Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 8
Pyoderma gangrenosum: challenges and solutions
Authors Gameiro A, Pereira N, Cardoso JC, Gonçalo M
Received 30 January 2015
Accepted for publication 27 February 2015
Published 28 May 2015 Volume 2015:8 Pages 285—293
DOI https://doi.org/10.2147/CCID.S61202
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jeffrey Weinberg
Ana Gameiro,1 Neide Pereira,2 José Carlos Cardoso,1 Margarida Gonçalo1
1Dermatology Department, Coimbra University Hospital, Coimbra, Portugal; 2Dermatology Department, Centro Hospitalar Cova da Beira, Covilhã, Portugal
Abstract: Pyoderma gangrenosum (PG) is a rare disease, but commonly related to important morbidity. PG was first assumed to be infectious, but is now considered an inflammatory neutrophilic disease, often associated with autoimmunity, and with chronic inflammatory and neoplastic diseases. Currently, many aspects of the underlying pathophysiology are not well understood, and etiology still remains unknown. PG presents as painful, single or multiple lesions, with several clinical variants, in different locations, with a non specific histology, which makes the diagnosis challenging and often delayed. In the classic ulcerative variant, characterized by ulcers with inflammatory undermined borders, a broad differential diagnosis of malignancy, infection, and vasculitis needs to be considered, making PG a diagnosis of exclusion. Moreover, there are no definitively accepted diagnostic criteria. Treatment is also challenging since, due to its rarity, clinical trials are difficult to perform, and consequently, there is no “gold standard” therapy. Patients frequently require aggressive immunosuppression, often in multidrug regimens that are not standardized. We reviewed the clinical challenges of PG in order to find helpful clues to improve diagnostic accuracy and the treatment options, namely topical care, systemic drugs, and the new emerging therapies that may reduce morbidity.
Keywords: pyoderma gangrenosum, neutrophilic dermatosis, treatment, biologics
Historical aspects
In 1908, the French dermatologist Louis Brocq reported a series of patients with typical features of pyoderma gangrenosum (PG), describing a new dermatological entity, and named it geometric phagedenism. Geometric was used to underline the geometric pattern of the lesion’s edge, and phagedenism, meaning food and consumption, from the Greek phageton, was used to emphasize the necrotic and rapidly progressive nature of the ulcer.1
Twenty years later, in 1930, Brunsting et al1 first introduced the term PG, describing five patients with recalcitrant ulcers, four with concomitant ulcerative colitis, and one with idiopathic chronic purulent pleurisy. These authors reproduced similar ulcers in experimental animals by injecting material cultured from patients’ lesions, and therefore, considered it as a bacterial infection. At that time, the term pyoderma was used in the context of purulent skin diseases due to infections agents, and the term gangrenosum was used to address the necrotic and destructive nature of the process.1
However, PG was not responsive to antibiotics, whereas corticosteroids and other immunosuppressive drugs, such as dapsone or clofazimine, consistently showed better results.2,3 The disease is currently recognized as a non-infectious inflammatory disease, but despite its inaccuracy, the term PG remains well entrenched in the medical literature.
Introduction
PG is a rare inflammatory skin disease with a chronic relapsing course, commonly associated with severe pain and tenderness that often regresses with cribriform mutilating scars.4
PG has been estimated to occur in three to ten individuals/million. However, it is difficult to determine the exact disease prevalence, as accurate epidemiological data are missing. PG is most commonly observed in young to middle-aged adults, with women being more affected.5 Cases in elderly people have been reported occasionally, and childhood PG accounts for approximately 4% of the cases.5,6
Classically, lesions begin as tender papules, papulopustules or vesicles, evolving into painful and rapidly enlarging ulcers. Healing frequently leaves a cribriform scar, which may lead to considerable disfiguring.4,7 On histology, PG lesions do not display specific features, being typically characterized by a prominent dermal neutrophilic infiltrate with abscess formation, with no signs of infection and usually without significant vasculopathy.
The disease usually has a devastating effect on a patient’s life: pain may alter the eating and sleeping patterns; may induce significant movement impairment depending on lesions’ location; and treatment usually implies numerous hospital appointments. Additionally, the characteristic features of the wound, such as malodor or exudate, may trigger or aggravate anxiety, depression, and social isolation.7
Pathophysiology
The etiology of PG is still unknown, and its pathophysiology is far from being completely understood. Inflammatory bowel disease (IBD), PAPA (pyogenic arthritis, PG, and acne) syndrome and PAPA-related syndromes, frequently evolve with PG; furthermore, other neutrophilic skin diseases, namely Sweet’s syndrome (SS) or Behçet’s disease, share clinicopathological aspects with PG. Based on the clinical, immunological, and genetic studies, concerning the diseases related to PG, we may assume that several factors may concur to PG physiopathology. A genetic background and an altered immune and inflammatory response are certainly involved, although triggers have not yet been identified.
