Back to Journals » Drug Design, Development and Therapy » Volume 20
Pterostilbene in Oxidative Stress–Related Diseases: Context-Dependent Regulation of Redox Homeostasis and Translational Challenges
Authors Zhang Y, Yu Y, Xiong Z, Bin W, Yao Y, Liu H ![]()
Received 11 March 2026
Accepted for publication 25 June 2026
Published 30 June 2026 Volume 2026:20 608538
DOI https://doi.org/10.2147/DDDT.S608538
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Yuyao Zhang, Yingying Yu, Zhengrong Xiong, Wenying Bin, Yuxuan Yao, Huan Liu
College of Integrated Traditional Chinese and Western Medicine, Southwest Medical University, Luzhou, Sichuan, 646000, People’s Republic of China
Department of Orthopedics, The Affiliated Traditional Chinese Medicine Hospital, Southwest Medical University, Luzhou, Sichuan, 646000, People’s Republic of China
Guangdong Provincial Key Laboratory of Medical Biomechanics, National Key Discipline of Human Anatomy, School of Basic Medical Sciences, Southern Medical University, Guangzhou, 510515, People’s Republic of China
Orthopaedic Research Center and Key Laboratory, The Affiliated Traditional Chinese Medicine Hospital, Southwest Medical University, Luzhou, Sichuan, 646000, People’s Republic of China
Correspondence: Huan Liu, Department of Orthopedics, The Affiliated Traditional Chinese Medicine Hospital, Southwest Medical University, Luzhou, Sichuan, 646000, People’s Republic of China, Email [email protected]
This narrative review synthesizes current evidence on pterostilbene in oxidative stress-related diseases, focusing on its chemical and pharmacokinetic basis, redox-related signaling responses, disease-specific evidence, and translational challenges. Most available evidence comes from preclinical models and suggests that the biological effects of pterostilbene cannot be explained solely by direct reactive oxygen species (ROS) scavenging. Instead, pterostilbene is associated with several redox-sensitive processes, including Nrf2-related antioxidant responses, redox–inflammatory crosstalk, mitochondria-associated stress responses, metabolic regulation, and cell death-related pathways. Evidence from osteoarthritis, neurodegenerative and cognitive dysfunction models, ischemia–reperfusion injury, metabolic diseases, and cancer indicates that these effects vary substantially across pathological contexts. In non-malignant degenerative, ischemic, and metabolic models, pterostilbene is generally associated with attenuation of oxidative stress-, inflammation-, or cellular stress-related injury markers, whereas in selected cancer models it may disrupt redox-adapted tumor-cell stress tolerance and promote apoptosis- or pyroptosis-related responses. Important translational barriers remain, including incomplete direct target validation, heterogeneous dosing and intervention designs, reliance on static oxidative stress endpoints, limited clinical validation, and insufficient integration of negative or context-dependent findings. By applying a redox homeostasis-centered framework, this review clarifies the contexts in which pterostilbene effects have been reported and identifies the evidence needed before disease-specific translational positioning can be established.
Keywords:
pterostilbene, oxidative stress, redox homeostasis, Nrf2/Keap1, NF-κB, mitochondria-associated stress, ischemia–reperfusion injury, metabolic diseases, cancer
Introduction
Oxidative stress is a central concept in redox biology and medicine and is widely involved in disease-related processes. It is commonly described as an imbalance between reactive oxygen species (ROS) production and endogenous antioxidant defense capacity; however, from a mechanistic perspective, it also reflects disruption of redox signaling and control.1,2 Importantly, ROS are not merely damaging by-products but can also function as physiological redox signaling molecules, with biological effects depending on concentration, subcellular location, timing, and cellular context.3,4 Persistent oxidative imbalance can contribute to oxidative damage to lipids, proteins, and DNA, while also engaging antioxidant defense, damage repair, stress adaptation, inflammatory responses, and apoptosis-related pathways.1,2,5 These considerations suggest that therapeutic strategies targeting oxidative stress should not be interpreted only as direct ROS elimination, but also in terms of broader regulation of redox homeostasis in specific pathological contexts. This review focuses on representative pathological contexts in which oxidative stress-related mechanisms are strongly implicated, including osteoarthritis, neurodegenerative and cognitive dysfunction models, ischemia–reperfusion injury, metabolic diseases, and cancer. These categories were selected not to provide an exhaustive catalogue of all pterostilbene-related disease models, but to represent biologically distinct redox environments.
Pterostilbene is a naturally occurring stilbene compound found in several edible and medicinal plants, including blueberries, grapes, and mulberries. Compared with resveratrol, pterostilbene contains two methoxy substitutions that increase its lipophilicity and have been associated with improved membrane permeability, metabolic stability, and oral bioavailability.6,7 These physicochemical and pharmacokinetic features provide a rationale for studying pterostilbene in preclinical models involving oxidative stress, inflammation, and cellular injury.7,8 Current evidence suggests that its biological activity is not limited to direct ROS scavenging, but may also involve modulation of endogenous antioxidant defenses, redox-sensitive inflammatory signaling, and mitochondria-associated or cellular stress responses.7,8 The representative natural sources, chemical structure, and key pharmacokinetic implications of pterostilbene are summarized in Figure 1.
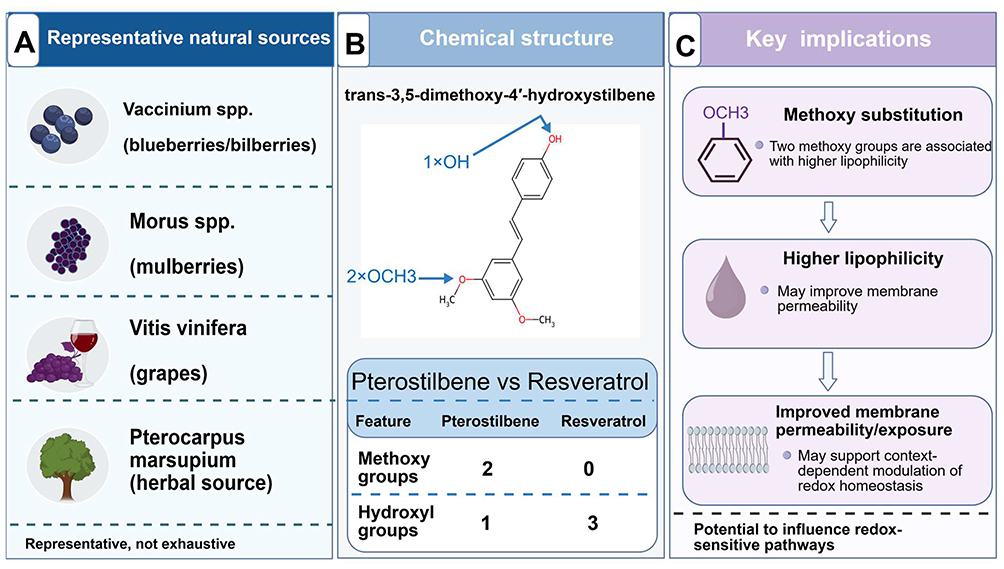 |
Figure 1 Representative natural sources, chemical structure, and pharmacokinetic implications of pterostilbene. This figure summarizes representative natural sources and structural features of pterostilbene. (A) shows selected plant sources in which pterostilbene has been reported, including Vaccinium spp. (blueberries/bilberries), Morus spp. (mulberries), Vitis vinifera (grapes), and Pterocarpus marsupium; these examples are representative and not exhaustive. (B) illustrates the chemical structure of pterostilbene, trans-3,5-dimethoxy-4′-hydroxystilbene, and compares key structural features with resveratrol. Compared with resveratrol, pterostilbene contains two methoxy groups and one phenolic hydroxyl group, a structural pattern associated with increased lipophilicity and altered pharmacokinetic behavior. (C) summarizes the main implication of methoxy substitution: higher lipophilicity may improve membrane permeability and systemic exposure, thereby supporting context-dependent modulation of redox-sensitive pathways. Figure artwork was created with BioGDP.com under academic license.9 Abbreviation: spp., species. |
This narrative review has three main aims. First, it summarizes current evidence on pterostilbene in oxidative stress-related pathological conditions and evaluates whether its reported effects are better interpreted as direct antioxidant activity or broader redox homeostasis modulation. Second, it organizes representative preclinical evidence according to distinct redox-related pathological contexts, including chronic degenerative and neural dysfunction models, ischemia–reperfusion injury, metabolic diseases, and cancer. Third, it discusses major evidence gaps and translational challenges, including limited clinical validation, uncertain direct molecular targets, heterogeneous dosing and intervention designs, incomplete dose–response information, and frequent reliance on downstream pathway markers rather than causal validation.
Relevant literature was identified from PubMed, Web of Science, and Scopus. The search covered publications from October 2000 to May 2026, with a final update conducted during the first revision of the manuscript in May 2026. English-language original experimental studies, mechanistic studies, pharmacokinetic studies, cell-based and animal studies, and relevant review articles were considered. Representative search strings used across databases included “pterostilbene” AND “oxidative stress”; “pterostilbene” AND “redox homeostasis”; “pterostilbene” AND (“reactive oxygen species” OR “ROS”); “pterostilbene” AND (“Nrf2” OR “Keap1” OR “ARE”); “pterostilbene” AND (“NF-κB” OR “NLRP3” OR “inflammation”); “pterostilbene” AND (“mitochondria” OR “mitochondrial dysfunction”); “pterostilbene” AND “osteoarthritis”; “pterostilbene” AND (“neurodegeneration” OR “neurodegenerative disease” OR “Alzheimer’s disease”); “pterostilbene” AND (“ischemia-reperfusion” OR “ischemia reperfusion” OR “I/R”); “pterostilbene” AND (“diabetes” OR “metabolic disease” OR “insulin resistance”); “pterostilbene” AND (“NAFLD” OR “hepatic steatosis” OR “steatohepatitis”); and “pterostilbene” AND “cancer.” Studies were selected when they addressed pterostilbene in relation to oxidative stress-, redox-, inflammatory-, mitochondrial-, metabolic-, or cell death-related outcomes, or when they provided relevant background on pterostilbene pharmacokinetics, redox biology, or disease-specific redox contexts. Studies were excluded when they were unrelated to pterostilbene, redox biology, or the disease contexts covered in this review, lacked disease- or mechanism-relevant biological endpoints, were not written in English, or provided insufficient information for contextual interpretation. Studies limited to chemical antioxidant assays were not used as disease-specific evidence, but were considered when relevant to the chemical antioxidant basis of pterostilbene. Because this is a narrative review rather than a systematic review, no PRISMA flow diagram, formal quality scoring, article-number screening, or meta-analysis was performed; evidence was interpreted qualitatively with emphasis on mechanistic relevance, disease context, and translational limitations.
To support this context-dependent interpretation, the review first introduces the chemical and pharmacokinetic basis of pterostilbene, then discusses redox-related signaling pathways and disease-specific evidence, and finally considers translational challenges and future research directions. The conceptual framework used in this review links pterostilbene-associated redox-related processes, including Nrf2/Keap1–ARE responses, mitochondria-associated stress responses, and NF-κB/NLRP3-related inflammatory signaling, with the pathological contexts and representative readouts discussed in subsequent sections (Figure 2).
