Back to Journals » Drug Design, Development and Therapy » Volume 8
(R)-(+)-α-Lipoic acid protected NG108-15 cells against H2O2-induced cell death through PI3K-Akt/GSK-3β pathway and suppression of NF-κβ-cytokines
Authors Kamarudin MNA, Mohd Raflee NA, Syed Hussein SS, Lo JY, Supriady H, Kadir H
Received 19 May 2014
Accepted for publication 9 July 2014
Published 8 October 2014 Volume 2014:8 Pages 1765—1780
DOI https://doi.org/10.2147/DDDT.S67980
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Muhamad Noor Alfarizal Kamarudin, Nur Afiqah Mohd Raflee, Sharifah Salwa Syed Hussein, Jia Ye Lo, Hadi Supriady, Habsah Abdul Kadir
Institute of Biological Sciences, Faculty of Science, University of Malaya, Kuala Lumpur, Malaysia
Abstract: Alpha-lipoic acid, a potent antioxidant with multifarious pharmacological benefits has been reported to be neuroprotective in several neuronal models and used to treat neurological disorders such as Alzheimer’s disease. Nonetheless, conclusive mechanisms of alpha-lipoic acid for its protective effects particularly in NG108-15 cells have never been investigated. In this study, the intricate neuroprotective molecular mechanisms by (R)-(+)-alpha-lipoic acid (R-LA) against H2O2-induced cell death in an in vitro model of neurodegeneration were elucidated. Pretreatment with R-LA (2 hours) significantly increased NG108-15 cell viability as compared to H2O2-treated cells and mitigated the induction of apoptosis as evidenced by Hoechst 33342/propidium iodide staining. R-LA (12.5–50 µM) aggrandized the reduced glutathione over glutathione disulfide ratio followed by a reduction in the intracellular reactive oxygen species level and an increase in mitochondrial membrane potential following H2O2 exposure. Moreover, pretreatment with R-LA stimulated the activation of PI3K-Akt through mTORC1 and mTORC2 components (mTOR, rictor and raptor) and production of antiinflammatory cytokine, IL-10 which led to the inactivation of glycogen synthase kinase-3β (GSK-3β) and reduction of both Bax/Bcl2 and Bax/Bcl-xL ratios, accompanied by inhibition of the cleaved caspase-3. Additionally, this observation was preceded by the suppression of NF-κβ p65 translocation and production of proinflammatory cytokines (IL-6 and TNF-α). The current findings accentuate new mechanistic insight of R-LA against apoptogenic and brain inflammatory factors in a neuronal model. These results further advocate the therapeutic potential of R-LA for the treatment of neurodegenerative diseases.
Keywords: alpha-lipoic acid (LA), cytokines, GSK-3β, neuroegenerative diseases, NF-κβ, PI3K-Akt
Introduction
The primary basis underlying neurodegenerative diseases is characterized mainly by typical aggregate-prone toxic proteins1,2 and aberrant neuronal apoptosis, which involves oxidative stress, mitochondrial dysfunction, perturbation in calcium homeostasis and trophic-factor withdrawal.3 Allied in a Pandora’s box, the pathogenesis of these diseases is largely akin to one another at the subcellular level. Notwithstanding, the principal etiology of these neurodegenerative diseases remains unclear; ergo, much research is centered on decoding their mystery, which aims to provide therapeutic advancement that could offer a better quality of life.
The PI3K-Akt pathway has risen to prominence since it is involved in neuronal survival and confers protection against drug-induced damage in neurons.4 PI3K protects neurons through its antiapoptotic effects mediated by its central downstream target, Akt, which regulates expression of apoptosis-related genes.5 The mammalian target of rapamycin (mTOR), one of the main downstream target proteins of PI3K-Akt, is a protein kinase that functions as a sensor of extracellular signals that positively regulates cell growth and protects cells from apoptosis.6 Glycogen synthase kinase-3β (GSK-3β) is a protein negatively regulated by the PI3K-Akt pathway,7 where its inhibition confers neuroprotection by interfering with the intrinsic apoptosis signaling of the upstream apoptosome and production of inflammatory factors.8,9
A reactive oxygen species (ROS) such as hydrogen peroxide (H2O2) is a highly reactive and diffusible free radical that can trigger various signaling pathways and redox-sensitive signal transduction that modulates cellular mechanisms for cell proliferation and survival, death, and immune responses by inducing the production of proinflammatory factors such as cytokines through the activation of NF-κβ.10–12 Furthermore, excessive secretion of proinflammatory cytokines has been established to be a crucial initiator of aberrant inflammatory responses in neurodegenerative diseases as they are transcriptionally and reciprocally regulated by NF-κβ.13,14 Cytokines are a group of proteins that originate from the immune system and are endogenously produced by brain cells, including microglia and neurons. In healthy adult brains, cytokines are expressed constitutively at low levels; however, their production can be augmented in response to neuronal injury or infection,15–17 making them key inflammatory factors in the brain.18 Cytokines exert their biological functions through interactions with receptors on plasma membrane expressed by neuronal and glial cells, which then activate diverse intracellular phosphorylative cascades responsible for apoptosis, cell survival, inflammatory responses, and neural pathfinding.19,20
Alpha-lipoic acid (LA), an organosulfur compound derived from octanoic acid, can be found naturally in fruits, vegetables, and synthesized in animals and humans; it acts as the key player in mitochondrial energy production.21 Numerous empirical evidence has supported the intake of LA as a nutraceutical since it is capable of evoking an array of cellular and molecular mechanisms in the prevention and treatment of chronic diseases. For instance, LA has been advocated as a potential antiinflammatory agent for Alzheimer’s disease (AD),22 neuroprotective in various neuronal models,23,24 and a modulator of various genes involved in cell survival, inflammation, and oxidative stress.25,26 Recently, (R)-(+)-alpha-lipoic acid (R-LA) was demonstrated to mitigate the accumulation of 4-hydroxy-2-nonenal (4HNE) and protect retinal neurons against oxidative stress by inducing the HO-1 activity-dependent mechanism.27 The antioxidative property of R-LA has been suggested to contribute to its neuroprotective activities; albeit, its conclusive molecular mechanisms are still not completely understood. In this study, R-LA was demonstrated to protect NG108-15 cells against H2O2-induced cell death through the PI3K-Akt/GSK-3β pathway, which then suppressed the production of proinflammatory cytokines following GSK-3β–mediated p65NF-κβ inactivation.
Materials and methods
Cell culture and materials
NG108-15 was obtained from the American Type Culture Collection (ATCC) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Sigma-Aldrich Co., St Louis, MO, USA) composed of 10% (volume per volume) heat-inactivated fetal bovine serum (FBS), 2% penicillin/streptomycin, 1% amphotericin B (all from PAA Laboratories GmbH, Pasching, Austria), and hypoxanthine-aminopterin-thymidine (Sigma-Aldrich Co.). NG108-15 cells were cultured and conditioned at 5% CO2 moist atmosphere at 37°C. Cells exposed to vehicle alone (10% FBS DMEM, dimethyl sulfoxide ≤0.5% volume per volume) were used as the control group. R-LA was purchased from Sigma-Aldrich Co. Morphological analysis was performed with a fluorescence microscope (Leica Inverted Fluorescence Microscope, DM16000B; Leica Microsystems, Wetzlar, Germany) and flow cytometric analysis was carried out with BD Accuri C6 Flow Cytometry and BD CFlow® Software.
Cell viability assay
The neuroprotective effects of R-LA against H2O2-induced apoptosis in NG108-15 cells were evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Cells were plated at a total density of 5×103 cells/well in a 96-well plate. The cells were left to adhere for 48 hours and treated with R-LA (2 hours) prior to H2O2 (400 μM) exposure for 24 hours. Twenty microliters of MTT solution (Sigma-Aldrich Co.) was added into each well and incubated at 37°C for another 4 hours. The absorbance was measured using a microplate reader (ASYS UVM340) at 570 nm (reference wavelength: 650 nm).
Nuclear morphological analysis
Cells were plated in a 60 mm culture dish and left to adhere. The cells were pretreated with R-LA (12.5–50 μM) for 2 hours prior to H2O2 exposure. The cells were then harvested and rinsed with phosphate-buffered saline (PBS). Hoechst 33342 (5 μg/mL, Sigma-Aldrich Co.) was added, followed by propidium iodide (PI) dye (2.5 μg/mL) into each sample. The cells were incubated for 15 minutes and observed by fluorescence microscope. Cells that exhibited intense blue fluorescence were considered to be at an early apoptosis stage. In contrast, cells that were dual stained with H33342 and PI were considered to be at the late apoptosis stage.
Determination of glutathione/glutathione disulfide (GSH/GSSG) ratio
Cells were plated into a 96-well plate and left to adhere. Cells were subjected to the designated treatments, and after 24 hours, the solution in the well plate was discarded. Fifty microliters of total glutathione lysis reagent or oxidized glutathione lysis reagent (Promega, Madison, WI, USA) were added to each well and the plate was shaken for 5 minutes at room temperature. Fifty microliters of Luciferin generation reagent was added to all wells and incubated at room temperature for 30 minutes. One hundred microliters of Luciferin detection reagent was added and the luminescence signal was then read. The GSH and GSSG concentrations in each sample were calculated based on the GSH and GSSG standard curve.
