1 from endosomes to the TGN, ultimately regulating production of Aβ.60 The retromer complex sorts APP and BACE, either through direct or indirect binding to the transmembrane adaptor protein SORLA.64 Interestingly, common variants in the gene that encodes SORLA have been associated with AD risk.65 Thus, genes that are involved in retromer function may influence AD pathogenesis and represent promising therapeutic targets.
Retromer complex dysfunction promotes APP accumulation in neurons and Aβ production. In addition, the retromer also appears to have a differential effect on Aβ isoforms, with evidence that retromer dysfunction alters Aβ40 secretion, which results in increasing the Aβ42/40 ratio.66 In mouse models, reduction of a single component within the retromer complex, Vps26, is sufficient to increase soluble Aβ and APP.67 Stabilization of the retromer in human-induced pluripotent stem cell-derived neurons through the use of the pharmacologic chaperone R33 has been shown to significantly reduce the phosphorylation of Tau in a manner that correlated with, but ultimately independent of, Aβ production.68 An RNAi screen of RABs that modify APP processing revealed that the silencing of RAB10 reduces Aβ without altering sAPPβ levels.36 The LRRK2-RAB10 signaling pathway also causes an overproduction of Aβ.38
Therapeutic implications for RAB10
Novel genes and pathways that contribute to the pathogenesis of AD provide insights into strategies to prevent or treat the disease. The amyloid hypothesis, which proposes that the accumulation of Aβ triggers the cascade of events that lead to AD, has been the focus of AD therapeutic development for decades. The development of anti-Aβ therapeutics remains the primary approach to treating AD based on our current understanding of the earliest features of this disease.69 In this review, we emphasize the importance of RAB10 in the processing of APP and the production of Aβ. The function of RAB10 in intracellular trafficking and evidence that a reduction of Rab10 results in a reduction of the Aβ42/40 ratio makes inhibition of RAB10 a potential therapeutic avenue.
There are several existing strategies for therapeutic inhibition of a gene product. Potential technologies for targeting of RAB10 are small molecules, antibodies, and antisense oligodeoxynucleotide technologies. For each of these approaches the challenge of delivery across the blood–brain barrier (BBB) is a significant one.70 A molecule with high lipid solubility and molecular mass <400 Da generally can diffuse across the BBB. These criteria limit the classes of small molecules that can be considered as potential therapeutic molecules for AD. Antibodies do not cross the BBB at therapeutic concentrations. Several approaches have been developed to deliver antibodies to the brain. Bispecific antibodies have been developed that leverage the transferrin receptor to successfully increase BBB penetration.71,72 In addition, antibody analogs that can cross the BBB are in development (Patent #EP3309171). Antisense oligonucleotides do not readily cross the BBB.73 However, at present, intrathecal bolus injection is an effective method for ensuring distribution in neurons and glial cells in the brain.74
Approaches to the development and delivery of small-molecule therapeutics are mature and the path and pitfalls are well established. Small molecules are effective enzyme inhibitors, receptor ligands, or allosteric modulators. The nature of small-molecule activity can make it difficult to find the balance between efficacy and side effects when treating humans.70 Despite these challenges, small-molecule drug development has seen many successes and >90% of existing therapeutics use small molecules. Small GTPases similar to RAB10 have been successfully targeted using small molecules in the past. One important use is the development of molecules to block oncogenic properties of the RAS protein to treat cancer.75 Recent discoveries about the structure of small GTPases and advances in targeting strategies have further increased efforts to use these important proteins as therapeutic targets.76 Targeting approaches, including interference with nucleotide binding, inactivation by irreversible covalent modification, inhibition of GTPase–GEF interactions, inhibition of GTPase–effector interactions, and stabilization of GTPase–protein complexes have been successful and are reviewed by Cromm et al.76 While challenges regarding specificity and crossing the BBB remain, these advances create some optimism about the use of small molecules to inhibit RAB10 and impact AD pathology.
