Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 21
Pulmonary-Intestinal Axis: Shared Genetic Basis and Mediating Factors Identified Through Multi-Omics Analysis
Authors Zhang Z, Liu H, Su Y, Wang C, Wang W, Li Y, Zhang X, Gao C, Tian X, Zhao C ![]()
Received 19 August 2025
Accepted for publication 9 March 2026
Published 7 April 2026 Volume 2026:21 561645
DOI https://doi.org/10.2147/COPD.S561645
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jill Ohar
Zhenzhu Zhang,1 Haoyue Liu,2 Yihang Su,3 Chengwen Wang,4 Wei Wang,1 Yinglin Li,1 Xiaobao Zhang,1 Chenguang Gao,5 Xue Tian,6 Chunqing Zhao7
1Graduate School, Chengde Medical University, Chengde, People’s Republic of China; 2Department of Cardiovascular Surgery, The Second Hospital of Jilin University, Changchun, People’s Republic of China; 3Department of Lung Cancer Surgery, Tianjin Medical University General Hospital, Tianjin, 300052, People’s Republic of China; 4Department of Cardiovascular Surgery & The Institute of Cardiovascular Diseases, TEDA International Cardiovascular Hospital, Tianjin University, Tianjin, People’s Republic of China; 5Department of Medical Laboratory Science, Jining Public Health Medical Center, Jining, People’s Republic of China; 6Department of Ultrasound, Jining Public Health Medical Center, Jining, People’s Republic of China; 7Department of General Surgery, The NO.2 Hospital of Baoding, Baoding, People’s Republic of China
Correspondence: Chunqing Zhao, Department of General Surgery, The NO.2 Hospital of Baoding, Baoding, People’s Republic of China, Email [email protected]
Background: Chronic obstructive pulmonary disease (COPD) is a systemic condition with comorbidities beyond the lung (eg, cardiovascular and metabolic disorders), and gastrointestinal (GI) disorders are also common. The shared genetic basis of COPD-GI comorbidity and its mediating factors remain unclear. We hypothesized that COPD and GI diseases share pleiotropic genetic architecture implicating lipid-metabolic pathways, with smoking mediating part of the association.
Methods: We analyzed publicly available European-ancestry GWAS summary statistics for COPD (Global Biobank Meta-analysis Initiative), 15 GI diseases (FinnGen), and smoking phenotypes (UK Biobank). Genetic correlation was estimated using linkage disequilibrium score regression (LDSC) and high-definition likelihood (HDL). Multi-trait analysis of GWAS (MTAG) boosted COPD discovery by leveraging genetically correlated GI traits. We integrated locus-to-gene mapping with multi-tissue expression quantitative trait loci (eQTL) and plasma protein quantitative trait loci (pQTL) evidence to prioritize shared loci, genes, and proteins. Bidirectional two-sample Mendelian randomization (MR) tested causal directions, and two-step mediation MR evaluated smoking.
Results: COPD showed significant genetic correlation with nine GI diseases. We identified six comorbidity-associated loci (three with CADD > 12.37) and 13 unique candidate pleiotropic genes; APOE was supported by proteomic evidence. Enrichment analyses highlighted lipid-metabolism pathways. MR suggested COPD increases risk of gastroesophageal reflux disease (GERD), irritable bowel syndrome (IBS), acute appendicitis, and gastric ulcer, while diverticular disease showed reverse causality toward COPD. Smoking partially mediated the COPD effect on GERD, acute appendicitis, and gastric ulcer.
Conclusion: COPD and multiple GI disorders share a distributed pleiotropic genetic basis within the broader systemic comorbidity spectrum of COPD. Multi-omics evidence supports a genomic pulmonary-intestinal axis in which lipid metabolism and smoking-related mechanisms contribute to COPD and GI comorbidity, providing targets for risk stratification and potential intervention.
Keywords: COPD, GWAS, genetic correlation, mediation analysis
Introduction
Chronic obstructive pulmonary disease (COPD) is characterized by persistent airway obstruction and chronic inflammation, leading to a progressive and largely irreversible decline in lung function, and currently affects approximately 544.9 million individuals worldwide.1,2 Increasing evidence indicates a close association between COPD and gastrointestinal (GI) disorders. A retrospective study of 1228 COPD patients reported that a substantial proportion experienced at least one GI symptom,3 most commonly gastroesophageal reflux disease (GERD) and irritable bowel syndrome (IBS).4 In parallel, structural and functional alterations of the intestinal barrier, including epithelial damage and increased permeability, have been observed in COPD patients,5 suggesting systemic involvement beyond the respiratory tract. Moreover, clinical trials have demonstrated that oral supplementation with probiotics such as Bifidobacterium and Lactobacillus can shorten the duration of respiratory symptoms and reduce antibiotic use compared with placebo,6 further supporting a functional link between the lung and gut.
These bidirectional interactions are thought to be mediated by the pulmonary-intestinal axis (PIA), which integrates immune signaling, autonomic nervous system activity, lipid metabolism, and the gut microbiota.7,8 Notably, the gut microbiota exerts a considerable influence on host lipid metabolism,9 providing a potential mechanistic bridge between COPD and GI disorders. Accordingly, lipid metabolic dysregulation may represent a critical component underlying their comorbidity.
Recent genome-wide association studies (GWAS) of specific GI diseases have highlighted the importance of shared genetic architecture across complex traits.10 Identifying common genetic variants or loci that contribute to genome-wide genetic correlations is essential for elucidating the molecular basis of PIA-related disorders. However, most previous studies have focused on single diseases in isolation, which may limit the detection of pleiotropic loci and shared regulatory mechanisms. Multi-trait analytical strategies offer an opportunity to expand the phenotypic scope, enhance statistical power, and systematically investigate shared genetic etiology across diseases.11 Cross-trait analyses leverage correlations among GWAS signals to identify pleiotropic variants or loci influencing multiple traits simultaneously,12,13 some of which may serve as potential therapeutic targets.
In addition, several pleiotropic loci identified in this study were consistently associated with smoking behavior, which is a well-recognized factor influencing gut microbiota composition and diversity. Evidence also suggests a bidirectional relationship between COPD and smoking behavior,14 while smoking exerts broad pathological effects on the GI tract.15 These observations underscore the complexity of the COPD–smoking–GI disease axis. To further clarify this relationship, we conducted mediation Mendelian randomization analyses to evaluate the extent to which smoking mediates the comorbidity between COPD and GI diseases.