Mitochondrial membrane potential (Δψm)
Alterations in Δψm were quantified by using a JC-1 kit according to the manufacturer’s protocol (Stratagene, La Jolla, CA, USA). The analysis of the R-LA effect on Δψm was evaluated as described previously.28 The green and red fluorescence signals were detected by flow cytometer. JC-1 aggregates, which emit red fluorescence signals within the intact mitochondria of healthy cells, were detected in the FL-2 channel, whereas JC-1 monomers with green fluorescence signals in the cytoplasm of apoptotic cells were detected in the FL-1 channel.
Measurement of intracellular ROS level
Cellular oxidative stress induced upon exposure to H2O2 and its modulation by R-LA were measured using the fluorescent probe 2′,7′-dichlorfluorescein-diacetate (DCFH-DA). Cells were plated and subjected to similar pretreatment of LA prior to the addition of H2O2. Cells were harvested, rinsed, and incubated with 10 μM of DCFH-DA for 30 minutes at 37°C in cell-loading medium. The fluorescence signal was measured using flow cytometer.
Western blot analysis
Total protein cell lysates of the treated NG108-15 cells were extracted with radioimmunoprecipitation assay (RIPA) buffer (Thermo Fisher Scientific, Waltham, MA, USA). The protein concentration was determined using the Bradford protein assay kit (Bio-Rad Laboratories Inc., Hercules, CA, USA). Twenty micrograms of each protein sample was electrophoresed on 10% SDS-PAGE. Proteins on the gel were transferred onto a nitrocellulose membrane and blocked with 5% bovine serum albumin (BSA). The membrane was then probed with the following primary antibodies: Bcl-2, Bcl-xL, Bax, caspase-3, cleaved caspase-3, phosphorylated Akt, Akt, phosphorylated mTOR, mTOR, Rictor, Raptor, and GSK-3β (Cell Signaling Technology, Danvers, MA, USA) at 4°C overnight followed by appropriate horseradish peroxidase (HRP)-conjugated secondary antibody and developed with enhanced chemiluminescence (ECL) reagent (Bio-Rad Laboratories Inc.). Proteins were quantified with Bio-1D software as a proportion of the signal of the housekeeping protein band (β-actin).
NF-κβ p65 translocation assay
Cells were plated onto coverslips and subject to designated treatments. After treatment, cells were gently rinsed and fixed with 4% paraformaldehyde in PBS. Cells were rinsed with PBS and blocked with blocking buffer (5% BSA, 0.5% Triton X-100 in PBS) for 1 hour. The cells were then incubated with rabbit anti-NF-κβ p65 (Cell Signaling Technology) overnight, followed by incubation with fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit secondary antibody (Pierce Antibodies, Rockford, IL, USA) for 2 hours, respectively. The cells were washed and then analyzed by using a fluorescence microscope.
Cytokines measurement
The production of cytokines was measured using Cytokine Bead Array (BD Biosciences, San Jose, CA, USA). In brief, following the designated treatment, 50 μL of culture medium was collected and mixed with the cytokine capture beads. The mixture was then mixed with phycoerythrin (PE)-conjugated detection antibodies to form sandwich complexes. The desired cytokines (IL-6, IL-10, and TNF-α) were then measured by flow cytometer and data were analyzed using FCAP Array™ software with comparison to mouse cytokines standard curves. To further validate the NF-κβ-cytokines regulation, the cells were pretreated with ethyl 3,4-dihydroxycinnamate (10 μM) prior to H2O2 exposure and the production of cytokines was then measured and compared with the H2O2-treated group.
Data analyses
All the experimental data are expressed as mean ± standard error (SE). Statistical differences between groups were analyzed by one-way analysis of variance (ANOVA), followed by Dunnett’s test. P-values <0.05 were considered to be statistically significant.
Results
R-LA–mitigated H2O2-induced cell death
The neuroprotective effects of R-LA (Figure 1A) against H2O2-induced cell death were evaluated with the MTT cell viability assay. In a prior study, the concentration-dependent effect of H2O2 on cell viability for 24hours was first optimized (data not shown). Exposure to H2O2 (400 μM) significantly reduced cell viability to 48.95%±2.82%; therefore, H2O2 at this concentration was selected for MTT cell viability study. Conversely, following pretreatment with R-LA (3.12–100 μM), NG108-15 cell viability was significantly increased in a dose-dependent manner. R-LA at 3.12 μM elicited the initial significant protection against H2O2 exposure and increased cell viability to 56.41%±1.87% as compared to H2O2-treated cells. It was observed that pretreatment with varying concentrations of R-LA (6.25, 12.5, 25, and 50 μM) significantly increased cell viability to 67.06%±2.77%, 69.89%±1.72%, 74.95%±2.15%, and 82.25%±2.97%, respectively. The highest protection level was achieved following pretreatment with R-LA at 100 μM, which significantly increased NG108-15 cell viability up to 89.37%±2.01% (Figure 1B). More importantly, R-LA was shown to protect the NG108-15 cells effectively against H2O2 exposure in a dose-dependent manner as compared to H2O2-treated cells.
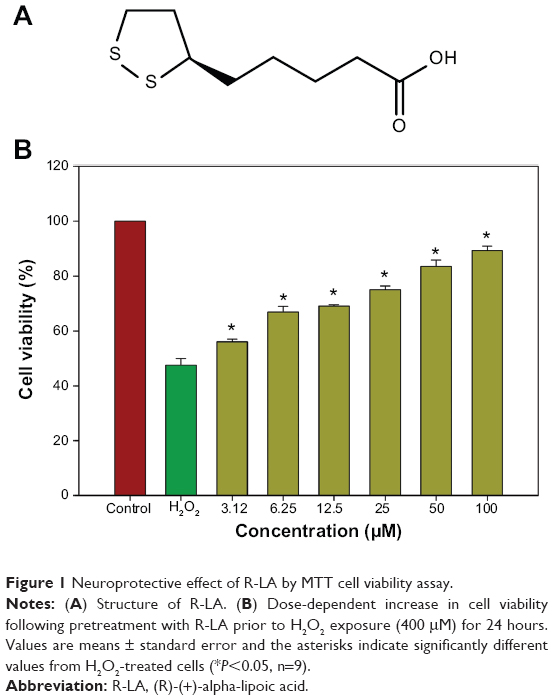 | Figure 1 Neuroprotective effect of R-LA by MTT cell viability assay. |
R-LA abated H2O2-induced nuclear morphological changes
Control untreated cells displayed evenly stained disseminated chromatin with intact nuclei with low blue fluorescence (Figure 2). H2O2-treated cells exhibited typical morphological characteristics of early apoptotic cells, which include condensed chromatin with fragmented DNA and the presence of apoptotic bodies with intense blue fluorescence (arrow 1, Figure 2). In addition, nuclei of NG108-15 cells were also dual-stained with both Hoechst 33342/PI, indicating late apoptotic cell population (arrow 2, Figure 2). However, pretreatment with R-LA notably abated the H2O2-induced nuclear morphological changes where the number of cells with nuclear condensation, fragmentation, and the presence of apoptotic bodies were significantly decreased.
