Back to Journals » Therapeutics and Clinical Risk Management » Volume 19
Quality by Design: Development of Safe and Efficacious Full-Thickness Acellular Dermal Matrix Based on EuroGTPII Methodologies
Authors López-Chicón P, Pérez ML, Castells-Sala C, Piteira AR ![]() , Fariñas O, Tabera J, Vilarrodona A
, Fariñas O, Tabera J, Vilarrodona A
Received 1 March 2023
Accepted for publication 5 June 2023
Published 4 July 2023 Volume 2023:19 Pages 567—578
DOI https://doi.org/10.2147/TCRM.S410574
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Patricia López-Chicón,1,2,* Maria Luisa Pérez,1,3,* Cristina Castells-Sala,1,2,* Ana Rita Piteira,1,2,* Oscar Fariñas,1,2 Jaime Tabera,1,2 Anna Vilarrodona1,3
1Barcelona Tissue Bank (BTB), Banc de Sang i Teixits (BST), Barcelona, Spain; 2Biomedical Research Institute (IIB-Sant Pau; SGR1113), Barcelona, Spain; 3Vall Hebron Institute of Research (VHIR), Barcelona, Spain
*These authors contributed equally to this work
Correspondence: Maria Luisa Pérez, Barcelona Tissue Bank (BTB), Banc de Sang i Teixits (BST, GenCAT), Passeig Taulat 116, Barcelona, E-08005, Spain, Tel +34 935 57 35 00, Email [email protected]
Background: The activities of tissue establishments are constantly and rapidly evolving. The development of a new type of allograft, full-thickness acellular dermal matrix, with high mechanical properties to be used in tendon repair surgeries and abdominal wall reconstruction, has determined the need for quality by design process in order to assess evidence of quality, safety and efficacy. The EuroGTPII methodologies were specifically tailored to perform the risk assessment, identify and suggest tests in order to mitigate the potential risk consequences of a novel tissue preparation implementation.
Methods: The new allograft and associated preparation processes were assessed using the EuroGTP methodologies and characterized to properly evaluate the novelty (Step 1), identify and quantify the potential risks and risk consequences (Step 2), and define the extent of pre-clinical and clinical assessments required to mitigate the risks identified in the assessment (Step 3).
Results: Four risk consequences associated with the preparation process were identified: (i) implant failure related with tissue procurement and the reagents used during the decellularization protocol; (ii) unwanted immunogenicity related with the processing; (iii) disease transmission linked with the processing, reagents used, reduction in the reliability of microbiology testing and the storage conditions; and (iv) toxicity related to the reagents used and handling of the tissue during clinical application. The outcome of the risk assessment was a low level of risk. Nevertheless, it determined the need for a series of risk mitigation strategies proposed to reduce each individual risk and to provide additional evidence of the safety and efficacy of full-thickness acellular dermal matrix grafts.
Conclusion: EuroGTPII methodologies allow us to identify the risks and ensure the correct definition of pre-clinical assessments required to address and mitigate the potential risk consequences, before proceeding with clinical use of the new allografts in patients.
Graphical Abstract:
Keywords: EuroGTPII methodology, acellular dermis, risk assessment, quality, safety
Graphical Abstract:

Introduction
The development of novel tissues and tissue-based therapies is driven by the need to improve treatment options for patients or to address unmet clinical needs. The development of new tissue preparations must comply with high quality and safety standards according to the requirements of the European Union Tissues and Cells Directives (EUTCD) to ensure a high level of health protection.1 The European Good Tissue Practices (EuroGTP) project developed in 2009, for the first time, the guidelines for tissue establishments (TE) on the recovery, processing and preservation of tissues, to ensure that all TE guarantee the highest level of quality and safety of tissues for human application.2
However, the tissue preparations are constantly evolving and there is always a risk that any change in the donation, processing or preservation procedures, or in clinical application, can result in harm to the recipient. It is therefore vital to evaluate every potential risk of a process whenever any significant change is considered.3 Evaluation of the risk resulting from all aspects of the preparation process (from donor to patient) allows the proper design of studies that ensure the safety and quality of the new product. In recognition of the need to perform a risk-benefit analysis, the European Good Tissue and Cells Practices II (EuroGTPII) set up a systematic methodology with regard to pre-clinical and clinical evaluation of Substances of Human Origin (SoHO).4 This consists in a risk-based mechanism and an Interactive Assessment Tool (IAT: http://tool.goodtissuepractices.site), which allows evaluating if a new or changed tissue preparation has significant novelty; determine the overall risk arising from the novelty; determine an appropriate level of pre-clinical and clinical evaluations to address and assess the risk; implement the result of risk assessment into routine practice and follow up the results.5
Since 2019, the EuroGTP II methodology has been applied by the different European TE as a tool to identify, quantify and mitigate the risks associated with the development of novel preparation processes,6 and changes in any process.7 This has allowed a harmonized quantification of the risk level associated with the novel SoHO, the adoption of standard strategies to reduce it, and the definition of suitable clinical evaluations, required to demonstrate safety and efficacy.8
Tissue banking programmes in Europe started in 1970 and 1980s in response to an expanding clinical need for preserved skin allografts to be used as a coverage in major burns. First activities took place in laboratories stated directly in centres where burned patients were treated.9 Now, accredited skin banking facilities comply with EU Directives (EU 2004/23/EC) as well as national legislation (in Spain, RD-L 9/2014, 2014), meeting standards in accordance with European Directives 2006/17/EC and 2006/86/EC, as well as the Guide to the Quality and Safety of Tissues and Cells for Human Application (EDQM, 5th Ed.).