Accordingly, lipid metabolism was defined as a central analytical focus, given its intersection with microbial regulation, smoking-related exposure, and systemic inflammatory processes relevant to both COPD and gastrointestinal disorders. From a genetic perspective, lipid-associated traits provide a biologically plausible framework for exploring shared susceptibility across organ systems. On this basis, subsequent analyses were designed to interrogate lipid-related pleiotropic genetic architecture linking COPD and GI diseases. The flowchart illustrates the general design of our study (Figure 1).
 |
Figure 1 Overall study design. |
Research Methods
Data Collection and Quality Control
In this study, gene analysis was conducted using a comprehensive collection of GWAS summary data. All data processing and statistical analyses were conducted at The No. 2 Hospital of Baoding, Baoding, China. The study was conducted between December 2024 and April 2025 using publicly available GWAS summary statistics. Owing to the limited availability of GWAS data from non-European ancestries, the summary data were primarily sourced from publicly accessible datasets of European descent. GWAS data for COPD were obtained from European cohorts within the Global Biobank Meta-analysis Initiative, encompassing 58,559 patients and 937,358 controls. The 15 prevalent GI diseases included in this study were all sourced from the Finnish database:16 acute gastritis, acute pancreatitis, alcoholic liver disease, chronic gastritis, chronic pancreatitis, celiac disease, diverticular disease, duodenal ulcer, functional gastrointestinal disorders, gastric ulcer, IBS, inflammatory bowel disease, GERD, acute appendicitis, and non-alcoholic fatty liver disease. Smoking phenotype data were retrieved from the UK Biobank (ukb-b-10831) (Supplementary Table S1). To elucidate the genetic architecture of COPD, quantitative trait locus (QTL) data were incorporated, including expression QTLs (eQTLs) from 54 distinct tissues, such as GI tissues and blood, as well as plasma protein QTLs (pQTLs) from three sources: Decode, UKBPP, and ARIC. The eQTL analysis included data from the eQTLGen consortium, which provided trait-associated SNPs in a cohort of 31,684 individuals.17 Additionally, plasma pQTL data were obtained from deCODE’s comprehensive protein measurements involving 35,559 Icelandic participants, focusing on 4907 plasma proteins,18 and the UKBPP project, which included 54,219 UK Biobank participants and examined 2923 plasma proteins.19 During the analysis, any SNVs within the major histocompatibility complex region on chromosome 6, specifically within the 25–35 MB region, were excluded to mitigate potential confounding effects. SNPs with a minor allele frequency < 0.01 were filtered out, and those with duplicate or missing rsIDs in the GWAS summary datasets were removed. Rigorous quality control procedures were applied to all data, including adjustments for genomic control values. To ensure consistency, data aligned with the GRCh38 reference were converted to the GRCh37 reference using the liftOver tool.20 The analytical workflow of this study consisted of the following steps: (1) collection of GWAS summary statistics; (2) quality control and harmonization; (3) genetic correlation analyses using LDSC and HDL; (4) multi-trait GWAS analysis (MTAG); (5) identification of independent and pleiotropic loci; (6) transcriptomic and proteomic prioritization; (7) drug–gene interaction analysis; and (8) causal and mediation analyses using Mendelian randomization.
Exploring the Genetic Interactions Between COPD and GI Diseases
The study employed linkage disequilibrium score regression (LDSC) analysis to examine the genetic interactions between COPD and 15 GI diseases individually. Specifically, linkage disequilibrium (LD) scores were computed utilizing data from individuals of European descent provided by the 1000 Genomes Project.21 This approach facilitates the consideration of residual confounding variables and assesses the potential for sample overlap in GWAS datasets. Subsequent to the LDSC analysis, genetic correlations were further validated using high-definition likelihood (HDL) analysis.22 To ensure the robustness and accuracy of the findings, all statistical tests were subjected to Benjamini-Hochberg (BH) correction for multiple comparisons, establishing the significance threshold at an adjusted P-value below 0.05.
Comprehensive Analysis of COPD and GI Disease Characteristics
Building upon the findings from the prior stage, an extensive multi-trait analysis was performed. The GI disease phenotypes—including GERD, appendicitis, IBS, diverticular disease, acute gastritis, chronic pancreatitis, and alcoholic liver disease—that exhibited genetic correlations with COPD in both LDSC and HDL analyses were analyzed through a multi-trait analysis of GWAS (MTAG). This approach enables the amalgamation of data across multiple GWAS, thereby augmenting the effective sample size, bolstering the detection power for genetic variants, and minimizing the likelihood of false-positive or false-negative results due to genetic correlations. Furthermore, MTAG incorporates LDSC adjustments to account for any overlapping GWAS samples among correlated traits.23 In this process, the GWAS data of GI disease phenotypes with confirmed genetic correlations to COPD were integrated to generate MTAG-COPD. For this analysis, the threshold for genome-wide significance was stringently established at a P-value below 5×10−8 to ensure the rigorous identification of associations.
Analysis of Genetic Risk Factors for COPD and GI Comorbidities
Identification of Independent Risk Loci
To further elucidate the genomic risks associated with COPD-GI comorbidity, distinct independent signals within genomic loci linked to MTAG-COPD were examined. This was achieved through conditional and joint association analyses using the stepwise model selection framework provided by GCTA-conditional and joint analysis (COJO).24 The analysis focused on multi-allelic variants displaying significant associations (P.mtag < 5×10−8) within recognized genomic risk loci, with additional signals being validated based on a joint P-value threshold of < 5×10−8. This investigation utilized the European ancestry reference cohort dataset from the third phase of the 1000 Genomes Project.21 For pairs of traits showing significant genetic correlation or overlap, pleiotropic analysis under the composite null hypothesis (PLACO)13 was employed to detect potential pleiotropic SNVs. In this analysis, squared Z scores were computed for each variant, and SNVs with exceedingly high Z2 values (> 80) were excluded. Furthermore, due to the potential correlation between the two diseases, the Z correlation matrix was estimated and integrated into the analysis. Subsequently, the hypothesis of no pleiotropy was assessed using the intersection-union test method with a horizontal α, leading to the determination of the final pleiotropic p-values. Significant pleiotropic variants were identified as SNVs with P-values less than 5×10−8. Following the PLACO results, the identified loci were mapped to neighboring genes to investigate the shared biological mechanisms underlying these pleiotropic loci. Additional annotation was performed using the Functional Mapping and Annotation (FUMA) platform.25 FUMA facilitated the identification of genomic risk loci, utilizing European ancestry data from the 1000 Genomes Project Phase 3 as a reference panel. Strict criteria were applied, setting the maximum P-value for lead SNVs at < 5 × 10−8, while adopting a more inclusive significance threshold of P < 0.05. Independent SNV identification was based on an r2 threshold of less than 0.6, with lead SNVs requiring an r2 of less than 0.1 within a 1 Mb radius. Criteria for defining genomic risk loci included merging regions with lead SNVs less than 250 kb apart. SNVs validated through COJO and FUMA analyses were classified as risk variants for COPD. These variants were annotated using ANNOVAR,26 and their potential deleterious impact was evaluated through combined annotation-dependent depletion (CADD) scores, with values exceeding 12.37, suggesting a higher likelihood of harmful effects.27 RegulomeDB (RDB)28 provides a classification score ranging from 1a to 7, indicating the regulatory function of SNPs based on eQTLs and chromatin marks. A score of 1a represents the strongest biological evidence for the SNP as a regulatory element.
