Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
PTPLAD2 and USP49 Involved in the Pathogenesis of Smoke-Induced COPD by Integrative Bioinformatics Analysis
Authors Zhang Q, Song W, Ayidaerhan N, He Z
Received 20 February 2020
Accepted for publication 21 July 2020
Published 15 October 2020 Volume 2020:15 Pages 2515—2526
DOI https://doi.org/10.2147/COPD.S250576
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Qiang Zhang,1 Wei Song,1 Nahemuguli Ayidaerhan,2 Zheng He3
1Department of Pulmonary and Critical Care Medicine, Shengjing Hospital of China Medical University, Shenyang, Liaoning Province 110042, People’s Republic of China; 2Department of Pulmonary and Critical Care Medicine, Tarbagatay Prefecture People’s Hospital, Tacheng, Xinjiang, People’s Republic of China; 3Department of Obstetrics and Gynecology Medicine, Shengjing Hospital of China Medical University, Shenyang, People’s Republic of China
Correspondence: Zheng He
Department of Obstetrics and Gynecology Medicine, Shengjing Hospital of China Medical University, Shenyang, Liaoning Province 110042, People’s Republic of China
Email [email protected]
Purpose: Chronic obstructive pulmonary disease (COPD) is a typical chronic disease, but its molecular pathogenesis remains unclear. This study aimed to investigate the expression of biomarkers during COPD development.
Methods: Markers significantly associated with COPD were screened using bioinformatics tools. qRT-PCR and Western blot were used to explore the expression of PTPLAD2 and USP49 in BEAS-2B cells. CCK-8 assay was used to determine the influence of PTPLAD2 and USP49 in BEAS-2B on cell proliferation.
Results: In this study, 86 DEGs were identified in GSE76925. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway analyses suggested that the phosphoinositide 3-kinase-Akt signaling pathway, ECM–receptor interaction, mRNA process, and viral transcription were all involved in the development of COPD. In addition, 14 hub genes were identified by WGCNA. PTPLAD2 and USP49 shared DEGs and hub genes and their expression levels were significantly reduced after CSE-treatment in BEAS-2B cells.
Conclusion: Our results suggest that PTPLAD2 and USP49 may be useful biomarkers of COPD.
Keywords: COPD, GEO, WGCNA, cigarette
Introduction
Chronic obstructive pulmonary disease (COPD) is a typical chronic disease with a global average prevalence of 13.1%.1 Its main features include persistent airflow limitation and recurrent airway inflammation.2,3 The main cause of COPD is exposure to harmful gases or particles, with smoking being one of the main risk factors.4 However, the molecular pathogenesis of COPD remains unclear and effective prevention and treatment methods for COPD are still lacking. Further studies of the molecular mechanisms of COPD are therefore needed to identify novel drug targets.
High-throughput sequencing has progressed rapidly over the past decade, while the development of bioinformatics has deepened our understanding of disease mechanisms and promoted the development of drug targets.5 Next-generation sequencing has been widely applied for studying numerous tumors, as well as COPD.6,7 Several studies8,9 have identified hundreds of differentially expressed genes (DEGs) based on mRNA gene expression profiles. However, these DEGs cannot fully explain the pathogenesis of COPD because of differences between individual samples and a lack of understanding of the relationships between the genes. However, integrative bioinformatics analysis based on multiple analytical methods may enable the identification of reliable and efficacious molecular markers.
Weighted gene co-expression network analysis (WGCNA) is a novel and effective method that has begun to be applied to the screening of biomarkers.10 It can analyze signal networks by transforming gene expression data into co-expression modules. WGCNA clusters genes with similar expression patterns and then groups them into different modules,11,12 with the genes connected at the center of the module considered as key genes. WGCNA is currently widely used to analyze various biological processes13,14 and to screen biomarkers and drug targets.
In this study, we used differential analysis and WCGNA and identified two key genes, PTPLAD2 and USP49, that were differentially expressed in COPD. We further confirmed that the expression levels of these genes were significantly reduced after cigarette smoke extract (CSE) treatment. Overall, these results suggest that PTPLAD2 and USP49 may be useful biomarkers of COPD.