Bio-substitutes are widely used, but their high cost and limited accessibility make them unaffordable for some public health systems and inaccessible for patients. Among them, extracellular matrices (ECM) such as acellular dermal matrices (ADM)10 from different origins are a natural and biocompatible alternative, with successful outcomes in different applications.11–13 TE have developed dermal matrices of human origin, enabling access to safe and efficacious alternatives for soft tissue regeneration.14–17 Our TE has previously developed a split-thickness acellular dermal matrix that represents an effective alternative for the treatment of soft tissue loss such as gingival retraction.18
Nevertheless, the need for grafts with higher thickness and a wide variety of graft sizes to be applied in tendon reinforcement and closure of large wounds, eg, abdominal wall repair, has prompted the search for full-thickness ADM (ftADM). On one hand, rotator cuff repair is considered successful when there is complete healing of the tear in order to withstand high-tension rates after the repair and avoid re-tears; the use of a scaffold to cover the tear (patch augmentation surgery) enhances the speed and quality of the healing to improve tendon strength.19,20 In addition to providing a scaffold to support tissue remodeling, augmentation surgery improves mechanical properties in comparison to frail and injured tissue.21 In this context, it is paramount to ensure appropriate mechanical properties as critical attributes of the graft according to its final application. The stiffness of a graft, for example, represents the resistance to stretching, and is more representative of the graft performance clinically than tensile modulus. In our experience, greater stiffness is achieved by thicker grafts, while, as reported elsewhere, thinner grafts tend to tear with suture tying.19 Moreover, for irreparable massive rotator cuff tears, superior capsule reconstruction has recently been described entailing the fixation of a thick graft, achieving pain relief and improving function postoperatively.22,23 On the other hand, the main objective of ftADM in abdominal wall repair surgeries is to reinforce the tissues. Contrary to ventral hernia repair with synthetic meshes, bioprosthetic meshes like human ADM must be placed on a great deal of tension. If the elasticity in human ADM is inadequately addressed during the repair, laxity of the repair may occur,24 making ftADM an ideal graft for this type of surgery. Although human ADM has a high number of elastin fibers and is likely to stretch over time, long-term studies conclude that ADM provide durable repair with a low rate of recurrence.25 Moreover, cells responsible for rejection are removed in the processing of acellular dermal matrices, making ADM a durable scaffold for cellular and vascular ingrowth, promoting tissue regeneration and eventual integration with surrounding tissue, rather than encapsulation. Due to its revascularization capacity and incorporation with surrounding tissue, ADM is associated with lower rates of infection, extrusion, erosion, and adhesion formation compared to synthetic mesh.26 Hernia reconstruction may also benefit from the use of dermal matrices, not only because of their size and biomechanical properties but also due to their ability to remove bacterial contamination when there is a history of surgical site infection.27,28
The development of a decellularization protocol for full-thickness skin is closely related to the maintenance of the structural characteristics of the tissue, requiring the implementation of significant changes in the protocols associated with procurement, processing, decellularization, preservation and clinical application of ADM, previously validated and authorized in our TE.18 The procedure for preparation of ftADM required adaptation of this protocol to the size, thickness and anatomy of the full-thickness skin starting material.
The present study encompasses the procedures required for the safe implementation of an innovative tissue in the routine practice of our TE, including evaluation of novelty, risk assessment, design and performance of studies to mitigate the potential risks identified, and the definition of a specific Clinical Follow-up Plan (CFUpP), necessary to monitor safety and assess the efficacy of the ftADM allografts in patients.
Materials and Methods
The evaluation of novelty, risk assessment and definition of studies required to safely implement the ftADM were performed using the EuroGTPII methodologies and interactive assessment tool (http://tool.goodtissuepractices.site/). Briefly, the new product was characterized to properly evaluate the novelty (Step 1); thereafter, the risks associated with the novelty were identified and quantified through a risk assessment (Step 2). The results of this assessment give a Final Risk Score that was used to define the extent of the pre-clinical and clinical evaluation (Step 3). Flowchart is included in the Supplementary Information (Supplementary Figure 1).
Evaluation of Novelty
The first part of the methodology was intended to identify any change that could significantly affect the quality of the product and/or the safety of the recipients. This evaluation was carried out by answering seven key questions related to all the processes and activities of the supply chain, from donation to clinical application of the novel tissue (Table 1, Step 1, questions A to G).