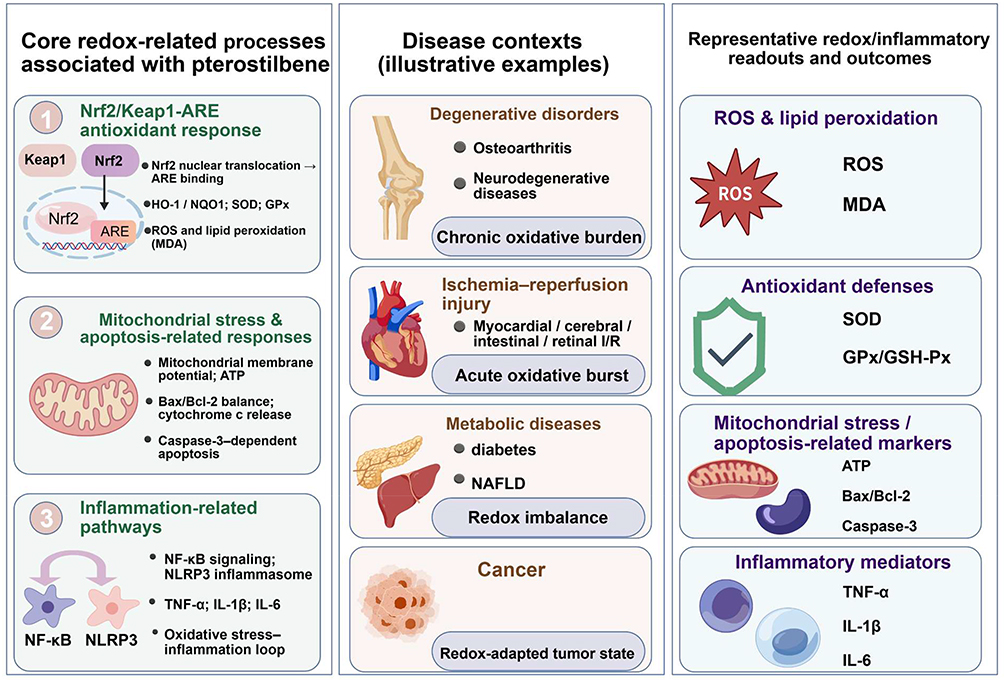 |
Figure 2 Conceptual framework linking pterostilbene-associated redox-related processes, disease contexts, and representative experimental readouts. This figure provides an overview of the redox homeostasis-centered framework used in this review. The left panel summarizes major redox-related processes associated with pterostilbene in preclinical studies, including Nrf2/Keap1–ARE-related antioxidant responses, mitochondria-associated stress and apoptosis-related responses, and inflammation-related pathways such as NF-κB signaling and NLRP3 inflammasome activation. The middle panel illustrates representative disease contexts discussed in the review, including chronic degenerative disorders such as osteoarthritis and neurodegenerative diseases, ischemia–reperfusion injury involving myocardial, cerebral, intestinal, and retinal models, metabolic diseases such as diabetes and non-alcoholic fatty liver disease, and cancer as a redox-adapted tumor state. The right panel shows representative experimental readouts and outcomes used to assess redox-, inflammatory-, mitochondria-associated, and cell death-related effects, including ROS, MDA, SOD, GPx/GSH-Px, ATP, Bax/Bcl-2, caspase-3, TNF-α, IL-1β, and IL-6. The figure is intended as a conceptual summary and does not imply that all pathways or readouts are uniformly affected in every disease model. Figure artwork was created with BioGDP.com under academic license.9 Abbreviations: ARE, antioxidant response element; ATP, adenosine triphosphate; Bax, BCL2-associated X protein; Bcl-2, B-cell lymphoma 2; GPx/GSH-Px, glutathione peroxidase; IL, interleukin; I/R, ischemia–reperfusion; MDA, malondialdehyde; NAFLD, non-alcoholic fatty liver disease; NF-κB, nuclear factor-κB; NLRP3, NOD-like receptor family pyrin domain-containing 3; Nrf2, nuclear factor erythroid 2-related factor 2; ROS, reactive oxygen species; SOD, superoxide dismutase; TNF-α, tumor necrosis factor-α. |
Previous reviews have summarized the synthesis, bioactivity, pharmacokinetic properties, anti-inflammatory and oncological effects, dietary sources, metabolism, and health-related activities of pterostilbene.7,8,10 These studies provide an important basis for understanding the biological activity of pterostilbene. However, the available literature remains largely organized around pharmacological activity categories or individual disease areas, rather than around the distinct redox environments in which pterostilbene is tested. The conceptual advance of this review is to integrate these dispersed findings into a disease-context-based redox framework, rather than to reclassify pterostilbene simply as another antioxidant compound. Within this framework, the selected disease contexts are distinguished by their dominant redox features: chronic oxidative burden and persistent redox–inflammatory stress in osteoarthritis and neurodegenerative/cognitive dysfunction models, acute reperfusion-associated redox and inflammatory amplification in ischemia–reperfusion injury, redox–metabolic imbalance in metabolic diseases, and redox-adapted malignant states in cancer. This organization helps clarify why similar experimental readouts, such as ROS, Nrf2, NF-κB, mitochondria-associated markers, or apoptosis-related proteins, may have different biological meanings across disease settings. By emphasizing disease-specific redox state, therapeutic aim, dose exposure, and biological threshold, this review provides a framework for interpreting context-dependent pterostilbene-related effects across different pathological settings.
Chemical and Pharmacokinetic Basis of Pterostilbene
Pterostilbene is a naturally occurring dimethoxylated stilbene analogue of resveratrol, containing two methoxy groups and one phenolic hydroxyl group. It has been reported in several edible and medicinal plant sources, including blueberries, grapes, and mulberries.7,10 Compared with resveratrol, the methoxy substitutions of pterostilbene increase lipophilicity and reduce polarity, properties associated with improved membrane permeability, metabolic stability, and oral bioavailability.6,7,10 Pharmacokinetic studies comparing resveratrol and pterostilbene have reported greater oral bioavailability, higher plasma exposure, and more favorable metabolic profiles for pterostilbene.6 This pharmacokinetic profile should be considered when interpreting pterostilbene-associated biological effects, rather than attributing those effects solely to intrinsic chemical antioxidant reactivity.
From a chemical standpoint, pterostilbene contains one phenolic hydroxyl group and has been reported to exhibit radical-scavenging activity in cell-free antioxidant assays. For example, pterostilbene showed activity against DPPH, ABTS, hydroxyl radicals, superoxide radicals, and hydrogen peroxide in vitro, and protected isolated biological macromolecules against free radical–mediated oxidative damage.11 Higher bioavailability, however, should not be equated with stronger intrinsic radical-scavenging capacity than resveratrol. Comparative evidence suggests that hydroxyl substitution patterns and molecular interactions, including protein binding, may influence measured antioxidant activity.12
Chemical antioxidant assays alone are insufficient to explain the diversity of pterostilbene-associated effects observed in cellular and animal models. Cell-based evidence suggests that pterostilbene-related antioxidant responses may involve not only direct radical-scavenging activity, but also intracellular ROS reduction and Nrf2/ARE-related gene regulation.13 In biological systems, ROS generation is dynamic and compartmentalized, and redox regulation is influenced by endogenous antioxidant defenses, inflammatory signaling, and stress-response pathways.1–4 For this reason, pterostilbene-associated redox effects are better interpreted as a combination of chemical antioxidant potential and modulation of cellular responses to oxidative stimuli, rather than as simple radical neutralization alone.7,8,11,13
Pterostilbene’s redox-related relevance, then, cannot be explained by a single chemical or pharmacokinetic property. Improved bioavailability may increase biological exposure, but it should not be directly translated into stronger intrinsic antioxidant activity or therapeutic efficacy. The following sections therefore discuss pterostilbene as a compound with both chemical antioxidant potential and the capacity to modulate cellular redox responses in disease-specific contexts.
Redox-Related Signaling and Cellular Stress Responses Associated with Pterostilbene
Nrf2-Related Endogenous Antioxidant Defense
Nuclear factor erythroid 2-related factor 2 (Nrf2) links cellular redox status with endogenous antioxidant defense, stress adaptation, and intermediary metabolism.14 The NRF2–KEAP1 axis also participates in redox, metabolic, protein-homeostasis, and inflammatory regulation and has been explored as a therapeutic target in several chronic disease contexts, although target specificity, pharmacodynamic properties, efficacy, and safety remain important challenges.15 For this reason, Nrf2-related findings in pterostilbene studies should be interpreted according to disease context and the strength of mechanistic validation, rather than as uniformly beneficial pathway activation. Pterostilbene-associated Nrf2/ARE responses have been reported in several oxidative stress, inflammatory, and metabolic models. Cell-based evidence links pterostilbene to intracellular ROS reduction and Nrf2/ARE-related gene regulation.13 Stronger mechanistic support comes from arsenic-induced human keratinocyte injury, where Nrf2 knockdown weakened the protective effect of pterostilbene,16 and from IL-1β-stimulated chondrocytes and rat osteoarthritis cartilage, where Nrf2 silencing partially reversed pterostilbene-associated reductions in ROS production and inflammatory mediator expression.17 Nrf2/heme oxygenase-1 (HO-1)- or Nrf2-mediated antioxidant signaling has also been reported in fructose-fed diabetic rats and streptozotocin (STZ)-induced diabetes models.18,19 Yet pterostilbene should not be described as a uniform Nrf2 activator across all contexts: in pancreatic cancer cells, it was reported to decrease glucocorticoid receptor- and Nrf2-dependent antioxidant signaling, thereby reducing tumor-cell antioxidant defenses.20 Nrf2-related evidence therefore supports a context-dependent mechanism of pterostilbene-associated redox modulation. Studies involving Nrf2 knockdown or silencing provide stronger causal support, whereas studies reporting only Nrf2 expression, nuclear translocation, HO-1 expression, or antioxidant enzyme changes should be interpreted as pathway-associated evidence rather than definitive proof that Nrf2 is a universal primary target of pterostilbene.
