Back to Journals » Drug Design, Development and Therapy » Volume 8
QSAR and docking studies on xanthone derivatives for anticancer activity targeting DNA topoisomerase IIα
Received 15 July 2013
Accepted for publication 10 September 2013
Published 31 January 2014 Volume 2014:8 Pages 183—195
DOI https://doi.org/10.2147/DDDT.S51577
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Sarfaraz Alam, Feroz Khan
Metabolic and Structural Biology Department, Central Institute of Medicinal and Aromatic Plants, Council of Scientific and Industrial Research, Lucknow, Uttar Pradesh, India
Abstract: Due to the high mortality rate in India, the identification of novel molecules is important in the development of novel and potent anticancer drugs. Xanthones are natural constituents of plants in the families Bonnetiaceae and Clusiaceae, and comprise oxygenated heterocycles with a variety of biological activities along with an anticancer effect. To explore the anticancer compounds from xanthone derivatives, a quantitative structure activity relationship (QSAR) model was developed by the multiple linear regression method. The structure–activity relationship represented by the QSAR model yielded a high activity–descriptors relationship accuracy (84%) referred by regression coefficient (r2=0.84) and a high activity prediction accuracy (82%). Five molecular descriptors – dielectric energy, group count (hydroxyl), LogP (the logarithm of the partition coefficient between n-octanol and water), shape index basic (order 3), and the solvent-accessible surface area – were significantly correlated with anticancer activity. Using this QSAR model, a set of virtually designed xanthone derivatives was screened out. A molecular docking study was also carried out to predict the molecular interaction between proposed compounds and deoxyribonucleic acid (DNA) topoisomerase IIα. The pharmacokinetics parameters, such as absorption, distribution, metabolism, excretion, and toxicity, were also calculated, and later an appraisal of synthetic accessibility of organic compounds was carried out. The strategy used in this study may provide understanding in designing novel DNA topoisomerase IIα inhibitors, as well as for other cancer targets.
Keywords: drug likeness, ADMET, regression model, HeLa cell line
Introduction
Drug discovery and development is not only a time-consuming process, but also a costly procedure. Therefore, we wanted to apply computational methods for lead generation and lead optimization in the drug discovery process. This emerging trend has immense importance in reducing the phase time, as well as in amplifying the design of small molecule-based leads with better biological activity and minimal side effects for a disease-specific target. After the development of the first peptide-based HIV protease inhibitors,1 followed by a target for antihypertension2 and inhibitors of the H5N1 avian influenza,3 scientists are paying more attention to the in silico approach. Even with such improvements, the design of a novel anticancer drug that works effectively on a patient is still out of reach. Cancer, which is the uncontrolled growth and proliferation of cells due to mutation of genes which accelerate cell division rates and evade the programmed cell death, is the leading cause of death in the world.4 The frequency of one particular manifestation of cancer, cervical cancer, is dramatically increasing. A link between cancer and human deoxyribonucleic acid (DNA) topoisomerase type IIα (Top2A) (enzyme commission number [EC]: 5.99.1.3)5 has already been ascribed, and there is an interest in developing a specific inhibitor as a new therapeutic regimen for the cancer.
Xanthones, which are used in this study, comprise a large number of oxygenated heterocycles which play an important role in medicinal chemistry. Their derivatives are widely distributed in various plants, and they have a variety of biological properties, such as antioxidant, hepato-protective, anti-inflammatory, anti-α-glucosidase, and anticancer properties.6 Due to their antitumor effect, xanthones are attracting more interest. Until now, there have been only a few computational studies on xanthone; also, the protein targets of xanthones have not yet received a great deal of attention.6
Traditionally it is difficult to select the best chemical moiety of compound that plays an effective role in treating or preventing cancer, so we used computational strategies that include quantitative structure activity relationship (QSAR) modeling, virtual screening, shape similarity screening, pharmacophore searching, molecular docking, and ADMET (absorption, distribution, metabolism, excretion, and toxicity properties of a molecule within an organism) studies to identify potential protein targets of xanthone and other phytochemicals.7 Using these computational methodologies, we demonstrate a multiple linear regression QSAR model for activity prediction that successfully predicts the anticancer activities of newly designed xanthone derivatives. In the QSAR model, the regression coefficient (r2), which indicates the relationship correlation, was 0.84, while the cross-validation regression coefficient (r2CV), which indicates the prediction accuracy of the model, was 0.82. The QSAR study indicates that dielectric energy, group count (hydroxyl), LogP, shape index basic (order 3), and solvent-accessible surface area were significantly correlated with anticancer activity. After successful validation of this model, it was then used to design and virtually screen 50 compounds and identify 39 with IC50 values of ≤20 μM. Lipinski’s rule of five was used to filter the compounds and was further accompanied by molecular docking studies, which were performed for predicting active compounds against highly promising anticancer drug targeting (Figure 1). Since in humans the drug target protein for doxorubicin (DrugBank ID: DB00997) is Top2A, we selected it as a target protein. This target is widely used for existing anticancer agents: eg, etoposide; anthracyclines (doxorubicin, daunorubicin); and mitoxantrone. These drugs work either through the poison of topoisomerase II cleavage complexes or by inhibiting the ATPase activity by acting as noncompetitive inhibitors of adenosine triphosphate (ATP).8 A docking study was carried out to identify the putative binding site of active xanthone derivatives (which could be helpful in explaining the underlying structure–activity relationship), by using a crystal structure of inhibitor-bound Top2A. Based on the QSAR model, molecular docking, ADMET, and synthesis accessibility, we then identified four inhibitors with IC50 values of 7.94 μM, 0.63 μM, 2.51 μM, and 0.16 μM, as potent inhibitors of Top2A (Figure 1). This study is a significant approach in the identification of hits compounds with structural diversity, which may provide further helpful insights to screening and designing novel anticancer compounds and their respective protein targets. Moreover, this study is also projected to explore the molecular mechanism by which xanthone derivatives can be further utilized with better activity by rational modifications.