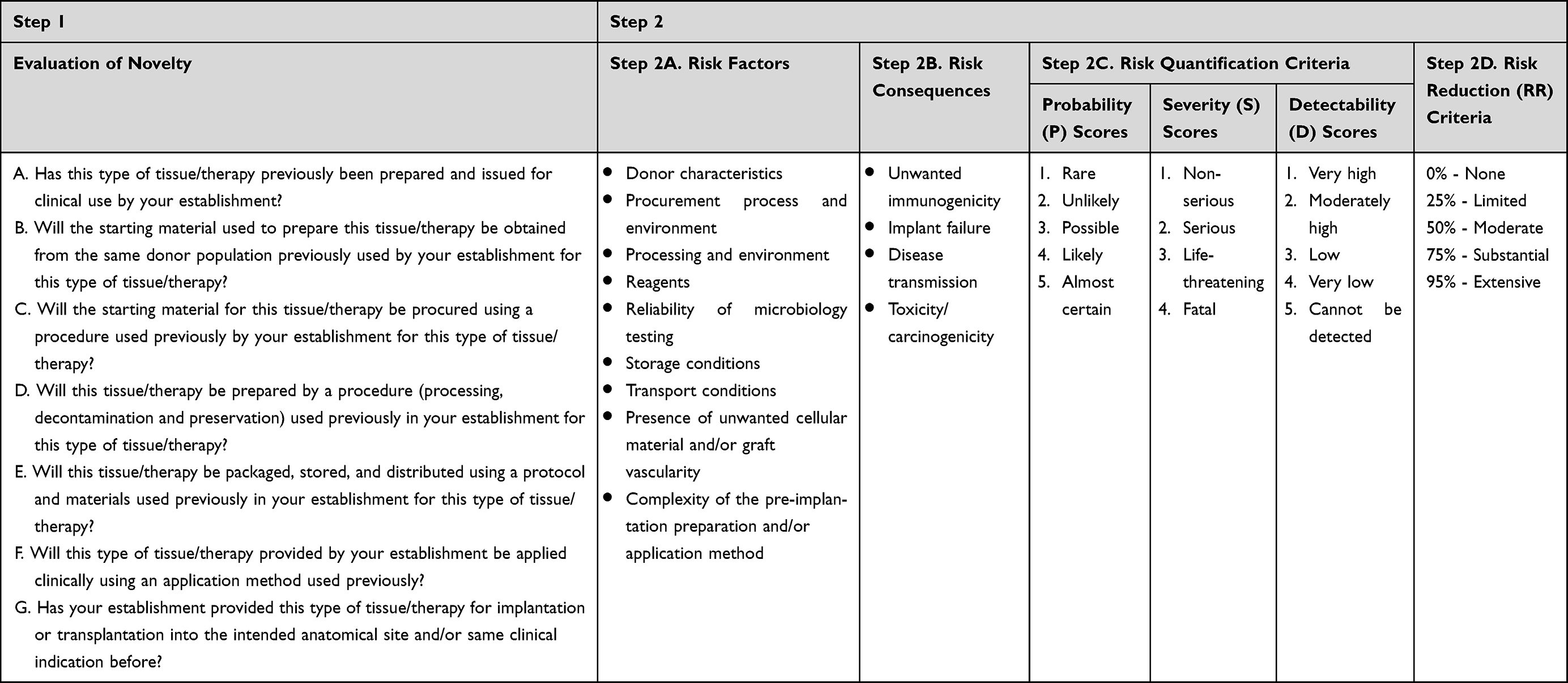 |
Table 1 Summary of EuroGTPII Methodology – Criteria and Scores Used for the Evaluation of Novelty and Risk Assessment3,4 |
Risk Assessment
The risk assessment was focused on identifying the risk factors and quantifying the risk consequences associated with the previously evaluated novelty. Again, all the processes from donor selection to clinical application were evaluated. The risk factors (Table 1, Step 2A) and their respective risk consequences (Table 1, Step 2B) were identified, evaluated in detail and quantified, awarding a score for each one (Table 1, Step 2).18 For each risk evaluated, a score (risk quantification criteria - Table 1, Step 2C) was defined for the probability of the risk occurring, the severity of the consequences and the ability to detect each individual risk consequence before clinical application. Any relevant data available to support reduction of the calculated risk scores were recorded and documented (Table 1, Step 2D).6 Supplementary Tables, are provided for the interpretation and quantification of the different parameters of the risk: assessment methodology: probability, severity, detectability and percentage of risk reduction (Supplementary Tables 1–4, respectively) following the definitions and instructions of the EuroGTPII guide – page 37−38.5
The outcome of the exercise was a single Final Risk Score, ie, a single overall risk score (ranging from 0 to 100). The possible categories were established: Negligible (0–2), Low (2–6), Moderate (6–22), and High (>22).
Estimation of the Final Risk Score was defined as follows:
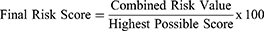
This Final Risk Score took into account the number of individual risks, defined as the Preliminary Score, and the Combined Risk Value as follows:
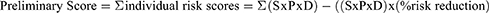
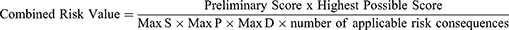
Where S is severity, P is probability, and D is detectability. The Applicable Number of risk consequences ranges from 1 to 45, and Highest Possible Risk Score is 4500.
Risk Reduction Strategies and Definition of the Extent of Clinical Evaluation
The Final Risk Score obtained from the risk assessment (Step 2) determined the corresponding extent of studies required to ensure the safety and efficacy of the novel ftADM. The definition of the extent of studies (Step 3) targeted the mitigation of each individual risk identified during the risk assessment exercise. The studies were designed following the strategies proposed in the EuroGTPII Guide5 to define the set of pre-clinical assays, including the validation studies, preparation process control strategy and key quality indicators required before clinical application of the newly developed grafts, and the definition of the CFUpP.
Results
Evaluation of Novelty
The evaluation of novelty was performed by answering the questions in the first step of the EuroGTPII methodologies (Table 2). The new procedures and ftADM specifications were compared with the previous experience of our TE with ADM and other skin products and procedures.18 This exercise identified four significant changes in the preparation process and clinical application of ftADM: i) use of a scalpel instead of a dermatome in the procurement procedure; ii) adaptation of the decellularization protocol from ADM, in terms of reagents and incubation times, to ensure the correct processing of skin with different anatomy, size, and thickness; iii) change in the concentration of glycerol as preservation medium; and iv) ftADM application at different anatomical sites, and for different clinical indications.