Insights into COPD and GI Comorbidity at the Transcriptome Level
In a comprehensive analysis targeting the genetic underpinnings of COPD-GI comorbidities, two distinct strategies were implemented. For each approach, P-values were corrected for the false discovery rate (FDR) using the BH method, with genes displaying an FDR < 0.05 and those consistently identified by both approaches being deemed significantly associated risk genes. The multi-marker analysis of genoMic annotation (MAGMA) and polygenic priority score (POPS) was applied to identify and prioritize relevant genes.29,30 The gene-centric analysis conducted in MAGMA was derived from an extensive catalog of protein-coding genes, and when combined with POPS, it enabled a more refined gene prioritization strategy. This methodology integrated summary GWAS data with comprehensive expression datasets and biological pathways, establishing a POPS score threshold greater than 1 for gene selection.
To investigate the biological relevance of genes associated with COPD-GI comorbidity, enrichment analyses were performed, encompassing both phenotypic and genomic evaluations. Phenotypic enrichment was assessed using the Mammalian Phenotype Ontology from Mouse Genome Informatics,31 comparing the specificity of mammalian phenotype (MP) genes against non-MP genes through phenotypic associations. Genomic enrichment involved data from the Molecular Signatures Database,32 with significant biological pathways identified by the ClusterProfiler tool after adjusting for multiple comparisons.33
Insights from Proteomics on COPD and GI
To unravel the complex proteomic profiles linked with COPD and GI diseases, the study utilized the “Biomarker Imputation from Summary Statistics (BLISS)” technique. This innovative methodology markedly differs from the conventional proteome-wide association study (PWAS) model training, which typically depends on detailed proteomic data at the individual level. Such reliance often constrains the capacity to exploit the vast array of publicly available summary-level pQTL data.34 BLISS introduces a novel approach for developing protein interpolation models directly from summary-level pQTL data, thereby enabling the creation of comprehensive European PWAS models. This was accomplished by employing extensive pQTL information from large repositories, including the UKB, deCODE, and ARIC studies.34 In this segment of the research, the BH procedure continued to be applied to ascertain the FDR of p-values. Proteins exhibiting an FDR of less than 0.05 were recognized as significant risk proteins, underscoring their potential critical role in the pathophysiology of COPD and GI disease comorbidity.
Establishing a Drug-Gene Interaction Network
Utilizing the drug prediction database DGIdb (http://www.dgidb.org/),35 the interactions between drugs and pleiotropic genes were further anticipated through the application of the “FDA approved” filter. Following this, a drug-gene interaction network was established employing Cytoscape software, grounded on the aforementioned prediction outcomes.
Two-Sample MR (TSMR) Analysis
The widely recognized instrumental variable technique, MR analysis, was employed to discern causal relationships between COPD and GI diseases. SNPs associated with exposure served as instruments,36 while data derived from GWAS summaries were utilized to pinpoint variants correlated with COPD and GI diseases, adhering to a P-value threshold of < 5.0e-8. The inverse-variance weighted (IVW) approach was primarily implemented, with LD and physical distance thresholds established at 0.001 and 10 MB, respectively. To ensure the robustness of the instrumental variables, the coefficient of determination (r2) and F-statistic were computed, incorporating only those SNPs with F-values exceeding 10. Furthermore, the MR-PRESSO method, employing 1000 iterations, was utilized to identify outliers.37 LDlink facilitated the exclusion of confounding SNPs, and the identified outliers were subsequently removed for reassessment.
Mediation MR Analysis
Drawing upon the TSMR findings and insights from pleiotropic loci, an assessment was conducted to determine whether smoking serves as a mediator in this association. A subsequent two-step mediation MR analysis was performed. The total effect (beta0) signifies the causal effect of COPD on GI diseases, including IBS, GERD, acute appendicitis, and gastric ulcer. The step1 effect (beta1) reflects the causal impact of COPD on smoking, while the step2 effect (beta2) denotes the causal influence of smoking on GI diseases such as IBS, GERD, acute appendicitis, and gastric ulcer. All values for beta0, beta1, and beta2 were derived using the IVW method. If beta0 was not significant, but both beta1 and beta2 were significant, the exposure-outcome association was regarded as fully mediated by the specified mediator. Conversely, if beta0, beta1, and beta2 were all significant, the association was deemed partially mediated. However, if beta0 was significant while beta1 and/or beta2 were not, the mediator was considered to exert no effect on the exposure-outcome relationship. Upon identifying a significant mediation effect, the proportion of mediation was calculated accordingly.
Research Results
Revealing the Genetic Interactions Between COPD and Multiple GI Disorders
Genetic correlation analyses revealed a non random comorbidity spectrum between COPD and gastrointestinal traits. We identified significant positive genetic correlations between COPD and multiple GI disorders, whereas several phenotypes showed negative or non significant correlations. These correlation signals provided the basis for downstream prioritization of shared loci, genes, and proteins, and for subsequent causal inference analyses described in the following sections (Figure 1). An LDSC analysis was conducted to explore the genetic interactions between COPD and 15 GI disorders, with HDL analysis employed for validation purposes. Following stringent BH correction, significant genetic correlations were consistently observed between COPD and 9 GI disorders, including GERD, appendicitis, IBS, diverticular disease, acute gastritis, acute pancreatitis, chronic pancreatitis, functional gastrointestinal disorder, and alcoholic liver disease, across both methods (Table 1).