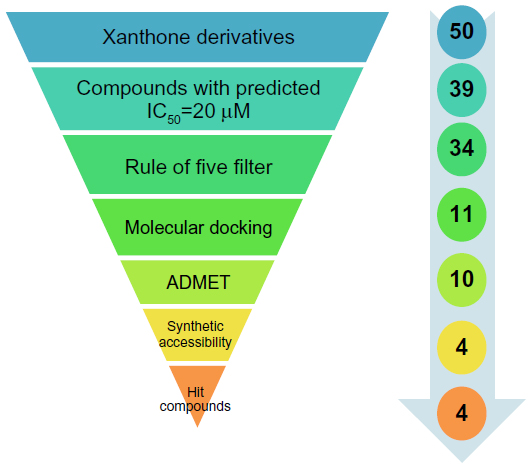 | Figure 1 Virtual screening protocol for the identification of novel DNA Top2A inhibitors. |
Methods and computational details
Structure cleaning
Drawing and geometry cleaning of compounds with anticancer activity was performed using ChemBioDraw Ultra version 12.0 (2010) software (PerkinElmer Informatics, Waltham, MA, USA). The two-dimensional (2D) structures were transformed into three-dimensional (3D) structures using the converter module of ChemBio3D Ultra. The 3D structures were then subjected to energy minimization, which was performed in two steps. The first step was energy minimization using molecular mechanics-2 (MM2) until the root mean square (RMS) gradient value became smaller than 0.100 kcal/mol/Å; then in a second step, minimized MM2 (dynamics) compounds were subjected to reoptimization through the MOPAC (Molecular Orbital Package, ChemBioDraw Ultra version 12.0 [2010] software; PerkinElmer Informatics, Waltham, MA, USA) method, until the RMS gradient attained a value smaller than 0.0001 kcal/mol/Å.
Parameters for QSAR model development
In the present study, cancer cell line-based QSAR modeling was performed. Initially, a total of 64 compounds with reported anticancer activity against the human cervical cancer cell line (HeLa) were used as a training data set while developing the QSAR model (Tables 1 and S1).6,9–16 The anticancer activity was in IC50 form. A total of 52 chemical descriptors (physicochemical properties) were calculated for each compound. The selection was made on the basis of structural/pharmacophore or chemical class similarity. Similarly, in order to select the best subset of descriptors, highly correlated descriptors were excluded. Finally, a model was developed based on the forward stepwise multiple linear regression method. The resulting QSAR model exhibited a high regression coefficient. The model was successfully validated using random test set compounds (Table S2), and was evaluated for the robustness of its predictions via the cross-validation coefficient.
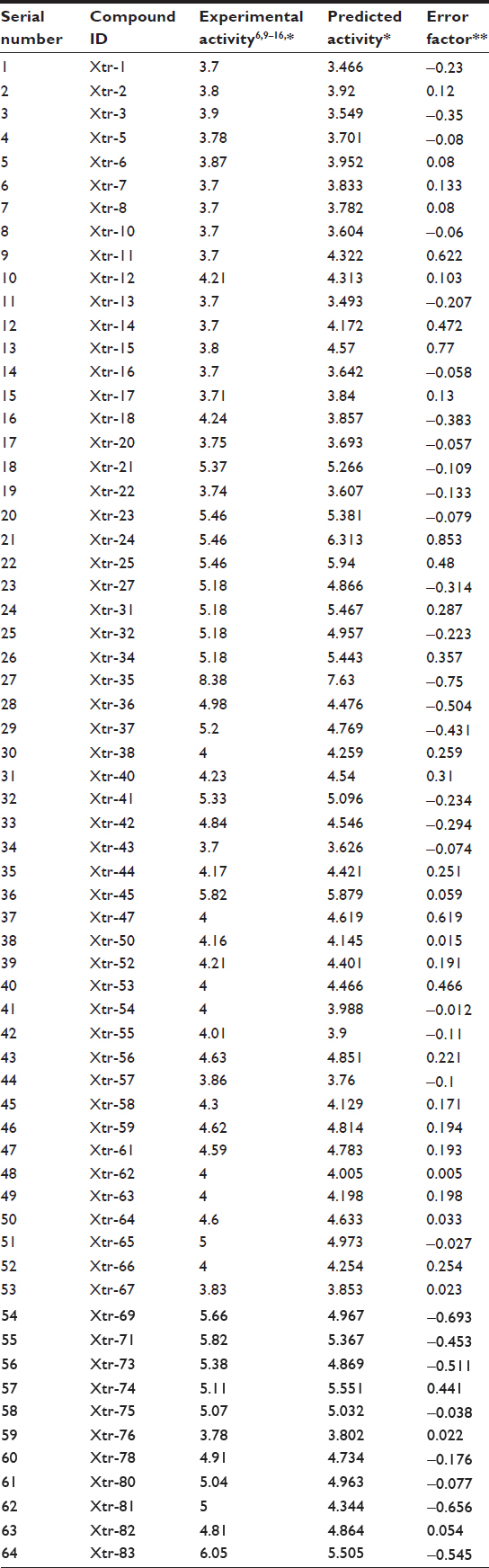 | Table 1 Comparison of experimental and predicted activities of training data set molecules based on QSAR model |
Various descriptors like steric, electronic, and thermodynamic were calculated by the Scigress Explorer software (Fujitsu, Tokyo, Japan). For the validation of QSAR models, the leave one out method was used;17 the best model was selected on the basis of various statistical parameters, such as a square of the correlation coefficient (R2), and the quality of each model was estimated from the cross-validated squared correlation coefficient (rCV2).
Statistical calculations used in QSAR modeling
The stepwise multiple linear regression method calculates QSAR equations by adding one variable at a time and testing each addition for significance. Only variables that are found to be significant are used in the QSAR equation. This regression method is especially useful when the number of variables is large and when the key descriptors are not known. In the forward mode, the calculation begins with no variables and builds a model by entering one variable at a time into the equation. In the backward mode, the calculation begins with all variables included and drops variables one at a time until the calculation is complete; however, backward regression calculations can lead to overfitting.