Materials and Methods
Gene Data
We searched the GEO database using the search term “Chronic obstructive pulmonary disease” as the key word and limited the search scope to “Expression profiling by array”. We also searched with the term “Homo sapiens”. The gene chip GSE76925 from the GPL570 chip platform (Illumina, HumanHT-12 V4.0 expression bead chip) was selected. The dataset GSE76925,15 including 111 samples from COPD patients and 40 control samples, was used for subsequent analysis.
Screening for DEGs
The data were downloaded using R and GEOquery. Genes cannot be mapped to chromosomal positions were removed. In the event of multiple probes corresponding to one gene, we took the average value as the expression level of the gene. DEGs between COPD cases and controls were identified using the limma package with difference analysis conditions of | logFC |> 1, P <0.05.
Construction of Coexpressing Gene Module for COPD
The WGCNA algorithm was used to construct a scale-free co-expression network. First, a gene co-expression similarity matrix was calculated composed of the absolute value of the correlation coefficient between genes. Common correlation coefficients included Pearson’s and Spearman correlation coefficients, and the range of correlation coefficient is. Pearson’s correlation coefficients are often used for continuous variables such as genes. Second, the correlation matrix was transformed into an adjacency matrix using the power function amn = | cmn | β. Finally, the most appropriate β value was selected to convert the adjacency matrix into a topological overlap matrix. Based on the gene dendrogram with a minimum size of 30, an average linkage hierarchical clustering was established to divide similar genes into modules.
Screening and Annotation of Clinically Meaningful Modules
Two methods were used to identify modules related to clinical characteristics. Gene significance (GS) was defined as a log10 transformation of the P value (GS = logP) based on a linear regression between the clinical characteristics and gene expression. The module significance (MS) was calculated using the average GS value of all genes in a module. The module with the largest absolute value was generally considered to be the module most closely related to the clinical features. The meaning of the module was defined as the correlation between the main components of the gene module and the clinical characteristics to identify the relevant modules. Modules that were highly relevant to a given clinical feature were selected for further analysis.
Screening Key Genes
A hub gene was defined as a gene with an absolute value of Pearson’s correlation >0.8 and absolute value of clinical trait relationship >0.2 in the hub gene module. The intersection of the hub gene and the DEGs was defined as the key gene.
Cell Culture
BEAS-2B human bronchial epithelial cells (American Type Culture Collection, USA) were cultured in DMEM supplemented with 10% fetal bovine serum in a humidified incubator under 5% CO2 at 37°C. Cells were treated with 2% CSE for 24 h. CSE was prepared by bubbling the smoke from two filterless cigarettes through 10 mL DMEM for 2 min per cigarette to produce 100% CSE. This solution was then passed through a 0.22-μM filter for sterilization and stored at −80°C.
Quantitative Real Time-Polymerase Chain Reaction (qRT-PCR)
Total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA) reagent. For mRNA detection, reverse transcription was performed using a PrimeScript® RT reagent Kit with gDNA Eraser (Takara, Japan). cDNA was generated from 1000 ng total RNA using SYBR® Premix EX Taq™ II (Tli RNaseH Plus, Takara). qRT-PCR was carried out using an Applied Biosystems® 7500 Real-Time PCR System (Thermo Fisher Scientific, USA). Transcripts of 18s in the same incubations were used as an internal control. The primer sequences were as follows: PTPLAD2 forward (5′-AGCAACCGTACTAAAGCAATTGTGA-3′) and reverse (5′-TCTGCAGAAGGCTTGGCATAA-3′); USP49 forward (5′-TCCCACAAAGGAAGTAACC-3′) and reverse (5′-TATGACAGCAGCAAGTAGG-3′); 18s forward (5′-CCCGGGGAGGTAGTGACGAAAAAT-3′) and reverse (5′-CGCCCGCCCGCTCCCAAGAT-3′).