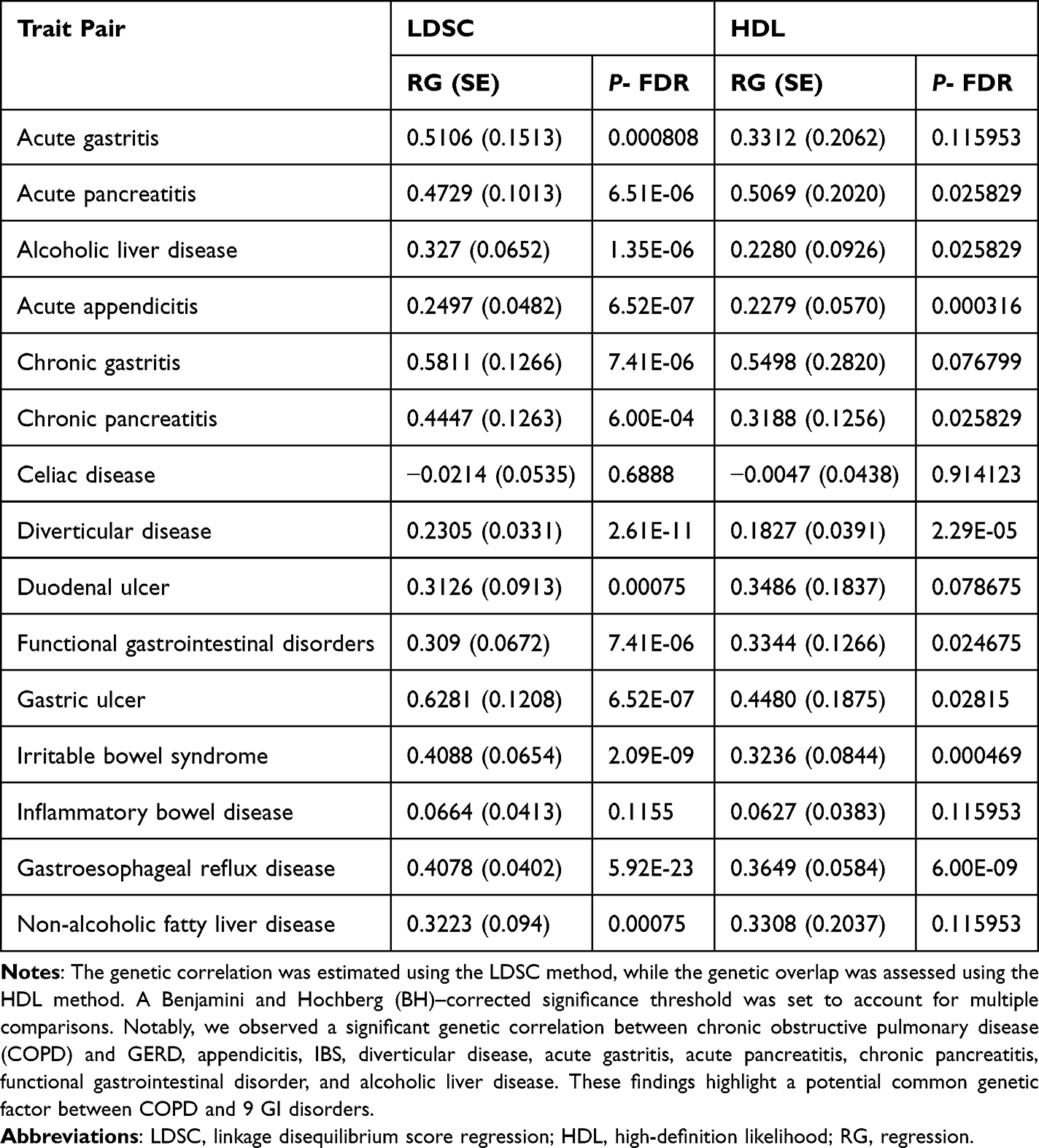 |
Table 1 Genetic Correlation Analysis Results |
Genetic Patterns of COPD and GI Comorbidities Through Multi-Trait Analysis
The scope of the investigation was expanded to explore the intricate genetic patterns of COPD through the application of multi-trait analysis, utilizing summary data from GWAS of COPD and 9 GI disease phenotypes (Figure 2). The MTAG method was employed to generate an enhanced dataset for COPD (MTAG-COPD), which included 10,426,094 SNPs. From this dataset, 729 variant SNPs were identified as meeting the established threshold criteria. Advanced GCTA-COJO tools were subsequently used to perform conditional and joint association analyses on the MTAG-COPD dataset, resulting in the identification of 21 SNVs (Supplementary Table S2). Following this, PLACO identified 6938 SNVs (5662 unique) as potential pleiotropic variants across 9 COPD-GI disease pairs. The PLACO results were further analyzed using the FUMA platform, which yielded 57 lead SNVs (Supplementary Table S3). Of particular note, 8 (6 unique) SNVs demonstrated consistency across both COJO and FUMA analyses, thus establishing them as independent genetic risk loci for COPD-GI disease comorbidity, encompassing 6 distinct chromosomal regions.
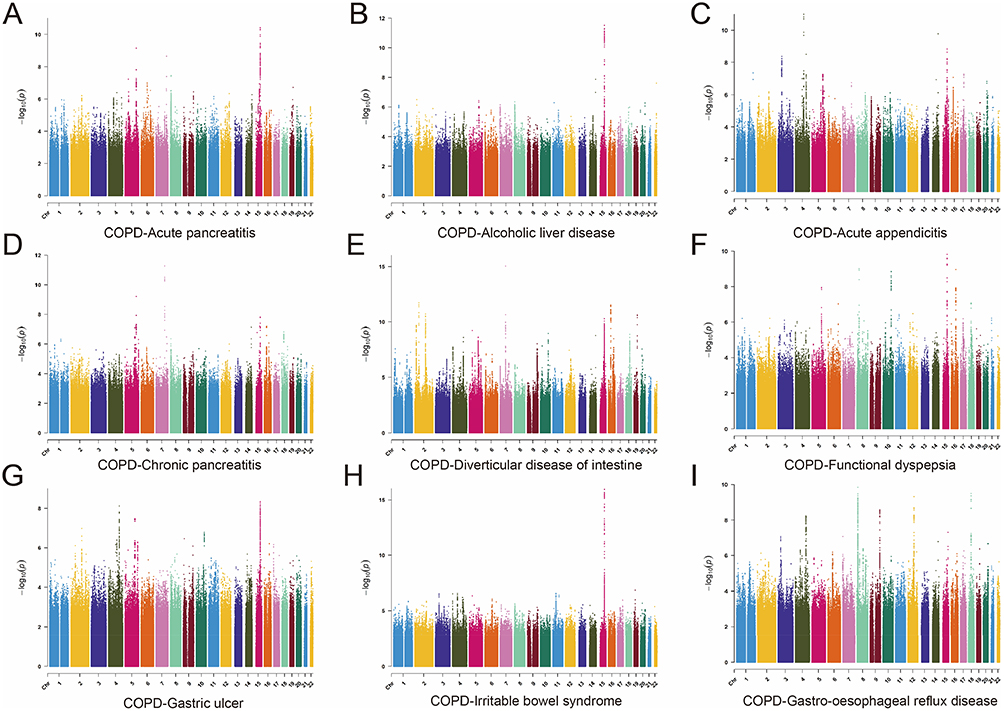 |
Figure 2 Manhattan plots of snp-based enrichments of PLACO associations with (A) Acute pancreatitis, (B) Alcoholic liver disease, (C) Acute appendicitis, (D) Chronic pancreatitis, (E) Diverticular disease of intestine, (F) Functional dyspepsia, (G) Gastric ulcer, (H) Irritable bowel syndrome, and (I) Gastro-oesophageal reflux disease. The x-axis shows chromosomal position, and the y-axis shows association p-values on a −log10 scale. |
ANNOVAR category annotation revealed that 6 (4 unique) of the 8 (6 unique) index SNVs were intergenic variants, with 1 classified as an intronic variant (rs1023518) and 1 as an exonic variant (rs8040868). Among these, the 14q32.13 region (mapped gene: SERPINA1) was recognized as a pleiotropic region across multiple trait pairs, indicating its broad pathogenic effects. Additionally, rs9964724 (CELF4) and rs13141641 (KRT18P51), both situated in intergenic regions, were distinguished by CADD scores exceeding 12.37, suggesting their potential pathogenic roles as SNVs.