PG is currently considered an inflammatory disease within the spectrum of neutrophilic disorders, which includes many diseases exhibiting an extraordinary clinical heterogeneity. Nevertheless, these disorders are linked by the presence of perivascular and diffuse neutrophilic infiltrates with no identifiable infectious agents.8 Additionally, neutrophilic diseases commonly occur in association with an underlying systemic condition such as malignancy, neutropenia, rheumatologic diseases, infections, auto-inflammatory syndromes, and immunodeficiency. Moreover, neutrophilic diseases are frequently triggered by medications and pathergy, the latter being a typical sign of PG that is certainly involved in post-surgical and peristomal PG.8–10
The similarities between PG and neutrophilic diseases suggest that underlying common inflammatory pathways probably converge to their pathophysiology, leading to abnormalities in polymorphonuclear neutrophils (PMN) recruitment or homeostasis.8 In PG and other neutrophilic diseases, elevated skin and/or circulating levels of the pro-inflammatory cytokines (IL1β, IL6, TNF-α, IFN-ϒ, G-CSF)10–12 and, particularly, of the potent PMN attracting chemokines, namely IL8/CXCL8 and CXCL1,2,3, along with skin infiltration by T cells, particularly at the edge of the PG ulcer, suggests an active recruitment of PMN to the skin.10,13 Also, in both PG and SS skin lesions, there is a high expression of MMPs (MMP-2, MMP-9, MMP-10), a family of Zn2+ endopeptidades that are not only major contributors to the breakdown and reconstitution of the extracellular matrix in wound repair, but also influence the production of neutrophilic chemokines in neutrophilic disorders. Furthermore, the increase of elafin, the neutrophil elastase inhibitor, expressed by injured epidermal keratinocytes, and the intensification of the FAS/FASL system, involved in tissue damage and apoptosis, may also contribute to the formation of ulcers in PG, and impairment of tissue remodeling and wound healing.10,13,14
IL17 produced by Th17 cells, shown to play an important role in IBD and other skin diseases that involve neutrophil recruitment, like psoriasis and acute generalized exanthematous pustulosis, has also been found to be elevated in skin lesions of PG.12,14–16 Th17 cells seem to be overrepresented in PG lesions together with a low level of regulatory T cells (Treg) and related cytokines, namely IL10.17 However, in a more recent case, IL17 was not significantly elevated in PG, whereas IL23, responsible for driving T cells into the Th17 phenotype, was elevated and was considered the support for therapy with ustekinumab, a monoclonal antibody that binds the shared p40 subunit of IL23 and IL12.17,18
The mechanisms responsible for the clinical heterogeneity of skin lesions in neutrophilic disorders – ulcers in PG, skin plaques in SS and folliculitis and aphthous ulcers in Behçet’s disease, as well as their associated manifestations remain unknown. Nevertheless, differences in the balance of Treg/Th17, the relative intensity of the expression of cytokines and MMPs, and a different genetic background may in part justify the different patterns of skin aggression in these diseases.14,17,19
PG also shares many features with auto-inflammatory disorders, including the chronic remitting course, the dysregulation of the innate immune system with neutrophil recruitment and activation, and a role for excessive cytokine production.8 Additionally, PG lesions are almost always observed in patients with PAPA syndrome, a rare inherited auto-inflammatory syndrome with excessive IL1 production.13,20
Genetic abnormalities have been found in patients with PG in the context of PAPA, PAPASH (initially described as PASS – pyogenic arthritis, PG, acne, and hidradenitis suppurativa), and PASH (PG, acne, and hidradenitis suppurativa) syndromes in genes that code for an aberrant production of IL1, namely PSTPIP1 gene,8,21,22 or in its promoters.20 In cases of PG associated with IBD, genetic abnormalities have occasionally been found concerning loci for IL8RA, and TIMP3 and TRAF31P2 genes, which code respectively for an MMP inhibitor and a protein that interacts with TNF receptor-associated factors, involved in the IL17 immune pathway.21
Rarely, PG appears during the treatment with infliximab, etanercept, or G-CSF. Nevertheless, most reports confirm the benefit of therapy with the different TNF inhibitors, which further support an underlying immunologic mechanism in PG pathogenesis.23,24
Clinical features and diagnosis
The diagnosis is commonly challenging and delayed, and misdiagnosis is also frequent, mostly by non-dermatologists who are less aware of this entity. The multiple clinical variants (classic ulcerative, pustular, bullous, and superficial granulomatous), and the overlapping forms with other neutrophilic diseases, frequently observed in clinical practice, make the diagnosis even more difficult.8,25 Moreover, there are countless causes for skin ulceration, including infections, tumors, vascular disorders, vasculitis, and trauma, which make PG a diagnosis of exclusion (Table 1).