Redox–Inflammatory Crosstalk and NF-κB-Related Signaling
In addition to Nrf2-related antioxidant responses, pterostilbene has been associated with attenuation of redox–inflammatory crosstalk, particularly through NF-κB-related signaling. In IL-1β-stimulated chondrocytes, pterostilbene reduced ROS production together with inflammatory mediators such as cyclooxygenase-2 (COX-2), inducible nitric oxide synthase (iNOS), prostaglandin E2 (PGE2), and nitric oxide (NO).17 In cerebral ischemia–reperfusion injury, pterostilbene attenuated astrocytic inflammation and neuronal oxidative injury in association with inhibition of NF-κB phosphorylation.21 In a 2,4,6-trinitrobenzenesulfonic acid (TNBS)-induced ulcerative colitis rat model, pterostilbene reduced serum inflammatory cytokines and oxidative stress-related indices, including TNF-α, IL-17, IL-6, IL-1β, malondialdehyde (MDA), and myeloperoxidase (MPO), while increasing superoxide dismutase (SOD) and glutathione peroxidase (GPx) activity; these effects were accompanied by suppressed NF-κB expression and increased peroxisome proliferator-activated receptor gamma (PPAR-γ) expression.22 These studies support an association between pterostilbene treatment, reduced oxidative stress-related markers, and attenuated NF-κB-related inflammatory signaling. Because many studies measure oxidative and inflammatory endpoints in parallel without time-course analysis or pathway-specific causal validation, the direction of the redox–inflammatory relationship remains incompletely defined. Pterostilbene is therefore better described as modulating redox–inflammatory crosstalk, rather than as a confirmed direct inhibitor of individual inflammatory mediators.
Mitochondria-Associated Redox and Cellular Stress Responses
Mitochondria are major sources and targets of intracellular ROS and are closely involved in cellular stress and apoptosis-related responses. Pterostilbene-associated mitochondrial findings, however, should not be generalized as uniform improvement of mitochondrial function across all models. In ischemic or oxidative injury models, pterostilbene has been linked to attenuation of mitochondria-associated oxidative injury or dysfunction. For example, in skeletal muscle ischemia–reperfusion injury, pterostilbene attenuated oxidative stress injury and mitochondrial dysfunction in association with SIRT1 activation, with dose-related effects reported under the experimental conditions tested.23 In cerebral ischemia–reperfusion injury, pterostilbene treatment attenuated mitochondrial oxidative damage in association with HO-1 signaling activation.24 In oxidative stress-induced intestinal injury models, pterostilbene was reported to improve mitochondrial redox homeostasis and function through SIRT1 signaling; SIRT1 depletion weakened the protective effects against mitochondrial ROS overproduction, mitochondrial dysfunction, and apoptosis in intestinal epithelial cells.25 In tumor-cell contexts, mitochondria-associated changes may reflect disruption rather than preservation of cellular homeostasis. In diffuse large B-cell lymphoma cells, pterostilbene induced dose-dependent inhibition of cell viability, accompanied by increased ROS levels, reduced mitochondrial membrane potential, increased apoptosis, and cell-cycle arrest.26 In breast cancer cells, pterostilbene induced mitochondrially derived apoptosis, involving Bax upregulation, redistribution of cytochrome c and Smac/DIABLO, cytosolic Ca2⁺ overload, and reduced cell survival.27 These findings support a context-dependent interpretation of mitochondria-associated responses to pterostilbene: such responses may contribute to cytoprotection in ischemic or oxidative injury models, but may also promote stress-induced cell death in cancer cells. Pterostilbene should therefore be described as modulating mitochondria-associated redox and cellular stress responses in a model-specific manner, rather than as generally improving mitochondrial function.
Mechanistic Evidence Limitations and Disease-Specific Interpretation
Current mechanistic evidence suggests that pterostilbene-associated redox effects may involve Nrf2-related antioxidant responses, NF-κB-related inflammatory signaling, and mitochondria-associated cellular stress pathways. The strength of this evidence differs across experimental models. Studies employing Nrf2 knockdown, Nrf2 silencing, SIRT1 depletion, or pathway-specific interventions provide stronger causal support, whereas studies reporting only changes in protein expression, nuclear translocation, enzyme activity, oxidative stress markers, inflammatory cytokines, or apoptosis-related proteins should be interpreted as pathway-associated evidence rather than direct target validation.This distinction matters because similar pathway changes may have different biological implications across pathological contexts. Nrf2 activation may be associated with cytoprotection in some oxidative injury models, whereas suppression of tumor-cell antioxidant defenses may contribute to anticancer effects. Similarly, mitochondria-associated responses may reflect attenuation of oxidative injury in ischemic or intestinal injury models, but disruption of mitochondrial homeostasis in cancer cells. Pterostilbene should not be described as acting through a single universal antioxidant mechanism.The following sections therefore organize the available evidence by pathological context rather than treating all antioxidant-related findings as mechanistically equivalent. This disease-specific organization helps avoid overgeneralization of pathway markers and allows pterostilbene-associated effects to be evaluated in relation to model dependence, causal evidence, and translational relevance.
Disease-Specific Evidence Across Pathological Contexts
Table 1 summarizes representative experimental evidence for pterostilbene across the major pathological contexts discussed in this section, including disease models, main findings, proposed redox- or stress-related mechanisms, and selected references.
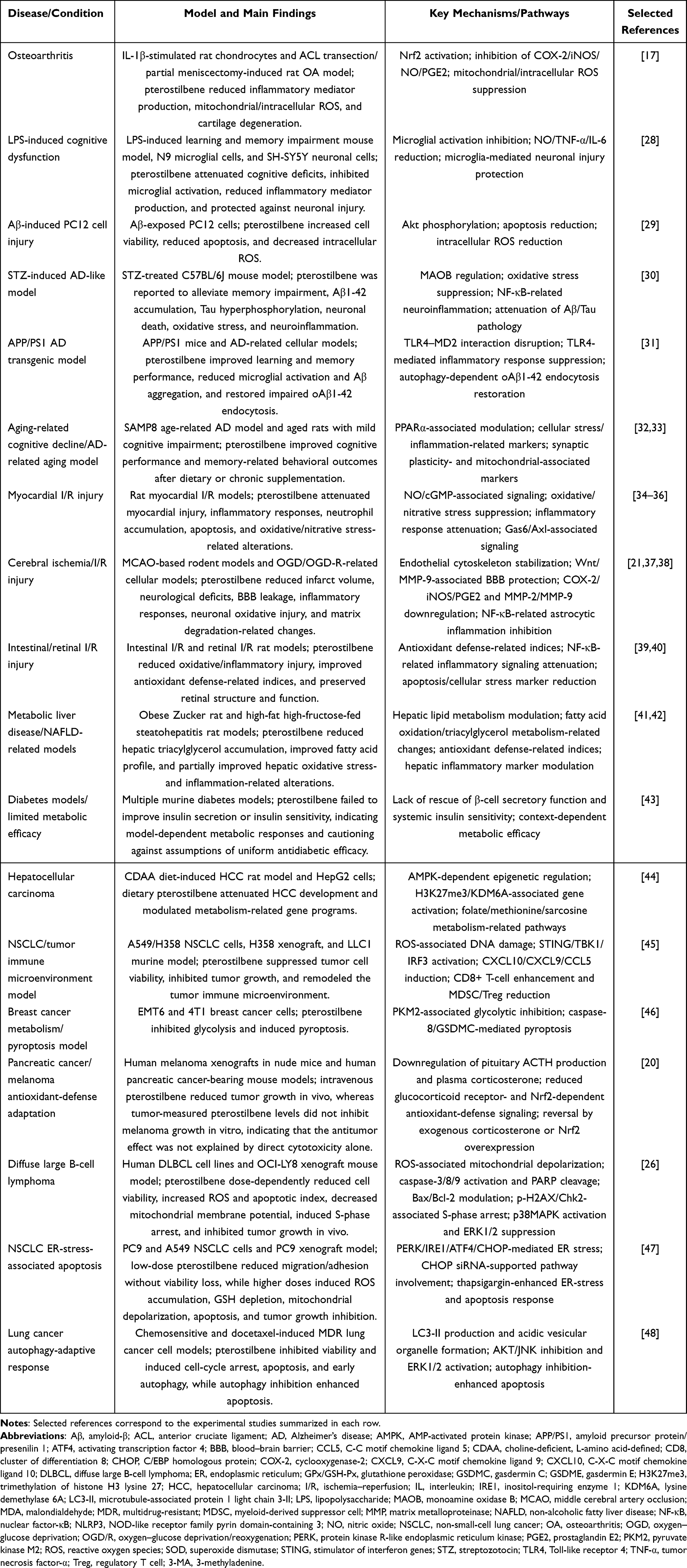 |
Table 1 Representative Experimental Evidence for Pterostilbene in Oxidative Stress-Related Pathological Conditions |
Chronic Oxidative Burden-Related Degenerative Diseases
Osteoarthritis
Osteoarthritis (OA) is a whole-joint degenerative disease involving articular cartilage, subchondral bone, synovium, and surrounding periarticular tissues. It is commonly characterized by pain, stiffness, functional impairment, progressive cartilage deterioration, and osteophyte formation, particularly in weight-bearing joints.49,50 From a redox-pathology perspective, OA is associated with persistent oxidative stress, impaired antioxidant defense, inflammatory signaling, and disruption of chondrocyte and joint-tissue homeostasis. Age-related imbalance between ROS production and antioxidant capacity has been linked to cartilage degradation and chondrocyte cell death, while mitochondrial dysfunction, reduced SOD2 activity, and altered Nrf2-related antioxidant defense may contribute to increased mitochondrial-derived ROS in aging cartilage.51 ROS/oxidative stress signaling is also involved in chondrocyte senescence and apoptosis, extracellular matrix synthesis and degradation, synovial inflammation, and subchondral bone dysfunction.52 Inflammatory mediators such as IL-1β, TNF-α, and IL-6 can further promote ROS production and matrix-degrading protease expression, reinforcing the interaction between oxidative stress and inflammation in OA pathogenesis.53 At the same time, ROS in chondrocytes also participate in physiological redox-regulated processes, including pH regulation, indicating that OA-related redox disturbance should not be interpreted simply as a need for indiscriminate ROS elimination.54
Direct pterostilbene-specific evidence in OA remains preclinical, but recent studies have linked pterostilbene to several OA-relevant redox and cellular stress-response processes. In IL-1β-stimulated chondrocytes and a rat OA model, pterostilbene reduced ROS production and inflammatory mediator expression, promoted Nrf2 nuclear translocation, and Nrf2 silencing partially reversed these effects, supporting an OA-relevant role for Nrf2-associated antioxidant defense and redox–inflammatory regulation.17 A subsequent in vitro and in vivo study reported that pterostilbene attenuated OA-associated chondrocyte senescence through inhibition of the phosphoinositide 3-kinase/protein kinase B (PI3K/AKT)/NF-κB signaling axis, suggesting that its effects may extend beyond acute inflammatory suppression to senescence-associated stress responses.55 Another recent study suggested that pterostilbene-mediated OA protection may involve NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome inactivation and gut microbiota improvement, indicating a possible connection between local inflammatory responses and gut microbiota-related regulation.56 In addition, a network-analysis and experimental-validation study suggested that pterostilbene may attenuate OA progression through p53-dependent autophagy activation, providing another potential cellular stress-adaptation mechanism.57
Current OA-related evidence therefore points to several disease-relevant stress-response processes, including antioxidant defense, inflammatory and senescence-related signaling, inflammasome-associated responses, gut microbiota-related changes, and autophagy-related adaptation. These findings come from different preclinical models and do not yet define a unified mechanism or establish clinical disease-modifying efficacy. At present, pterostilbene is better framed as a candidate redox- and stress-response modulator requiring further OA-specific validation, rather than as an established OA therapeutic agent.