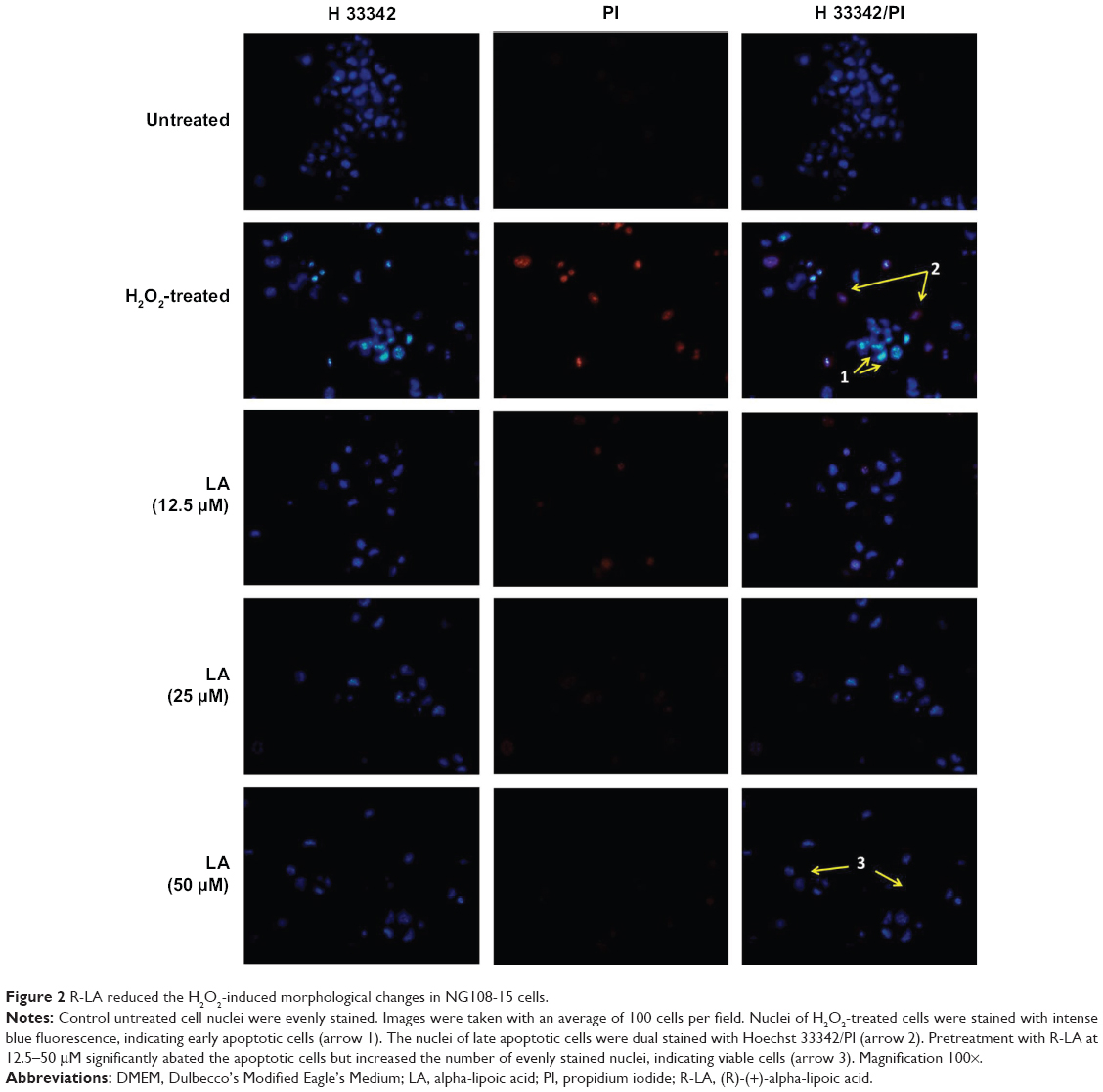 | Figure 2 R-LA reduced the H2O2-induced morphological changes in NG108-15 cells. |
R-LA augmented the GSH/GSSG ratio and suppressed the intracellular ROS level
Diffusion of exogenous H2O2 into the cell nucleus leads to elevated generation of the hydroxyl radical (·OH), which can directly injure the DNA of culture cells. GSH is an important intracellular antioxidant in the mammalian redox system that detoxifies H2O2 and keeps the thiol groups of proteins in the reduced state. Following oxidative stress, GSH will be oxidized into a dimer of two of the peptide elements linked by a disulfide bond between the cysteines known as oxidized glutathione (GSSG). The GSH/GSSG ratio has been used as an indicator of cell health and oxidative stress in neurodegenerative studies. Hence, the ratio of GSH/GSSG in the cells was investigated following pretreatment with R-LA and exposure to H2O2. The intracellular GSH level was determined from a GSH standard curve (0.05–1,000 nM) while the GSSG level was calculated based on the GSSG standard curve derived from the GSH standard curve according to the manufacturer’s protocol. H2O2 exposure significantly decreased (Figure 3A) the reduced GSH level but elevated the GSSG level, which led to reduction of the GSH/GSSG ratio to 1.15-fold as compared to 60.13-fold in untreated cells (Figure 3B). However, pretreatment with R-LA (12.5–50 μM) significantly increased the reduced GSH level and decreased the GSSG level, yielding 2.5-, 5.09-, and 27.14-fold increases of GSH/GSSG in a dose-dependent manner when compared to H2O2-treated cells (Figure 3B). Following this, the intracellular ROS level in the NG108-15 cells was determined by flow cytometry analysis. It was demonstrated that exposure of NG108-15 cells to H2O2 increased the intracellular ROS production significantly as illustrated by the right shift in the histogram (Figures 3C and D); 4.52-fold when compared to untreated cells. Notably, since pretreatment with R-LA was shown to elevate the GSH/GSSG ratio, it concomitantly suppressed the increase of intracellular ROS levels by H2O2 in NG108-15 cells as evidenced by the shift to the left in the histogram (Figures 3C and D).
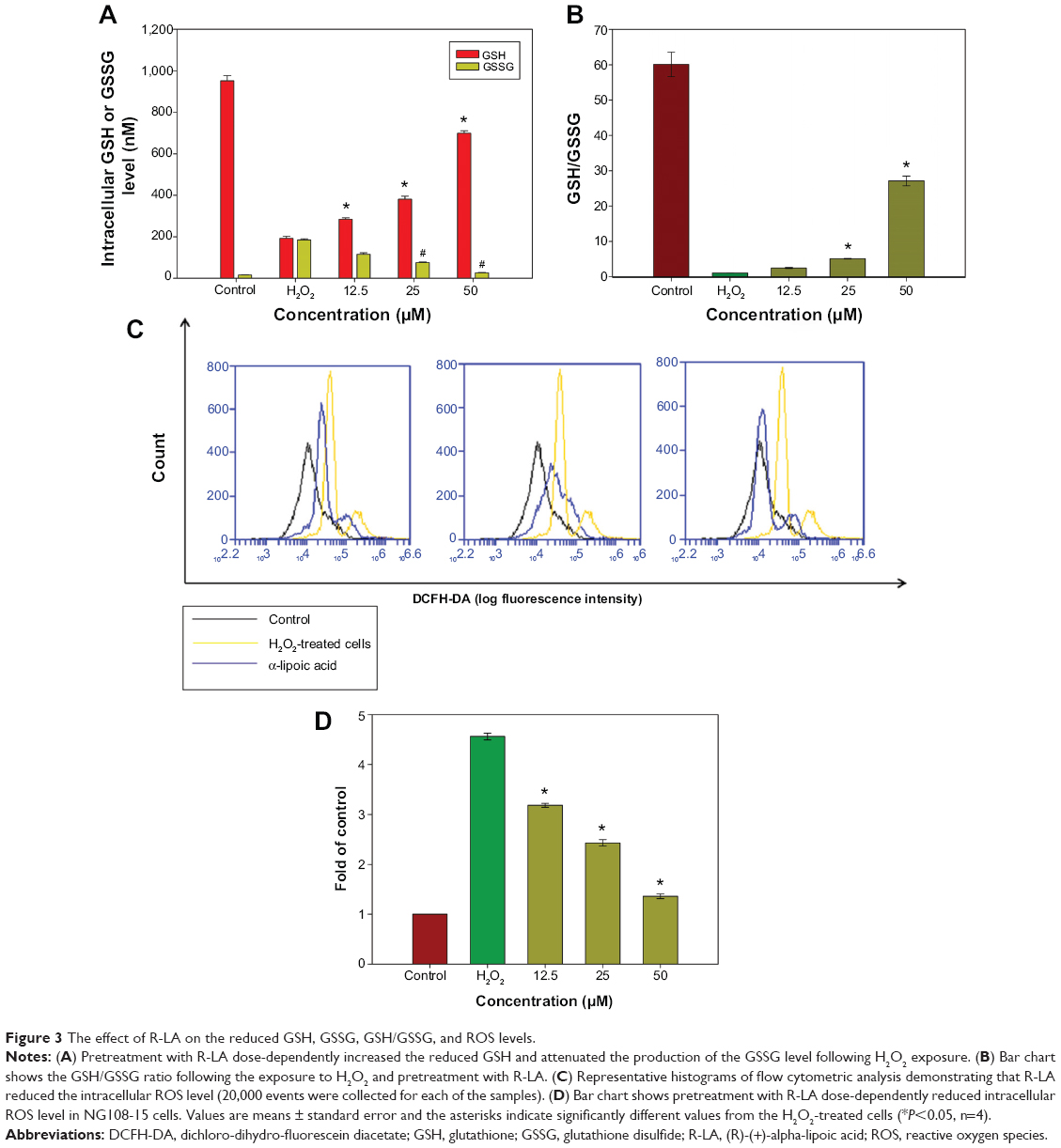 | Figure 3 The effect of R-LA on the reduced GSH, GSSG, GSH/GSSG, and ROS levels. |
R-LA ameliorated Δψm
Changes in Δψm reflect the alteration of the formation of a proton gradient across the inner mitochondrial membrane and is considered to be one of the key indicators of cellular viability. H2O2-treated cells exhibited changes in the fluorescence signal from the upper right quadrant to the lower quadrant, which leads to a lower red fluorescence signal (23.09%±2.57%) and higher green fluorescence signal (77.91%±1.47%), indicating the dissipation in Δψm (Figure 4A). Treatment with R-LA significantly reversed dissipation in Δψm as shown in Figure 4B. Pretreatment with R-LA at 50 μM suppressed the effect of H2O2-induced Δψm dissipation, shifting the fluorescence signal from the lower quadrant (37.14%±1.74%), resulting in an increase of the upper quadrant to 62.86%±1.75% (Figure 4A). These results indicated that pretreatment with R-LA attenuated the H2O2-induced apoptosis through the mitochondrial-mediated pathway.