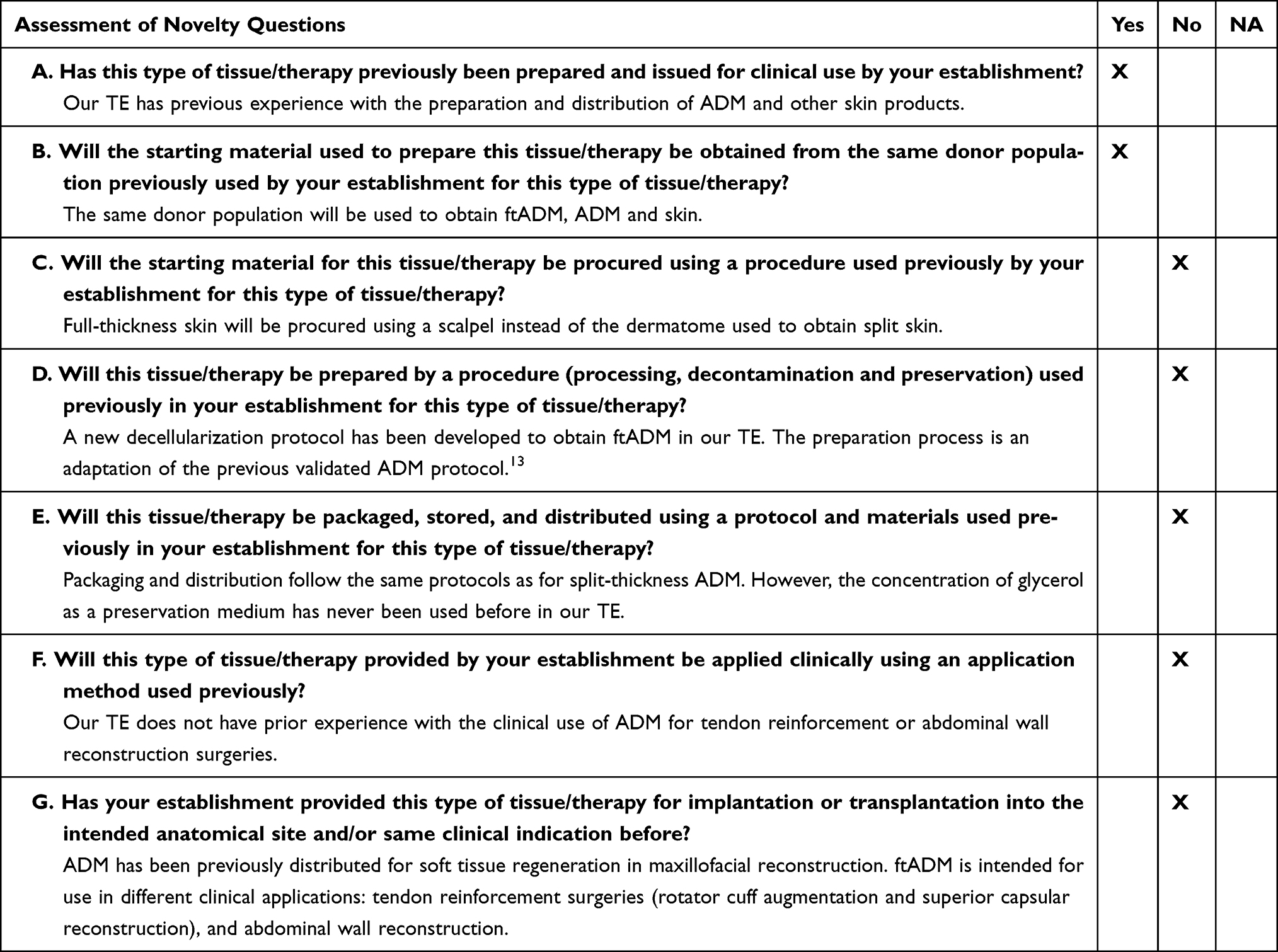 |
Table 2 Assessment of Novelty of the ftADM Grafts, According to Step 1 of the EuroGTPII Methodologies |
Risk Assessment
After evaluation of the novelty, the exercise proposed for Step 2 of the EuroGTPII methodologies was performed to evaluate the risks and their associated consequences for recipients. Table 3 shows the rationale and scoring obtained in this exercise. The risk assessment was performed considering the newly designed decellularization protocol for two different clinical applications: 1) tendon reinforcement and 2) hernia repair/abdominal wall reconstruction. As a result of the algorithm used in the interactive tool, the assessment indicated a low level of risk (Final Risk Score = 5), and that the ftADM grafts were safe and efficacious for clinical use and unlikely to cause harm to recipients.