 | Table 1 Differential diagnosis of the unspecific ulcerative lesions of pyoderma gangrenosum |
There are no specific laboratory tests, and although PG characteristically shows a neutrophilic inflammation, histopathology is nonspecific and may change according to different PG types and evolution.5
The pretibial area is the most commonly affected site, but PG can occur anywhere in the body including breast, hands, abdomen, head and neck, scrotum, penis, and peristomal skin.5,26
PG ulcers often demonstrate pathergy, ie, worsening in response to a minor trauma or surgical debridement, and can be triggered by a surgical procedure, namely breast surgery, cesarean section, or colostomy.9,10 In fact, parastomal PG comprises 15% of all cases.27
The disease onset is extremely variable, some patients present with one or two slowly growing ulcers, while others experience an abrupt appearance of multiple rapidly enlarging ulcerations. Associated symptoms may be present and include fever, malaise, myalgia, and arthralgia. Systemic symptoms may result from IL1β elevation due to inflammasome activation, and consequent signal cascades maintaining systemic inflammation.28
Very rarely, as in SS, extracutaneous neutrophilic infiltrates can be observed, namely in the bones, liver, lungs, pancreas, spleen, kidneys, and central nervous system.29,30
Particularly, in the classic ulcerative type, two distinct stages are described: the active ulcerative stage and the wound healing stage. At the ulcerative stage, the wound has a peripheral erythematous inflammatory halo and the edges are erythematous, raised, and sometimes necrotic, with an undermining border, whose size expresses how rapidly the ulcer edge will evolve. Alternatively, the ulcer can extend through the appearance of surrounding pustules. In contrast, at the healing stage, the edges show string-like projections of epithelium, which straddle the border between the ulcer bed and the normal surrounding skin – Gulliver’s sign (Figures 1 and 2).31,32 Classic healing results in an atrophic cribriform scar, described also as “cigarette paper-like” scar (Figure 3).32,33
 | Figure 1 (A) Ulcerative stage of a pyoderma gangrenosum lesion, showing the typical violaceous, raised undermined border, with a granulation base, surrounded by a erythematous halo. (B) Healing stage, after 4 days therapy with systemic corticosteroids and cyclosporine, showing a decrease in the surrounding inflammation. |
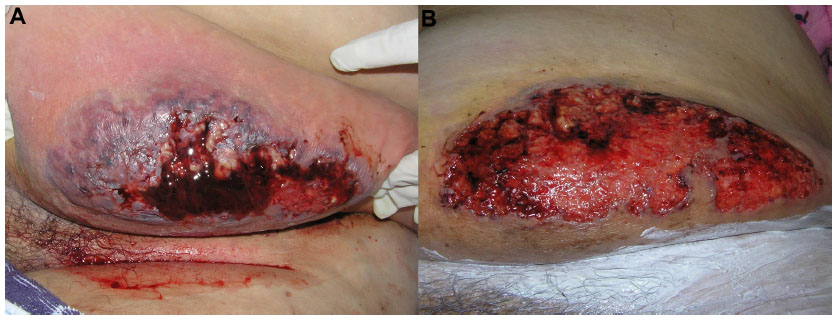 | Figure 2 (A) Ulcerative stage of classical and rapidly progressive pyoderma gangrenosum ulcer, showing an elevated, violaceous, undermined border, with a necrotic and hemorrhagic base. (B) The healing stage ulcer, 3 months after systemic therapy, presenting the “Gulliver” sign; ulcer base containing granulation tissue, and necrotic tissue in a lesser extent. |
 | Figure 3 Characteristic cribriform scar after healing of pyoderma gangrenosum. |
Clinical variants
There are four recognized clinical variants of PG – classical or ulcerative, pustular, bullous, and vegetative or granulomatous. Some patients develop only a particularly subtype, whereas others may have more than one subtype simultaneously.33,34 Some subtypes show preferential associations with underlying diseases, eg, pustular variant in patients with IBD.8
- Classical PG, also known as ulcerative type, is characterized by rapidly progressive painful ulcers, which typically have undermined, overhanging, dusky purple edges, with surrounding induration and erythema. The base of the ulcer commonly has granulation tissue, and occasionally, necrotic tissue and a purulent exudate. Classic PG ulcers are aseptic, although superinfection may occur.29,35
- Pustular PG variant is characterized by multiple sterile pustules with a surrounding erythematous halo, generally arising on the trunk and extensor aspect of the limbs. Histopathology reveals a dermal neutrophilic infiltrate and subcorneal neutrophilic micropustules. This variant is commonly associated with IBD, and has a tendency to remit with IBD control.29,34
- Bullous PG typically presents with grouped vesicles that rapidly spread and coalesce to form large bullae, and further develop into ulcerations, showing central necrosis and a peripheral halo of erythema. This variant generally occurs at atypical sites, on the dorsal surface of the hands, extensor aspects of the arms, or on the head, and is mostly seen in patients with lymphoproliferative diseases, as a paraneoplastic phenomenon. In patients with hematologic disease, the presence of PG may signify malignant transformation, and is suggestive of poor prognosis or aggressive disease (Figure 4).33