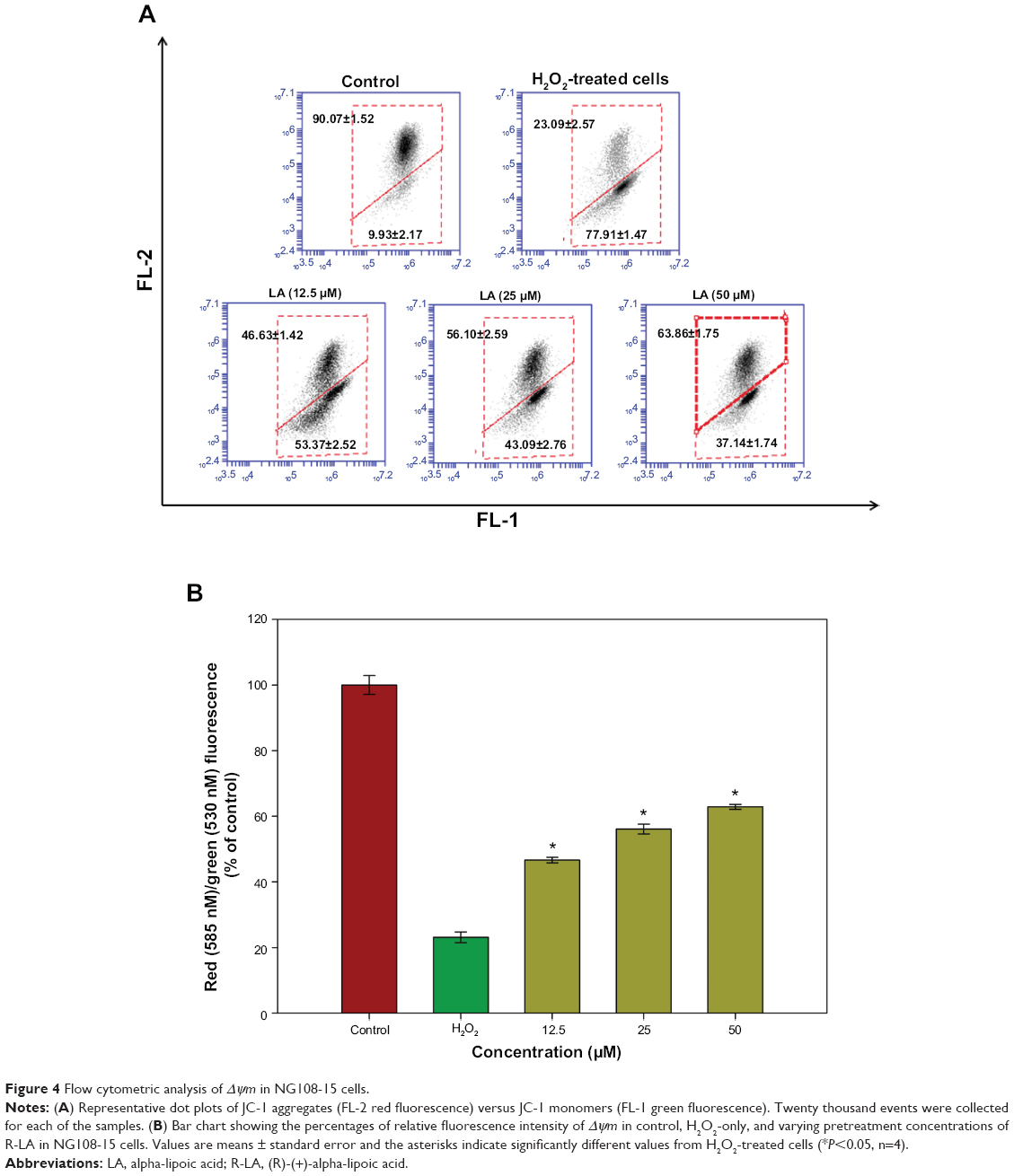 | Figure 4 Flow cytometric analysis of Δψm in NG108-15 cells. |
R-LA modulated the expression of apoptotic proteins and caspase-3
The increase in the ratio of proapoptotic protein to antiapoptotic protein is known to initiate a cascade of events leading to the activation of caspases, which in turn triggers apoptosis. The data in Figure 5A shows that Bax protein expression increases significantly in H2O2-treated NG108-15 cells. In addition, H2O2 reduced both Bcl-2 and Bcl-xL protein expression (Figure 5A), leading to a significant increase in both Bax/Bcl-2 and Bax/Bcl-xL ratios (Figures 5B and C), eventually resulting in apoptosis-favored conditions. Conversely, Bcl-2 and Bcl-xL protein expression in R-LA–treated NG108-15 cells were elevated (Figure 5A), while Bax protein expression was suppressed. This led to a significant reduction in Bax/Bcl-2 ratio from ~5.23 to ~1.69 (Figure 5B) and also Bax/Bcl-xL ratio from ~4.20 to ~1.75 (Figure 5C). To examine the involvement of the caspase-dependent pathway, the expression of procaspase-3 and cleaved caspase-3 were analyzed. The results showed that R-LA–treated cells expressed higher procaspase-3 as compared to H2O2-treated cells (Figures 5A and D). Moreover, the increase of cleaved caspase-3 in H2O2-treated cells was reduced following treatment with R-LA (Figures 5A and D).
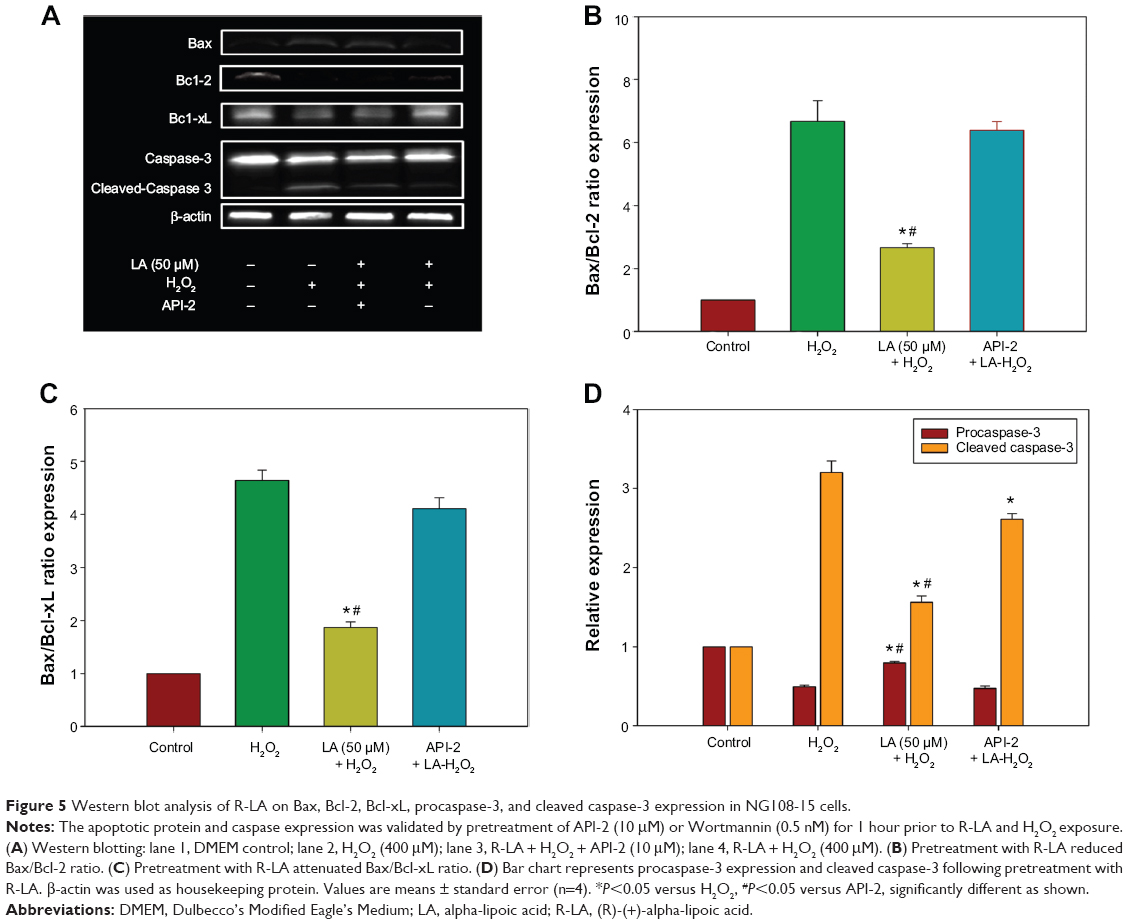 | Figure 5 Western blot analysis of R-LA on Bax, Bcl-2, Bcl-xL, procaspase-3, and cleaved caspase-3 expression in NG108-15 cells. |
R-LA–induced activation of PI3K-Akt via mTORC1 and mTORC2
The PI3K-Akt pathway plays vital roles in the regulation of neuronal survival. There was a significant reduction in the activation/phosphorylation of Akt (Thr308 and Ser473) in H2O2-treated cells when compared to untreated cells, but this change was prevented by pretreatment with R-LA (Figure 6A). The increase in Akt (Thr308 and Ser473) phosphorylation level (Figure 6A) led to an increase of both pAkt(Thr308)/Akt and pAkt(Ser473)/Akt protein ratios (Figures 6B and C). Full activation of Akt can be achieved via phosphorylation by the mammalian target of rapamycin complex 2 (mTORC2), which contains the regulatory proteins Rictor and mTOR. R-LA treatment was shown to augment phosphorylated mTOR protein expression (Figure 6D) significantly as compared to H2O2-treated cells, resulting in an increase of the pmTOR/mTOR protein ratio (Figure 6E). Furthermore, R-LA treatment upregulated Rictor expression (Figure 6F) as compared to H2O2-treated cells. More interestingly, R-LA also elevated the Raptor expression, which implied the activation of the mTORC1 complex following Akt activation (Figure 6G). To validate PI3K-Akt activation, an inhibitor of PI3K (Wortmannin) and Akt (API-2) was added prior to R-LA addition. Addition of Wortmannin and API-2 significantly attenuated neuroprotective activity of R-LA as shown by the MTT cell viability assay (Figure 6H) and reduced pAkt protein expression (Figures 6A–C), which suggests the involvement of the PI3K-Akt pathway.