Western Blot Analysis
Total protein was extracted from BEAS-2B cells and lysed, and the concentration was determined using a BCA kit (Thermo, USA). Equal amounts of protein from each sample were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane (Millipore, USA). After blocking with non-fat milk, the membrane was incubated with specific primary antibody at 4°C overnight, washed with TBST, and then incubated with secondary antibody at room temperature for 1 h. The primary antibodies for PTPLAD2, USP49 and β-actin were obtained from Cell Signaling Technology (Danvers, USA). Immunoreactive signals were quantified using Image Lab (Bio-Rad, USA).
Plasmid Transfections
The overexpression plasmids containing whole coding sequence of PTPLAD2, USP49 and pcDNA 3.1 vector served as the NC were purchased from GeneChem (Shanghai, China). Cells were seeded at 2 × 105 cells/well in six-well plates overnight and then transfected by Lipofectamine 2000 reagent with plasmid.
Gene Set Enrichment Analysis
Gene set enrichment analysis (GSEA) was performed using the JAVA program (http://www.broadinstitute.org/gsea) and GSE76925 datasets by comparing the expression of genes in the PTPLAD2/USP49‐high/low groups divided by the median expression level of PTPLAD2/USP49. The MSigDB “c2: curated gene sets (KEGG)” gene set were used as a reference in this step to evaluate the pathways that PTPLAD2 and USP49 may modulate.
Statistical Analysis
Statistical analysis was carried out using GraphPad Prism 5 and R version 3.5.3. Data were presented as mean ± standard deviation and compared between groups using unpaired Student’s t-tests. A value of P < 0.05 was considered to indicate a statistically significant difference.
Results
Identification of DEGs in COPD
A total of 86 DEGs were identified using the limma package based on the GSE76925 database (corrected P < 0.05; |log2FC| > 1). A Volcano map (Figure 1A) and heatmap (Figure 1B) of the DEGs are shown in Figure 1.
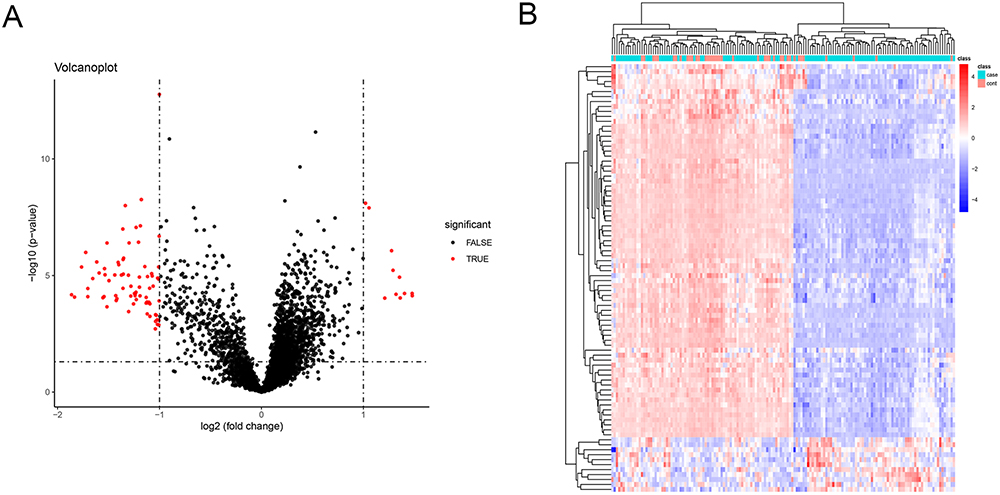 |
Figure 1 Identification of DEGs in COPD and normal lung tissues. (A) Volcano plot of all DEGs. (B) Heatmap of all DEGs. |
Gene Function and Annotation Enrichment Analysis of DEGs
We applied the clusterprofiler package to compare gene clusters according to their enriched biological processes. In KEGG analysis (Figure 2A), DEGs were mostly enriched in the phosphoinositide 3-kinase-Akt signaling pathway and ECM–receptor interaction, etc. In GO analysis (Figure 2B), the DEGs were mostly enriched in mRNA process and viral transcription, etc. The above enrichment analyses can help us to understand the roles of DEGs in COPD.