Multiple regression correlation coefficient
Variations in the data are quantified by the correlation coefficient (r), which measures how closely the observed data track the fitted regression line. This is a measure of how well the equation fits the data (ie, it measures how good the correlation is). A perfect relation has r=+1 (positively correlated) or −1 (negatively correlated); no correlation has r=0. The regression coefficient, r2, is sometimes quoted, and this gives the fraction of the variance (in percentage) that is explained by the regression line. The more scattered the data points, the lower the value of r. A satisfactory explanation of the data is usually indicated by an r2. Errors in either the model or in the data will lead to a bad fit. This indicator of fit to the regression line is calculated as:

where the regression variance is defined as the original variance minus the variance around the regression line. The original variance is the sum of the squares of the distances of the original data from the mean.
Validating QSAR equations and data
The cross-validation coefficient, rCV2, can be calculated as
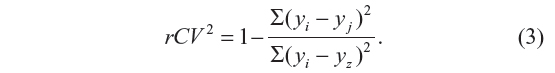
Here, yi and yj are the measured and predicted values of dependent variables, respectively. yz is the averaged value of dependent variable of the training set.
Leave one out cross-validation
Leave one out cross-validation (LOOCV) is one of the most effective methods for validation of a model with a small training dataset. Here, training is done with a data size of (N–1) and tested the remaining one, where N represents the complete set of data. In the LOOCV method, the training and testing are repeated for N amount of time, so as to pass each individual data through the testing process.
Virtual designing of novel xanthone derivatives
The 50 compounds (Table S3) were virtually designed and then validated. The QSAR model was used to predict the biological responses to these chemical structures.
Rule of five filters
All the chemical structures are evaluated for good oral bioavailability in order to be an effective drug-like compound, subject to Lipinski’s rule of five.18 According to this rule, a drug-like molecule should have not more than one of the following violations: no more than five hydrogen bond donors; no more than ten hydrogen bond acceptors; molecular weight no more than 500; and LogP no more than 5.
Protein preparation
The protein preparation protocol is used to perform tasks such as inserting missing atoms in incomplete residues, deleting alternate conformations (disorder), removing waters, standardizing the names of the atoms, modeling missing loop regions, and protonating titratable residues by using predicted pKs (negative logarithmic measure of acid dissociation constant). CHARMM (Chemistry at HARvard Macromolecular Mechanics; Cambridge, MA, USA) is used for protein preparation with an energy of −31.1116, initial RMS gradient energy of 181.843, and grid spacing of 0.5 angstrom (Å). The hydrogen atoms were added before the processing. Protein coordinates from the crystal structure of Top2A (PDB [Protein Data Bank] ID: 1ZXM) Chain A determined at a resolution of 1.87 Å were used (Figure 2).
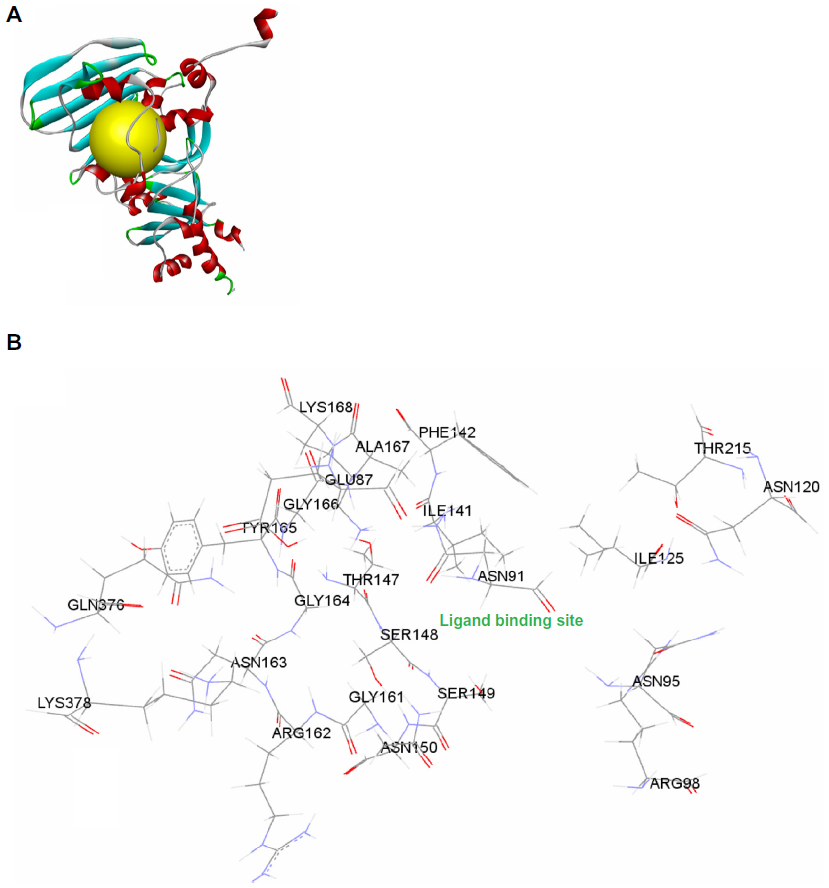 | Figure 2 (A) Structural model of human DNA Top2A (PDB ID: 1ZXM) with ATP binding site (yellow); (B) ATP binding site pocket residues. |
Protein–ligand docking
Molecular docking studies were performed to generate the bioactive binding poses of inhibitors in the active site of enzymes by using the LibDock program from Discovery Studio, version 3.5 (Accelrys, San Diego, CA, USA). LibDock uses protein site features, referred to as hot spots, consisting of two types (polar and apolar). The ligand poses are placed into the polar and apolar receptor interactions site. In the current study, the Merck Molecular Force Field was used for energy minimization of the ligands. The binding sphere was primarily defined as all residues of the target within 5 Å from the first binding site. Here, the ATP binding site was used to define the active site, referred to as the hot spots (Figure 2). Conformer Algorithm based on Energy Screening And Recursive build-up (CAESAR) was used for generating conformations. Then, the smart minimizer was used for in situ ligand minimization. All other docking and consequent scoring parameters used were kept at their default settings.
We also analyzed the protein ligand complexes to better understand the interactions between protein residues and bound ligands, along with the binding site residues of the defined receptor. The 2D diagrams helped to identify the binding site residue, including amino acid residues, waters, and metal atoms.