Identification of Genes Associated with the Risk of COPD and GI Comorbidities
Through MAGMA analysis, a total of 301 genes were identified as being associated with shared risk SNVs for comorbidity (Supplementary Table S4). Following this, POPS screening identified 15 genes with POPS scores exceeding 1, marking them as potential pleiotropic genes linked to comorbidity (Supplementary Table S5). Genome enrichment analysis revealed significant enrichment within biological processes related to lipid metabolism, such as lipid transport involved in lipid storage, regulation of lipid metabolic processes, triglyceride metabolic process, and cholesterol transport, which indicates the involvement of lipid metabolism in COPD and its associated GI diseases (Supplementary Table S6). Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis further underscored the importance of cholesterol metabolism and the cyclic Adenosine Monophosphate signaling pathway, among others (Supplementary Table S7). Significant phenotypic correlations were observed within the categories of respiratory system phenotypes and cardiovascular system phenotypes, highlighting the complex nature of COPD-GI comorbidity and its systemic effects (Supplementary Table S8).
Identification of Proteins Associated with the Risk of COPD and GI Comorbidities
The BLISS method, in conjunction with MTAG COPD data, was employed to assess plasma proteins within the UKB-PP, Decode, and ARIC databases. This extensive analysis led to the identification of 210 proteins (178 unique) linked to the risk of COPD and GI comorbidities (Supplementary Table S9). Notably, the gene encoding apolipoprotein E (APOE) was found among the previously identified pleiotropic genes, thereby further affirming the validity of the pleiotropic gene identification.
Prediction of Drug-Gene Interactions
Drawing from the drug-gene interaction data of pleiotropic genes provided by the DGIdb database, 184 drug-gene interaction pairs were identified, encompassing 6 pleiotropic genes (MAML3, APOE, DRD2, CRHR1, PDE4B, and APOC1) and 180 small drug molecules. A drug-gene interaction network was subsequently constructed using Cytoscape (Supplementary Table S10), potentially aiding in the identification of new therapeutic targets for treating COPD-GI disease comorbidity.
TSMR
A bidirectional MR analysis was performed to explore the potential causal links between COPD and nine associated GI diseases. The findings demonstrated causal associations between COPD and GERD [OR: 1.32; 95% CI: 1.17–1.49], IBS [OR: 1.25; 95% CI: 1.09–1.42], acute appendicitis [OR: 1.11; 95% CI: 1.01–1.21], and gastric ulcer [OR: 1.18; 95% CI: 1.00–1.39], identifying COPD as a risk factor for these gastrointestinal conditions (Figure 3). Visual representations, including scatter plots, funnel plots, and leave-one-out sensitivity analyses, are provided in Supplementary Figures S1–S3. Conversely, reverse MR analysis uncovered a positive causal link between diverticular disease [OR: 1.04; 95% CI: 1.01–1.07] and the occurrence of COPD (Figure 4). Visual representations, including scatter plots, funnel plots, and leave-one-out sensitivity analyses, are provided in Supplementary Figures S4–S6.
 |
Figure 3 Causal correlations of 9 gastrointestinal diseases (GERD, appendicitis, IBS, diverticular disease, acute gastritis, acute pancreatitis, chronic pancreatitis, functional gastrointestinal disorder, and alcoholic liver disease) on COPD. Estimates and 95% confidence intervals (CI) are shown using square plots and error bars. |
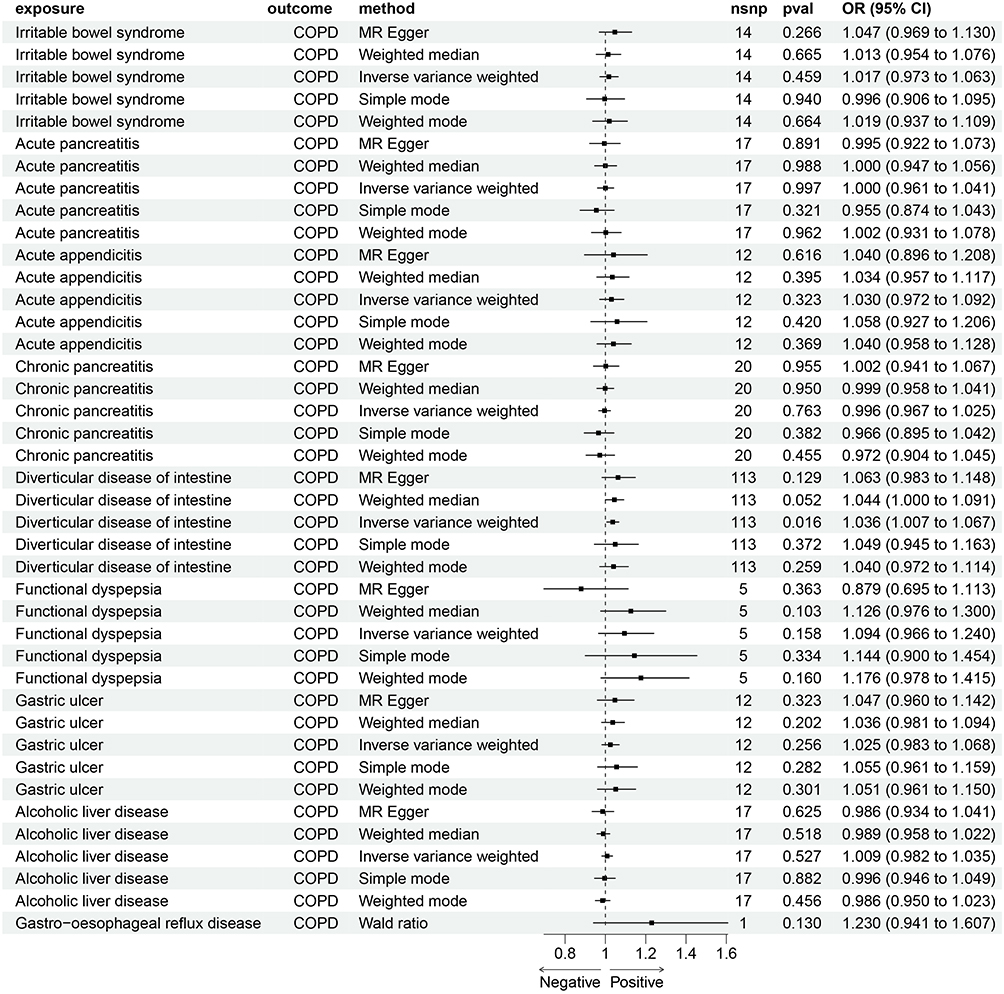 |
Figure 4 Causal correlations of COPD on 9 gastrointestinal diseases (GERD, appendicitis, IBS, diverticular disease, acute gastritis, acute pancreatitis, chronic pancreatitis, functional gastrointestinal disorder, and alcoholic liver disease). Estimates and 95% confidence intervals (CI) are shown using square plots and error bar. |
Smoking Mediates MR in COPD-GI Comorbidity
In light of previous findings that identified pleiotropic loci linked to smoking phenotypes, a mediation MR study was undertaken, with smoking serving as the mediator. Initially, the causal impact of COPD on smoking was assessed, with results indicating a positive causal relationship, thereby suggesting that COPD elevates the risk of smoking. Following this, MR analyses were carried out between smoking and four GI diseases (GERD, IBS, acute appendicitis, and gastric ulcer). The outcomes demonstrated that smoking exerts a positive causal effect on GERD, acute appendicitis, and gastric ulcers, functioning as a risk factor for these conditions. Consequently, the findings imply that smoking can act as a mediator for GERD, acute appendicitis, and gastric ulcers. The mediation effects and rates are presented in Table 2.