- Vegetative or superficial granulomatous PG is characterized by a solitary, erythematous, ulcerated plaque, lacking the violaceous border that is typically present in the classical variant.26,29 Histologically, it is characterized by granulomas with a three-layered structure: the center contains the neutrophilic inflammation, surrounded by a palisade of histiocytes, which is in turn rimmed by a lymphocytic infiltrate. This is the most uncommon and benign subtype, usually shows a good response to less aggressive treatments, and it is less frequently associated with underlying systemic disorders.35,36
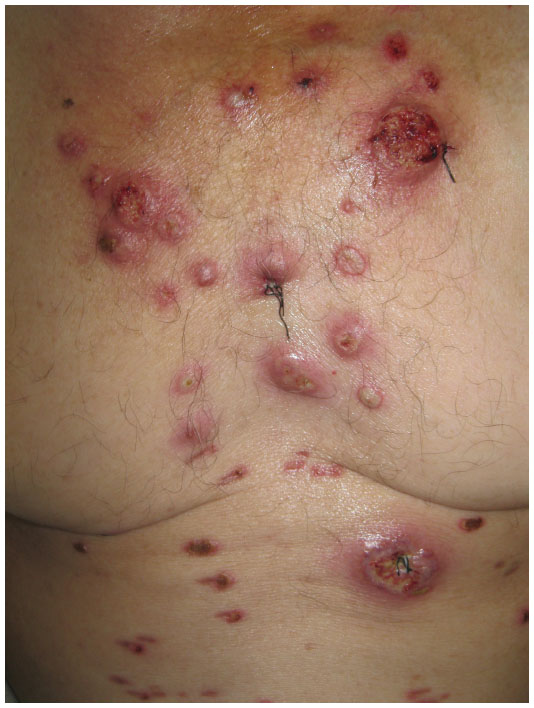 | Figure 4 Bullous variant of pyoderma gangrenosum, in a patient with acute myeloid leukemia, presenting overlap features with bullous variant of Sweet’s syndrome. |
Diagnosis
The work-up starts with a cutaneous biopsy that preferentially includes the border of the ulcer and the adjacent skin. Histology is essential to exclude vasculitis and malignancy, and special stains and tissue cultures may also be used to rule out infection. Complete work-up should include extensive blood and urine screening (complete blood cell count, chemistry and liver function tests, erythrocyte sedimentation rate and CRP, urine and serum protein electrophoresis or immunofixation, anti-nuclear antibodies and anti-neutrophil cytoplasmatic antibodies) and colonoscopy, in order to exclude underlying coexisting conditions, namely IBD, inflammatory, autoimmune, and neoplastic diseases.
Pathological skin changes depend on the type of lesion (ulcerative, pustular, bullous, and superficial granulomatous), stage of evolution, and the site from where the specimen is obtained. The active untreated expanding lesions typically show dense neutrophilic dermal infiltrates, often forming micro-abscesses and occasionally extending to the subcutis. Vasculitis is not the main observation, but it may occur along with leukocytoclasia. The fully developed ulcers may only present marked tissue necrosis and surrounding mononuclear cell infiltrates.25 However, none of these histological features is pathognomonic.
In order to increase PG recognition and improve diagnostic specificity, diagnostic criteria have been proposed, first by von den Driesch, and later by Su et al4,32 (Table 2). They are quite similar and require the same two major criteria (a characteristic PG classical type ulcer, and the exclusion of other causes of cutaneous ulceration), combined with at least two minor criteria: suggestive history of pathergy or a cribriform scar (item only presented in the latest diagnostic criteria), presence of a relevant associated disease, compatible histopathological findings, or response to systemic steroid treatment.9,10
 | Table 2 Proposed diagnostic criteria for classic ulcerative pyoderma gangrenosum, according to Su et al,4 diagnosis would require two major and at least two minor criteria |
Associated diseases
PG can involve only the skin, but in more than half the cases it is associated with an underlying systemic disease, which can occur before, coincidently, or after the diagnosis.26,37,38
Once considered a pathognomonic feature of ulcerative colitis, PG is now known to occur in association with several other distinct diseases. The most common are arthritis (namely: seronegative arthritis, ankylosing spondylitis, and rheumatoid arthritis), Crohn’s disease, and myeloproliferative disorders or monoclonal gammopathy. The inflammasome activation and IL1β overproduction, have probably a role in PG physiopathology, and can also be responsible for systemic inflammatory symptoms such as arthralgia. Furthermore, the inflammatory cascades activated by IL1β, can be responsible for chronic inflammation and sterile neutrophilic inflammation, which may result in arthritis, a common feature in auto-inflammatory diseases.28,39
PG has also been associated with HIV, hepatitis, systemic lupus erythematous, PAPA syndrome and the associated syndromes (PAPASH and PASH), Takayasu’s arteritis, solid tumors, and pregnancy.23,29,33 Furthermore, drugs such as propylthiouracil, isotretinoin, pegfilgrastim, TNF inhibitors, and gefitinib have been reported to induce PG.4,6,24,37
Treatment
Outside dermatology, the diagnosis of PG is frequently considered only after multiple and unsuccessful treatment attempts, with antibiotics and surgical debridement. Lesions usually progress when treated as infection, and due to pathergy, worsen after surgical procedures.6
Treatment is challenging; there is no universally accepted “gold standard” and although a small trial has been published, comparing infliximab to placebo, no randomized controlled trials have been performed for the usual systemic therapies.40,41 Little evidence supports a rational therapeutic approach to this clinical entity, and currently, treatments are mostly trying to target a broad spectrum of immunologic mediators and inflammatory cells, shown to be involved in PG, including neutrophils, lymphocytes, and cytokines.