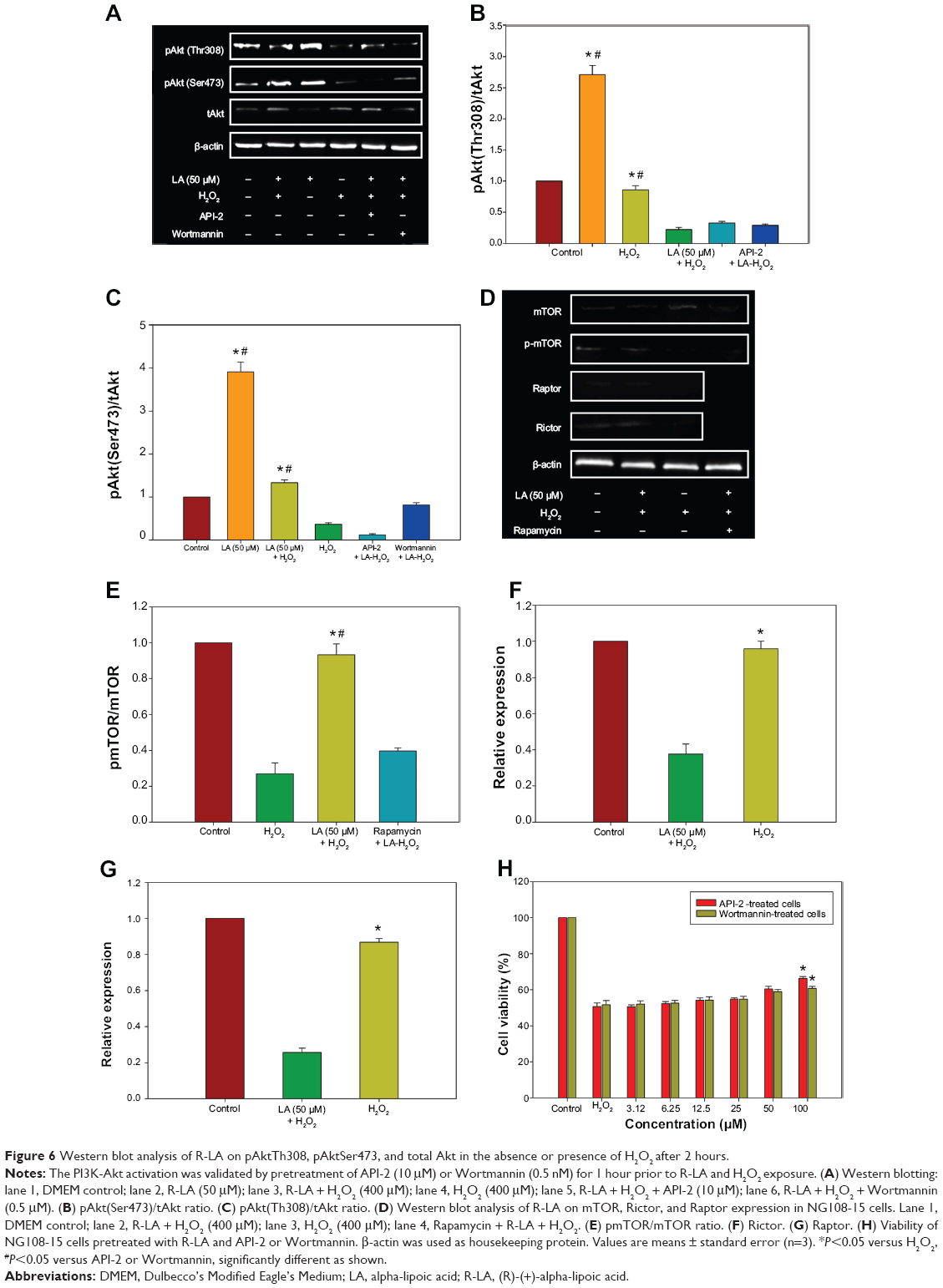 | Figure 6 Western blot analysis of R-LA on pAktTh308, pAktSer473, and total Akt in the absence or presence of H2O2 after 2 hours. |
R-LA modulated the production of cytokines by inactivating GSK-3β and NF-κβ p65 translocation
To further corroborate the participation of Akt downstream targets other than mTORC2, the regulation of GSK-3β, an important Tau kinase protein that plays a role in neuronal apoptosis was then determined. GSK-3β is reported to be essential in regulating NF-κβ activity since it can promote rapid NF-κβ p65 translocation. Based on this, the activation of PI3K-Akt by R-LA was postulated to mediate the inhibition of GSK-3β, which will be followed by suppression of NF-κβ p65 translocation. A significant increase in GSK-3β expression accompanied by a reduction of pGSK-3β (Ser9) was observed in H2O2-treated cells (Figure 7A), but these changes were prevented when the NG108-15 cells were pretreated with LA (Figure 7B). In fact, pretreatment with R-LA (50 μM) was shown to suppress the translocation of NF-κβ p65. Similarly, the immunofluorescence data substantiated that treatment with R-LA inhibited the translocation and activation of NF-κβ p65 in NG108-15 cells that were exposed to H2O2 (Figure 7C). Next, the effects of R-LA on the production of major inflammatory cytokines (TNF-α, IL-6, and IL-10) were analyzed since GSK-3β and NF-κβ activation is reciprocally regulated with the production of cytokines. Following H2O2 exposure, TNF-α and IL-6 production was augmented significantly as compared to control untreated cells (Figures 7D and E). Moreover, these changes were accompanied by the significant reduction of the antiinflammatory cytokine, IL-10. Intriguingly, upon inhibition of GSK-3β and suppression of NF-κβ p65, R-LA treatment aggrandized the production of IL-10, which then resulted in further diminution of TNF-α and IL-6 as compared to H2O2-treated cells. To further validate the regulation of NF-κβ-cytokines modulated by R-LA, the NG108-15 cells were also pretreated with ethyl 3,4-dihydroxycinnamate, a specific NF-κβ inhibitor and its subsequent mediators upon exposure to H2O2. It was observed that pretreatment with ethyl 3,4-dihydroxycinnamate suppressed the production of IL-6 and TNF-α as well as IL-10 when compared to the control and H2O2-treated groups. Unlike the R-LA–treated group, the production of IL-10 was significantly greater following pretreatment with R-LA, which may suggest that R-LA induced neuroprotection against H2O2 by modulating both anti- and proinflammatory cytokines through their reciprocal regulation with NF-κβ.
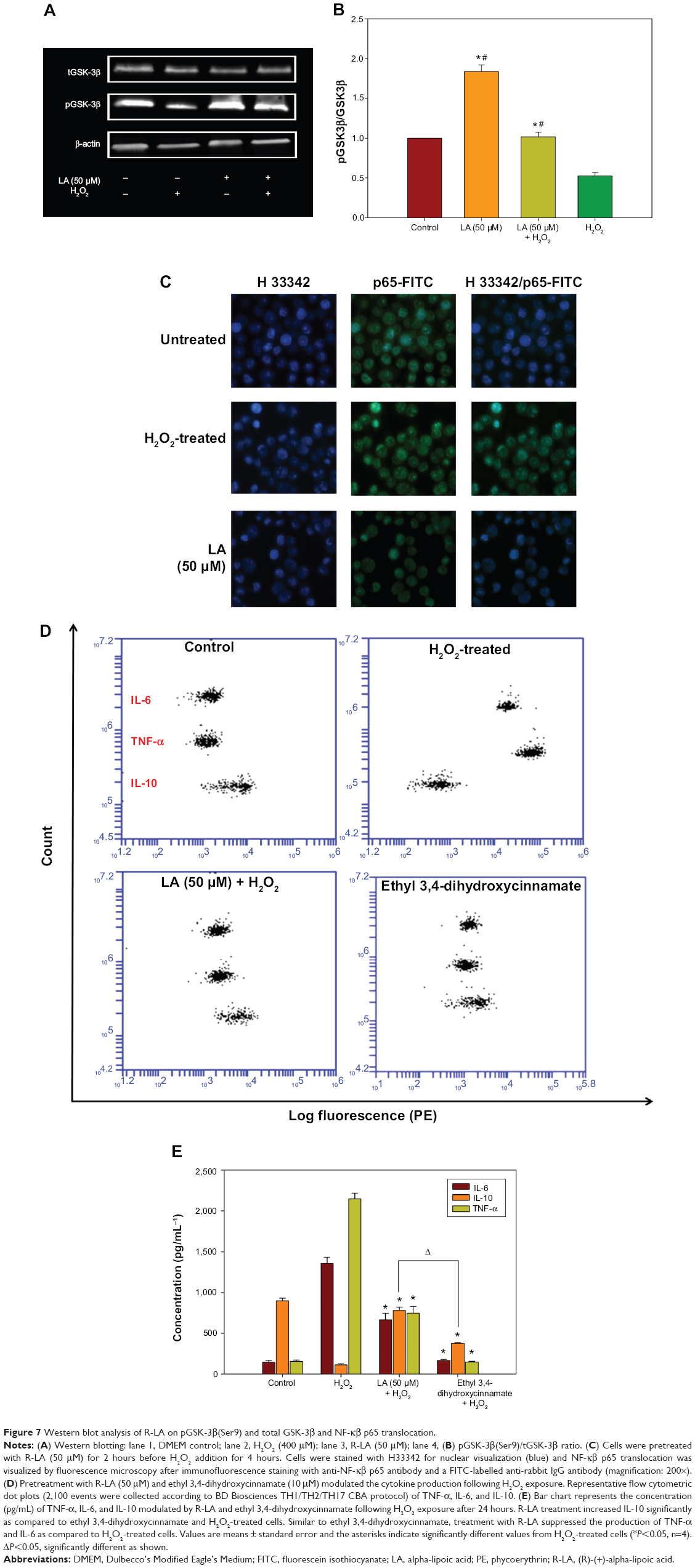 | Figure 7 Western blot analysis of R-LA on pGSK-3β(Ser9) and total GSK-3β and NF-κβ p65 translocation. |
Discussion
Cogent evidence has supported the multitude use of LA for metabolic syndrome29 and its associated-diseases,30 as a potential cognitive enhancer,31 and an important modulator of various inflammatory signaling pathways,26 making it a beneficial nutraceutical with multifarious properties. Additionally, LA has been proposed as a potential antiinflammatory drug since it interferes with the pathogenesis of AD.22,32 MTT cell viability assay revealed that R-LA exhibited a dose-dependent neuroprotective effect against H2O2-induced cell death in NG108-15 cells (Figure 1). Induction of apoptosis was detected using H33342/PI fluorescence morphological analysis. Morphological changes in H2O2-induced apoptosis in NG108-15 cells were characterized by the condensation of chromatin, the shrinkage of cell nuclei, and apoptotic bodies being stained with intense blue fluorescence. Conversely, these morphological alterations were evidently abrogated following R-LA treatment as compared to the H2O2-treated cells. These findings first suggested the evasion of cell death by R-LA through mitigation of apoptosis in NG108-15 cells.