Antibodies function in extra- and intracellular immunity to recognize foreign or abnormal agents and target them for destruction with very high specificity. The production of monoclonal antibodies (mAbs) has resulted in their use for the treatment of a wide range of human diseases.71 There are currently >60 approved mAbs for human therapy and >50 in late-stage clinical trials.77
RNase H-dependent antisense oligonucleotides technology is a popular method for knockdown in cell culture. It offers specific and efficient knockdown and is a powerful tool for functional studies of genes with unknown function.78 Antisense technology has challenges such as site specificity, toxicity at high concentrations, and the difficulty of targeting specific cell types. However, it is a mature technology and antisense oligonucleotides offer a means to manipulate specific steps in mRNA processing, for example, splicing.69 As discussed previously, in vitro reduction of Rab10/RAB10 expression using shRNA and RNAi has been successful.36 Expansion of antisense oligodeoxynucleotide from in vitro to in vivo represents a promising therapeutic avenue given recent successes with spinal muscular atrophy and Huntington’s disease.79,80
Conclusion
Here we have outlined the potential role of RAB10 in AD pathology and the case for RAB10 inhibition as a therapeutic intervention for AD. While small GTPases like RAB10 present significant challenges for therapeutic development, recent work in small molecules, antibodies, and antisense oligonucleotides suggests that success can be achieved.
Acknowledgment
This work was supported by grants from the National Institutes of Health (K01 AG046374, U01 AG052411, R01 AG062359, and RF1 AG054052).
Disclosure
The authors report no conflicts of interest in this work.
References
Holtzman DM, Morris JC, Goate AM. Alzheimer’s disease: the challenge of the second century. Sci Transl Med. 2011;377(77):77sr1. | ||
Jack CR, Holtzman DM. Biomarker modeling of Alzheimer’s disease. Neuron. 2013;80(6):1347–1358. | ||
Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. 2015;77(1):43–51. | ||
Ryman DC, Acosta-Baena N, Aisen PS, et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology. 2014;83(3):253–260. | ||
Cairns NJ, Perrin RJ, Franklin EE, et al. Neuropathologic assessment of participants in two multi-center longitudinal observational studies: the Alzheimer Disease Neuroimaging Initiative (ADNI) and the Dominantly Inherited Alzheimer Network (DIAN). Neuropathology. 2015;35(4):390–400. | ||
Ridge PG, Hoyt KB, Boehme K, et al. Assessment of the genetic variance of late-onset Alzheimer’s disease. Neurobiol Aging. 2016;41:200.e13–200.e20. | ||
Ridge PG, Mukherjee S, Crane PK, Kauwe JS. Alzheimer’s disease genetics C. Alzheimer’s disease: analyzing the missing heritability. PLoS One. 2013;8(11):e79771. | ||
Kauwe JS, Ridge PG, Foster NL, Cannon-Albright LA. Strong evidence for a genetic contribution to late-onset Alzheimer’s disease mortality: a population-based study. PLoS One. 2013;8(10):e77087. | ||
Cannon-Albright LA, Dintelman S, Maness T, et al. Population genealogy resource shows evidence of familial clustering for Alzheimer disease. Neurol Genet. 2018;4(4):e249. | ||
Ridge PG, Karch CM, Hsu S, et al. Linkage, whole genome sequence, and biological data implicate variants in RAB10 in Alzheimer’s disease resilience. Genome Med. 2017;9(1):100. | ||
Strittmatter WJ, Weisgraber KH, Huang DY, et al. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(17):8098–8102. | ||
Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(3):a006312. | ||
Castellano JM, Kim J, Stewart FR, et al. Human apoE isoforms differentially regulate brain amyloid-peptide clearance. Sci Transl Med. 2011;3(89):89ra57. | ||
Verghese PB, Castellano JM, Garai K, et al. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc Natl Acad Sci U S A. 2013;110(19):E1807–E1816. | ||
Shi Y, Yamada K, Liddelow SA, et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549(7673):523–527. | ||
Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923. | ||
Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45(12):1452–1458. | ||
Cruchaga C, Karch CM, Jin SC, et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature. 2014;505(7484):550–554. | ||
Wetzel-Smith MK, Hunkapiller J, Bhangale TR, et al. A rare mutation in UNC5C predisposes to late-onset Alzheimer’s disease and increases neuronal cell death. Nat Med. 2014;20(12):1452–1457. | ||
Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368(2):117–127. | ||
Jin SC, Benitez BA, Karch CM, et al. Coding variants in TREM2 increase risk for Alzheimer’s disease. Hum Mol Genet. 2014;23(21):5838–5846. | ||
Jin SC, Carrasquillo MM, Benitez BA, et al. TREM2 is associated with increased risk for Alzheimer’s disease in African Americans. Mol Neurodegener. 2015;10(1):19. | ||
Maxwell TJ, Corcoran C, del-Aguila JL, et al. Genome-wide association study for variants that modulate relationships between cerebrospinal fluid amyloid-beta 42, tau, and p-tau levels. Alzheimer’s Res Ther. 2018;10(1):86. | ||
Kauwe JS, Bailey MH, Ridge PG, et al. Genome-wide association study of CSF levels of 59 Alzheimer’s disease candidate proteins: significant associations with proteins involved in amyloid processing and inflammation. PLoS Genet. 2014;10(10):e1004758. | ||
Deming Y, Li Z, Kapoor M, et al. Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathol. 2017;133(5):839–856. | ||
Ridge PG, Maxwell TJ, Foutz SJ, et al. Mitochondrial genomic variation associated with higher mitochondrial copy number: the Cache County Study on Memory Health and Aging. BMC Bioinformatics. 2014;15(Suppl 7):S6. | ||
Kring SI, Brummett BH, Barefoot J, et al. Impact of psychological stress on the associations between apolipoprotein E variants and metabolic traits: findings in an American sample of caregivers and controls. Psychosom Med. 2010;72(5):427–433. | ||
Ostergaard SD, Mukherjee S, Sharp SJ, et al. Associations between potentially modifiable risk factors and alzheimer disease: a mendelian randomization study. PLoS Med. 2015;12(6):e1001841; discussion e1001841. | ||
Lorenzi M, Altmann A, Gutman B, et al. Susceptibility of brain atrophy to TRIB3 in Alzheimer’s disease, evidence from functional prioritization in imaging genetics. Proc Natl Acad Sci U S A. 2018;115(12):3162–3167. | ||
Ridge PG, Kauwe JSK, Mitochondria KJSK. Mitochondria and Alzheimer’s disease: the role of mitochondrial genetic variation. Curr Genet Med Rep. 2018;6(1):1–10. | ||
Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63(3):287–303. | ||
Kim J, Eltorai AE, Jiang H, et al. Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Aβ amyloidosis. J Exp Med. 2012;209(12):2149–2156. | ||
Huynh TV, Davis AA, Ulrich JD, Holtzman DM. Apolipoprotein E and Alzheimer’s disease: the influence of apolipoprotein E on amyloid-β and other amyloidogenic proteins. J Lipid Res. 2017;58(5):824–836. | ||
Corder EH, Saunders AM, Risch NJ, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7(2):180–184. | ||