Topical and systemic corticosteroids are considered the first therapeutic option, whereas other immunosuppressors and cytostatics can be used as steroid-sparing agents. Multidrug regimens have not been well described in the literature, but they represent a good alternative for patients with refractory disease.42
Treatment of the underlying disease
Treatments differ between patients with idiopathic disease and those who have an underlying disorder. In the latter case, the fundamental approach is the control of the associated condition, which is, nevertheless, not always possible. Treatments serving this purpose may include: colectomy in patients with chronic ulcerative colitis, plasmapheresis or granulocyte apheresis in patients with leukemia, or thalidomide in patients with myelodysplastic syndromes.43,44
Anti-inflammatory and immunosuppressive drugs can be effective both in PG and the underlying disease, as the example of the IL1 antagonist anakinra for PG in the context of PAPA syndrome,45,46 and of infliximab and other anti-TNF agents in the context of Crohn’s disease and ulcerative colitis.30,42 Among the anti-TNF biologics, etanercept is the only one reported not to be effective in IBD, which should be taken into consideration when treating these patients. Nevertheless, in many case reports describing successful treatment of PG, it is not possible to determine whether therapy had a direct effect on PG, or an indirect effect by controlling the associated disorder.47
Topical treatment
Patients with mild disease and, particularly, those with the vegetative/superficial granulomatous form of PG have been reported to respond well to topical therapy and wound dressing. Topical corticosteroids, tacrolimus, sodium cromoglicate, nicotine, 5-aminosalicylic acid, and intralesional corticosteroids and cyclosporine have been successfully used.30,48 However, in most cases, the preference is for topical corticosteroids or tacrolimus. More recently, intralesional activated protein C and timolol gel, already used in chronic venous and diabetic ulcers, showed promising results.49,50
It is important to highlight the equal importance of treating the inflammatory state, balanced with optimized wound care.51 Therefore, wound dressing is particularly important, since most ulcers show heavy exudates. Occlusion is not recommended in highly active progressing ulcers with inflammatory borders but may be beneficial in slow healing ulcers after the resolution of inflammatory component.
Surgical intervention can worsen PG through pathergy, and similar to surgical debridement, topical debriding agents are partially contraindicated. To summarize: compression, occlusive wound dressings, debridement, and skin grafting (autologous and bio-engineered), should only be performed in ulcers devoid of an inflammatory border and in conjunction with immunosuppression.47,51,52 Furthermore, in case of parastomal PG, relocation of stoma is contraindicated, except for other reasons, eg, herniation or dysfunction.27
Systemic treatment
For a more widespread or rapidly progressive disease, systemic treatment is mandatory. Corticosteroids in moderate to high doses (eg, prednisolone, 0.5–2 mg/kg/day) and cyclosporine (3–6 mg/kg/day) are the most frequently used drugs, and also the best documented in literature.31,41,42 Other oral and intravenous immunosuppressors have been reported to successfully treat PG, or to be good corticosteroid sparing agents, namely azathioprine, sulfasalazine, dapsone, thalidomide, minocycline, clofazimine, methotrexate, mycophenolate mofetil, tacrolimus, intravenous immunoglobulin, and cyclophosphamide.53,54 Systemic antibiotics including tetracyclines, vancomycin, rifampicin, and mezlocillin, besides their role in inhibiting secondary bacterial infection, may also modulate the course of PG through anti-inflammatory mechanisms.6
Over the past decade, PG has been reported to respond to many different biologic medications, most commonly, to anti-TNF drugs such as etanercept, adalimumab, certolizumab, and infliximab.6,55,56 Infliximab (5 mg/kg) represents the biological drug most widely studied in PG, and was demonstrated to be superior to placebo in a randomized control trial.23,41,57 Furthermore, ustekinumab (anti-IL12/IL23) has been recently used in the management of PG with good results, and anakinra (anti-IL1) induces a very good response in patients with PAPA syndrome.11,44,46 However, since biologic therapies are relatively new, their unknown long-term side effects should be taken into consideration.
Combination therapy
Although multidrug therapy has not been well described in the literature, it should be considered for refractory disease. The combination of systemic steroids and cyclosporine is commonly used for controlling the inflammatory progressive stage, with further slow tapering.42
When this combination fails, less conventional treatments are chosen, generally based on immunosuppressive drug combinations used in other immune diseases, which are considered relatively safe. The common combinations are: 1) methotrexate and infliximab, and 2) cyclosporine, mycophenolate mofetil, and prednisone.