The score ligand poses protocol was used for the scoring functions, such as LibDock score, Jain, LigScore 1, LigScore 2, piecewise linear potential (PLP) and potential of mean force (PMF) 04, to evaluate ligand binding in a receptor cavity.
Validation using AutoDock Vina
AutoDock Vina19 software (Scripps Research Institute, La Jolla, CA, USA) was also used for molecular docking studies to validate the LibDock score. For this, the designed compounds were optimized and then used for docking experiments. The same binding site and receptor used in the LibDock program are used for this study. The docking program takes the PDBQT file format of ligands and receptor, a modified PDB file, which has added polar hydrogens and partial charges. Other docking parameters were set to the software’s default values.
Pharmacokinetics parameters
ADMET refers to the absorption, distribution, metabolism, excretion, and toxicity properties of a molecule within an organism, and were predicted using ADMET descriptors in Discovery Studio 3.5 (Accelrys). In this module, six mathematical models (aqueous solubility, blood–brain barrier penetration, cytochrome P450 2D6 inhibition, hepatotoxicity, human intestinal absorption, and plasma protein binding) are used to quantitatively predict properties of a set of rules that specify ADMET characteristics of the chemical structure of the molecules. These ADMET descriptors allow us to eliminate compounds with unfavorable ADMET characteristics early on to avoid expensive reformulation, preferably before synthesis, and also help to evaluate proposed structural refinements that are designed to improve ADMET properties.
Validation of synthetic accessibility for hit compounds using SYLVIA
Synthetic accessibility scores for hit compounds were used to validate the synthetic possibilities. For this, the SYLVIA-XT 1.4 program (Molecular Networks, Erlangen, Germany) was used to calculate the synthetic accessibility of these optimized compounds.20 The appraisal of synthetic accessibility of organic compounds using SYLVIA provides a score on a scale from 1 (very easy to synthesize) to 10 (complex and challenging to synthesize). A number of criteria, such as complexity of the ring system, complexity of the molecular structure, number of stereo centers, similarity to commercially available compounds, and potential for using powerful synthetic reactions have been independently weighted to provide a single value for synthetic accessibility.
Toxicity
To predict a variety of toxicities that are often used in drug development, the models in Table 2 are used and calculated through TOPKAT parameters/protocols using Accelrys DS 3.5. These predictions help in optimizing therapeutic ratios of lead compounds for further development and assessing their potential safety concerns. They will help in evaluating intermediates, metabolites, and pollutants, along with setting dose range for animal assays.
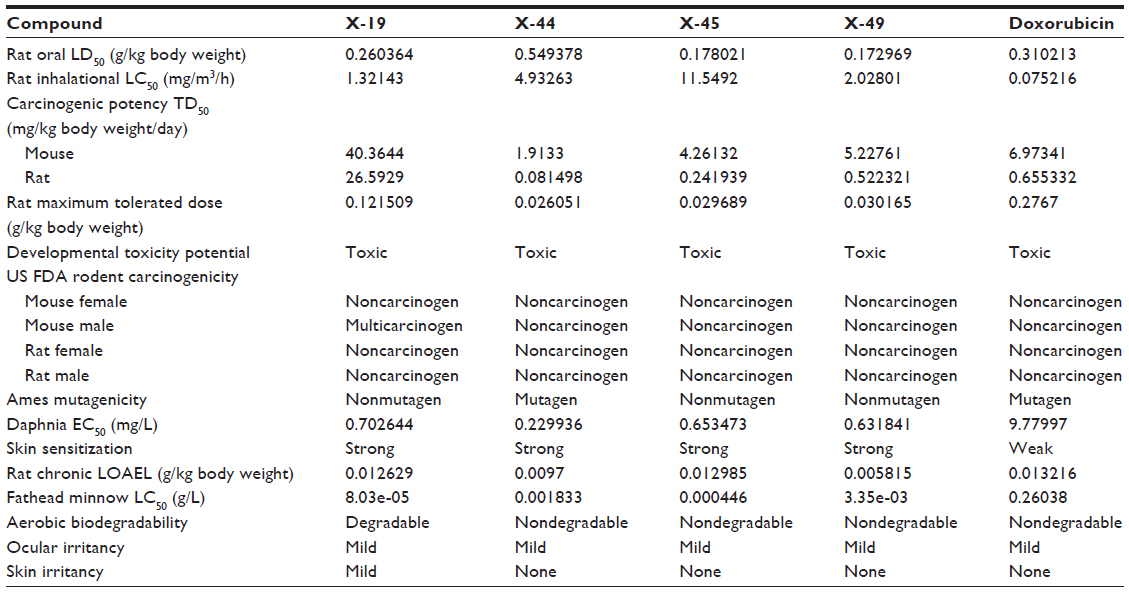 | Table 2 In silico screening of xanthone derivatives for toxicity risk assessment |
Results and discussion
Predicting anticancer activity with the QSAR model
Prior studies of xanthone showed its promising role in the development of novel anticancer compounds.6 In the present work, we studied the structure activity relationship of xanthone. The structure–activity relationship denoted by the QSAR model yielded a high activity–descriptors relationship accuracy of 84% referred by regression coefficient (r2=0.84) and a high activity prediction accuracy of 82%. Five molecular descriptors – dielectric energy, group count (hydroxyl), LogP, shape index basic (order 3), and the solvent-accessible surface area – were significantly correlated with anticancer activity. The QSAR model equation is given below, showing the relationship between experimental activity in vitro (ie, the inhibitory concentration to 50% of the population [IC50]) as the dependent variable and five independent variables (chemical descriptors):

Here, the rCV2 is 0.82, which indicates that the newly derived QSAR model has a prediction accuracy of 82%, and the r2 is 0.84, which indicates that the correlation between the activity (dependent variable) and the descriptors (independent variables) for the training data set compounds was 84% (Figure 3); the LOOCV r2 is 0.79. It is evident from the above equation that among the molecular descriptors, dielectric energy, group count (hydroxyl), and solvent-accessible surface area are positively correlated, meaning the biological activity increases when the values of these descriptors is positively increased. On other hand, the descriptors LogP and Shape index basic (order 3), are both negatively correlated with activity; the activity decreases when the values of these descriptors increases. Thus, we successfully developed a QSAR model for prediction of in vitro anticancer activity. A multiple linear regression QSAR mathematical model was developed for activity prediction that successfully and accurately predicted the anticancer activities of newly designed xanthone derivatives.