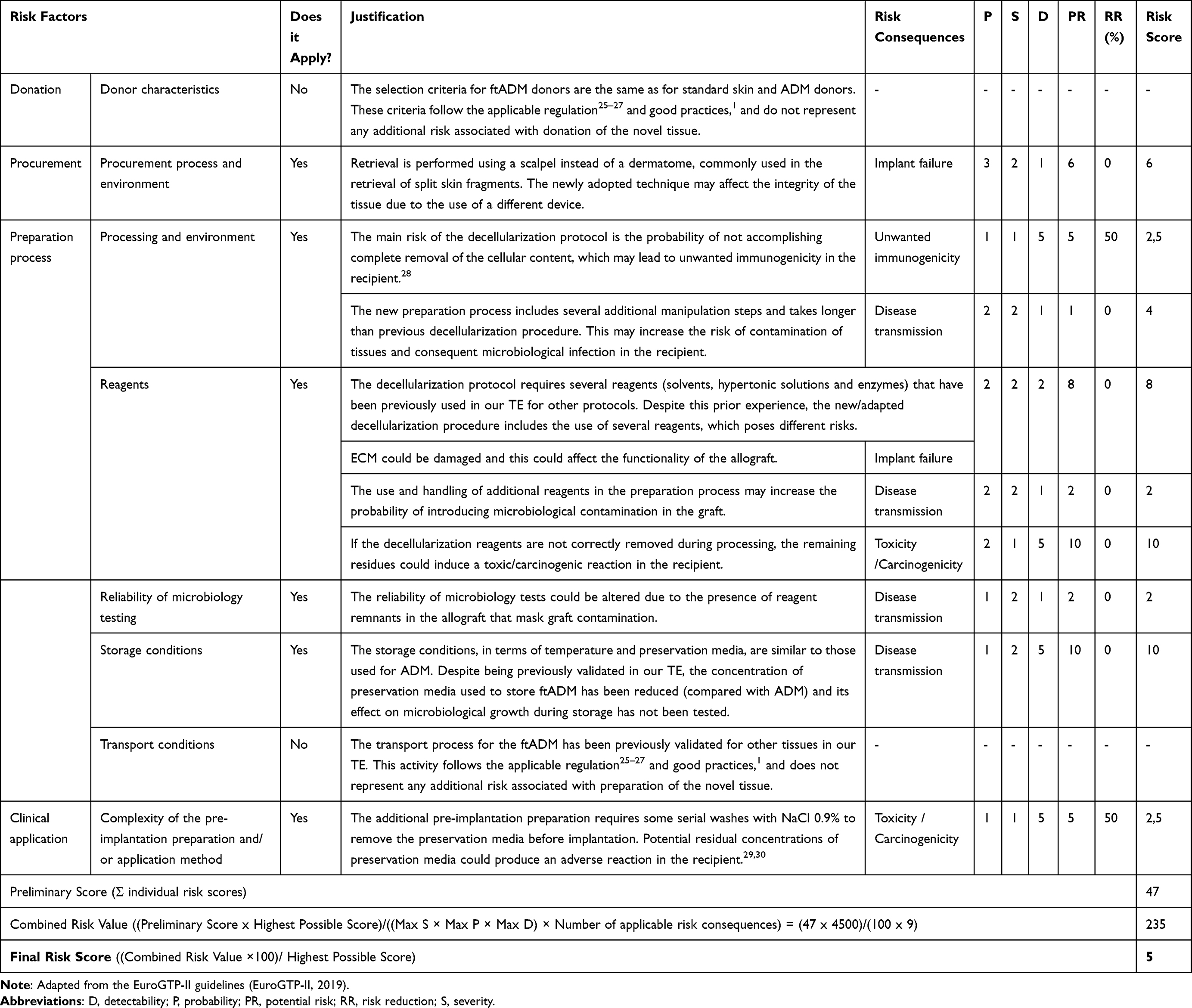 |
Table 3 Assessment of Risk Associated with the Implementation and Clinical Use of ftADM |
Risk Mitigation
The low level of risk calculated determined the need to perform an extensive validation of all new preparation procedures adopted to prepare ftADM grafts. Moreover, to mitigate the potential risk consequences identified, a set of specific pre-clinical studies were established in the following step (Table 4).
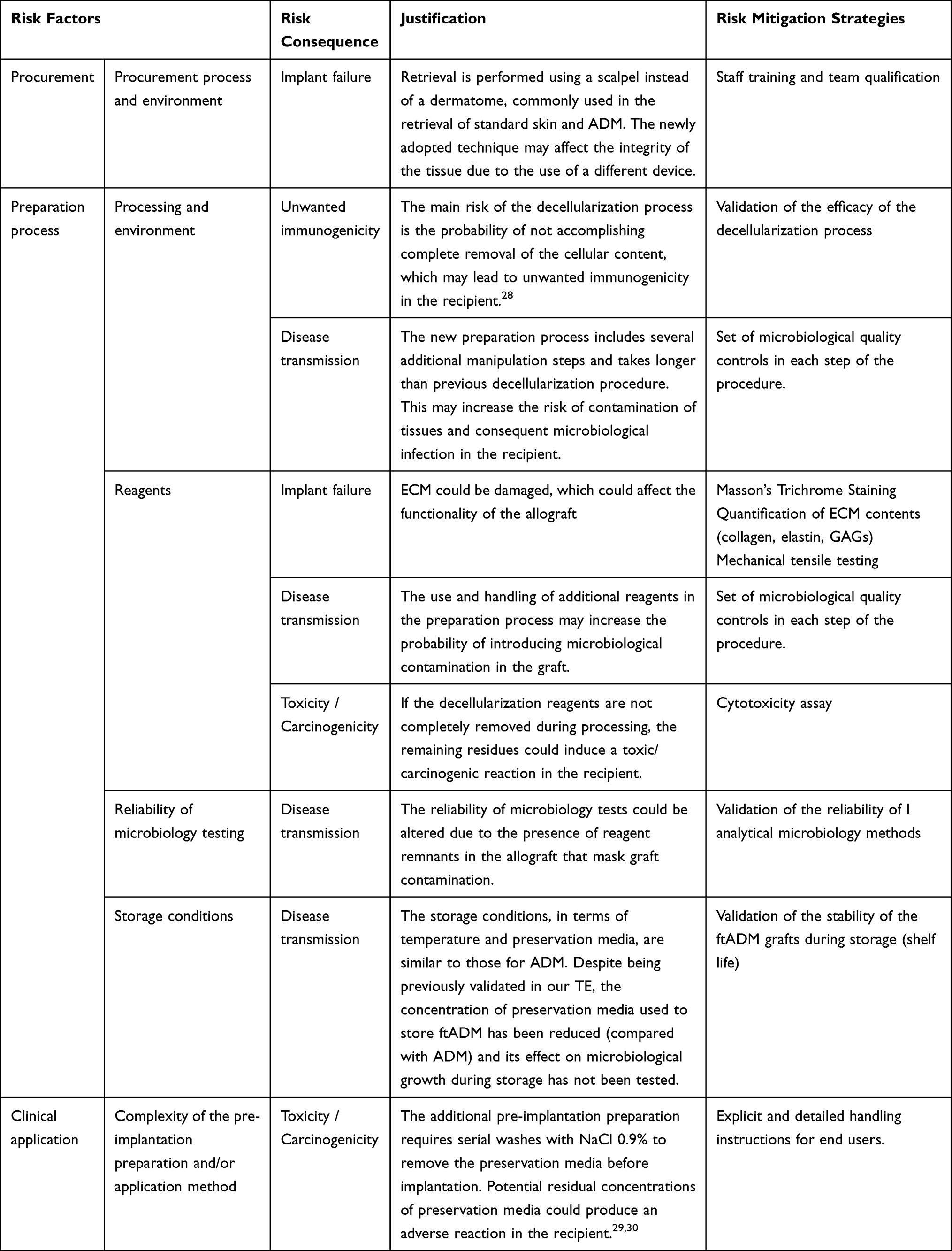 |
Table 4 Risk Mitigation Strategies Associated with the Implementation and Clinical Use of ftADM |
Discussion
EuroGTPII methodologies have identified the processes that represent new or unknown risks associated with clinical application of the newly developed ftADM grafts for tendon reinforcement and abdominal wall reconstruction. The risk factors were associated with each significant change in our original ADM protocol and allowed us to estimate the risk consequences and the overall level of risk. After identifying and quantifying the risks, different mitigation strategies were proposed to reduce each individual risk.
Four significant changes were identified through the use of EuroGTP methodologies, which led to the identification of four risk consequences: implant failure, unwanted immunogenicity, disease transmission, and toxicity/carcinogenicity. The outcome of the risk assessment exercise had a Final Risk Score of 5, corresponding to a low level of risk, and determined the extent of studies required to ensure the safety and efficacy of the ftADM grafts in terms of pre-clinical and clinical evaluation. According to EuroGTPII methodologies, after identifying and quantifying the risks, different mitigation strategies are proposed to reduce each individual risk.
The risk of implant failure was perceived as a risk consequence due to two risk factors: procurement and reagents. During procurement, the retrieval of full thickness skin fragments will be performed using scalpels instead of the dermatomes commonly used in the retrieval of split thickness skin. The newly adopted technique may affect the integrity of the tissue due to the use of a different device. To mitigate this risk, all the staff involved in the procurement of full-thickness skin will be trained and qualified to perform the new retrieval procedure to avoid blade lesions on skin. With regard to the reagents, it has been reported that ECM could be damaged, which may affect the functionality of the allograft. To reduce this risk, the integrity of the ECM will be validated by performing a set of in vitro tests (biochemical quantifications, Masson’s Trichrome histological staining) to evaluate the preservation of major ECM biomolecules (collagen, elastin and GAGs) in the ftADM. Moreover, a uniaxial biomechanical test will be performed to assess the suitability of mechanical properties in the final graft. The results of this set of tests will demonstrate the integrity of the ECM after the decellularization treatment.