Another approach is to treat patients with combination therapy initially, and attempt to maintain remission with a less toxic monotherapy regimen after the patient has been controlled for several months.42
Other treatment considerations
In case of malignancy, immunosuppression should only be performed at the minimum doses. PG may be associated with malignancy, which sometimes precedes the diagnosis; therefore, immunosuppressors, as well as biological agents, can be risky.18
It is not uncommon for PG to have secondary wound infection, documented by a positive culture from smear, and clinically suspected by CRP elevation and signs such as erythema and swelling. In these cases, antibiotic treatment should be promptly initiated, and except in sepsis, immunosuppression should be maintained to prevent progression.27
A persistent ulcer does not necessarily mean treatment failure. The ulcers gradually heal after a successful treatment with suppression of the pathogenic inflammatory response. Therefore, being able to distinguish a residual poorly healing, but non-inflammatory ulcer, from an active inflammatory ulcer is essential, because in the former, immunosuppression can be tapered.53
Pain is a major complaint and should be carefully controlled, to improve patients’ quality of life. Analgesia can be tapered gradually after clinical evidence of improvement, since pain tends to decrease with ulcer healing.26,47
Prognosis and outcome
Factors that are considered to be associated with a poor prognosis include disease severity, older age at diagnosis, the ulcerative and bullous variants, unresponsiveness to treatment of the associated disease, secondary infection, and sepsis.47
The disease often shows a chronic relapsing course; therefore, long-term prognosis of PG is unpredictable, and the decision on the treatment duration can be difficult. Importantly, careful management of chronic underlying disorders is critical to prevent rebound flares.8 Relapses may occur in patients who responded rapidly and completely to the chosen drug regimen.28 Unfortunately, despite advances in the therapeutic management, PG is still a potentially life-threatening disease, and the risk of death was shown to be three times higher than the general population.32,55
Conclusion
PG is a rare challenging disease, described more than a 100 years ago as an infection, and that is now included within the spectrum of neutrophilic diseases or auto-inflammatory syndromes.
The diagnosis can be difficult, particularly for non-dermatologists who seldom recognize this entity, but also for dermatologists, as there is no pathognomonic biomarker, clinical or histological feature. Moreover, there are no definite diagnostic criteria or severity parameters established, that could facilitate multicenter randomized clinical trials to improve therapy of this condition.
Further clarification of the underlying pathogenic mechanisms may help to achieve better therapies, namely those targeting IL17 and, consequently, be able to offer a more effective management to these patients.18,21
Disclosure
The authors have no conflicts of interest to disclose.
References
Farhi D. The clinical and histopathological description of geometric phagedenism (pyoderma gangrenosum) by Louis Brocq one century ago. Arch Dermatol. 2008;144(6):755. | |
Gold S. Pyoderma Gangrenosum. Proc R Soc Med. 1956;49(4):234–235. | |
Muende I. Pyoderma Gangrenosum. Proc R Soc Med. 1960;53(4):296–297. | |
Su WP, Davis MD, Weenig RH, Powell FC, Perry HO. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43(11):790–800. | |
Wollina U. Pyoderma gangrenosum – a review. Orphanet J Rare Dis. 2007;2:19. | |
Wines N, Wines M, Ryman W. Understanding pyoderma gangrenosum: a review. MedGenMed. 2001;3(3):6. | |
Hemp L, Hall S. Pyoderma gangrenosum: from misdiagnosis to recognition, a personal perspective. J Wound Care. 2009;18(12):521–526. | |
Alavi A, Sajic D, Cerci FB, Ghazarian D, Rosenbach M, Jorizzo J. Neutrophilic dermatoses: an update. Am J Clin Dermatol. 2014;15(5):413–423. | |
Cardoso JC, Coelho S, Gonçalo M, Oliveira H, Brites MM, Figueiredo A. Pioderma gangrenoso da parede abdominal após cesariana [Pyoderma gangrenosum of the abdominal wall after cesarean]. Trab Soc Port Dermatol Venereol. 2008;66:245–250. | |