 |
Figure 2 KEGG analysis and GO analysis of DEGs. (A) KEGG analysis; (B) GO analysis. |
Weighted Gene Co-Expression Network Construction
We then performed WGCNA according to the sample grouping. We included all the genes, and simultaneously, in the WGCNA pretreatment stage, we used 75% of the genes with a median absolute deviation (MAD), and at least those with MAD greater than 0.01 were included in WGCNA analysis. A total of 3699 genes were selected for the study. Clustering to detect outliers suggested that the samples clustered well (Figure 3). In this study, we chose β = 3 (scale-free R2 = 0.91) as the soft-threshold to ensure a scale-free network (Figure 4A-D)
 |
Figure 3 Sample clustering of GSE17025 to detect outliers. |
 |
Figure 4 Determination of soft-thresholding power in the WGCNA. (A) The correlation coefficients between log (K) and log (P (k)) corresponding to different soft thresholds; (B) A genetic network corresponding to different soft thresholds. The average value of the gene adjacency coefficient in the medium, which reflects the average connection level of the network; for the selected soft threshold β = 3 to test whether the network conforms to the scale-free network distribution; (C) The distribution of the connectivity of each node in the network; (D) A scatter plot of log (K) and log (P (k)). The linear regression results show that the correlation coefficient of 0.86 is consistent with the characteristics of degreeless networks. |
Identification of Modules and Key Genes Related to COPD
After determining the soft threshold, we proceeded with network construction. According to the basic idea of WGCNA, the correlation matrix and the adjacency matrix of the expression spectrum were calculated successively and then transformed into a topological matrix. A systematic clustering tree of genes was then obtained according to the dissimilarity between the genes. First, the minimum number of genes for each gene module was defined as 30, a moderate degree of classification was selected to find the core gene cluster, and the genes not classified into any gene cluster in the previous step were then assigned to different gene clusters according to the correlation. A total of 10 gene modules were obtained (Figure 5).
 |
Figure 5 Gene cluster tree classification diagram. A total of 10 gene modules were obtained. Different colors indicate different gene modules, and gray indicates genes that do not belong to any known module. |
We then screened the modules related to COPD using two methods. Modules with larger MS were considered to be more closely related to disease development. We found that the ME in the red module also showed the highest GS (Figure 6A and B). In addition, the ME in the red module showed a higher correlation with disease development than other disease modules. The red module was therefore identified as a clinically significant module and was selected for further analysis.
 |
Figure 6 Identification of modules associated with clinical traits of COPD. (A) Distribution of average gene significance and errors in different modules. (B) Heatmap of correlation between module eigengenes and clinical traits of COPD. |
Selection of Key Genes
Using a threshold module connectivity (cor. gene Module Membership) >0.8 and clinical trait relationship (cor. gene Trait Significance) >0.1, we identified 17 mRNAs (ASB1, CLDN14, COX5B, DRG1, ELP4, ERGIC1, ERLIN2, LOC644250, LOC646786, PPLAD2, RGS10, RIT1, SEC22C, TAF12, TOPBP1, USP49, ZNF14) as key mRNAs (Figure 7A). We then intersected these with 86 DEGs. Two mRNAs, PTPLAD2 and USP49, were identified as shared genes and used as the focus of the next step (Figure 7B).