Jonsson T, Atwal JK, Steinberg S, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488(7409):96–99. | ||
Udayar V, Buggia-Prévot V, Guerreiro RL, et al. A paired RNAi and RabGAP overexpression screen identifies Rab11 as a regulator of β-amyloid production. Cell Rep. 2013;5(6):1536–1551. | ||
Zhang Y, Sloan SA, Clarke LE, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron. 2016;89(1):37–53. | ||
Yan T, Wang L, Gao J, et al. Rab10 phosphorylation is a prominent pathological feature in Alzheimer’s disease. J Alzheimers Dis. 2018;63(1):157–165. | ||
Eguchi T, Kuwahara T, Sakurai M, et al. LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc Natl Acad Sci U S A. 2018;115(39):E9115–E9124. | ||
Barr F, Lambright DG, Gefs R. Rab GEFs and GAPs. Curr Opin Cell Biol. 2010;22(4):461–470. | ||
Chua CEL, Tang BL. Rab 10-a traffic controller in multiple cellular pathways and locations. J Cell Physiol. 2018;233(9):6483–6494. | ||
Lv P, Sheng Y, Zhao Z, et al. Targeted disruption of Rab10 causes early embryonic lethality. Protein Cell. 2015;6(6):463–467. | ||
English AR, Voeltz GK. Rab10 GTPase regulates ER dynamics and morphology. Nat Cell Biol. 2013;15(2):169–178. | ||
Chang J, Blackstone C. Rab10 joins the ER social network. Nat Cell Biol. 2013;15(2):135–136. | ||
Schuldt A. Membrane dynamics: ER trailblazing by RAB10. Nat Rev Mol Cell Biol. 2013;14(2):63. | ||
Liu Y, Xh X, Chen Q, et al. Myosin Vb controls biogenesis of post-Golgi Rab10 carriers during axon development. Nat Commun. 2005;2013:4. | ||
Zou W, Yadav S, Devault L, Jan YN, Sherwood DR. RAB-10-dependent membrane transport is required for dendrite arborization. PLoS Genet. 2015;11(9):e1005484. | ||
Deng CY, Lei WL, Xu XH, Ju XC, Liu Y, Luo ZG. JIP1 mediates anterograde transport of Rab10 cargos during neuronal polarization. J Neurosci. 2014;34(5):1710–1723. | ||
Glodowski DR, Chen CCH, Schaefer H, Grant BD, Rongo C. RAB-10 regulates glutamate receptor recycling in a cholesterol-dependent endocytosis pathway. Mol Biol Cell. 2007;18(11):4387–4396. | ||
Chen CCH, Schweinsberg PJ, Vashist S, Mareiniss DP, Lambie EJ, Grant BD. RAB-10 is required for endocytic recycling in the Caenorhabditis elegans intestine. Mol Biol Cell. 2006;17(3):1286–1297. | ||
Shi A, Chen CC, Banerjee R, et al. EHBP-1 functions with RAB-10 during endocytic recycling in Caenorhabditis elegans. Mol Biol Cell. 2010;21(16):2930–2943. | ||
Pant S, Sharma M, Patel K, Caplan S, Carr CM, Grant BD. AMPH-1/Amphiphysin/Bin1 functions with RME-1/Ehd1 in endocytic recycling. Nat Cell Biol. 2009;11(12):1399–1410. | ||
Shi A, Liu O, Koenig S, et al. RAB-10-GTPase-mediated regulation of endosomal phosphatidylinositol-4,5-bisphosphate. Proc Natl Acad Sci U S A. 2012;109(35):E2306–E2315. | ||
Encarnação M, Espada L, Escrevente C, et al. A Rab3a-dependent complex essential for lysosome positioning and plasma membrane repair. J Cell Biol. 2016;213(6):631–640. | ||
Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell. 2001;106(2):157–169. | ||
Sato S, Fukasawa M, Yamakawa Y, et al. Proteomic profiling of lipid droplet proteins in hepatoma cell lines expressing hepatitis C virus core protein. J Biochem. 2006;139(5):921–930. | ||
Li Z, Schulze RJ, Weller SG, et al. A novel Rab10-EHBP1-EHD2 complex essential for the autophagic engulfment of lipid droplets. Sci Adv. 2016;2(12):e1601470. | ||
Babbey CM, Ahktar N, Wang E, Chen CCH, Grant BD, Dunn KW. Rab10 regulates membrane transport through early endosomes of polarized Madin-Darby canine kidney cells. Mol Biol Cell. 2006;17(7):3156–3175. | ||