The primary etiology of hereditary and sporadic neurodegenerative diseases remains vague, albeit growing evidence implicating the role of oxidative stress in the process. Neurons are considered to be highly susceptible to oxidative stress as they are intrinsically ill-equipped to protect against an increase in ROS due to low levels of antioxidants relative to other mammalian cell types.33 ROS plays various roles in cell signaling, including apoptosis, cell proliferation, gene expression, and the activation of cell signaling cascades.34,35 Excessive production of ROS may lead to oxidative stress, loss of cell functions, and ultimately, cell death.36 H2O2 exposure caused a shift to the right in the histogram (Figure 3C), indicating an elevation in intracellular ROS production. In contrast, pretreatment with LA caused a reduction in intracellular ROS formation, which suggests that R-LA protected the NG108-15 cells through its antioxidative activity. The current paradigm advocates that a decline in intracellular GSH below a threshold level constitutes an apoptotic signal that will initiate neuronal cell death signaling.37 H2O2 exposure caused a significant reduction in the reduced GSH level and elevated the GSSG level in NG108-15 cells (Figure 3A), leading to a significant diminution in the ratio of GSH/GSSG. The reduction in the ratio of GSH/GSSG indicated oxidative stress in the cellular redox system, which eventually led to neuronal cell death. Notwithstanding, pretreatment with R-LA dose-dependently aggrandized the reduced GSH level and markedly increased the ratio of GSH/GSSG (Figure 3B), which further corroborated that LA protects the NG108-15 cells by ameliorating the cellular redox state via its antioxidant activity. GSH depletion exacerbates the dissipation of Δψm, which leads to mitochondrial release of apoptogenic factors such as cytochrome c and hence, initiating the activation of caspases.34 However, R-LA treatment significantly mitigated the dissipation of Δψm (Figure 4), which further established the ability of R-LA to ameliorate the Δψm after consequential loss of potential due to H2O2 exposure. Collectively, through its ability to preserve the mitochondrial membrane integrity, R-LA directly suppressed cytochrome c release and formation of apoptosome, which eventually led to disruption of the apoptotic signaling cascades.
Research in recent times has seen unprecedented advanced understanding of PI3K-Akt signaling, which plays a central role in cell survival and proliferation by mediating the antiapoptosis and antiinflammatory mechanisms.38 This includes the activation of mTOR and its downstream effectors, which affect a number of cellular processes including proliferation and motility.39,40 The involvement of the PI3K-Akt signaling pathway by R-LA was further examined and it was observed that treatment with R-LA suppressed H2O2-induced inhibition of Akt (Thr308/Ser473) phosphorylation, thereby resulting in an increased pAkt/Akt ratio (Figures 6B and C), suggesting that R-LA exerts its neuroprotective effect through the activation of the PI3K-Akt pathway. The addition of both API-2 and Wortmannin further reduced the LA-treated cell viability (Figure 6H) and thereby abrogated the neuroprotective effect of R-LA in NG108-15 cells, which further substantiated the involvement of PI3K-Akt. Furthermore, Western blot analysis demonstrated that addition of API-2 and Wortmannin suppressed R-LA–induced Akt (Thr308/Ser473) protein phosphorylation (Figures 6A–C), thus indicating the neuroprotective effect of R-LA through activation of the PI3K-Akt pathway.
As the ability of R-LA to activate Akt signaling has been demonstrated, the intracellular molecular events has since been evaluated. The full activation of Akt requires phosphorylation at Ser473 with the aid of mTORC2.41 R-LA–treated cells exhibited an elevation of both mTOR and Rictor expression (Figures 6E and F), suggesting that administration with R-LA induced full activation of Akt protein. The activated Akt in turn phosphorylates to activate its diverse downstream protein substrates, including mTORC1 component, mTOR, and Raptor (Figure 6G). The suppression of neuronal apoptosis through mTOR mainly depends on Akt activation, which occurs in several stages such as fostering cell survival through preserving Δψm, caspase inactivation, and promoting inflammatory cell inactivation.42 Moreover, the ability of mTORC1 to inhibit apoptotic neuronal cell death is associated with Akt activation,43 and inflammatory cells have been shown to capitulate apoptosis during oxidative stress following Akt and mTOR inhibition.44,45 In this context, our data indicated that LA induced full activation of Akt through its regulation of mTORC1 and mTORC2 components that confer protection against H2O2 insult. In addition, PI3K-Akt protects neurons from damage38,46 and regulates survival in neurons by regulating the Bcl-2 family of proteins.47 H2O2 exposure elevated the expression of Bax protein and attenuated Bcl-2 and Bcl-xL, which resulted in the increase of Bax/Bcl-2 and Bax/Bcl-xL ratios (Figures 5B and C), and thus led to conditions that favor apoptosis. However, this was prevented by R-LA treatment, which increased both Bcl-2 and Bcl-xL protein expression, leading to the decrease in the Bax/Bcl-2 and Bax/Bcl-xL ratios. This then suppressed the release of cytochrome c and formation of apoptosome with concomitant suppression of caspase activation.
Furthermore, Akt confers neuronal survival by inactivating its other target, GSK-3β, through phosphorylation. GSK-3β is a multitasked kinase that plays imperative roles in various signaling pathways since it targets and regulates important metabolic and signaling proteins, structural proteins, and transcription factors.48 GSK-3β is tightly regulated by pAkt through phosphorylation of its Ser9, rendering it inactive. Inhibition of GSK-3β was reported to be protective against a plethora of neuronal insults,9 thus implying that its suppression confers neuronal survival. In accordance with this, activation of Akt was accompanied by increased phosphorylation of GSK-3β when compared to H2O2-treated cells (Figures 7A and B), suggesting that R-LA prevented cell death through inactivation of GSK-3β. GSK-3β is a well-known upstream mediator of caspase-3, although the precise mechanism by which GSK-3β facilitates neuronal death remains unclear.49 Caspase-3 is activated in the apoptotic cell both by extrinsic and intrinsic pathways. Following a decrease in Bax/Bcl-2 and Bax/Bcl-xL ratios and inhibition of GSK-3β, R-LA suppressed the cleavage of procaspase-3 into cleaved caspase-3 (Figure 5D). These findings highlighted that R-LA protected the NG108-15 cells via activation of PI3K-Akt, which led to subsequent inhibition of proapoptotic Bax and GSK-3β accompanied by activation of antiapoptotic Bcl-2 and Bcl-xL, which concomitantly suppressed caspase-3 activation.