The second risk consequence identified was potential unwanted immunogenic reactions in the recipient, caused by incomplete removal of the cellular content during the decellularization process. The efficacy of the decellularization procedure will be assessed through a set of in vitro tests (DNA quantification and Hematoxylin-Eosin histological staining) to determine the presence of cellular remnants in ftADM after decellularization. The results of this mitigation strategy must confirm the absence of cell nuclei in the histology testing, and that the DNA content is below 50 ng/mg in the dry tissue.29
The third risk consequence identified was potential disease transmission, related to the new steps in the preparation process, use of reagents, reliability of microbiology testing and the storage conditions. Due to the characteristics of full thickness skin, the ADM preparation process was modified, increasing the length of the procedure by one day, and including several additional manipulation steps. The use of additional reagents during the process may increase the probability of introducing microbiological contamination in the graft. Furthermore, the thickness of the skin may be an obstacle for removal of reagents used during processing, thereby potentially reducing the reliability of microbiology testing due to the presence of remnants that could mask graft contamination in routine quality controls. Moreover, although the storage conditions (temperature and preservation media) are similar to those previously validated for ADM, the concentration of preservation media used to store ftADM will be reduced with respect to ADM and could increase the chance of microbiological growth during storage. These factors may increase the risk of tissue contamination and consequent microbiological infection in the recipient. To avoid these risks, the decontamination efficacy of the entire process will be validated through (i) validation of the efficacy of the antibiotic/antimitotic decontamination cocktail, (ii) validation of the analytical method used as a microbiology test, (iii) implementation of new microbiology controls at different stages of the process, and (iv) implementation of a final filtration step of the reagents involved in the process. In addition, all reagents will be prepared in a closed system and a microbiology test will be performed on each one by the end of the aliquot process. The absence of microbiological growth during storage will be confirmed through accelerated and ongoing in vitro stability assays. To accept the mitigation of disease transmission risk, all validation and implementation procedures must demonstrate that the tests are able to detect each microorganism previously inoculated. Moreover, the results of microbiology tests performed in each reagent used during the preparation, and the results of the stability tests, must be negative for both accelerated and ongoing tests.