Neurodegenerative and Cognitive Dysfunction Models
Neurodegenerative diseases and related cognitive dysfunction models are frequently associated with redox-related disturbances, although the initiating pathological drivers differ across models. Oxidative stress and mitochondrial dysfunction have been implicated in neuronal injury and neuronal loss, and oxidative stress has been associated with the progression of several neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis.58,59 In Alzheimer’s disease (AD)-related models, oxidative stress is also closely linked to synaptic dysfunction, amyloid-β (Aβ)- and Tau-related pathology, and cognitive decline.60 More broadly, neurodegeneration involves crosstalk among oxidative stress, protein aggregation, neuroinflammation, and cellular stress responses.61 This pathological background supports evaluating pterostilbene in relation to cognitive or behavioral outcomes, neuroinflammatory changes, oxidative stress-related markers, and disease-specific cellular stress responses across different neural dysfunction models.
Early evidence for pterostilbene in aging-related neural dysfunction came from a study comparing resveratrol analogues in cellular and aged-animal models. Joseph et al examined several stilbene analogues in an oxidative stress-sensitive cellular system and then tested pterostilbene in 19-month-old Fischer 344 rats, where pterostilbene feeding improved cognitive behavioral deficits and working memory was correlated with pterostilbene levels in the hippocampus.62 In inflammation-driven neural dysfunction, pterostilbene was evaluated in a lipopolysaccharide (LPS)-induced learning and memory impairment model together with microglial and neuronal cell models; pterostilbene attenuated cognitive deficits, inhibited microglial activation, reduced NO, TNF-α, and IL-6 production, and protected against microglia-mediated neuronal injury.28 In Aβ-exposed PC12 cells, pterostilbene increased cell viability, reduced apoptosis, and decreased intracellular ROS, with the observed effects linked to Akt phosphorylation and anti-apoptotic signaling.29 These studies indicate that pterostilbene has been examined across aging-, inflammation-, and Aβ-associated neural stress models, involving behavioral, inflammatory, oxidative stress-related, and neuronal survival outcomes.
More disease-oriented evidence comes from AD-like animal models. In an STZ-treated C57BL/6J mouse model, pterostilbene alleviated memory impairment and was associated with reductions in Aβ1-42 accumulation, Tau hyperphosphorylation, neuronal death, oxidative stress, and neuroinflammation, with monoamine oxidase B (MAOB) regulation implicated in the observed effects.30 In amyloid precursor protein/presenilin 1 (APP/PS1) mice and AD-related cellular models, pterostilbene improved learning and memory, reduced microglial activation and Aβ aggregation, disrupted the Toll-like receptor 4–myeloid differentiation factor 2 (TLR4–MD2) interaction, suppressed TLR4-mediated inflammatory responses, and restored impaired oligomeric amyloid-β1-42 (oAβ1-42) endocytosis in an autophagy-dependent manner.31 These findings connect pterostilbene treatment with AD-relevant pathological readouts, including Aβ/Tau-related pathology, microglial inflammatory activation, neuronal injury, and cognitive performance.
Aging-related cognitive decline has also been examined in senescence-accelerated mouse prone 8 (SAMP8) age-related AD models and aged rats with mild cognitive impairment. Dietary or chronic pterostilbene supplementation improved cognitive performance, learning and memory, or memory consolidation, accompanied by changes in cellular stress-, inflammation-, synaptic plasticity-, mitochondrial marker-, or PPARα-associated pathways.32,33 Beyond AD-like and aging-related models, pterostilbene has also been evaluated in a ketamine-induced schizophrenia-like behavioral model, where behavioral alterations and oxidative stress-related markers were assessed.63
Current preclinical studies show a recurring but model-dependent pattern: pterostilbene is associated with improved cognitive or behavioral outcomes, reduced neuroinflammatory responses, and changes in oxidative stress-, Aβ/Tau pathology-, neuronal survival-, or cellular stress-related markers.28–33,62,63 Yet LPS-induced neuroinflammation, Aβ-exposed cells, STZ-induced AD-like injury, APP/PS1 transgenic models, aging-related cognitive decline, and ketamine-induced behavioral alterations represent distinct experimental contexts rather than interchangeable models of neurodegeneration. The current evidence therefore supports pterostilbene as a candidate redox–inflammatory and cellular stress-response modulator in selected neural dysfunction models, while its disease-modifying relevance in neurodegenerative disorders requires further validation.
Evidence Limitations and Knowledge Gaps
Evidence from OA and neural dysfunction models shows overlapping but mechanistically heterogeneous patterns of pterostilbene-associated effects. In OA, reported mechanisms include Nrf2-related antioxidant defense, PI3K/AKT/NF-κB-associated senescence signaling, NLRP3 inflammasome inactivation, gut microbiota-related changes, and p53-dependent autophagy activation.17,55–57 In neural models, pterostilbene has been examined across LPS-induced neuroinflammation, Aβ-related cellular injury, STZ-induced AD-like pathology, APP/PS1-associated amyloid and inflammatory responses, aging-related cognitive decline, and ketamine-induced behavioral alterations.28–33,62,63 These findings support the relevance of redox–inflammatory and cellular stress-response processes, but they do not yet define a unified mechanism of action across chronic degenerative models.
Several evidence gaps remain. In OA, direct evidence is still concentrated in chondrocyte- and rodent-based models, whereas clinical joint-level outcomes and long-term structural modification have not been established. In neural models, behavioral improvement, Aβ/Tau-related markers, neuroinflammation, and oxidative stress endpoints are not measured consistently across models. Future work would benefit from broader dose–response designs, early- versus late-intervention comparisons, time-course analyses, pathway inhibition or knockdown, rescue experiments, and standardized behavioral, histological, oxidative stress, and inflammatory endpoints.
Ischemia–Reperfusion Injury and Acute Redox Disruption
In contrast to chronic degenerative conditions, ischemia–reperfusion (I/R) injury represents an acute and time-sensitive redox-disruptive context. During ischemia, interruption of blood flow restricts oxygen and nutrient supply, leading to ATP depletion, intracellular acidosis, ionic imbalance, calcium overload, and activation of cellular injury pathways. Reperfusion restores oxygen and blood flow, but can also amplify ischemia-initiated injury through rapid ROS generation, inflammatory cell recruitment, mitochondrial permeability transition pore opening, and necrotic, apoptotic, or other regulated forms of cell death.64,65
Unlike chronic oxidative burden, I/R injury is defined by abrupt changes in oxygen availability, redox state, and inflammatory activation. In redox biology, low-level or spatially controlled ROS can participate in physiological signaling, whereas excessive or poorly controlled ROS may damage lipids, proteins, and DNA and trigger pathological signaling responses.3,66 In I/R injury, ROS can arise from multiple enzymatic and cellular sources, including mitochondria, NADPH oxidases, xanthine oxidase, and uncoupled nitric oxide synthase.64 Their relative contribution may differ across tissues; for example, mitochondrial ROS has been emphasized in metabolically active organs such as the heart and brain, whereas other enzymatic sources may be more prominent in different organs.64 Such tissue-specific differences are important for interpreting pterostilbene-related findings, because protective outcomes in different organs may involve distinct redox and inflammatory mechanisms.
Myocardial and cerebral I/R are representative settings in which acute redox disruption is closely linked to organ-specific injury patterns. In myocardial reperfusion injury, restoration of coronary blood flow after ischemia can paradoxically produce additional myocardial damage beyond the initial ischemic insult, involving mitochondrial injury, calcium overload, inflammation, and cell-death signaling.65,67 In ischemic brain injury, ROS can directly damage cellular macromolecules and also participate in signaling pathways involving mitochondria, DNA repair enzymes, and transcription factors that may contribute to apoptosis.68 These differences support evaluating pterostilbene-associated effects according to reperfusion-phase oxidative and inflammatory amplification, organ-specific vulnerability, and acute cell-death responses.
In myocardial I/R models, pterostilbene has been evaluated in relation to infarct injury, inflammatory infiltration, oxidative/nitrative stress, apoptosis, and reperfusion-associated signaling changes. In one rat myocardial I/R model, pterostilbene reduced infarct area and lowered serum creatine kinase (CK) and lactate dehydrogenase (LDH) levels, consistent with attenuation of acute myocardial injury. The same study reported decreased myocardial MPO activity and neutrophil accumulation, together with reduced serum and myocardial TNF-α levels. The protective effects were attenuated by L-NAME and methylene blue, suggesting involvement of nitric oxide/cyclic guanosine monophosphate (NO/cGMP) signaling in this model.34
Another rat myocardial I/R study reported that pterostilbene treatment improved cardiac function and reduced myocardial infarct size and apoptosis after 30 min of ischemia followed by 3 h of reperfusion. This protection was accompanied by reduced myocardial peroxynitrite content, superoxide generation, and MDA levels, increased SOD activity, decreased p38 MAPK activation, lower iNOS and gp91phox expression, increased phosphorylated eNOS expression, and reduced myocardial TNF-α, IL-1β, and MPO activity. The observation that peroxynitrite suppression by either pterostilbene or uric acid reduced myocardial injury supports the relevance of oxidative/nitrative stress attenuation in this model.35 A further rat myocardial I/R study specifically investigated Gas6/Axl signaling and associated pterostilbene-mediated protection with activation of this pathway.36
In cerebral I/R models, pterostilbene-associated protection has been evaluated across vascular, inflammatory, and neuronal injury endpoints. In a middle cerebral artery occlusion (MCAO) model and oxygen–glucose deprivation (OGD)-based in vitro system, pterostilbene reduced cerebral infarct volume, improved neurological deficits, increased cerebral microcirculation, and alleviated blood–brain barrier (BBB) leakage. The protective effect was associated with early inhibition of endothelial cytoskeleton reorganization and later reduction of basement membrane degradation, involving ADF-related cytoskeletal regulation, Wnt pathway activation, MMP-9 inhibition, and increased junction protein expression.37 Another MCAO study reported reduced neurological injury, brain edema, infarct volume, and Evans blue leakage, together with decreased COX-2, iNOS, PGE2, inflammatory cytokines, and MMP-2/MMP-9 expression, supporting an anti-inflammatory and matrix-protective profile in cerebral ischemic injury.38 In an MCAO/R model, post-reperfusion pterostilbene treatment attenuated infarct volume, brain edema, neuronal apoptosis, and long-term neurological deficits, accompanied by reduced ROS and MDA production, increased SOD and glutathione peroxidase (GPx) activities, decreased TNF-α, IL-1β, and IL-6 levels, and inhibition of NF-κB phosphorylation and nuclear translocation.21
Evidence from other organ-specific I/R models further supports the relevance of oxidative and inflammatory injury modulation, although the measured endpoints differ by tissue. In an intestinal I/R model, pterostilbene reduced ROS, lipid peroxidation, and protein carbonyl content, restored antioxidant status and membrane-bound ATPase activities, and downregulated NF-κB, COX-2, TNF-α, and IL-1β, indicating protection associated with antioxidant defense and inflammatory suppression.39 In a retinal I/R model, pterostilbene improved retinal function, reflected by increased electroretinography (ERG) b-wave amplitude, reduced retinal thinning, preserved retinal structure, and decreased glial fibrillary acidic protein (GFAP), heat shock protein 70 (HSP70), poly(ADP-ribose) polymerase 1 (PARP1), and NF-κB expression. These findings suggest attenuation of glial activation, cellular stress, apoptosis-associated signaling, and inflammation after retinal reperfusion injury.40
Across myocardial, cerebral, intestinal, and retinal I/R models, pterostilbene-related effects are best interpreted in relation to the dominant injury compartment of each organ. In myocardial I/R, the evidence centers on infarct injury, oxidative/nitrative stress, inflammatory infiltration, apoptosis, and reperfusion-associated signaling; in cerebral I/R, it shifts toward BBB stability, microcirculation, endothelial cytoskeleton and basement membrane protection, astrocytic inflammation, neuronal oxidative injury, and neurological outcomes; in intestinal and retinal I/R, it is reflected mainly by antioxidant defense, inflammatory mediators, membrane enzyme activity, tissue structure, cellular stress markers, apoptosis-associated signaling, and functional readouts.21,34–40 This cross-organ pattern supports a context-dependent interpretation of pterostilbene-associated protection in I/R models, centered on attenuation of reperfusion-related injury amplification rather than a single organ-independent mechanism. The apparent protective profile is shaped by organ vulnerability, vascular or barrier involvement, treatment timing, and selected endpoints.