Suppression of GSK-3β can result in both activation and suppression of an array of transcription factors that are critical in regulating the production of pro- and antiinflammatory cytokines8 by enhancing the binding of cAMP response element-binding protein (CREB) while inhibiting the binding of NF-κβ p65 to the nuclear coactivator CREB-binding protein.50 Inhibition of GSK-3β was reported to suppress toll-like receptor-mediated inflammatory responses. For instance, phosphorylation of GSK-3β(Ser9) inhibited the translocation of NF-κβ p65 into cell nuclei and the production of inflammatory factors in various neuronal models.8,51 NF-κβ is an important regulator of DNA transcription that regulates the expression of genes involved in innate and adaptive immune responses, inflammatory responses, cell survival, and cell proliferation.52,53 Furthermore, NF-κβ and proinflammatory cytokines’ reciprocal activation play a major role in inflammatory processes;8,54 nonetheless, aberrant activation of NF-κβ can result in excessive neuroinflammation that may lead to neuronal cell death.13,14
H2O2 escalated the intracellular ROS level, thereby inducing oxidative stress, which was followed by NF-κβ p65 activation in NG108-15 cells. Moreover, oxidative stress was reported to promote NF-κβ p65 activation through phosphorylation of Iκβ55,56 and addition of ROS scavenger compound subsequently abated the transcriptional activity and translocation of NF-κβ p65,57 which concomitantly suppressed the production of TNF-α and IL-6. Our current findings demonstrated that H2O2 exposure significantly induces NF-κβ p65 nuclear translocation (Figure 7C), as well as production of proinflammatory cytokines (Figures 7D and E) as compared to untreated cells. However, the increase in the production of proinflammatory cytokines was clearly suppressed following pretreatment with the NF-κβ specific inhibitor, ethyl 3,4-dihydroxycinnamate, as compared to both control untreated and H2O2-treated cells. Following GSK-3β inhibition, R-LA suppressed the NF-κβ p65 nuclear translocation and subsequently decreased the production of proinflammatory cytokines. It was observed that R-LA inhibited H2O2-induced production of TNF-α and IL-6, which are NF-κβ–regulated cytokines (Figures 7D and E); this observation was similar to pretreatment with ethyl 3,4-dihydroxycinnamate. Cytokines are well-known major regulators of inflammatory responses in the brain since they constitute a cardinal role in the nerve cell response towards brain infection or injury.58 Although neuronal release of cytokines aims to eradicate threats through microglial activation, nevertheless, when they are secreted in imbalanced fashion, the homeostatic balance will be broken and the damage will persist leading to excessive production of proinflammatory cytokines that promote progressive neuronal cell death. TNF-α and IL-6 are usually among the first proinflammatory cytokines to be expressed following neuronal injury and their overproduction is highly detrimental to neuronal cells.59 TNF-α and IL-6 are major components of neuroinflammation in AD and Parkinson’s disease since direct intraparenchymal injection of TNF-α induced dopaminergic neuron degeneration and inhibition of both TNF-α and IL-6 prevented preplaque amyloid aggregation, thereby preventing the acceleration of AD-like pathology.60,61 Furthermore, various findings suggest that soluble inflammatory factors such as TNF-α possess complex injury-promoting properties as well as protective effects on neurons by modulating the electrophysiological membrane properties of neurons and glial cells. For instance, chronic inhibition of soluble TNF ameliorated dopaminergic neuron death by interfering with PI3K-Akt signaling and activation of caspase-3.62 Moreover, TNF-α was shown to promote neuronal cell death in neuroblastoma cells through the activation of Fas ligand/Fas receptor (FasL/Fas) in the absence of glial cells.63
More importantly, in a more intricate inflammatory network, various anti- and proinflammatory cytokines such as TNF-α, IL-1β, IL-2, IL-6, and IL-10 are produced simultaneously and may stimulate or suppress each other during inflammation.64 Inhibition of GSK-3(α/β) mediated by mTORC1 through PI3K-Akt was reported to increase the production of a potent antiinflammatory cytokine, IL-10, which is capable of inhibiting the production of proinflammatory cytokines such as TNF-α, IL-1, IL-6, and IL-12.51,65 Furthermore, IL-10 has been reported to suppress neuroinflammation and the production of proinflammatory cytokines and inflammatory mediators through its inhibitory activity on the NF-κβ pathway activation in various cellular models.66–69 Likewise, in the present study, R-LA was demonstrated to increase IL-10 production in NG108-15 cells, thereby decreasing the production of induced TNF-α and IL-6 following H2O2 exposure, resulting in the suppression of caspase-dependent neuronal apoptosis. Moreover, pretreatment with R-LA was shown to increase the production of IL-10 as compared to ethyl 3,4-dihydroxycinnamate. Even though both ethyl 3,4-dihydroxycinnamate and R-LA suppressed H2O2-induced neuroinflammation in NG108-15 cells through inactivation of NF-κβ p65 translocation, which subsequently inhibited the production of IL-6 and TNF-α, R-LA was observed to significantly increase the IL-10 level significantly higher than both H2O2- and ethyl 3,4-dihydroxycinnamate–treated cells. This observation may suggest that R-LA induced the production of IL-10, which exerted its antiinflammatory effect by inhibiting NF-κβ activation with concomitant IL-6 and TNF-suppression. The production of cytokines can be either detrimental or advantageous for neuronal survival depending on multiple factors and the signaling pathway involved. The current study clearly demonstrates that R-LA significantly attenuated H2O2-induced overproduction of proinflammatory cytokines by reciprocally stimulating the production of antiinflammatory cytokines by modulating NF-κβ-cytokines regulation. Thus, a potential therapeutic therapy of various nutraceuticals against oxidative stress should also consider the modulation of neuroinflammation-mediated neurodegenerative diseases by targeting both anti- and proinflammatory cytokines.
Conclusion
The contribution of the PI3K-Akt/GSK-3β pathway and its coregulation with NF-κβ-cytokines in neuroprotection remains controversial and thus, illustrates the complexity of these pathways. Nevertheless, it is unraveled at least in part in this study. R-LA inhibition of GSK-3β resulted in the suppression of proinflammatory cytokines with subsequent mitigation of mitochondrial-mediated caspase-dependent neuronal apoptosis. Nevertheless, it is noteworthy that targeting GSK-3β through PI3K-Akt would confer neuronal survival in NG108-15 cells owing to GSK-3β diverse mechanisms that include NF-κβ-cytokines–specific reciprocal regulation. Based on these findings, R-LA mitigated the H2O2-induced neuronal cell death in NG108-15 cells by suppressing both mitochondrial-mediated caspase-dependent apoptosis and production of proinflammatorry cytokines via inhibition of GSK-3β through the PI3K-Akt pathway. More importantly, these findings exhibited the ability of R-LA to modulate NF-κβ-cytokines expression in the neuronal model and highlighted the rational use of R-LA in the intervention of neurodegenerative diseases with a potential new molecular target site.
Acknowledgments
This research is supported by High Impact Research Chancellory Grant UM.C/625/1/HIR/175 from the University of Malaya and University of Malaya Postgraduate Research Fund (PG124-2012B).
Author contributions
MNAK contributed to the overall experimental design, performed the experiments, statistical data analysis, data interpretation, and the writing of the manuscript. NAMR, SSSH, and LJY performed the experiments and contributed to the writing of the manuscript. HS provided his assistance in repeating the experiments and data analysis prior to resubmission and also revised the manuscript. HAK contributed to the overall experimental design, data interpretation, and critical manuscript review. All authors read and approved the final manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443(7113):780–786. | ||
Dasuri K, Zhang L, Keller JN. Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic Biol Med. 2012;62:170–185. | ||
Brady S, Morfini G. A perspective on neuronal cell death signaling and neurodegeneration. Mol Neurobiol. 2010;42(1):25–31. | ||
Wang X, Zhang X, Cheng Y, et al. Alpha-lipoic acid prevents bupivacaine-induced neuron injury in vitro through a PI3K/Akt-dependent mechanism. Neurotoxicology. 2010;31(1):101–112. | ||
Franke TF. PI3K/Akt: getting it right matters. Oncogene. 2008;27(50):6473–6488. | ||
Wrighton KH. Cell signalling: where the mTOR action is. Nat Rev Mol Cell Biol. 2013;14(4):191. | ||
Medina M, Garrido JJ, Wandosell FG. Modulation of GSK-3 as a therapeutic strategy on tau pathologies. Front Mol Neurosci. 2011;4:24. | ||
Wang MJ, Huang HY, Chen WF, Chang HF, Kuo JS. Glycogen synthase kinase-3β inactivation inhibits tumor necrosis factor-α production in microglia by modulating nuclear factor κβ and MLK3/JNK signaling cascades. J Neuroinflammation. 2010;7:99. | ||
Lei P, Ayton S, Bush AI, Adlard PA. GSK-3 in neurodegenerative diseases. Int J Alzheimers Dis. 2011;2011:189246. | ||
Nakano H, Nakajima A, Sakon-Komazawa S, Piao JH, Xue X, Okumura K. Reactive oxygen species mediate crosstalk between NF-kappaB and JNK. Cell Death Differ. 2006;13(5):730–737. | ||
Siomek A. NF-κβ signaling pathway and free radical impact. Acta Biochim Pol. 2012;59(3):323–331. | ||