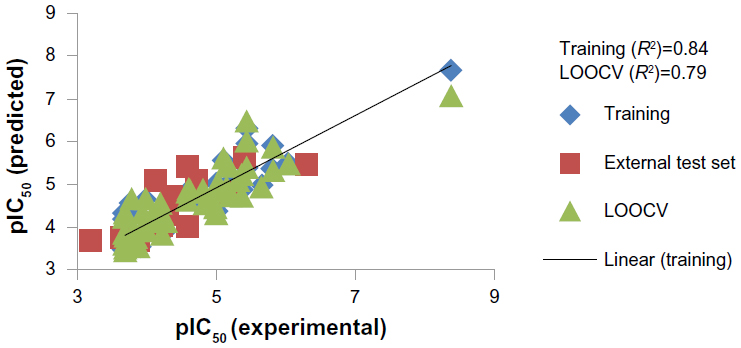 | Figure 3 Regression plot representing training, testing, and cross-validation of model. |
Experimental validation of QSAR model
The multiple linear regression-based QSAR model for the inhibitory activity of xanthone derivatives against HeLa cell lines has been validated with four compounds, Xan-1, Xan-2, Xan-3, and Xan-4 (Table 3).21 It was found that the predicted results through the QSAR model show compliance with their experimental results.
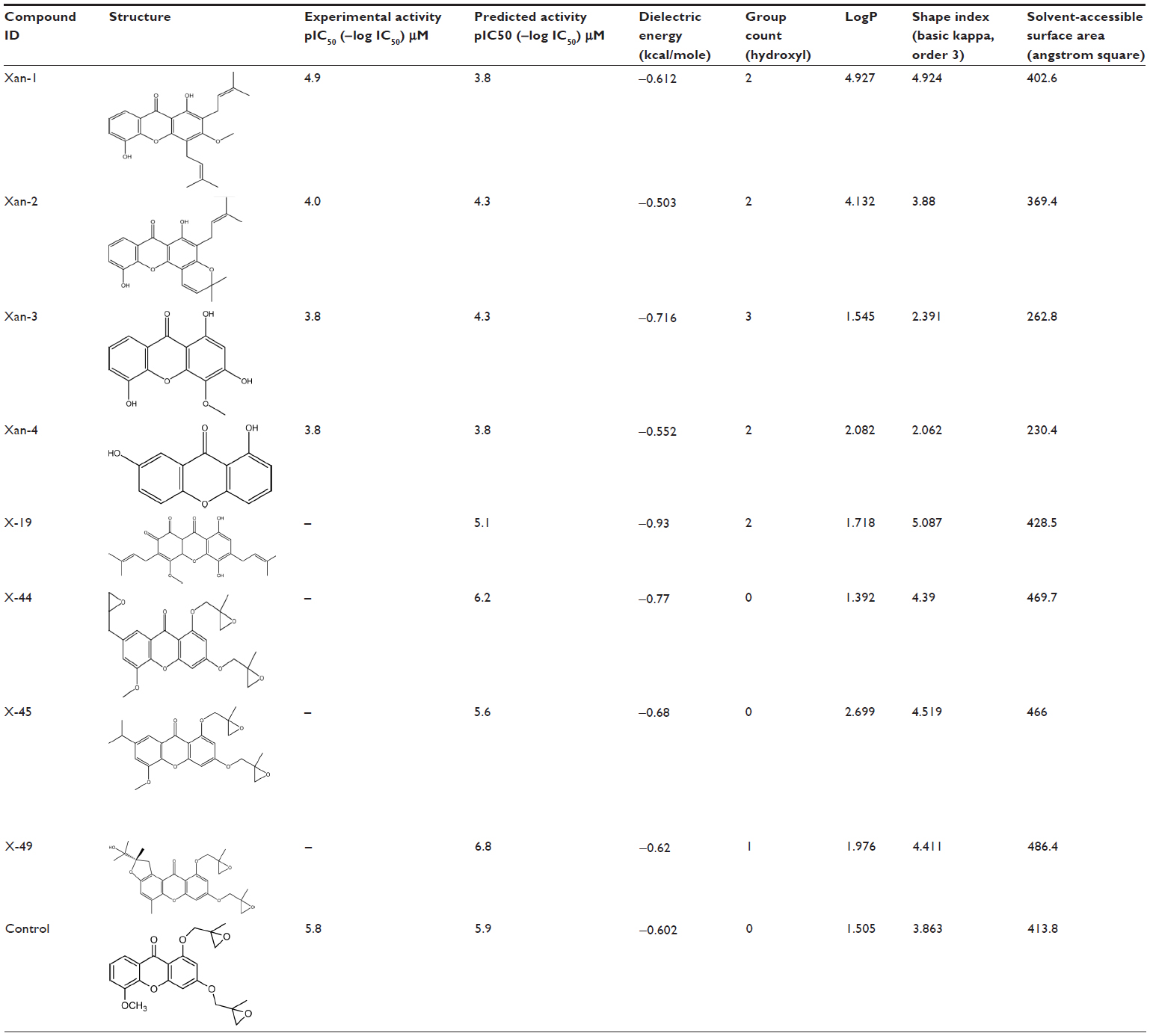 | Table 3 Screened out xanthone derivatives targeting/inhibiting Top2A and HeLa human cancer cell line |
Virtually designing and filtering of novel xanthone derivatives
Using this multiple linear regression QSAR mathematical model, which was developed for activity prediction against HeLa cell line, we predicted the anticancer activities of some newly designed xanthone derivatives (Table S3). The predicted IC50 value of final hit compounds X-19, X-44, X-45, and X-49 are 7.94 μM, 0.63 μM, 2.51 μM, and 0.16 μM, respectively. The QSAR model quantified the activity-dependent chemical descriptors and predicted the inhibitory concentration (log IC50) of each derivative, thus indicating its potential range of inhibition (Table 3).