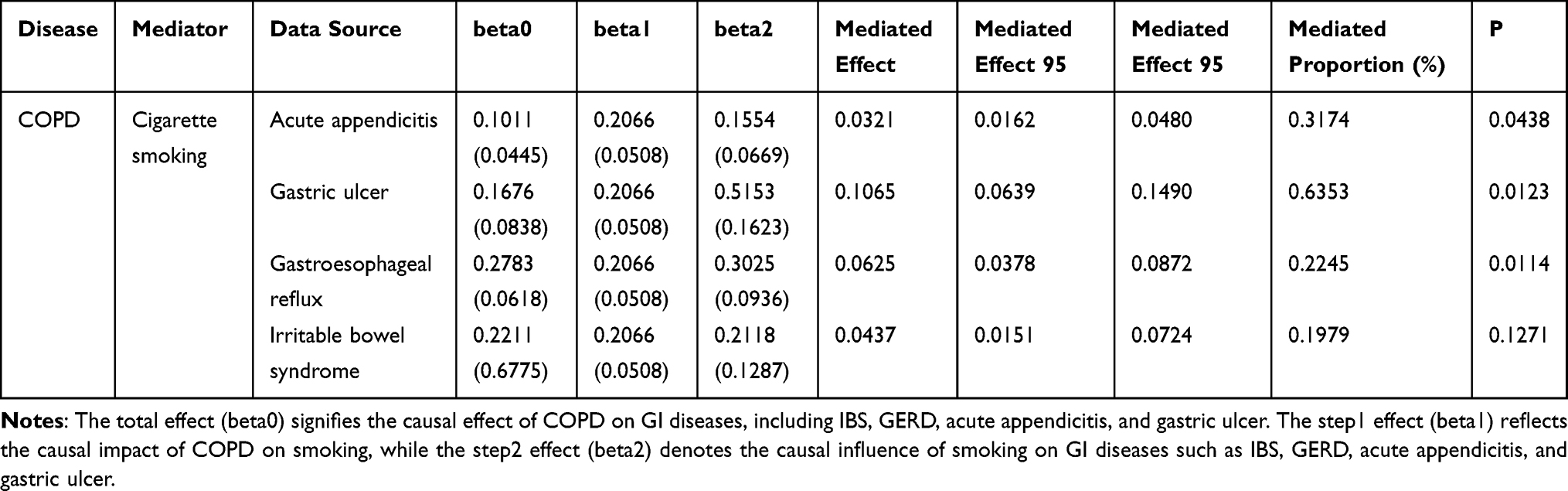 |
Table 2 The MR Effect of Mediators on Gastrointestinal Diseases and Mediate Proportion |
Discussion
This study presents the first comprehensive evidence of genome-wide genetic correlations and genetic overlap between COPD and various GI diseases. Through genetic correlation analysis conducted using LDSC and HDL across COPD and 15 GI diseases, significant genetic correlations were detected between COPD and 9 specific GI diseases, including GERD, appendicitis, IBS, diverticular disease, acute gastritis, chronic pancreatitis, and alcoholic liver disease. MR analysis further established positive causal relationships between COPD and IBS, GERD, appendicitis, and gastric ulcer, in addition to confirming a positive causal relationship between diverticular disease and COPD. Moreover, smoking was identified as a mediating factor in the associations between COPD and GERD, appendicitis, and gastric ulcers. These findings substantially advance our understanding of the comorbidity and causal relationships linking COPD and GI diseases.
Several mechanisms may account for the heightened risk of GERD associated with COPD. Firstly, the increased expiratory pressure in COPD patients is likely to elevate the risk of reflux.38 Additionally, the development of barrel chests in severe cases of COPD can compress the lower esophageal sphincter (LES), thus facilitating esophageal reflux.39 Moreover, smoking has been implicated, as nicotine can induce relaxation of the LES circular muscle and diminish esophageal clearance.40 In COPD patients, a high smoking index and a greater number of pack-years have been identified as independent risk factors for GERD (OR 1.015 [95% CI 1.004–1.025]),41 indicating that smoking may contribute to the onset of GERD in these individuals. Consistent with our findings, research has shown that COPD may substantially increase the risk of gastric ulcer bleeding (HR = 1.93, [95% CI: 1.73–2.17]).42 Concurrently, gastric ulcers may reduce the FEV1/FVC ratio and potentially serve as a risk factor for COPD.43 Smoking, recognized as a risk factor for gastric ulcers, could further exacerbate this vicious cycle.44 Additionally, chronic systemic inflammation, a hallmark of COPD, disrupts the intestinal immune system and gut microbiome environment,45 while chronic hypoxia associated with COPD increases intestinal epithelial permeability and promotes ecological imbalance,46 potentially heightening the risk of IBS. The BODE cohort, which included 1664 COPD patients, reported 79 comorbidities,47 underscoring the systemic impact of COPD. Therefore, the findings of this study suggest that the positive causal relationship between COPD and acute appendicitis may be linked to COPD itself and the potential for multimorbidity. However, the relationship between diverticular disease and COPD has not been extensively documented in the literature and warrants further investigation.
Through the integration of COPD with 9 associated GI diseases into a comprehensive multi-trait analysis, six independent risk loci significantly linked to COPD-GI comorbidities were identified using PLACO and COJO. Notably, the 14q32.13 locus was identified across various phenotypes, including alcoholic liver disease, acute appendicitis, and diverticular disease. Previous research has substantiated the role of 14q32.13 in COPD pathogenesis. For instance, several rare genetic polymorphisms mapped to the SERPINA1 gene have been shown to significantly reduce α1-antitrypsin levels in the blood, thereby increasing the risk of early-onset COPD.48 Additionally, rs8040868, situated in the exonic region of CHRNA3 at 15q21.1, with a CADD score of 15.25 and an RDB score of 1f, demonstrates strong pathogenicity and transcriptional regulatory effects. Variants in this gene are linked to an increased risk of smoking initiation and a heightened susceptibility to emphysema. For example, rs8040868 has been identified as a significant indicator of emphysema and the severity of chest computed tomography manifestations in a Chinese population cohort,49 thereby reinforcing the reliability of our findings.