Nakajima S, Kitamura M. Bidirectional regulation of NF-κβ by reactive oxygen species: a role of unfolded protein response. Free Radic Biol Med. 2013;65:162–174. | ||
Minami M, Katayama T, Satoh M. Brain cytokines and chemokines: roles in ischemic injury and pain. J Pharmacol Sci. 2006;100(5):461–470. | ||
Starossom SC, Mascanfroni ID, Imitola J, et al. Galectin-1 deactivates classically activated microglia and protects from inflammation-induced neurodegeneration. Immunity. 2012;37(2):249–263. | ||
Verma S, Nakaoke R, Dohgu S, Banks WA. Release of cytokines by brain endothelial cells: A polarized response to lipopolysaccharide. Brain Behav Immun. 2006;20(5):449–455. | ||
Helmy A, De Simoni MG, Guilfoyle MR, Carpenter KL, Hutchinson PJ. Cytokines and innate inflammation in the pathogenesis of human traumatic brain injury. Prog Neurobiol. 2011;95(3):352–372. | ||
Steinman L. Inflammatory cytokines at the summits of pathological signal cascades in brain diseases. Sci Signal. 2013;6(258):pe3. | ||
Cardona AE, Li M, Liu L, Savarin C, Ransohoff RM. Chemokines in and out of the central nervous system: much more than chemotaxis and inflammation. J Leukoc Biol. 2008;84(3):587–594. | ||
Deverman BE, Patterson PH. Cytokines and CNS development. Neuron. 2009;64(1):61–78. | ||
Rubio-Perez JM, Morillas-Ruiz JM. A review: inflammatory process in Alzheimer’s disease, role of cytokines. Scientific World Journal. 2012;2012:756357. | ||
Nagesh Babu G, Kumar A, Singh RL. Chronic pretreatment with acetyl-L-carnitine and ± DL-α-lipoic acid protects against acute glutamate-induced neurotoxicity in rat brain by altering mitochondrial function. Neurotox Res. 2011;19(2):319–329. | ||
Maczurek A, Hager K, Kenklies M, et al. Lipoic acid as an anti-inflammatory and neuroprotective treatment for Alzheimer’s disease. Adv Drug Deliv Rev. 2008;60(13–14):1463–1470. | ||
Fujita H, Shiosaka M, Ogino T, et al. Alpha-lipoic acid suppresses 6-hydroxydopamine-induced ROS generation and apoptosis through the stimulation of glutathione synthesis but not by the expression of heme oxygenase-1. Brain Res. 2008;1206:1–12. | ||
Zaitone SA, Abo-Elmatty DM, Shaalan AA. Acetyl-l-carnitine and α-lipoic acid affect rotenone-induced damage in nigral dopaminergic neurons of rat brain, implication for Parkinson’s disease therapy. Pharmacol Biochem Behav. 2012;100(3):347–360. | ||
Bitar MS, Ayed AK, Abdel-Halim SM, Isenovic ER, Al-Mulla F. Inflammation and apoptosis in aortic tissues of aged type II diabetes: amelioration with alpha-lipoic acid through phosphatidylinositol 3-kinase/Akt-dependent mechanism. Life Sci. 2010;86(23-24):844–853. | ||
Salinthone S, Yadav V, Bourdette DN, Carr DW. Lipoic acid: a novel therapeutic approach for multiple sclerosis and other chronic inflammatory diseases of the CNS. Endocr Metab Immune Disord Drug Targets. 2008;8(2):132–142. | ||
Koriyama Y, Nakayama Y, Matsugo S, Kato S. Protective effect of lipoic acid against oxidative stress is mediated by Keap1/Nrf2-dependent heme oxygenase-1 induction in the RGC-5 cellline. Brain Res. 2013;1499:145–157. | ||
Chan G, Kamarudin MN, Wong DZ, et al. Mitigation of H(2)O(2)-induced mitochondrial-mediated apoptosis in NG108-15 cells by novel mesuagenin C from Mesua kunstleri (King) Kosterm. Evid Based Complement Alternat Med. 2012;2012:156521. | ||
Ziegler D, Ametov A, Barinov A, et al. Oral treatment with alpha-lipoic acid improves symptomatic diabetic polyneuropathy: the SYDNEY 2 trial. Diabetes Care. 2006;29(11):2365–2370. | ||
Cakatay U, Kayali R, Sivas A, Tekeli F. Prooxidant activities of α-lipoic acid on oxidative protein damage in the aging rat heart muscle. Arch Gerontol Geriatr. 2005;40(3):231-240. | ||
Manda K, Ueno M, Anzai K. Memory impairment, oxidative damage and apoptosis induced by space radiation: ameliorative potential of alpha-lipoic acid. Behav Brain Res. 2008;187(2):387–395. | ||
Shay KP, Moreau RF, Smith EJ, Smith AR, Hagen TM. Alpha-lipoic acid as a dietary supplement: molecular mechanisms and therapeutic potential. Biochim Biophys Acta. 2009;1790(10):1149–1160. | ||
Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. 2009;7(1):65–74. | ||
Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48(6):749–762. | ||
Gough DR, Cotter TG. Hydrogen peroxide: a Jekyll and Hyde signalling molecule. Cell Death Dis. 2011;2:e213. | ||
Preiser JC. Oxidative stress. JPEN J Parenter Enteral Nutr. 2012;36(2):147–154. | ||
Johnson WM, Wilson-Delfosse AL, Mieyal JJ. Dysregulation of glutathione homeostasis in neurodegenerative diseases. Nutrients. 2012;4(10):1399–1440. | ||
Matsuda S, Nakanishi A, Wada Y, Kitagishi Y. Roles of PI3K/AKT/PTEN pathway as a target for pharmaceutical therapy. Open Med Chem J. 2013;7:23–29. | ||
Yap TA, Garrett MD, Walton MI, Raynaud F, de Bono JS, Workman P. Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr Opin Pharmacol. 2008;8(4):393–412. | ||
Betz C, Hall M. Where is mTOR and what is it doing there? J Cell Biol. 2013;203(4):563–574. | ||
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–1101. | ||
Bhaskar K, Miller M, Chludzinski A, Herrup K, Zagorski M, Lamb BT. The PI3K-Akt-mTOR pathway regulates Abeta oligomer induced neuronal cell cycle events. Mol Neurodegener. 2009;4:14. | ||
Guertin DA, Stevens DM, Thoreen CC, et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11(6):859–871. | ||
Shang YC, Chong ZZ, Wang S, Maiese K. Prevention of β-amyloid degeneration of microglia by erythropoietin depends on Wnt1, the PI3-K/mTOR pathway, Bad, and Bcl-xL. Aging. 2012;4(3):187–201. | ||
Maiese K, Chong ZZ, Shang YC, Wang S. mTOR: on target for novel therapeutic strategies in the nervous system. Trends Mol Med. 2013;19(1):51–60. | ||
Shao JL, Wan XH, Chen Y, et al. H2S protects hippocampal neurons from anoxia-reoxygenation through cAMP-mediated PI3K/Akt/p70S6K cell-survival signaling pathways. J Mol Neurosci. 2011;43(3):453–460. | ||
Malla R, Gopinath S, Alapati K, et al. Downregulation of uPAR and cathepsin B induces apoptosis via regulation of Bcl-2 and Bax and inhibition of the PI3K/Akt pathway in gliomas. PLoS One. 2010;5(10):e13731. | ||
Kaidanovich-Beilin O, Woodgett JR. GSK-3: Functional insights from cell biology and animal models. Front Mol Neurosci. 2011;4:40. | ||
Jo J, Whitcomb DJ, Olsen KM, et al. Aβ(1-42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3β. Nat Neurosci. 2011;14(5):545–547. | ||
Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6(8):777–784. | ||
Huang WC, Lin YS, Wang CY, et al. Glycogen synthase kinase-3 negatively regulates anti-inflammatory interleukin-10 for lipopolysaccharide-induced iNOS/NO biosynthesis and RANTES production in microglial cells. Immunology. 2009;128(1 Suppl):e275–e286. | ||
Baltimore D. NF-κβ is 25. Nat Immunol. 2011;12(8):683–685. | ||
Hayden MS, Ghosh S. NF-κβ in immunobiology. Cell Res. 2011;21(2):223–244. | ||
Tourniaire F, Romier-Crouzet B, Lee JH, et al. Chemokine expression in inflamed adipose tissue is mainly mediated by NF-κβ. PLoS One. 2013;8(6):e66515. | ||
Kabe Y, Ando K, Hirao S, Yoshida M, Handa H. Redox regulation of NF-κβ activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal. 2005;7(3–4):395–403. | ||
Morgan MJ, Liu Z. Crosstalk of reactive oxygen species and NF-κβ signaling. Cell Res. 2011;21(1):103–115. | ||
Fang IM, Yang CH, Yang CM, Chen MS. Chitosan oligosaccharides attenuates oxidative-stress related retinal degeneration in rats. PLoS One. 2013;8(10):e77323. | ||
Kriz J. Inflammation in ischemic brain injury: timing is important. Crit Rev Neurobiol. 2006;18(1–2):145–157. | ||
Correale J, Villa A. The neuroprotective role of inflammation in nervous system injuries. J Neurol. 2004;251(11):1304–1316. | ||
McAlpine FE, Lee JK, Harms AS, et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol Dis. 2009;34(1):163–177. | ||
Chen SY, Chen TF, Lai LC, et al. Sequence variants of interleukin 6 (IL-6) are significantly associated with a decreased risk of late-onset Alzheimer’s disease. J Neuroinflammation. 2012;9:21. | ||
Martinez TN, Chen X, Bandyopadhyay S, Merrill AH, Tanseyet MG. Ceramide sphingolipid signaling mediates Tumor Necrosis Factor (TNF)-dependent toxicity via caspase signaling in dopaminergic neurons. Mol Neurodegener. 2012;7:45. | ||
Álvarez SÁ, Blanco A, Fresno M, Muñoz-Fernández A. TNF-α contributes to Caspase-3 independent apoptosis in Neuroblastoma cells: role of NFAT. PLoS One. 2011;6(1):e16100. | ||
Guyon A, Massa F, Rovère C, Nahon JL. How cytokines can influence the brain: a role for chemokines? J Neuroimmunol. 2008;198(1-2):46–55. | ||
Wang H, Brown J, Gu Z, et al. Convergence of the mammalian target of rapamycin complex 1- and glycogen synthase kinase 3-β-signaling pathways regulates the innate inflammatory response. J Immunol. 2011;186(9):5217–5226. | ||
Heyen JR, Ye S, Finck BN, Johnson RW. Interleukin (IL)-10 inhibits IL-6 production in microglia by preventing activation of NF-kappaB. Brain Res Mol Brain Res. 2000;77(1):138–147. | ||
Loscher CE, Draper E, Leavy O, Kelleher D, Mills KH, Roche HM. Conjugated linoleic acid suppresses NF-kappa B activation and IL-12 production in dendritic cells through ERK-mediated IL-10 induction. J Immunol. 2005;175(8):4990–4998. | ||
Dhingra S, Sharma AK, Arora RC, Slezak J, Singal PK. IL-10 attenuates TNF-alpha-induced NF kappaB pathway activation and cardiomyocyte apoptosis. Cardiovasc Res. 2009;82(1):59–66. | ||
Hovsepian E, Penas F, Siffo S, Mirkin GA, Goren NB. IL-10 Inhibits the NF-κβ and ERK/MAPK-mediated production of pro-inflammatory mediators by up-regulation of SOCS-3 in Trypanosoma cruzi-infected cardiomyocytes. PLoS One. 2013;8(11):e79445. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.