The fourth risk consequence is toxicity/carcinogenicity of the final product. This risk consequence is related to the reagents used and the complexity of the handling procedures before clinical application. If the decellularization reagents are not completely removed during processing, the remaining residues could induce a toxic/carcinogenic reaction in the recipient. To mitigate this risk, a series of rinse steps were included in the procedure to eliminate the remaining residues. The cytotoxicity study will be performed following the cell culture model defined in ISO directive 10,993–5. To accept the results of this mitigation strategy, the cell viability must be ≥70%. Furthermore, glycerol preservation requires serial washes with NaCl 0.9% to remove the preservation media before implantation. Potential residual concentrations of preservation media could produce an adverse reaction in the recipient.30–32 Although our TE cannot directly mitigate this risk, explicit handling instructions will be sent to the clinicians/end users and the document will be added to the ftADM packaging.
The risk reduction process is supported by our prior experience in the preparation and manipulation of ADM, as well as a significant amount of relevant literature.14,16–18 Nevertheless, despite the low level of risk obtained for ftADM, the use of new procedures and reagents and the graft characteristics determined the need for intensive validation of our internal protocols in order to ensure the correct specifications and the safety of the allograft before its clinical use. The numerous pre-clinical investigations performed aimed either to reduce the probability of risk consequences occurring or to increase the detectability in case of process deviations, namely tissue contamination during the preparation process.
The risk assessment exercise and the results subsequently obtained will support the submission of the preparation process dossier to our competent authority, demonstrating the safety of our newly developed graft. Further CFUpP will address potential risk consequences that were not fully mitigated in the pre-clinical assessments and will focus mainly on the mandatory reporting of serious adverse reactions and the long-term efficacy of the grafts, as suggested by the EuroGTPII methodologies for grafts/therapies with a low level of risk.
Conclusion
EuroGTPII methodologies allowed us to identify and quantify the risks associated with the introduction of innovation in our activities. In addition, the use of this standard methodology generates a complete report on the rationale followed during the development and validation of a novel therapy and documents the studies required to address and mitigate the risks, thereby promoting transparency and expediting authorization procedures by competent authorities.
Despite the low level of risk determined for the novel ftADM preparation process, a set of pre-clinical assessments were needed to address and mitigate the potential risk consequences and to guarantee a high level of safety for the clinical application of ftADM. The development of novel grafts and preparation processes through the quality-by-design methodologies proposed by the EuroGTPII tools will lead to optimized and continuous improvement of the products and therapies developed by our TE.
Data Sharing Statement
No new data were generated or analyzed in support of this research.
Acknowledgments
We would like to thank to all donors and their relatives for the altruistic act of donation. We would also like to thank transplant coordinators, the Donor Centre and recovery teams for their daily efforts.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the European Union’s Health Programme (2014–2020), Grant Agreement number: 709567 – EuroGTP II – HP-PJ-2015. This study represents the views of the authors only and is their sole responsibility; it cannot be considered to reflect the views of the European Commission and/or the Consumers, Health, Agriculture and Food Executive Agency or any other body of the European Union. The European Commission and the Agency do not accept any responsibility for use that may be made of the information it contains.
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. EDQM; Council of Europe. Guide to the Quality and Safety of Tissues and Cells for Human Application. EDQM; 2019.
2. EuroGTP. (2007 207) - Good tissue practices; 2007. Available from: http://eurogtps.com.
3. [VISTART] [676969]; Vigiliance and Inspection for the Safety of Transfusion AR and T. Principles for competent authorities for the evaluation and approval of clinical follow up protocols for blood, tissues and cells prepared with newly developed and validated processing methods; 2018:1–32.
4. Trias E, Lomas R, Tabera J, et al. EuroGTP II: a tool to assess risk, safety and efficacy of substances of human origin. Int J Qual Heal Care. 2019;32(1):80–84. doi:10.1093/intqhc/mzz048
5. EuroGTPII (709567)- Good Practices for demonstrating safety and quality through recipient follow-up. Euro GTP II Guide - good practices for evaluating safety, quality and efficacy of tissue and cellular therapies and products; 2020.
6. Trias E, Gallon P, Ferrari S, et al. Banking of corneal stromal lenticules: a risk-analysis assessment with the EuroGTP II interactive tool. Cell Tissue Bank. 2019;21(2):189–204. doi:10.1007/s10561-020-09813-8
7. Trias E, Nijs M, Rugescu IA, et al. Evaluating risk, safety and efficacy of novel reproductive techniques and therapies through the EuroGTP II risk assessment tool. Hum Reprod. 2020;35(8):1821–1838. doi:10.1093/humrep/deaa146
8. Lierman S, Bus A, Andries S, Trias E, Bols PEJ, Tilleman K. Passive slow freezing is an efficacious and cost-effective alternative to controlled slow freezing for ovarian tissue cryopreservation. Cryobiology. 2021;100(2020):164–172. doi:10.1016/j.cryobiol.2021.01.013
9. Tognetti L, Pianigiani E, Ierardi F, et al. Current insights into skin banking: storage, preservation and clinical importance of skin allografts. J Biorepository Sci Appl Med. 2017;5:41–56. doi:10.2147/bsam.s115187
10. Sobti N, Ji E, Brown RL, et al. Evaluation of acellular dermal matrix efficacy in prosthesis-based breast reconstruction. Plast Reconstr Surg. 2018;141(3):541–549. doi:10.1097/PRS.0000000000004109
11. Barber FA, Burns JP, Deutsch A, Labbé MR, Litchfield RB. A prospective, randomized evaluation of acellular human dermal matrix augmentation for arthroscopic rotator cuff repair. Arthrosc J Arthrosc Relat Surg. 2012;28(1):8–15. doi:10.1016/j.arthro.2011.06.038
12. Gelber PE, Erquicia JI, Ramírez-Bermejo E, Fariñas O, Monllau JC. Fresh osteochondral and meniscus allografting for post-traumatic tibial plateau defects. Arthrosc Tech. 2018;7(6):e661–e667. doi:10.1016/j.eats.2018.02.010
13. Asaad M, Kapur SK, Baumann DP, Liu J, Butler CE. Acellular dermal matrix provides durable long-term outcomes in abdominal wall reconstruction. Ann Surg. 2020. doi:10.1097/sla.0000000000004454
14. Richters CD, Pirayesh A, Hoeksema H, et al. Development of a dermal matrix from glycerol preserved allogeneic skin. Cell Tissue Bank. 2008;9(4):309–315. doi:10.1007/s10561-008-9073-4
15. Ghetti M, Bondioli E, Purpura V, Cenacchi G, Ruscelli P, Melandri D. Decellularized human dermal matrix produced by a skin bank A new treatment for abdominal wall defects. Ann Ital Chir. 2017;5:443–448.