Evidence Limitations and Knowledge Gaps
The available I/R studies point to a tissue-dependent protective profile, but several issues limit mechanistic interpretation. Treatment schedules vary substantially across studies, including pretreatment, administration during ischemia, and delivery after reperfusion. This timing difference is particularly important in I/R injury because the effect of an intervention may depend on whether it is applied during early ROS/nitrative stress amplification, inflammatory cell recruitment, endothelial or barrier disruption, or later cell-death responses. Without time-course designs that distinguish these phases, it remains difficult to determine which stage of reperfusion-associated injury is most affected by pterostilbene treatment.
Many studies also rely on endpoint measurements of oxidative stress, inflammation, and cell-death markers, such as ONOO−, ROS, MDA, SOD, MPO, TNF-α, IL-1β, NF-κB, MMP-9, COX-2, iNOS, p-eNOS, and apoptosis-associated proteins. These readouts are useful for describing injury attenuation, but they do not by themselves establish direct molecular targets or causal pathway hierarchy. For example, changes in NF-κB activation, MMP-9 expression, or Gas6/Axl-related signaling could represent direct pathway modulation, compensatory tissue protection, or secondary consequences of reduced injury severity. More informative designs would combine broader dose–response curves, early-versus-delayed treatment comparisons, reperfusion time-course analyses, pathway inhibition or rescue experiments, and organ-specific cellular models, including cardiomyocytes, endothelial cells, astrocytes, neurons, intestinal epithelial cells, and retinal cells. Such approaches would help clarify whether pterostilbene preferentially limits acute redox/nitrative amplification, vascular or barrier dysfunction, inflammatory propagation, or downstream cell-death responses in specific I/R contexts.
Metabolic Diseases Characterized by Redox–Metabolic Imbalance
Obesity, type 2 diabetes, and related metabolic disorders represent chronic redox–metabolic contexts in which nutrient overload, insulin resistance, mitochondrial redox signaling, and low-grade inflammation are closely interconnected. Chronic overnutrition and physical inactivity are major risk factors for insulin resistance and type 2 diabetes, and mitochondrial hydrogen peroxide (H2O2) emission has been proposed as a redox-linked signal connecting fuel overload to impaired insulin sensitivity.69 In diabetes, hyperglycemia-induced oxidative stress and mitochondrial disturbances are closely associated with diabetic tissue injury and complications, involving pathways such as protein kinase C (PKC) activation, advanced glycation end-product formation, polyol pathway flux, and impaired antioxidant defense.70 Nutrient excess also affects β-cell insulin secretion and insulin action in target tissues such as skeletal muscle, adipose tissue, and liver, while altered lipid metabolism and inflammatory activation contribute to metabolic syndrome progression.71
In metabolic disease models, redox changes are embedded within broader metabolic adaptation or maladaptation rather than occurring as isolated oxidative injury. In skeletal muscle from rodents and humans, high-fat intake increased mitochondrial H2O2-emitting potential, shifted the cellular redox environment toward a more oxidized state, and reduced redox-buffering capacity without necessarily reducing mitochondrial respiratory function.72 Immune and metabolic pathways are also closely integrated, with chronic inflammatory activation contributing to obesity, insulin resistance, type 2 diabetes, and related cardiometabolic disorders.73,74 Metabolic redox changes can have context-dependent roles: in humans, antioxidant supplementation blunted exercise-induced improvements in insulin sensitivity and endogenous antioxidant defense, suggesting that some exercise-associated oxidative signals contribute to adaptive metabolic responses.75 Pterostilbene-related findings in metabolic disease models should therefore be interpreted by integrating oxidative stress markers with metabolic and tissue-specific outcomes, including insulin sensitivity, glycolipid metabolism, hepatic lipid accumulation, inflammatory responses, and mitochondrial or redox-related indices where directly measured.
Within this redox–metabolic framework, pterostilbene has been examined in models of diet-induced metabolic stress, obesity-associated lipid remodeling, hepatic steatosis or steatohepatitis, diabetic nephropathy, and diabetes-associated cognitive impairment. Its effects on glucose homeostasis appear inconsistent across models. In sucrose-fed rats, pterostilbene improved glycemic and lipid-related indices and enhanced hepatic antioxidant status.76 By contrast, in multiple murine diabetes models, pterostilbene failed to rescue insulin secretion or insulin sensitivity.43 This discrepancy argues against a general glucose-lowering interpretation and instead points to a context-dependent metabolic profile shaped by nutritional challenge, species or strain background, disease model, dosing strategy, and selected endpoints.
In obesity-associated and hepatic metabolic models, the evidence is more concentrated on lipid handling, adiposity, and liver injury. In obese or obesogenic diet models, pterostilbene reduced fat accumulation and was associated with changes in energy metabolism or adipose tissue remodeling.77–79 In obese Zucker rats, six-week pterostilbene administration reduced serum insulin and homeostatic model assessment of insulin resistance (HOMA-IR), lowered hepatic triacylglycerol content, and improved hepatic fatty acid profile; the reported delipidating effect was linked to reduced fatty acid availability and triacylglycerol synthesis, together with increased very-low-density lipoprotein (VLDL) assembly and fatty acid oxidation.41 In a high-fat high-fructose diet-induced steatohepatitis model, pterostilbene partially prevented oxidative stress, inflammation, and ballooning degeneration, mainly at 30 mg/kg/day, although hepatocarcinoma-related gene alterations were not prevented.42 The evidence for glucose homeostasis remains mixed, whereas hepatic studies more consistently support pterostilbene-associated effects on lipid handling, hepatic steatosis, and steatohepatitis-related oxidative or inflammatory injury.
Evidence from metabolic complication models extends this tissue-specific pattern. In a high-fat diet plus STZ-induced diabetic cognitive impairment model, pterostilbene improved cognitive dysfunction and glycolipid metabolic disorder, while reducing neuronal injury, β-amyloid accumulation, oxidative/carbonyl stress, astrocyte and microglial activation, and dopaminergic neuronal loss.80 The same study connected these effects with microbiota–gut–brain regulation, including modulation of colon and brain Toll-like receptor 4 (TLR4)/NF-κB signaling, intestinal microbiota homeostasis, short-chain fatty acid-related changes, intestinal tight junction preservation, and systemic inflammation.80 In diabetic renal injury models, pterostilbene was reported to ameliorate nephropathy-related injury and was associated with suppression of hyperglycemia, inflammatory responses, and fibrotic changes.81,82 The metabolic disease evidence does not support a uniform claim that pterostilbene broadly improves metabolism across all models. The available findings are more consistent with tissue- and endpoint-specific effects, including lipid handling in obesity and liver disease, inflammatory and barrier-related regulation in diabetic cognitive impairment, and inflammatory or fibrotic responses in diabetic renal injury.
Evidence Limitations and Context-Specific Gaps
The mixed glycemic findings discussed above highlight a key unresolved issue in metabolic disease models: pterostilbene-related effects appear to depend strongly on experimental context rather than reflecting a uniform glucose-lowering or insulin-sensitizing action. Relevant contextual variables include nutritional background, species or strain, baseline insulin resistance, tissue lipid burden, inflammatory tone, dose regimen, and intervention duration. Metabolic evidence should be separated by endpoint category—glycemic control, hepatic lipid handling, inflammatory regulation, gut–brain interaction, and diabetic organ injury—rather than summarized as a uniform metabolic benefit. Without systematic variation of these factors, it remains difficult to determine whether the observed effects reflect correction of metabolic stress, modulation of inflammatory–redox crosstalk, tissue-specific lipid handling, or secondary consequences of improved systemic metabolic status.
Mechanistic interpretation is also limited by reliance on downstream readouts. Changes in HOMA-IR, hepatic triacylglycerol content, oxidative stress indices, inflammatory mediators, TLR4/NF-κB signaling, gut microbiota-related markers, or renal fibrotic markers are informative, but they do not identify direct molecular targets or establish a causal hierarchy across liver, adipose tissue, gut–brain, and kidney contexts. More informative studies would combine broader dose–response designs, time-course analyses, and tissue-specific validation in liver, adipose tissue, skeletal muscle, pancreatic β-cells, kidney, gut, and brain. Direct comparisons across models with different basal redox states and metabolic phenotypes would help clarify whether pterostilbene acts preferentially under nutrient-overload, inflammatory, lipotoxic, or diabetic complication settings.
Cancer: Context-Dependent Perturbation of Tumor Redox and Stress-Adaptive States
Cancer represents a distinct redox-related pathological context in which malignant cells may both generate and exploit elevated redox pressure while maintaining sufficient antioxidant and stress-response capacity to avoid lethal oxidative injury. ROS can influence cancer evolution in concentration-dependent and apparently contradictory ways, contributing to tumor initiation and proliferation at some levels while triggering cell death when oxidative stress exceeds adaptive capacity.83 During progression, tumor cells can adapt to oxidative stress by remodeling NADPH generation, antioxidant transcriptional programs, and metabolic pathways.83–85 NRF2-related signaling further illustrates this duality, because it may suppress tumor initiation under some conditions but promote tumor survival, metabolic adaptation, progression, metastasis, or therapy resistance in established cancers.84 In this review, cancer is therefore treated as a redox-adapted malignant context rather than as a simple oxidative injury model; pterostilbene-related findings are interpreted according to tumor redox state, antioxidant-buffering capacity, metabolic wiring, dose, exposure duration, and host microenvironment.