Tanaka N, Fujioka A, Tajima S, Ishibashi A, Hirose S. Elafin is induced in epidermis in skin disorders with dermal neutrophilic infiltration: interleukin-1 beta and tumour necrosis factor-alpha stimulate its secretion in vitro. Br J Dermatol. 2000;143(4):728–732. | |
Kluger N. Ustekinumab for pyoderma gangrenosum. Arch Dermatol. 2012;148(5):655. | |
Kabashima R, Sugita K, Sawada Y, Hino R, Nakamura M, Tokura Y. Increased circulating Th17 frequencies and serum IL-22 levels in patients with acute generalized exanthematous pustulosis. J Eur Acad Dermatol Venereol. 2011;25(4):485–488. | |
Marzano AV, Cugno M, Trevisan V, et al. Role of inflammatory cells, cytokines and matrix metalloproteinases in neutrophil-mediated skin diseases. Clin Exp Immunol. 2010;162(1):100–107. | |
Marzano AV, Fanoni D, Antiga E, et al. Expression of cytokines, chemokines and other effector molecules in two prototypic autoinflammatory skin diseases, pyoderma gangrenosum and Sweet’s syndrome. Clin Exp Immunol. 2014;178(1):48–56. | |
Keller M, Spanou Z, Schaerli P, et al. T Cell-regulated neutrophilic inflammation in autoinflammatory diseases. J Immunol. 2005;175(11):7678–7686. | |
Lowes MA, Russell CB, Martin DA, Towne JE, Krueger JG. The IL-23/T17 pathogenic axis in psoriasis is amplified by keratinocyte responses. Trends Immunol. 2013;34(4):174–181. | |
Caproni M, Antiga E, Volpi W, et al. The Treg/Th17 cell ratio is reduced in the skin lesions of patients with pyoderma gangrenosum. Brit J Dermatol. Epub January 20, 2015. | |
Guenova E, Teske A, Fehrenbacher B, et al. Interleukin 23 expression in pyoderma gangrenosum and targeted therapy with ustekinumab. Arch Dermatol. 2011;147(10):1203–1205. | |
Marzano AV, Cugno M, Trevisan V, et al. Inflammatory cells, cytokines and matrix metalloproteinases in amicrobial pustulosis of the folds and other neutrophilic dermatoses. Int J Immunopathol Pharmacol. 2011;24(2):451–460. | |
Duchatelet S, Miskinyte S, Join-Lambert O, et al. First Nicastrin mutation in PASH syndrome. Br J Dermatol. Epub January 20, 2015. | |
Defilippis EM, Feldman SR, Huang WW. The genetics of pyoderma gangrenosum and implications for treatment: a systematic review. Br J Dermatol. Epub October 28, 2014. | |
Bruzzese V. Pyoderma gangrenosum, acne conglobata, suppurative hidradenitis, and axial spondyloarthritis: efficacy of anti-tumor necrosis factor α therapy. J Clin Rheumatol. 2012;18(8):413–415. | |
Goodarzi H, Sivamani RK, Garcia MS, et al. Effective Strategies for the Management of Pyoderma Gangrenosum. Adv Wound Care (New Rochelle). 2012;1(5):194–199. | |
Kowalzick L, Bertolini J, Baumann C, Walther B, Truhm B, Eickenscheidt L. Paradoxical reaction to etanercept: development of pyoderma gangraenosum during therapy of psoriasis arthritis. J Dtsch Dermatol Ges. 2013;11(5):447–449. | |
Ruocco E, Sangiuliano S, Gravina AG, Miranda A, Nicoletti G. Pyoderma gangrenosum: an updated review. J Eur Acad Dermatol Venereol. 2009; 23(9):1008–1017. | |
Pereira N, Brites MM, Gonçalo M, Tellechea O, Figueiredo A. Pyoderma gangrenosum – a review of 24 cases observed over 10 years. Int J Dermatol. 2013;52(8):938–945. | |
Wu XR, Shen B. Diagnosis and management of parastomal pyoderma gangrenosum. Gastroenterol Rep (Oxf). 2013;1(1):1–8. | |
Nguyen TV, Cowen EW, Leslie KS. Autoinflammation: From monogenic syndromes to common skin diseases. J Am Acad Dermatol. 2013;68(5):834–853. | |
Tüzün Y, Maraşoğlu O. Pyoderma Gangrenosum. J Turk acad Dermatol. 2007;1(3):71301r. | |
Reichrath J, Bens G, Bonowitz A, Tilgen W. Treatment recommendations for pyoderma gangrenosum: an evidence-based review of the literature based on more than 350 patients. J Am Acad Dermatol. 2005;53(2):273–283. | |
Landis ET, Taheri A, Jorizzo JL. Gulliver’s sign: A recognizable transition from inflammatory to healing stages of pyoderma gangrenosum. J Dermatolog Treat. In press 2014. | |
Von den Driesch P. Pyoderma gangrenosum: a report of 44 cases with follow-up. Br J Dermatol. 1997;137(6):1000–1005. | |
Patel F, Fitzmaurice S, Duong C, et al. Effective Strategies for the Management of Pyoderma Gangrenosum: A Comprehensive Review. Acta Derm Venereol. In press 2014. | |
Omiya W, Ujiie H, Akiyama M, et al. Coexistence of pustular and vegetative pyoderma gangrenosum in a patient with myelodysplastic syndrome. Eur J Dermatol. 2012;22(5):711–712. | |
Feliciani C, De Simone C, Amerio P. Dermatological signs during inflammatory bowel diseases. Eur Rev Med Pharmacol Sci. 2009;13 Suppl 1:15–21. | |