Protein–ligand docking studies
Following development of the model and filtering through Lipinski’s rule of five, we first analyzed Top2A, and five active sites were obtained. We chose one of these with ATP binding sites, shown in Figure 2. In order to understand the ligand recognition in Top2A, we initially carried out docking with the known Top2A inhibitor/anticancer drug doxorubicin, and later with the most active 34 among the designed and filtered compounds. Out of 34, 20 failed to dock and three showed lower scores than the control. The docking program produces several poses with different orientations within the defined active site. All poses produce a different LibDock score. The best score was taken into account for further study. The compounds X-12, X-19, X-29, X-32, X-35, X-39, X-40, X-44, X-45, X-48, and X-49 (Table S3) were selected as candidate compounds based on their high docking score compared to doxorubicin. The analysis of the protein ligand complexes revealed binding site residue, including amino acid residues, waters, and metal atoms. A 2D diagram showing various interactions, such as hydrogen bonds, atomic charge interactions, and Pi-sigma interactions between the surrounding residues and the ligand, was also displayed. Different interactions were represented by different colors: eg, pink indicates electrostatic interaction; purple indicates covalent bond; and green indicates van der–Waals molecular interaction. Solvent accessibility of the ligand atom and the amino acid residues are shown in light blue shading surrounding the atom or residue. High shading indicates more exposure to solvent. The inhibitory activity of xanthone has been explained by two major factors: H-bond and pi-sigma interactions (Figure 4).
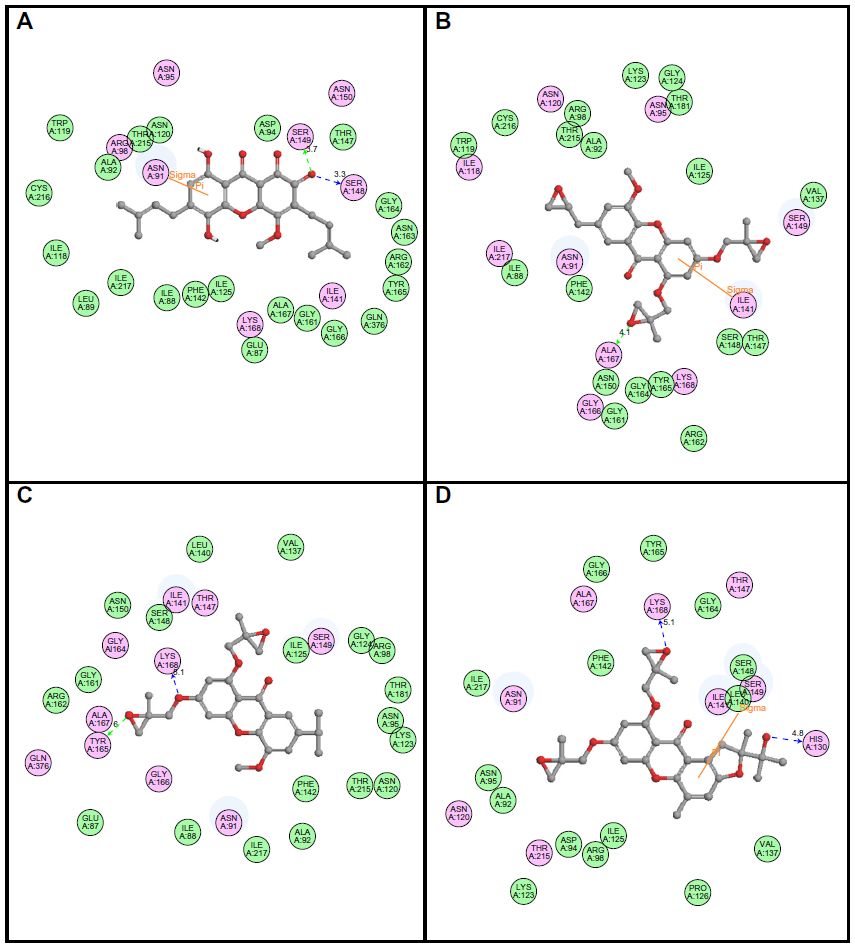 | Figure 4 2D diagrams illustrating protein–ligand interactions: (A) Compound X-19; (B) Compound X-44; (C) Compound X-45; (D) Compound X-49. |
To evaluate ligand binding in a receptor cavity, the score ligand poses protocol was used for the scoring functions for LibDock score, Jain, LigScore 1, LigScore 2, PLP, and PMF 04. The H-bond and pi-sigma interactions residues are also provided (Table 4).