 |
Figure 7 Screening of key genes. (A) Scatter plot of module eigengenes in the hub gene module. (B) Venn plot showing two genes shared by hub genes and DEGs. |
Expression of PTPLAD2 and USP49 in CSE-Induced BEAS-2B Cells
First, we check gene expression levels of all these 17 key genes in response to 2% CSE stimuli. Due to the gene names corresponding to LOC644250 and LOC646786 could not be found, we tested the remaining 15 genes. Figure 8A shows that ASB1, DRG1, ERGIC1, ERLIN2, RIT1 and SEC22C expression were significantly increased after CSE-treatment. CLDN14, COX5B, PPLAD2, TAF12, TOPBP1 and USP49 expression were significantly decreased after CSE-treatment. In addition, ELP4, RGS10 and ZNF14 expression did not show significant change after CSE-treatment. Next, we further examined the roles of PTPLAD2 and USP49 expression in COPD using BEAS-2B cells treated with 2% CSE. As shown in Figure 8B and C, CSE significantly inhibited the expression of PTPLAD2 and USP49 in a concentration- and time-dependent manner. The protein (Figure 8D) levels of PTPLAD2 and USP49 decreased after treatment with 2% CSE. In addition, we measure the influence of PTPLAD2 and USP49 to cell viabilities of BEAS-2B cells under CSE stimulation via CCK-8 test. Overexpressed PTPLAD2 and USP49 could significantly reduce CSE-induced decrease in cell viability (Figure 8E). These results further confirmed the important roles of PTPLAD2 and USP49 in the process of COPD.
 |
Figure 8 Relative gene and protein expression levels of PTPLAD2 and USP49 in cigarette smoke extract (CSE)-induced BEAS-2B cells. (A) qRT-PCR analysis of 15 key gene gene expression levels; (B and C) qRT-PCR analysis of PTPLAD2 and USP49 gene expression levels; (D) Western blot analysis of protein level of PTPLAD2 and USP49 protein levels; (E) CCK8 analysis of overexpressed PTPLAD2 and USP49 BEAS-2B cells with CSE-treatment; *p < 0.05 (vs control group). |
Underlying Mechanisms of PTPLAD2 and USP49 in COPD
Next, we check the expression of PTPLAD2 and USP49 in another GEO dataset (GSE112260). We downloaded the GSE112260 data set from GEO website, which contains 4 COPD samples and 4 normal lung samples. We used the same analysis method and found that PTPLAD2 and USP49 were low expression in COPD and high expression in normal samples. At the same time, we used box plots to show the expression of these two genes in GSE76925 and GSE112260 (Figure 9A-D) and found that they have similar expression trends. In addition, we try to explore the underlying mechanisms of PTPLAD2 and USP49 in COPD via GSEA analysis. We extracted the expression data of 111 COPD patients in the GSE76925 data set, grouped the two genes according to the median expression value, and then downloaded “c2: curated gene sets (KEGG)” from the msigdb database for GSEA Enrichment analysis. And we found that PTPLAD2 and USP49 could all be enriched to “KEGG_ALZHEIMERS_DISEASE”, “KEGG_BASAL_CELL_CARCINOMA”, “KEGG_INTESTINAL_IMMUNE_NETWORK_FOR_IGA_PRODUCTI”, “KEGG_RIBOSOME”, “KEGG_T_CELL_RECEPTOR_SIGNALING_PATHWAY” (Figure 9E-F). It suggested that PTPLAD2 and USP49 may function via these signaling pathways.
 |
Figure 9 The expression of PTPLAD2 and USP49 and GSEA analysis. (A–B) Box plots to show the expression of these PTPLAD2 and USP49 in GSE76925; (C–D) Box plots to show the expression of these PTPLAD2 and USP49 in GSE112260; (E-F) GSEA analysis of PTPLAD2 (E) and USP49 (F) in GSE76925. |
Discussion
COPD affects millions of people worldwide and is associated with high morbidity and mortality.16 This incurable lung disease is characterized by progressive airflow obstruction, including emphysema, destruction of parenchymal emphysema, and excessive mucus secretion due to bronchiolitis.17 COPD is estimated to become the third most common cause of death by 2030.18 However, a lack of effective biomarkers and disease remission therapies present challenges for the control and treatment of COPD.