Speeckaert R, De Smet L, De Schepper S, et al. Pyoderma gangrenosum with granuloma formation: not always a benign disorder. J Eur Acad Dermatol Venereol. Epub August 29, 2014. | |
Dasanu CA, Bockorny B, Alexandrescu DT. Pyoderma gangrenosum due to lenalidomide use for multiple myeloma. J Oncol Pharm Pract. In press 2014. | |
Duchnowska R, Ziajka E, Góralska A, Grala B. Recurrent pyoderma gangrenosum precipitated by breast cancer: a case report and review of the literature. J Med Case Rep. 2014;8:226. | |
Doria A, Zen M, Bettio S, et al. Autoinflammation and autoimmunity: bridging the divide. Autoimmun Rev. 2012;12(1):22–30. | |
Craig FF, Thomas KS, Mitchell EJ, et al. UK Dermatology Clinical Trials Network’s STOP GAP trial (a multicentre trial of prednisolone versus ciclosporin for pyoderma gangrenosum): protocol for a randomised controlled trial. Trials. 2012;28;13:51. | |
Brooklyn TN, Dunnill MG, Shetty A, et al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006;55(4):505–509. | |
Sharon V, Burrall B, Patel F, et al. Multimodal therapy of idiopathic pyoderma gangrenosum. Dermatol Online J. 2014;15;20(6). | |
Andrade P, Brites MM, Figueiredo A. Synchronous pyoderma gangrenosum and inflammatory bowel disease, healing after total proctocolectomy. An Bras Dermatol. 2012;87(4):637–639. | |
Cosgarea I, Lovric Z, Körber A, Dissemond J. Successful treatment of refractory pyoderma gangrenosum with ustekinumab only after excision of renal cell carcinoma. Int Wound J. Epub October 8, 2014. | |
Pazyar N, Feily A, Yaghoobi R. An overview of interleukin-1 receptor antagonist, anakinra, in the treatment of cutaneous diseases. Curr Clin Pharmacol. 2012;7(4):271–275. | |
Naik HB, Cowen EW. Autoinflammatory pustular neutrophilic diseases. Dermatol Clin. 2013;31(3):405–425. | |
Ye MJ, Ye JM. Pyoderma gangrenosum: a review of clinical features and outcomes of 23 cases requiring inpatient management. Dermatol Res Pract. Epub October 8, 2014. | |
DeMartyn LE, Faller NA, Miller L. Treating peristomal pyoderma gangrenosum with topical crushed prednisone: a report of three cases. Ostomy Wound Manage. 2014;60(6):50–54. | |
Kapila S, Reid I, Dixit S, et al. Use of dermal injection of activated protein C for treatment of large chronic wounds secondary to pyoderma gangrenosum. Clin Exp Dermatol. 2014;39(7):785–790. | |
Liu DY, Fischer R, Fraga G, Aires DJ. Collagenase ointment and topical timolol gel for treating idiopathic pyoderma gangrenosum. J Am Acad Dermatol. 2014;71(5):e225–e226. | |
Imus G, Golomb C, Wilkel C, Tsoukas M, Nowak M, Falanga V. Accelerated healing of pyoderma gangrenosum treated with bioengineered skin and concomitant immunosuppression. J Am Acad Dermatol. 2001;44(1):61–66. | |
Sick I, Trautner B, Ruzicka T. [Surgical management of Pyoderma gangrenosum]. Hautarzt. 2012;63(7):577–583. German. | |
Cafardi J, Sami N. Intravenous immunoglobulin as salvage therapy in refractory pyoderma gangrenosum: report of a case and review of the literature. Case Rep Dermatol. 2014;23;6(3):239–244. | |
Sinnya S, Hamza S. Pyoderma gangrenosum of the breast treated with intravenous immunoglobulin. J Dermatol Case Rep. 2013;30;7(2):64–68. | |
Campanati A, Brisigotti V, Ganzetti G, et al. Finally, recurrent pyoderma gangrenosum treated with Adalimumab: case report and review of the literature. J Eur Acad Dermatol Venereol. In press 2014. | |
Cinotti E, Labeille B, Perrot JL, Pallot-Prades B, Cambazard F. Certolizumab for the treatment of refractory disseminated pyoderma gangrenosum associated with rheumatoid arthritis. Clin Exp Dermatol. 2014;39(6):750–751. | |
Staub J, Pfannschmidt N, Strohal R, et al. Successful treatment of PASH syndrome with infliximab, cyclosporine and dapsone. J Eur Acad Dermatol Venereol. In press 2014. | |
Langan SM, Groves RW, Card TR, Gulliford MC. Incidence, mortality, and disease associations of pyoderma gangrenosum in the United Kingdom: a retrospective cohort study. J Invest Dermatol. 2012;132(9):2166–2170. | |
Bennett ML, Jackson JM, Jorizzo JL, Fleischer AB Jr, White WL, Callen JP. Pyoderma gangrenosum. A comparison of typical and atypical forms with an emphasis on time to remission. Case review of 86 patients from 2 institutions. Medicine (Baltimore). 2000;79(1):37–46. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.