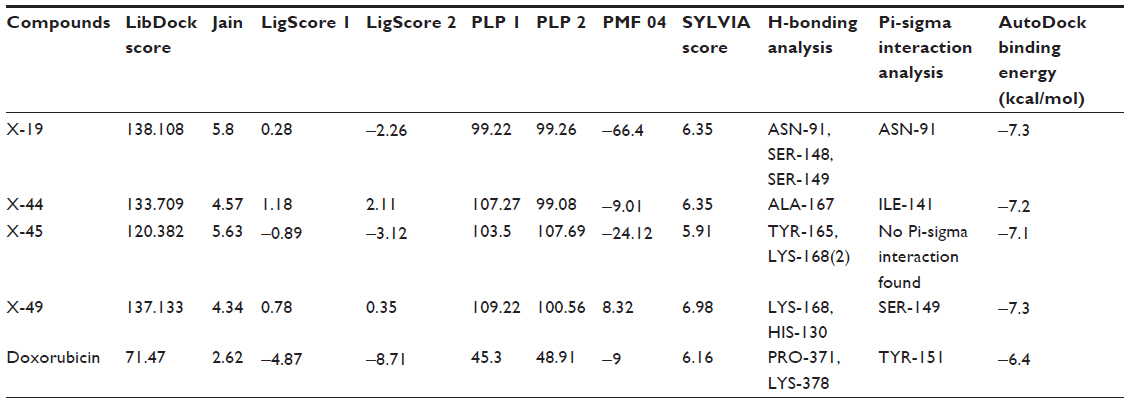 | Table 4 LibDock scoring functions, SYLVIA synthetic accessibility scores, and AutoDock binding affinity of identified potential xanthone derivatives inhibitors for DNA Top2A |
Assessment through pharmacokinetic parameters
Since the docking studies were found to be promising, the chemical descriptors for the pharmacokinetic properties were also calculated, so as to check the compliance of study compounds with standard range. For this, the aqueous solubility, blood–brain barrier penetration, cytochrome P450 2D6 binding, hepatotoxicity, intestinal absorption, and plasma protein binding were calculated. Calculating these properties was intended as the first step toward analyzing the novel chemical entities in order to check the failure of lead candidates, which may cause toxicity or be metabolized by the body into an inactive form or one unable to cross the membranes. The results of this analysis are reported in Table 5, together with a biplot (Figure 5). The pharmacokinetic profiles of all the compounds under investigation were predicted by means of six precalculated ADMET models provided by the Accelrys Discovery Studio 3.5 program. The biplot shows the two analogous 95% and 99% confidence ellipses for the blood–brain barrier penetration and human intestinal absorption models, respectively. The polar surface area (PSA) was shown to have an inverse relationship with percent human intestinal absorption, and thus cell wall permeability, although a relationship between PSA and permeability has been demonstrated. Moreover, when we calculated the PSA as a chemical descriptor for passive molecular transport through membranes, the results showed a lower PSA value of hit compounds than doxorubicin, but still within the limit; ie, <140 Å2. The aqueous solubility predictions (defined in water at 25°C) show that hit compounds are soluble in water. LogP value, which is a measure of lipophilicity and is the ratio of the solubility of the compound in octanol compared to its solubility in water, was found to be in range of the hit compounds and follows Lipinski’s rule of five, implicating a better oral bioavailability. The excretion process that eliminates the compound from the human body also depends on LogP. The hit compounds are highly (≥90%) bound to carrier proteins in the blood. This binding shows the efficiency of drugs. The drugs which are orally administered must be absorbed by the intestine; here the predicted result shows that all the compounds can be easily absorbed by the intestine, in comparison to doxorubicin (Table 5). The hit compounds are found to be noninhibitors of cytochrome P450 2D6 (CYP2D6), which indicates that all compounds may be well metabolized in Phase I metabolism. The CYP2D6 enzyme is one of the important enzymes involved in drug metabolism.22 Obtained results (Table 5) were cross-checked with the standard levels listed in Table S4.
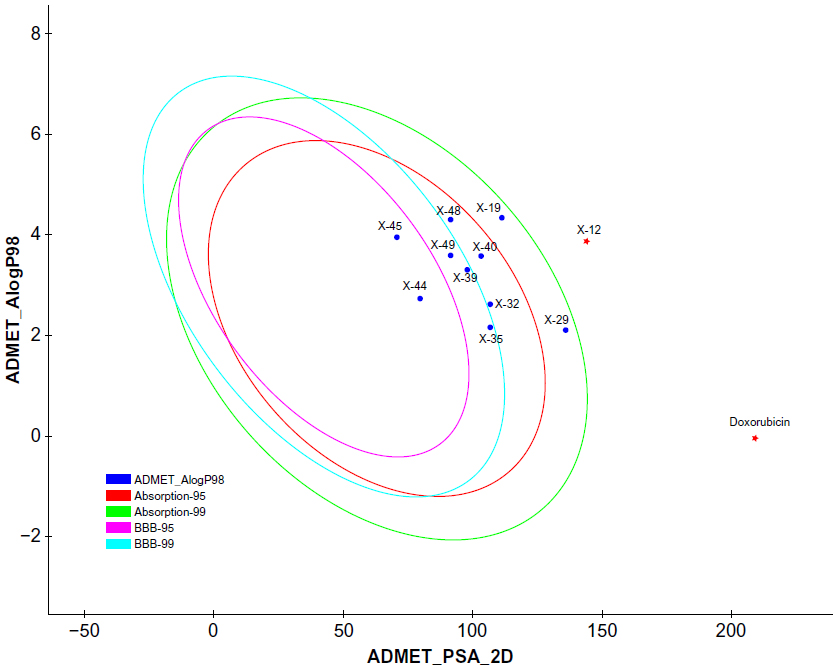 | Figure 5 Plot of PSA versus LogP for candidate compounds showing the 95% and 99% confidence limit ellipses corresponding to the blood–brain barrier and intestinal absorption models. |
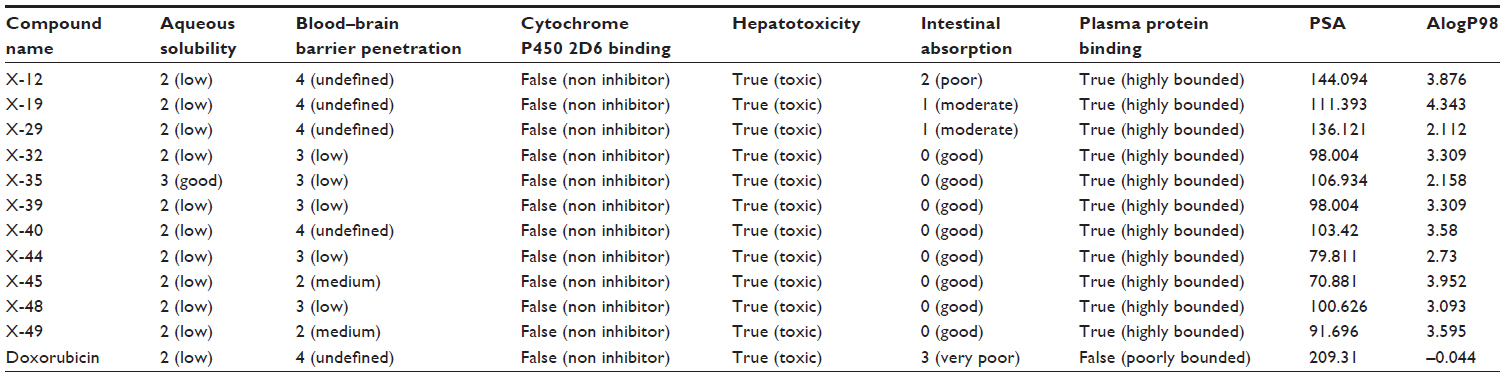 | Table 5 Compliance of compounds to the theoretical parameters of oral bioavailability and drug likeness properties |
Molecular docking validation
To validate the LibDock score, a further docking study through AutoDock Vina was completed. The docking study with DNA Top2A (PDB:1ZXM) revealed that the final hit compounds have shown a high binding affinity, as compared to the standard anticancer drug doxorubicin (Table 4).