The development of bioinformatics has allowed researchers to identify a series of genes related to COPD and to establish potential molecular targets for treatment through differential analysis. In addition, WGCNA has been widely used to explore potential biomarkers for various diseases.19,20 In this study, we screened 82 DEGs from GSE76925 and used WGCNA to analyze the expression profile of GSE76925. We identified 17 hub genes, of which PTPLAD2 and USP49 were shared hub genes and DEGs. These results suggested that PTPLAD2 and USP49 may play important roles in the progression of COPD.
PTPLAD2 encodes protein tyrosine phosphatase-like A domain-containing 2 protein, which is a member of the PTPLAD family21 and acts as tumor suppressor in glioblastoma22 and esophageal squamous cell carcinoma.23 USP49 encodes ubiquitin-specific protease 49, which is a deubiquitinating enzyme. Previous studies indicated that USP49 played important roles in cardiomyocytes,24 pancreatic cancer,25 and lung cancer.26 However, the roles of PTPLAD2 and USP49 in COPD are unclear. The results of the current bioinformatics and cell experiments suggest that PTPLAD2 and USP49 may also play important roles in the progression of COPD.
This study had some limitations. First, the specific mechanisms of PTPLAD2 and USP49 in COPD need to be investigated in further studies both in vitro and in vivo. In addition, further comprehensive and in-depth analyses will be needed to support the clinical application of PTPLAD2 and USP49 as potential biomarkers in COPD.
Conclusion
Using comprehensive bioinformatics analysis, this study provided novel insights into the complex pathogenesis of COPD. In addition, we demonstrated that CSE exposure significantly reduced the expression levels of PTPLAD2 and USP49 in BEAS-2B cells, suggesting that these genes may play key roles in CSE-induced COPD. Further studies are needed to determine the detailed mechanisms responsible for these effects.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Blanco I, Diego I, Bueno P, Casas-Maldonado F, Miravitlles M. Geographic distribution of COPD prevalence in the world displayed by geographic information system maps. Eur Respir J. 2019;54:1. doi:10.1183/13993003.00610-2019
2. Decramer M, Janssens W, Miravitlles M. Chronic obstructive pulmonary disease. Lancet (London, England). 2012;379(9823):1341–1351. doi:10.1016/S0140-6736(11)60968-9
3. Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol. 2009;4:435–459. doi:10.1146/annurev.pathol.4.110807.092145
4. Wang C, Xu J, Yang L, et al. Prevalence and risk factors of chronic obstructive pulmonary disease in China (the China Pulmonary Health [CPH] study): a national cross-sectional study. Lancet (London, England). 2018;391(10131):1706–1717. doi:10.1016/S0140-6736(18)30841-9
5. Cheng F, Desai RJ, Handy DE, et al. Network-based approach to prediction and population-based validation of in silico drug repurposing. Nat Commun. 2018;9(1):2691. doi:10.1038/s41467-018-05116-5
6. Tsai M-J, Chang W-A, Jian S-F, Chang K-F, Sheu C-C, Kuo P-L. Possible mechanisms mediating apoptosis of bronchial epithelial cells in chronic obstructive pulmonary disease - a next-generation sequencing approach. Pathol Res Pract. 2018;214(9):1489–1496. doi:10.1016/j.prp.2018.08.002
7. Tsai M-J, Tsai Y-C, Chang W-A, et al. Deducting MicroRNA-mediated changes common in bronchial epithelial cells of asthma and chronic obstructive pulmonary disease-a next-generation sequencing-guided bioinformatic approach. Int J Mol Sci. 2019;20:3. doi:10.3390/ijms20030553
8. Lin Y-Z, Zhong X-N, Chen X, Liang Y, Zhang H, Zhu D-L. Roundabout signaling pathway involved in the pathogenesis of COPD by integrative bioinformatics analysis. Int J Chron Obstruct Pulmon Dis. 2019;14:2145–2162. doi:10.2147/COPD.S216050
9. Wei L, Xu D, Qian Y, et al. Comprehensive analysis of gene-expression profile in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2015;10:1103–1109.
10. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9:559. doi:10.1186/1471-2105-9-559
11. Liu X, Hu A-X, Zhao J-L, Chen F-L. Identification of key gene modules in human osteosarcoma by co-expression analysis weighted gene co-expression network analysis (WGCNA). J Cell Biochem. 2017;118(11):3953–3959. doi:10.1002/jcb.26050
12. Shi Z, Derow CK, Zhang B. Co-expression module analysis reveals biological processes, genomic gain, and regulatory mechanisms associated with breast cancer progression. BMC Syst Biol. 2010;4:74. doi:10.1186/1752-0509-4-74
13. Zhang H, Zhao X, Wang M, Ji W. Key modules and hub genes identified by coexpression network analysis for revealing novel biomarkers for larynx squamous cell carcinoma. J Cell Biochem. 2019;120(12):19832–19840. doi:10.1002/jcb.29288
14. Yao Q, Song Z, Wang B, Qin Q, Zhang J-A. Identifying key genes and functionally enriched pathways in Sjögren’s syndrome by weighted gene co-expression network analysis. Front Genet. 2019;10:1142. doi:10.3389/fgene.2019.01142
15. Morrow JD, Zhou X, Lao T, et al. Functional interactors of three genome-wide association study genes are differentially expressed in severe chronic obstructive pulmonary disease lung tissue. Sci Rep. 2017;7:44232. doi:10.1038/srep44232
16. Rabe KF, Watz H. Chronic obstructive pulmonary disease. Lancet (London, England). 2017;389(10082):1931–1940. doi:10.1016/S0140-6736(17)31222-9
17. Busch R, Qiu W, Lasky-Su J, Morrow J, Criner G, DeMeo D. Differential DNA methylation marks and gene comethylation of COPD in African-Americans with COPD exacerbations. Respir Res. 2016;17(1):143. doi:10.1186/s12931-016-0459-8
18. McLean S, Hoogendoorn M, Hoogenveen RT, et al. Projecting the COPD population and costs in England and Scotland: 2011 to 2030. Sci Rep. 2016;6:31893. doi:10.1038/srep31893
19. Chou W-C, Cheng A-L, Brotto M, Chuang C-Y. Visual gene-network analysis reveals the cancer gene co-expression in human endometrial cancer. BMC Genomics. 2014;15:300. doi:10.1186/1471-2164-15-300
20. Wang Y, Chen L, Ju L, et al. Novel biomarkers associated with progression and prognosis of bladder cancer identified by co-expression analysis. Front Oncol. 2019;9:1030. doi:10.3389/fonc.2019.01030
21. Strausberg RL, Feingold EA, Grouse LH, et al. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc Natl Acad Sci U S A. 2002;99(26):16899–16903.
22. Nord H, Hartmann C, Andersson R, et al. Characterization of novel and complex genomic aberrations in glioblastoma using a 32K BAC array. Neuro-Oncology. 2009;11(6):803–818. doi:10.1215/15228517-2009-013
23. Zhu S, Wang Z, Zhang Z, et al. PTPLAD2 is a tumor suppressor in esophageal squamous cell carcinogenesis. FEBS Lett. 2014;588(6):981–989. doi:10.1016/j.febslet.2014.01.058
24. Zhang W, Zhang Y, Zhang H, Zhao Q, Liu Z, Xu Y. USP49 inhibits ischemia-reperfusion-induced cell viability suppression and apoptosis in human AC16 cardiomyocytes through DUSP1-JNK1/2 signaling. J Cell Physiol. 2019;234(5):6529–6538. doi:10.1002/jcp.27390
25. Luo K, Li Y, Yin Y, et al. USP49 negatively regulates tumorigenesis and chemoresistance through FKBP51-AKT signaling. EMBO J. 2017;36(10):1434–1446. doi:10.15252/embj.201695669
26. Shen W-M, Yin J-N, Xu R-J, Xu D-F, Zheng S-Y. Ubiquitin specific peptidase 49 inhibits non-small cell lung cancer cell growth by suppressing PI3K/AKT signaling. Kaohsiung J Med Sci. 2019;35(7):401–407.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.