Mitochondria-associated apoptotic responses provide one line of evidence for dose-dependent pterostilbene effects in tumor cells. In diffuse large B-cell lymphoma cells, pterostilbene showed a dose-dependent cytotoxic profile: 20–60 μM reduced cell viability and induced S-phase cell-cycle arrest after 24 h, accompanied by increased p-H2A.X and Chk2 and decreased Cdc25A, CDK2, and Cyclin A2. At 60 μM for 24 h, pterostilbene markedly increased intracellular ROS and reduced mitochondrial membrane potential, while 20–60 μM exposure for 48 h increased caspase activation and PARP cleavage, downregulated Bcl-2, and upregulated Bax. The pan-caspase inhibitor Z-VAD-FMK attenuated pterostilbene-induced growth inhibition, supporting caspase-dependent apoptotic involvement in this model.26 In breast cancer cells, pterostilbene also produced concentration-related mitochondria-associated apoptotic changes. In MCF-7 and MDA-MB-231 cells, intracellular Bax increased in a concentration-dependent manner, and Bax silencing reduced apoptosis and improved cell survival after pterostilbene treatment.27 At 50 and 75 μM, pterostilbene promoted cytosolic redistribution of cytochrome c and Smac/DIABLO from mitochondria in MCF-7 cells, accompanied by increased cytosolic Ca2⁺ and, in MDA-MB-231 cells, increased MnSOD activity.27 These two studies link pterostilbene-associated tumor-cell injury to dose-dependent mitochondrial stress and apoptotic signaling, with functional support from caspase inhibition or Bax silencing rather than marker changes alone.
Pancreatic cancer and melanoma models illustrate a different pattern, in which pterostilbene-related antitumor activity was not explained simply by direct cytotoxicity in cultured tumor cells. In these models, pterostilbene reduced glucocorticoid receptor- and NRF2-dependent antioxidant-defense signaling, thereby weakening the stress-buffering capacity that aggressive tumor cells use to tolerate elevated ROS. The study reported tumor-suppressive effects under in vivo conditions, whereas direct in vitro cytotoxicity was limited, indicating that pterostilbene-related cancer effects may depend on host–tumor context and antioxidant-defense adaptation rather than direct tumor-cell killing alone. This finding is relevant to the context-dependent framework of this review: in non-malignant injury models, enhancement of antioxidant defense is often interpreted as cytoprotective, whereas in redox-adapted tumor cells, reduction of antioxidant-buffering capacity may increase vulnerability to oxidative stress.20
Autophagy-related evidence further illustrates the model-dependent nature of pterostilbene-induced tumor-cell stress responses. In lung cancer cells, pterostilbene induced both autophagy and apoptosis, but inhibition of autophagy did not weaken apoptosis. Instead, pharmacological inhibition with 3-methyladenine or bafilomycin A1, as well as Beclin-1 silencing, enhanced pterostilbene-triggered apoptotic responses.48 This finding indicates that autophagy may function as a stress-adaptive survival response in this model, helping tumor cells buffer pterostilbene-induced injury rather than directly mediating cell death. Autophagy should not be described as a uniformly pro-death mechanism in pterostilbene-treated cancer cells; its biological meaning depends on tumor type, treatment intensity, and whether autophagy acts as a protective adaptation or as part of a cell-death program.
In a hepatocellular carcinoma (HCC) model involving a choline-deficient, L-amino acid-defined diet-induced rat model and HepG2 cells, dietary pterostilbene attenuated HCC development and modulated metabolism-related gene programs in association with AMPK-dependent epigenetic regulation, further supporting the relevance of tumor metabolic wiring in pterostilbene-associated anticancer effects.44
Metabolic and inflammatory cell-death mechanisms provide another layer of cancer-specific evidence beyond mitochondrial or endoplasmic reticulum (ER)-stress-associated apoptosis. In EMT6 and 4T1 breast cancer cells, pterostilbene was reported to suppress glycolytic activity and induce pyroptosis through a pyruvate kinase M2 (PKM2)/caspase-8/gasdermin C (GSDMC) signaling axis. This study is relevant to the redox–metabolic framework because PKM2 links glycolytic regulation to pyroptotic cell death, suggesting that pterostilbene-related tumor-cell injury may depend on the metabolic wiring of specific breast cancer models rather than on apoptosis alone.46 A related mouse breast cancer study further showed dose-dependent pyroptosis in EMT6 and 4T1 cells after pterostilbene exposure and identified a caspase-3/gasdermin E (GSDME)-dependent mechanism.86 Mechanistically, 40 μM pterostilbene for 24 h increased caspase-3 and GSDME-related pyroptosis markers, whereas GSDME silencing and the caspase-3 inhibitor Z-DEVD-FMK reduced pterostilbene-induced pyroptotic features, supporting a functional role for the caspase-3/GSDME axis. In EMT6 breast tumor-bearing mice, pterostilbene suppressed tumor growth in a dose-dependent manner and was associated with immune-system-related transcriptomic enrichment, increased antitumor immune-cell proportions, and reduced immunosuppressive populations in peripheral blood, spleen, and tumors.86
Evidence Limitations and Context-Specific Gaps
Cancer models provide some of the clearest examples of dose- and context-dependent pterostilbene effects, but several important gaps remain. The transition from sub-cytotoxic modulation to overt tumor-cell injury has not been systematically defined across tumor types. In non-small-cell lung cancer (NSCLC) cells, low concentrations of pterostilbene affected migratory and adhesive behaviors without markedly reducing viability, whereas higher concentrations induced ROS generation, mitochondrial depolarization, ER-stress signaling, apoptosis, and tumor growth inhibition.47 More recent NSCLC evidence further suggests that pterostilbene-induced redox perturbation may extend to tumor-immunity-related mechanisms. Kang et al reported ROS-associated DNA damage, stimulator of interferon genes (STING)/TANK-binding kinase 1 (TBK1)/interferon regulatory factor 3 (IRF3) activation, and enhanced antitumor CD8+ T-cell responses after pterostilbene treatment, suggesting a possible link between tumor-cell stress and immune microenvironment remodeling; this evidence remains model-specific and requires further validation before being generalized across cancer types.45 Similarly, diffuse large B-cell lymphoma (DLBCL) and breast cancer studies support dose-related mitochondrial stress and apoptotic signaling.26,27 These findings do not establish a general concentration threshold at which pterostilbene shifts from signal modulation to cytotoxic stress. This distinction is particularly important in cancer, where moderate ROS signaling may support malignant adaptation, while excessive redox stress may promote tumor-cell death.
In vitro cytotoxicity also does not fully predict in vivo antitumor activity. The pancreatic cancer and melanoma study showed that pterostilbene-related tumor suppression was linked to disruption of glucocorticoid receptor- and NRF2-dependent antioxidant-defense regulation, rather than being explained only by direct cytotoxicity in cultured tumor cells.20 This suggests that host factors, tumor antioxidant-buffering capacity, endocrine or immune context, and microenvironmental stress may influence whether pterostilbene produces measurable antitumor effects. Studies reporting no direct cytotoxicity, sub-cytotoxic effects, or context-dependent activity should not be treated as weak or irrelevant evidence; instead, they help define the boundary conditions under which pterostilbene may or may not be effective.
Most cancer studies still identify pathway-associated changes rather than direct molecular targets. Evidence involving C/EBP homologous protein (CHOP) knockdown, Beclin-1 silencing, caspase inhibition, GSDME silencing, or ER-stress enhancement provides stronger causal support than studies based only on ROS, Bax/Bcl-2, caspase cleavage, microtubule-associated protein 1 light chain 3 (LC3), NF-κB, Ki67, terminal deoxynucleotidyl transferase dUTP Nick-End labeling (TUNEL), or cytokine readouts.46–48 Even these intervention-based studies do not necessarily prove direct target engagement by pterostilbene. More informative studies would combine dose–response and time-course designs with genetic rescue, pathway-specific inhibition, target-engagement assays, and comparison across cancer cells with different basal ROS levels, NRF2/GSH status, mitochondrial dependency, ER-stress sensitivity, glycolytic activity, and immune microenvironmental features. Parallel testing in non-malignant cells and long-term low-dose exposure models will also be necessary to clarify whether pterostilbene disrupts tumor adaptation, has limited effect, or in some contexts risks reinforcing stress tolerance.
Translational Challenges and Opportunities
On the basis of the disease-specific evidence reviewed above, current evidence for pterostilbene remains largely preclinical and heterogeneous across pathological contexts. Although these studies repeatedly report redox-, inflammatory-, metabolic-, and stress-related changes, they do not yet establish validated targets, defined dose windows, or disease-specific therapeutic efficacy.
Most mechanistic evidence remains marker-based or pathway-level rather than target-validated. Marker changes in Nrf2, NF-κB, AMPK, SIRT1, inflammatory mediators, apoptosis-related proteins, or metabolic indices describe pathway-associated responses but do not establish primary target engagement.14,15 Loss-of-function approaches, including Nrf2 knockdown/silencing, SIRT1 depletion, and Beclin-1 silencing, provide stronger support for pathway involvement than marker-only studies.16,17,25,48 Affinity-based chemoproteomics has identified syntaxin/soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE)-complex-related proteins as potential pterostilbene interactors in pancreatic β-cell insulin secretion models.87 In OA-related chondrocytes, recent cellular thermal shift assay (CETSA)-based evidence suggests p53 engagement in the context of pterostilbene-associated autophagy activation.57 These findings remain disease- or cell-context specific and do not define a universal molecular target across oxidative stress-related diseases. Future work should combine pathway perturbation, rescue designs, and target-engagement approaches to distinguish causal pathway involvement or direct molecular interactions from secondary responses to reduced injury.
The effective dose window of pterostilbene remains poorly defined, partly because dose effects are usually assessed with static endpoint markers rather than dynamic redox readouts. The same exposure may have different implications in chronic degenerative or metabolic disorders, acute I/R injury, and redox-adapted tumors, where therapeutic goals range from mild redox–inflammatory modulation to disruption of tumor stress-adaptive thresholds. Commonly used endpoints, including ROS, MDA, SOD, GSH, cytokines, apoptosis-related proteins, and metabolic indices, mainly describe selected injury states and do not fully capture dynamic redox adaptation, transient signaling, or the transition from adaptive to injurious oxidative responses.1–4,66 Study comparability is further limited by differences in controls, sample size, blinding or randomization where applicable, intervention timing, endpoint selection, and mechanistic validation. The apparent consistency of benefit should also be interpreted cautiously, because weak or null findings are less likely to be emphasized in the literature. Negative findings in some metabolic models show that pterostilbene does not produce uniform benefits across all pathological settings.43 More informative studies should define dose ranges and intervention windows more explicitly, using time-course designs and standardized redox–inflammatory readouts rather than relying only on single endpoint measurements.