Validation of final hits using SYLVIA
To further validate our compounds, the synthetic accessibility of the compounds was also measured using the SYLVIA-XT 1.4 program. The synthetic accessibility of known drug doxorubicin was also calculated for comparison purposes. The SYLVIA score of hit compounds and doxorubicin is given in Table 4 for comparison. The SYLVIA score for the final hits illustrates that these compounds may be synthesized easily.
Toxicity risk assessment screening
Toxicity risk assessment screening was performed for all the hit compounds. Results showed that all the compounds are noncarcinogenic. There is mild ocular irritancy for all the compounds. Likewise, there is no skin irritancy, with the exception of X-19, which has mild irritancy. The other properties, such as rat oral LD50, Ames mutagenicity, developmental toxicity potential, rat inhalational LC50, rat maximum tolerated dose, fathead minnow LC50, and aerobic biodegradability, are also provided in Table 2.
Conclusion
Xanthones are natural constituents of plants which contain a variety of biological activities, along with anticancer effects. The present study deals with the multiple linear regression-QSAR modeling for xanthone derivatives against human cancer cell line HeLa and anticancer target Top2A. Four compounds (X-19, X-44, X-45, and X-49) were screened out through the QSAR model, docking, ADMET screening, and synthetic accessibility. The screened leads can be used for further analysis and drug development. Aside from this, this study also provided a significant approach in the identification of novel and potent anticancer compounds from xanthone derivatives, and can be utilized as a guide for future studies for screening and designing the structurally diverse compounds from the xanthone family.
Acknowledgments
We acknowledge the Council of Scientific and Industrial Research, New Delhi and the Council of Science and Technology, Uttar Pradesh, Lucknow for financial support through the GAP247 project at CSIR-Central Institute of Medicinal and Aromatic Plants, Lucknow, India.
Disclosure
The authors report no conflicts of interest in this work.
References
Roberts NA, Martin JA, Kinchington D, et al. Rational design of peptide-based HIV proteinase inhibitors. Science. 1990;248(4953):358–361. | |
Blundell T, Sibanda BL, Pearl L. Three-dimensional structure, specificity and catalytic mechanism of renin. Nature. 1983;304(5923):273–275. | |
von Itzstein M, Wu WY, Kok GB, et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature. 1993; 363(6428):418–423. | |
Centers for Disease Control and Prevention. Cancer data and Statistics (webpage on the Internet). Available from http://www.cdc.gov/cancer/dcpc/data/. Accessed December 5, 2013. | |
Fortune JM, Osheroff N. Topoisomerase II as a target for anticancer drugs: when enzymes stop being nice. Prog Nucleic Acid Res Mol Biol. 2000;64:221–253. | |
Su QG, Liu Y, Cai YC, Sun YL, Wang B, Xian LJ. Anti-tumour effects of xanthone derivatives and the possible mechanisms of action. Invest New Drugs. 2011;29(6):1230–1240. | |
Yadav DK, Meena A, Srivastava A, Chanda D, Khan F, Chattopadhyay SK. Development of QSAR model for immunomodulatory activity of natural coumarinolignoids. Drug Des Devel Ther. 2010;4:173–186. | |
Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol. 2010;17(5):421–433. | |
Pouli N, Marakos P. Fused xanthone derivatives as antiproliferative agents. Anticancer Agents Med Chem. 2009;9(1):77–98. | |
Kuete V, Wabo HK, Eyong KO, et al. Anticancer activities of six selected natural compounds of some Cameroonian medicinal plants. PLoS One. 2011;6(8):e21762. | |
Koizumi Y, Tomoda H, Kumagai A, Zhou XP, Koyota S, Sugiyama T. Simaomicin α, a polycyclic xanthone, induces G1 arrest with suppression of retinoblastoma protein phosphorylation. Cancer Sci. 2009;100(2):322–326. | |
Woo S, Kang DH, Nam JM, et al. Synthesis and pharmacological evaluation of new methyloxiranylmethoxyxanthone analogues. Eur J Med Chem. 2010;45(9):4221–4228. | |
Woo S, Jung J, Lee C, Kwon Y, Na Y. Synthesis of new xanthone analogues and their biological activity test – cytotoxicity, topoisomerase II inhibition, and DNA cross-linking study. Bioorg Med Chem Lett. 2007;17(5):1163–1166. | |
Cho HJ, Jung MJ, Woo S, et al. New benzoxanthone derivatives as topoisomerase inhibitors and DNA cross-linkers. Bioorg Med Chem. 2010;18(3):1010–1017. | |
Gao XM, Yu T, Cui MZ, et al. Identification and evaluation of apoptotic compounds from Garcinia oligantha. Bioorg Med Chem Lett. 2012;22(6):2350–2353. | |
Jun KY, Lee EY, Jung MJ, et al. Synthesis, biological evaluation, and molecular docking study of 3-(3′-heteroatom substituted-2′-hydroxy-1′-propyloxy) xanthone analogues as novel topoisomerase IIα catalytic inhibitor. Eur J Med Chem. 2011;46(6):1964–1971. | |
Qidwai T, Yadav DK, Khan F, Dhawan S, Bhakuni RS. QSAR, docking and ADMET studies of artemisinin derivatives for antimalarial activity targeting plasmepsin II, a hemoglobin-degrading enzyme from P. falciparum. Curr Pharm Des. 2012;18(37):6133–6154. | |
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. | |
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. | |
Boda K, Seidel T, Gasteiger J. Structure and reaction based evaluation of synthetic accessibility. J Comput Aided Mol Des. 2007;21(6):311–325. | |
Ee GC, Lim CK, Rahmat A, Lee HL. Cytotoxic activities of chemical constituents from Mesua daphnifolia. Trop Biomed. 2005;22(2):99–102. | |
Lynch T, Price A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician. 2007;76(3):391–396. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.