A comprehensive genetic association analysis identified 15 pleiotropic genes linked to COPD-GI comorbidity, with APOE validated at the protein level in the BLISS analysis. The APOE gene is integral to several lipid metabolism pathways, acting as one of the primary lipid carriers responsible for transporting lipids to cells and tissues to regulate blood lipid levels.50 Moreover, APOE interacts with various cell surface receptors, including low-density lipoprotein receptors and hepatic chylomicron remnant receptors, facilitating receptor-mediated endocytosis and thereby contributing to lipoprotein metabolism.51 Additionally, enrichment analysis revealed that the pleiotropic genes associated with COPD-GI comorbidity are enriched in multiple lipid metabolism pathways, such as cholesterol metabolism, lipid transport involved in lipid storage, regulation of lipid metabolic processes, triglyceride metabolic processes, and cholesterol transport. This underscores the role of lipid metabolism in COPD and its related GI diseases and supports the potential of APOE as a target for therapeutic intervention. Consequently, lipid metabolism pathways are highlighted as promising therapeutic targets for addressing COPD and GI disease comorbidity. This integrative perspective aligns with broader cross-organ disease interaction models. For example, within the cardiovascular–reproductive axis, pregnancy has been proposed as a physiological stress test that unmasks latent cardiovascular vulnerability and metabolic risk, illustrating how organ-specific perturbations can reveal systemic susceptibility.52 By analogy, framing COPD–GI comorbidity through the pulmonary–intestinal axis may strengthen the translational rationale for early risk stratification and targeted prevention at a system level. Future functional validation studies are needed to further elucidate the biological mechanisms underlying these genetic findings. Integrating transcriptome or single-cell RNA sequencing data from lung and gastrointestinal tissues will clarify whether the identified pleiotropic genes exhibit coordinated expression patterns across different organs.
Limitations
The participants in our study were all of European ancestry, which may limit the applicability of our findings to other populations. Replication in other cohorts is necessary. Because the underlying GWAS data were case–control based, we could not evaluate whether COPD stage or GI disease severity modifies these relationships, future studies integrating severity phenotypes and longitudinal cohorts are warranted.
Conclusion
Our study provides genome-wide evidence for shared genetic architecture between COPD and multiple GI disorders, identifying pleiotropic loci, candidate genes and proteins, and enrichment of lipid metabolism–related pathways. Bidirectional and mediation MR analyses further support smoking as a key mediator for selected COPD–GI comorbidities, offering a mechanistic link consistent with cross-organ interaction models. Clinically, these findings suggest that patients with COPD may benefit from proactive screening and integrated management of GI symptoms, alongside intensified smoking-cessation strategies, particularly in those with high genetic susceptibility. In addition, the prioritized loci and pathways may inform risk stratification, biomarker development, and potential therapeutic targeting or repurposing aimed at shared metabolic and inflammatory mechanisms.
Data Sharing Statement
The data presented in this study are openly available in gwascatlog at https://www.ebi.ac.uk/gwas/home. Further data and codes are available from the corresponding author.
Ethics Statement
The study titled “Pulmonary-Intestinal Axis:Shared Genetic Basis and Mediating Factors Identified through Multi-Omics Analysis” relied exclusively on genome wide association study GWAS summary statistics and publicly accessible eQTL and pQTL resources No individual level data were used The study did not recruit any new participants and did not collect or analyze any personally identifiable information or human biospecimens.
As this research is based solely on lawfully obtained, publicly accessible, fully de-identified data and involves no intervention or interaction with human subjects, it complies with Article 32 (Items 1 and 2) of the Measures for the Ethical Review of Life Sciences and Medical Research Involving Humans issued on February 18, 2023. Accordingly, the Ethics Committee of The NO.2 Hospital of Baoding has determined that this study is exempt from ethical review and that the requirement for informed consent is waived.
Consent for Publication
All authors consented to the submission and publication of this study.
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Wang Y, Xu J, Meng Y, et al. Role of inflammatory cells in airway remodeling in COPD. Int J Chron Obstruct Pulmon Dis. 2018;13:3341–14. doi:10.2147/COPD.S176122
2. Soriano JB, Kendrick PJ, Paulson KR, et al. Prevalence and attributable health burden of chronic respiratory diseases, 1990-2017: a systematic analysis for the global burden of disease study 2017. Lancet Respir Med. 2020;8(6):585–596.
3. Rutten EP, Spruit MA, Franssen FM, et al. GI symptoms in patients with COPD. Chest. 2014;145(6):1437–1438. doi:10.1378/chest.14-0285
4. Budden KF, Gellatly SL, Wood DL, et al. Emerging pathogenic links between microbiota and the gut-lung axis. Nat Rev Microbiol. 2017;15(1):55–63. doi:10.1038/nrmicro.2016.142
5. Rutten EPA, Lenaerts K, Buurman WA, et al. Disturbed intestinal integrity in patients with COPD: effects of activities of daily living. Chest. 2014;145(2):245–252. doi:10.1378/chest.13-0584
6. Heinken A, Khan MT, Paglia G, et al. Functional metabolic map of Faecalibacterium prausnitzii, a beneficial human gut microbe. J Bacteriol. 2014;196(18):3289–3302. doi:10.1128/JB.01780-14
7. Hagihara M, Kuroki Y, Ariyoshi T, et al. Clostridium butyricum modulates the microbiome to protect intestinal barrier function in mice with antibiotic-induced dysbiosis. iScience. 2020;23(1):100772. doi:10.1016/j.isci.2019.100772
8. Ariyoshi T, Hagihara M, Tomono S, et al. Clostridium butyricum MIYAIRI 588 modifies bacterial composition under antibiotic-induced dysbiosis for the activation of interactions via lipid metabolism between the gut microbiome and the host. Biomedicines. 2021;9(8):1065. doi:10.3390/biomedicines9081065
9. Brown EM, Clardy J, Xavier RJ. Gut microbiome lipid metabolism and its impact on host physiology. Cell Host Microbe. 2023;31(2):173–186. doi:10.1016/j.chom.2023.01.009
10. Liu B, Chen M, You J, et al. The causal relationship between gastroesophageal reflux disease and chronic obstructive pulmonary disease: a bidirectional two-sample mendelian randomization study. Int J Chron Obstruct Pulmon Dis. 2024;19:87–95. doi:10.2147/COPD.S437257
11. Lee CH, Shi H, Pasaniuc B, et al. PLEIO: a method to map and interpret pleiotropic loci with GWAS summary statistics. Am J Hum Genet. 2021;108(1):36–48. doi:10.1016/j.ajhg.2020.11.017
12. Bayardo M, Punzi F, Bondar C, et al. Transglutaminase 2 expression is enhanced synergistically by interferon-γ and tumour necrosis factor-α in human small intestine. Clin Exp Immunol. 2012;168(1):95–104. doi:10.1111/j.1365-2249.2011.04545.x
13. Ray D, Chatterjee N. A powerful method for pleiotropic analysis under composite null hypothesis identifies novel shared loci between Type 2 diabetes and prostate cancer. PLoS Genet. 2020;16(12):e1009218. doi:10.1371/journal.pgen.1009218
14. Floyd J, Mallow J, Wang K, et al. Differences in smoking behaviors and readiness to change for patients with COPD and differing categories of depressive symptoms: a descriptive cross-sectional design. BMC Pulm Med. 2023;23(1):335. doi:10.1186/s12890-023-02621-2
15. Ohlsson B. The role of smoking and alcohol behaviour in management of functional gastrointestinal disorders. Best Pract Res Clin Gastroenterol. 2017;31(5):545–552. doi:10.1016/j.bpg.2017.09.006
16. Kurki MI, Karjalainen J, Palta P, et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature. 2023;613(7944):508–518. doi:10.1038/s41586-022-05473-8
17. Võsa U, Claringbould A, Westra HJ, et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat Genet. 2021;53(9):1300–1310. doi:10.1038/s41588-021-00913-z
18. Ferkingstad E, Sulem P, Atlason BA, et al. Large-scale integration of the plasma proteome with genetics and disease. Nat Genet. 2021;53(12):1712–1721. doi:10.1038/s41588-021-00978-w
19. Sun BB, Chiou J, Traylor M, et al. Plasma proteomic associations with genetics and health in the UK Biobank. Nature. 2023;622(7982):329–338. doi:10.1038/s41586-023-06592-6
20. Genovese G, Rockweiler NB, Gorman BR, et al. BCFtools/liftover: an accurate and comprehensive tool to convert genetic variants across genome assemblies. Bioinformatics. 2024;40(2). doi:10.1093/bioinformatics/btae038