Current evidence remains insufficient to define pterostilbene as a stand-alone antioxidant therapy for oxidative stress-related diseases. Its relatively favorable pharmacokinetic profile and preclinical activity justify continued investigation, but these features should not be equated with therapeutic efficacy or disease-specific applicability. In selected chronic non-malignant conditions, pterostilbene may be better explored as an adjunctive modulator of redox–inflammatory or redox–metabolic networks than as an acute ROS-eliminating intervention. In I/R injury and cancer, potential application is even more context-dependent and will require clearer definition of intervention timing, dose thresholds, tissue context, tumor redox dependency, and safety boundaries. These considerations should guide future studies aimed at determining whether pterostilbene has disease-specific translational value as a redox-modulating compound.
Conclusion
The evidence reviewed in this article indicates that pterostilbene is best understood not as a compound whose value rests on antioxidant activity alone, but as a modulator of redox-sensitive stress responses whose effects depend on pathological context. Across degenerative, ischemic, metabolic, and malignant settings, the same categories of readouts—ROS-related indices, inflammatory mediators, antioxidant-defense pathways, mitochondria-associated stress markers, and cell-death signals—do not necessarily reflect the same biological process. In chronic degenerative or metabolic models, they generally point to partial modulation of sustained redox–inflammatory or redox–metabolic imbalance; in I/R injury, to attenuation of reperfusion-associated injury amplification; and in cancer, to disruption of tumor-cell stress tolerance rather than conventional cytoprotection. Pterostilbene should therefore not be treated as a uniformly protective antioxidant, but as a context-dependent redox modulator whose relevance is determined by disease-specific redox state, therapeutic aim, and biological threshold.
The main theoretical contribution of this review is to make this context dependence explicit by repositioning pterostilbene from a general antioxidant candidate to a modulator of redox-sensitive stress responses. This distinction provides a clearer basis for interpreting heterogeneous preclinical findings. Similar changes in ROS-related indices, Nrf2/NF-κB signaling, mitochondria-associated markers, or cell-death proteins may indicate partial modulation of sustained redox–inflammatory or redox–metabolic imbalance in chronic degenerative and metabolic models, attenuation of reperfusion-associated injury amplification in I/R injury, or disruption of redox-adapted tumor-cell stress tolerance in cancer. Differences among studies may therefore reflect variations in basal redox state, tissue compartment, disease stage, dose exposure, and therapeutic aim, rather than simply stronger or weaker antioxidant activity. This framework helps prevent overgeneralization from positive cell and animal data and shifts future research toward identifying the conditions under which pterostilbene is beneficial, neutral, or stress-enhancing.
Future work should move beyond the accumulation of additional positive disease-model data and focus on defining the biological boundaries of pterostilbene action. Its translational value will depend on whether specific redox states, cell populations, disease stages, dose windows, intervention timing, and therapeutic goals can be matched to measurable and reproducible effects. Such work will help determine whether pterostilbene can be evaluated as a context-specific redox-modulating strategy rather than as a general antioxidant candidate.
Author Contributions
Yuyao Zhang: Conceptualization, investigation, data curation, writing – original draft, and writing – review & editing. Yingying Yu: Investigation, data curation, writing – original draft, and writing – review & editing. Zhengrong Xiong: Resources, supervision, and writing – review & editing. Wenying Bin: Data curation, investigation, and writing – review & editing. Yuxuan Yao: Investigation, data curation, and writing – review & editing. Huan Liu: Conceptualization, resources, supervision, project administration, and writing – review & editing. All authors made a significant contribution to the work reported, whether in the conception, literature investigation, data curation, interpretation, drafting, revision, or critical review of the manuscript; approved the final version to be published; agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Key Research and Development Project of Sichuan Science and Technology Plan Project (2024YFFK0135) and the Fujian Provincial Natural Science Foundation of China (2024J011450).
Disclosure
The authors have declared that no competing interest exists.
References
1. Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biol. 2015;4:180–20. doi:10.1016/j.redox.2015.01.002
2. Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006;8(9–10):1865–1879. doi:10.1089/ars.2006.8.1865
3. Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24(10):R453–62. doi:10.1016/j.cub.2014.03.034
4. Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. 2020;21(7):363–383. doi:10.1038/s41580-020-0230-3
5. Davies KJ. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life. 2000;50(4–5):279–289. doi:10.1080/713803728
6. Kapetanovic IM, Muzzio M, Huang Z, Thompson TN, McCormick DL. Pharmacokinetics, oral bioavailability, and metabolic profile of resveratrol and its dimethylether analog, pterostilbene, in rats. Cancer Chemother Pharmacol. 2011;68(3):593–601. doi:10.1007/s00280-010-1525-4
7. Liu Y, You Y, Lu J, Chen X, Yang Z. Recent advances in synthesis, bioactivity, and pharmacokinetics of pterostilbene, an important analog of resveratrol. Molecules. 2020;25(21). doi:10.3390/molecules25215166
8. Liu P, Tang W, Xiang K, Li G. Pterostilbene in the treatment of inflammatory and oncological diseases. Front Pharmacol. 2023;14:1323377. doi:10.3389/fphar.2023.1323377
9. Jiang S, Li H, Zhang L, et al. Generic Diagramming Platform (GDP): a comprehensive database of high-quality biomedical graphics. Nucleic Acids Res. 2025;53(D1):D1670–d1676. doi:10.1093/nar/gkae973
10. Nagarajan S, Mohandas S, Ganesan K, Xu B, Ramkumar KM. New insights into dietary pterostilbene: sources, metabolism, and health promotion effects. Molecules. 2022;27(19). doi:10.3390/molecules27196316
11. Acharya JD, Ghaskadbi SS. Protective effect of Pterostilbene against free radical mediated oxidative damage. BMC Complement Altern Med. 2013;13:238. doi:10.1186/1472-6882-13-238
12. Yan M, Zhao Y, Feng S, Zheng J, Diao M, Zhang T. Hydroxyl group-induced enhancement of antioxidant activity of resveratrol over pterostilbene by binding to lactoferrin. Food Chem. 2024;441:138356. doi:10.1016/j.foodchem.2024.138356
13. Saw CL, Guo Y, Yang AY, et al. The berry constituents quercetin, kaempferol, and pterostilbene synergistically attenuate reactive oxygen species: involvement of the Nrf2-ARE signaling pathway. Food Chem Toxicol. 2014;72:303–311. doi:10.1016/j.fct.2014.07.038
14. Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. 2014;39(4):199–218. doi:10.1016/j.tibs.2014.02.002
15. Cuadrado A, Rojo AI, Wells G, et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat Rev Drug Discov. 2019;18(4):295–317. doi:10.1038/s41573-018-0008-x
16. Zhou J, Ci X, Ma X, et al. Pterostilbene activates the Nrf2-dependent antioxidant response to ameliorate arsenic-induced intracellular damage and apoptosis in human keratinocytes. Front Pharmacol. 2019;10:497. doi:10.3389/fphar.2019.00497
17. Xue EX, Lin JP, Zhang Y, et al. Pterostilbene inhibits inflammation and ROS production in chondrocytes by activating Nrf2 pathway. Oncotarget. 2017;8(26):41988–42000. doi:10.18632/oncotarget.16716
18. Kosuru R, Kandula V, Rai U, Prakash S, Xia Z, Singh S. Pterostilbene decreases cardiac oxidative stress and inflammation via activation of AMPK/Nrf2/HO-1 pathway in fructose-fed diabetic rats. Cardiovasc Drugs Ther. 2018;32(2):147–163. doi:10.1007/s10557-018-6780-3
19. Elango B, Dornadula S, Paulmurugan R, Ramkumar KM. Pterostilbene ameliorates streptozotocin-induced diabetes through enhancing antioxidant signaling pathways mediated by Nrf2. Chem Res Toxicol. 2016;29(1):47–57. doi:10.1021/acs.chemrestox.5b00378
20. Benlloch M, Obrador E, Valles SL, et al. Pterostilbene decreases the antioxidant defenses of aggressive cancer cells in vivo: a physiological glucocorticoids- and Nrf2-dependent mechanism. Antioxid Redox Signal. 2016;24(17):974–990. doi:10.1089/ars.2015.6437
21. Liu H, Wu X, Luo J, et al. Pterostilbene attenuates astrocytic inflammation and neuronal oxidative injury after ischemia-reperfusion by inhibiting NF-κB phosphorylation. Front Immunol. 2019;10:2408. doi:10.3389/fimmu.2019.02408
22. Shi L, Liu Q, Tang JH, Wen JJ, Li C. Protective effects of pterostilbene on ulcerative colitis in rats via suppressing NF-κB pathway and activating PPAR-γ. Eur J Inflammation. 2019;17. doi:10.1177/2058739219840152
23. Cheng Y, Di S, Fan C, et al. SIRT1 activation by pterostilbene attenuates the skeletal muscle oxidative stress injury and mitochondrial dysfunction induced by ischemia reperfusion injury. Apoptosis. 2016;21(8):905–916. doi:10.1007/s10495-016-1258-x
24. Yang Y, Wang J, Li Y, et al. HO-1 signaling activation by pterostilbene treatment attenuates mitochondrial oxidative damage induced by cerebral ischemia reperfusion injury. Mol Neurobiol. 2016;53(4):2339–2353. doi:10.1007/s12035-015-9194-2
25. Chen Y, Zhang H, Ji S, et al. Resveratrol and its derivative pterostilbene attenuate oxidative stress-induced intestinal injury by improving mitochondrial redox homeostasis and function via SIRT1 signaling. Free Radic Biol Med. 2021;177:1–14. doi:10.1016/j.freeradbiomed.2021.10.011
26. Kong Y, Chen G, Xu Z, et al. Pterostilbene induces apoptosis and cell cycle arrest in diffuse large B-cell lymphoma cells. Sci Rep. 2016;6:37417. doi:10.1038/srep37417
27. Moon D, McCormack D, McDonald D, McFadden D. Pterostilbene induces mitochondrially derived apoptosis in breast cancer cells in vitro. J Surg Res. 2013;180(2):208–215. doi:10.1016/j.jss.2012.04.027