16. Hogg P, Rooney P, Ingham E, Kearney JN. Development of a decellularised dermis. Cell Tissue Bank. 2013;14(3):465–474. doi:10.1007/s10561-012-9333-1
17. Dragúňová J, Kabát P, Babál P, et al. Development of a new method for the preparation of an acellular allodermis, quality control and cytotoxicity testing. Cell Tissue Bank. 2017;18(2):153–166. doi:10.1007/s10561-017-9625-6
18. Pérez ML, Castells-sala C, López-chicón P, Nieto-nicolau N, Aiti A. Fast protocol for the processing of split-thickness skin into decellularized human dermal matrix. Tissue Cell. 2021;72:101572. doi:10.1016/j.tice.2021.101572
19. Chalmers PN, Tashjian RZ. Patch augmentation in rotator cuff repair. Curr Rev Musculoskelet Med. 2020;13(5):561–571. doi:10.1007/s12178-020-09658-4
20. Avanzi P, Dei Giudici L, Capone A, et al. Prospective randomized controlled trial for patch augmentation in rotator cuff repair: 24-month outcomes. J Shoulder Elb Surg. 2019;28(10):1918–1927. doi:10.1016/j.jse.2019.05.043
21. Barber FA, Aziz-Jacobo J. Biomechanical testing of commercially available soft-tissue augmentation materials. J Arthrosc Relat Surg. 2009;25(11):1233–1239. doi:10.1016/j.arthro.2009.05.012
22. Wall KC, Toth AP, Garrigues GE. How to use a graft in irreparable rotator cuff tears: a literature review update of interposition and superior capsule reconstruction techniques. Curr Rev Musculoskelet Med. 2018;11(1):122–130. doi:10.1007/s12178-018-9466-3
23. Hirahara AM, Adams CR. Arthroscopic superior capsular reconstruction for treatment of massive irreparable rotator cuff tears. Arthrosc Tech. 2015;4(6):e637–e641. doi:10.1016/j.eats.2015.07.006
24. Diaz JJ, Conquest AM, Ferzoco SJ, et al. Multi-institutional experience using human acellular dermal matrix for ventral hernia repair in a compromised surgical field. Arch Surg. 2009;144(3):209–215. doi:10.1001/archsurg.2009.12
25. Maxwell DW, Hart AM, Keifer OP, Halani SH, Losken A. A comparison of acellular dermal matrices in abdominal wall reconstruction. Ann Plast Surg. 2019;82(4):435–440. doi:10.1097/SAP.0000000000001692
26. Garvey PB, Giordano SA, Baumann DP, Liu J, Butler CE. Long-term outcomes after abdominal wall reconstruction with acellular dermal matrix. J Am Coll Surg. 2017;224(3):341–350. doi:10.1016/j.jamcollsurg.2016.11.017
27. Espinosa-De-Los-Monteros A, De La Torre JI, Marrero I, Andrades P, Davis MR, Vásconez LO. Utilization of human cadaveric acellular dermis for abdominal hernia reconstruction. Ann Plast Surg. 2007;58(3):264–267. doi:10.1097/01.sap.0000254410.91132.a8
28. Albo D, Awad SS, Berger DH, Bellows CF. Decellularized human cadaveric dermis provides a safe alternative for primary inguinal hernia repair in contaminated surgical fields. Am J Surg. 2006;192(5):12–17. doi:10.1016/j.amjsurg.2006.08.029
29. Crapo PM, Gilbert TW, Badylak SF. An Overview of tissue and whole organ decellularisation processes. Biomaterials. 2011;32(12):3233–3243. doi:10.1016/j.biomaterials.2011.01.057.An
30. Saegeman VSM, Ectors NL, Lismont D, Verduyckt B, Verhaegen J. Short- and long-term bacterial inhibiting effect of high concentrations of glycerol used in the preservation of skin allografts. Burns. 2008;34(2):205–211. doi:10.1016/j.burns.2007.02.009
31. Khoo TL, Halim AS, Saad AZM, Dorai AA. The application of glycerol-preserved skin allograft in the treatment of burn injuries: an analysis based on indications. Burns. 2010;36(6):897–904. doi:10.1016/j.burns.2009.03.007
32. Becker LC, Bergfeld WF, Belsito DV, et al. Safety assessment of glycerin as used in cosmetics. Int J Toxicol. 2019;38(3_suppl):6S–22S. doi:10.1177/1091581819883820
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Donor Safety, Discrepancies Between Practice and Theory: Analysis of the Polish Supreme Audit Office’s Report
Patryn R, Zagaja A, Drozd M
The Application of Clinical Genetics 2023, 16:1-10
Published Date: 20 January 2023