Bonifacino JS, Hurley JH. Retromer. Curr Opin Cell Biol. 2008;20(4):427–436. | ||
Zhang QY, Tan MS, Yu JT, Tan L. The role of retromer in Alzheimer’s disease. Mol Neurobiol. 2016;53(6):4201–4209. | ||
Huse JT, Pijak DS, Leslie GJ, Lee VM, Doms RW. Maturation and endosomal targeting of beta-site amyloid precursor protein-cleaving enzyme. The Alzheimer’s disease beta-secretase. J Biol Chem. 2000;275(43):33729–33737. | ||
Kinoshita A, Fukumoto H, Shah T, Whelan CM, Irizarry MC, Hyman BT. Demonstration by FRET of BACE interaction with the amyloid precursor protein at the cell surface and in early endosomes. J Cell Sci. 2003;116(16):3339–3346. | ||
Choy RWY, Cheng Z, Schekman R. Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid β (Aβ) production in the trans-Golgi network. Proc Natl Acad Sci U S A. 2012;109(30):E2077–E2082. | ||
Gandy S, Odce S, Edce S, Suzuki T, Ehrlich M, Small S. Amyloid precursor protein sorting and processing: transmitters, hormones, and protein phosphorylation mechanisms. Intracellular Traffic and Neurodegenerative Disorders. Peter H St George-Hyslop and William C. Mobley, Springer Berlin Heidelberg: 2008:1–9. | ||
Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45(12):1452–1458. | ||
Sullivan CP, Jay AG, Stack EC, et al. Retromer disruption promotes amyloidogenic APP processing. Neurobiol Dis. 2011;43(2):338–345. | ||
Muhammad A, Flores I, Zhang H, et al. Retromer deficiency observed in Alzheimer’s disease causes hippocampal dysfunction, neurodegeneration, and a accumulation. Proc Natl Acad Sci U S A. 2008;105(20):7327–7332. | ||
Young JE, Fong LK, Frankowski H, Petsko GA, Small SA, Goldstein LSB. Stabilizing the retromer complex in a human stem cell model of Alzheimer’s disease reduces TAU phosphorylation independently of amyloid precursor protein. Stem Cell Reports. 2018;10(3):1046–1058. | ||
Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. | ||
Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2(1):3–14. | ||
Atwal JK, Chen Y, Chiu C, et al. A therapeutic antibody targeting BACE1 inhibits amyloid-β production in vivo. Sci Transl Med. 2011;3(84):84ra43. | ||
Yj Y, Zhang Y, Kenrick M, et al. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci Transl Med. 2011;384(84):ra44. | ||
Phillips JA, Craig SJ, Bayley D, Christian RA, Geary R, Nicklin PL. Pharmacokinetics, metabolism, and elimination of a 20-mer phosphorothioate oligodeoxynucleotide (CGP 69846A) after intravenous and subcutaneous administration. Biochem Pharmacol. 1997;54(6):657–668. | ||
Rigo F, Chun SJ, Norris DA, et al. Pharmacology of a central nervous system delivered 2′-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J Pharmacol Exp Ther. 2014;350(1):46–55. | ||
Gibbs JB, Oliff A, Kohl NE. Farnesyltransferase inhibitors: Ras research yields a potential cancer therapeutic. Cell. 1994;77(2):175–178. | ||
Cromm PM, Spiegel J, Grossmann TN, Waldmann H. Direct modulation of small GTPase activity and function. Angew Chem Int Ed Engl. 2015;54(46):13516–13537. | ||
Reichert JM. Antibodies to watch in 2017. MAbs. 2017;9(2):167–181. | ||
Devos SL, Miller TM. Antisense oligonucleotides: treating neurodegeneration at the level of RNA. Neurotherapeutics. 2013;10(3):486–497. | ||
Southwell AL, Kordasiewicz HB, Langbehn D, et al. Huntingtin suppression restores cognitive function in a mouse model of Huntington’s disease. Sci Transl Med. 2018;10(461):eaar3959. | ||
Passini MA, Bu J, Richards AM, et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. 2011;3(72):72ra18. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.