21. Auton A, Brooks LD, Durbin RM, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74.
22. Ning Z, Pawitan Y, Shen X. High-definition likelihood inference of genetic correlations across human complex traits. Nat Genet. 2020;52(8):859–864. doi:10.1038/s41588-020-0653-y
23. Turley P, Walters RK, Maghzian O, et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat Genet. 2018;50(2):229–237. doi:10.1038/s41588-017-0009-4
24. Yang J, Ferreira T, Morris AP, et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet. 2012;44(4):369–75,s1–3. doi:10.1038/ng.2213
25. Watanabe K, Taskesen E, van Bochoven A, et al. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8(1):1826. doi:10.1038/s41467-017-01261-5
26. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi:10.1093/nar/gkq603
27. Rentzsch P, Witten D, Cooper GM, et al. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–d894. doi:10.1093/nar/gky1016
28. Boyle AP, Hong EL, Hariharan M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22(9):1790–1797. doi:10.1101/gr.137323.112
29. De leeuw CA, Mooij JM, Heskes T, et al. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol. 2015;11(4):e1004219. doi:10.1371/journal.pcbi.1004219
30. Weeks EM, Ulirsch JC, Cheng NY, et al. Leveraging polygenic enrichments of gene features to predict genes underlying complex traits and diseases. Nat Genet. 2023;55(8):1267–1276. doi:10.1038/s41588-023-01443-6
31. Blake JA, Baldarelli R, Kadin JA, et al. Mouse genome database (MGD): knowledgebase for mouse-human comparative biology. Nucleic Acids Res. 2021;49(D1):D981–d987. doi:10.1093/nar/gkaa1083
32. Liberzon A, Birger C, Thorvaldsdóttir H, et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417–425. doi:10.1016/j.cels.2015.12.004
33. Yu G, Wang LG, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
34. Wu C, Zhang Z, Yang X, et al. Large-scale imputation models for multi-ancestry proteome-wide association analysis. 2023.
35. Cannon M, Stevenson J, Stahl K, et al. DGIdb 5.0: rebuilding the drug-gene interaction database for precision medicine and drug discovery platforms. Nucleic Acids Res. 2024;52(D1):D1227–d1235. doi:10.1093/nar/gkad1040
36. Gusev A, Ko A, Shi H, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. 2016;48(3):245–252. doi:10.1038/ng.3506
37. Verbanck M, Chen CY, Neale B, et al. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693–698. doi:10.1038/s41588-018-0099-7
38. Siboni S, Bonavina L, Rogers BD, et al. Effect of increased intra-abdominal pressure on the Esophagogastric junction: a systematic review. J Clin Gastroenterol. 2022;56(10):821–830. doi:10.1097/MCG.0000000000001756
39. Mittal R, Vaezi MF. Esophageal motility disorders and gastroesophageal reflux disease. N Engl J Med. 2020;383(20):1961–1972. doi:10.1056/NEJMra2000328
40. Pandolfino JE, Kahrilas PJ. Smoking and gastro-oesophageal reflux disease. Eur J Gastroenterol Hepatol. 2000;12(8):837–842. doi:10.1097/00042737-200012080-00002
41. Kim SW, Lee JH, Sim YS, et al. Prevalence and risk factors for reflux esophagitis in patients with chronic obstructive pulmonary disease. Korean J Intern Med. 2014;29(4):466–473. doi:10.3904/kjim.2014.29.4.466
42. Huang KW, Luo JC, Leu HB, et al. Chronic obstructive pulmonary disease: an independent risk factor for peptic ulcer bleeding: a nationwide population-based study. Aliment Pharmacol Ther. 2012;35(7):796–802. doi:10.1111/j.1365-2036.2012.05028.x
43. Siva R, Birring SS, Berry M, et al. Peptic ulceration, Helicobacter pylori seropositivity and chronic obstructive pulmonary disease. Respirology. 2013;18(4):728–731. doi:10.1111/resp.12075
44. Aro P, Storskrubb T, Ronkainen J, et al. Peptic ulcer disease in a general adult population: the Kalixanda study: a random population-based study. Am J Epidemiol. 2006;163(11):1025–1034. doi:10.1093/aje/kwj129
45. Bowerman KL, Rehman SF, Vaughan A, et al. Disease-associated gut microbiome and metabolome changes in patients with chronic obstructive pulmonary disease. Nat Commun. 2020;11(1):5886. doi:10.1038/s41467-020-19701-0
46. Sprooten RTM, Lenaerts K, Braeken DCW, et al. Increased small intestinal permeability during severe acute exacerbations of COPD. Respiration. 2018;95(5):334–342. doi:10.1159/000485935
47. Divo MJ, Casanova C, Marin JM, et al. COPD comorbidities network. Eur Respir J. 2015;46(3):640–650. doi:10.1183/09031936.00171614
48. Thun GA, Imboden M, Ferrarotti I, et al. Causal and synthetic associations of variants in the SERPINA gene cluster with alpha1-antitrypsin serum levels. PLoS Genet. 2013;9(8):e1003585. doi:10.1371/journal.pgen.1003585
49. Zhao Z, Jiang C, Zhao D, et al. Two CHRN susceptibility variants for COPD are genetic determinants of emphysema and chest computed tomography manifestations in Chinese patients. Int J Chron Obstruct Pulmon Dis. 2017;12:1447–1455. doi:10.2147/COPD.S134010
50. Yamazaki Y, Zhao N, Caulfield TR, et al. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019;15(9):501–518. doi:10.1038/s41582-019-0228-7
51. Martins IJ, Hone E, Chi C, et al. Relative roles of LDLr and LRP in the metabolism of chylomicron remnants in genetically manipulated mice. J Lipid Res. 2000;41(2):205–213. doi:10.1016/S0022-2275(20)32054-X
52. Mattioli AV, Coppi F, Bucciarelli V, et al. Cardiovascular risk stratification in young women: the pivotal role of pregnancy. J Cardiovasc Med. 2023;24(11):793–797. doi:10.2459/JCM.0000000000001557
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.