28. Hou Y, Xie G, Miao F, et al. Pterostilbene attenuates lipopolysaccharide-induced learning and memory impairment possibly via inhibiting microglia activation and protecting neuronal injury in mice. Prog Neuropsychopharmacol Biol Psychiatry. 2014;54:92–102. doi:10.1016/j.pnpbp.2014.03.015
29. Fu Z, Yang J, Wei Y, Li J. Effects of piceatannol and pterostilbene against β-amyloid-induced apoptosis on the PI3K/Akt/Bad signaling pathway in PC12 cells. Food Funct. 2016;7(2):1014–1023. doi:10.1039/c5fo01124h
30. Li Q, Li X, Tian B, Chen L. Protective effect of pterostilbene in a streptozotocin-induced mouse model of Alzheimer’s disease by targeting monoamine oxidase B. J Appl Toxicol. 2022;42(11):1777–1786. doi:10.1002/jat.4355
31. Xu J, Liu J, Li Q, et al. Pterostilbene participates in TLR4- mediated inflammatory response and autophagy-dependent Aβ(1-42) endocytosis in Alzheimer’s disease. Phytomedicine. 2023;119:155011. doi:10.1016/j.phymed.2023.155011
32. Chang J, Rimando A, Pallas M, et al. Low-dose pterostilbene, but not resveratrol, is a potent neuromodulator in aging and Alzheimer’s disease. Neurobiol Aging. 2012;33(9):2062–2071. doi:10.1016/j.neurobiolaging.2011.08.015
33. La Spina M, Sansevero G, Biasutto L, et al. Pterostilbene improves cognitive performance in aged rats: an in vivo study. Cell Physiol Biochem. 2019;52(2):232–239. doi:10.33594/000000017
34. Lv M, Liu K, Fu S, Li Z, Yu X. Pterostilbene attenuates the inflammatory reaction induced by ischemia/reperfusion in rat heart. Mol Med Rep. 2015;11(1):724–728. doi:10.3892/mmr.2014.2719
35. Yu Z, Wang S, Zhang X, Li Y, Zhao Q, Liu T. Pterostilbene protects against myocardial ischemia/reperfusion injury via suppressing oxidative/nitrative stress and inflammatory response. Int Immunopharmacol. 2017;43:7–15. doi:10.1016/j.intimp.2016.11.018
36. Wu M, Lu S, Zhong J, Huang K, Zhang S. Protective effects of pterostilbene against myocardial ischemia/reperfusion injury in rats. Inflammation. 2017;40(2):578–588. doi:10.1007/s10753-016-0504-2
37. Yang ZH, Liu YJ, Ban WK, et al. Pterostilbene alleviated cerebral ischemia/reperfusion-induced blood-brain barrier dysfunction via inhibiting early endothelial cytoskeleton reorganization and late basement membrane degradation. Food Funct. 2023;14(18):8291–8308. doi:10.1039/d3fo02639f
38. Yan W, Ren D, Feng X, et al. Neuroprotective and anti-inflammatory effect of pterostilbene against cerebral ischemia/reperfusion injury via suppression of COX-2. Front Pharmacol. 2021;12:770329. doi:10.3389/fphar.2021.770329
39. Sun Y, Xu Y, Wang G-N. Pterostilbene prevents intestinal ischemia reperfusion injury in Wistar rats via modulation of antioxidant defense and inflammation. Trop J Pharm Res. 2015;14(8). doi:10.4314/tjpr.v14i8.9
40. Pelles-Taskó B, Szekeres R, Takács B, et al. From nature to treatment: the impact of pterostilbene on mitigating retinal ischemia-reperfusion damage by reducing oxidative stress, inflammation, and apoptosis. Life. 2024;14(9). doi:10.3390/life14091148
41. Aguirre L, Palacios-Ortega S, Fernández-Quintela A, Hijona E, Bujanda L, Portillo MP. Pterostilbene reduces liver steatosis and modifies hepatic fatty acid profile in obese rats. Nutrients. 2019;11(5). doi:10.3390/nu11050961
42. Gómez-Zorita S, González-Arceo M, Trepiana J, et al. Comparative effects of pterostilbene and its parent compound resveratrol on oxidative stress and inflammation in steatohepatitis induced by high-fat high-fructose feeding. Antioxidants (Basel). 2020;9(11). doi:10.3390/antiox9111042
43. Damgaard MV, Jepsen SL, Ashcroft SP, Holst JJ, Treebak JT. Pterostilbene fails to rescue insulin secretion and sensitivity in multiple murine models of diabetes. Nutrients. 2022;14(18). doi:10.3390/nu14183741
44. Boycott C, Beetch M, Lubecka-Gajewska K, et al. AMPK-dependent epigenetic regulation of metabolism mediates the anti-cancer action of pterostilbene in hepatocellular carcinoma. Mol Nutr Food Res. 2025;69(23):e70217. doi:10.1002/mnfr.70217
45. Kang LP, Xie H, Huang HJ, et al. Pterostilbene inhibits non-small cell lung cancer progression by activating the STING pathway and enhancing antitumor immune response. Front Immunol. 2025;16:1622284. doi:10.3389/fimmu.2025.1622284
46. Pan T, Peng L, Dong J, Li L. Pterostilbene induces pyroptosis in breast cancer cells through pyruvate Kinase 2/Caspase-8/Gasdermin C signaling pathway. Int J Mol Sci. 2024;25(19). doi:10.3390/ijms251910509
47. Ma Z, Yang Y, Di S, et al. Pterostilbene exerts anticancer activity on non-small-cell lung cancer via activating endoplasmic reticulum stress. Sci Rep. 2017;7(1):8091. doi:10.1038/s41598-017-08547-0
48. Hsieh MJ, Lin CW, Yang SF, et al. A combination of pterostilbene with autophagy inhibitors exerts efficient apoptotic characteristics in both chemosensitive and chemoresistant lung cancer cells. Toxicol Sci. 2017;156(2):549. doi:10.1093/toxsci/kfx050
49. Brody LT. Knee osteoarthritis: clinical connections to articular cartilage structure and function. Phys Ther Sport. 2015;16(4):301–316. doi:10.1016/j.ptsp.2014.12.001
50. Loeser RF. Aging and osteoarthritis: the role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthritis Cartilage. 2009;17(8):971–979. doi:10.1016/j.joca.2009.03.002
51. Bolduc JA, Collins JA, Loeser RF. Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic Biol Med. 2019;132:73–82. doi:10.1016/j.freeradbiomed.2018.08.038
52. Lepetsos P, Papavassiliou AG. ROS/oxidative stress signaling in osteoarthritis. Biochim Biophys Acta. 2016;1862(4):576–591. doi:10.1016/j.bbadis.2016.01.003
53. Ansari MY, Ahmad N, Haqqi TM. Oxidative stress and inflammation in osteoarthritis pathogenesis: role of polyphenols. Biomed Pharmacother. 2020;129:110452. doi:10.1016/j.biopha.2020.110452
54. Milner PI, Wilkins RJ, Gibson JS. The role of mitochondrial reactive oxygen species in pH regulation in articular chondrocytes. Osteoarthritis Cartilage. 2007;15(7):735–742. doi:10.1016/j.joca.2007.01.008
55. Wang Y, Zhao H, Jia S, et al. Senomorphic agent pterostilbene ameliorates osteoarthritis through the PI3K/AKT/NF-κB axis: an in vitro and in vivo study. Am J Transl Res. 2022;14(8):5243–5262.
56. Lee YC, Chang YT, Cheng YH, et al. Pterostilbene protects against osteoarthritis through NLRP3 inflammasome inactivation and improves gut microbiota as evidenced by in vivo and in vitro studies. J Agric Food Chem. 2024;72(16):9150–9163. doi:10.1021/acs.jafc.3c09749
57. Wu J, Zhang C, Qin Y, Zhu L, Liu L, Xu F. Pterostilbene attenuates osteoarthritis progression through p53-dependent autophagy activation: evidence from network analysis and experimental validation. Front Pharmacol. 2026;17:1686555. doi:10.3389/fphar.2026.1686555
58. Trushina E, McMurray CT. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience. 2007;145(4):1233–1248. doi:10.1016/j.neuroscience.2006.10.056
59. Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov. 2004;3(3). doi:10.1038/nrd1330
60. Tönnies E, Trushina E. Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J Alzheimers Dis. 2017;57(4):1105–1121. doi:10.3233/jad-161088
61. Michalska P, León R. When it comes to an end: oxidative stress crosstalk with protein aggregation and neuroinflammation induce neurodegeneration. Antioxidants (Basel). 2020;9(8). doi:10.3390/antiox9080740
62. Joseph JA, Fisher DR, Cheng V, Rimando AM, Shukitt-Hale B. Cellular and behavioral effects of stilbene resveratrol analogues: implications for reducing the deleterious effects of aging. J Agric Food Chem. 2008;56(22):10544–10551. doi:10.1021/jf802279h
63. Bakshi V, Palsa D, Begum N, Kommidi J, Singh K, Yellu M. Neuroprotective effect of pterostilbene on ketamine induced schizophrenia in mice. Int J Appl Pharm Sci Res. 2016;1(03). doi:10.21477/ijapsr.v1i3.11336
64. Granger DN, Kvietys PR. Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol. 2015;6:524–551. doi:10.1016/j.redox.2015.08.020
65. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. 2012;298:229–317. doi:10.1016/b978-0-12-394309-5.00006-7
66. Forman HJ, Ursini F, Maiorino M. An overview of mechanisms of redox signaling. J Mol Cell Cardiol. 2014;73:2–9. doi:10.1016/j.yjmcc.2014.01.018
67. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357(11). doi:10.1056/NEJMra071667
68. Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21(1):2–14. doi:10.1097/00004647-200101000-00002
69. Fisher-Wellman KH, Neufer PD. Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol Metab. 2012;23(3):142–153. doi:10.1016/j.tem.2011.12.008
70. Rolo AP, Palmeira CM. Diabetes and mitochondrial function: role of hyperglycemia and oxidative stress. Toxicol Appl Pharmacol. 2006;212(2):167–178. doi:10.1016/j.taap.2006.01.003
71. Newsholme P, Cruzat V, Arfuso F, Keane K. Nutrient regulation of insulin secretion and action. J Endocrinol. 2014;221(3):R105–20. doi:10.1530/joe-13-0616
72. Anderson EJ, Lustig ME, Boyle KE, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009;119(3):573–581. doi:10.1172/jci37048
73. Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867. doi:10.1038/nature05485
74. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116(7):1793–1801. doi:10.1172/jci29069
75. Ristow M, Zarse K, Oberbach A, et al. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci U S A. 2009;106(21):8665–8670. doi:10.1073/pnas.0903485106
76. de Morais JMB, Cruz EMS, da Rosa CVD, et al. Pterostilbene influences glycemia and lipidemia and enhances antioxidant status in the liver of rats that consumed sucrose solution. Life Sci. 2021;269:119048. doi:10.1016/j.lfs.2021.119048
77. Nagao K, Jinnouchi T, Kai S, Yanagita T. Pterostilbene, a dimethylated analog of resveratrol, promotes energy metabolism in obese rats. J Nutr Biochem. 2017;43:151–155. doi:10.1016/j.jnutbio.2017.02.009
78. Gómez-Zorita S, Fernández-Quintela A, Lasa A, Aguirre L, Rimando AM, Portillo MP. Pterostilbene, a dimethyl ether derivative of resveratrol, reduces fat accumulation in rats fed an obesogenic diet. J Agric Food Chem. 2014;62(33):8371–8378. doi:10.1021/jf501318b
79. La Spina M, Galletta E, Azzolini M, et al. Browning effects of a chronic pterostilbene supplementation in mice fed a high-fat diet. Int J Mol Sci. 2019;20(21). doi:10.3390/ijms20215377
80. Zhang ZT, Deng SM, Chen C, et al. Pterostilbene could alleviate diabetic cognitive impairment by suppressing TLR4/NF-кB pathway through microbiota-gut-brain axis. Phytother Res. 2023;37(8):3522–3542. doi:10.1002/ptr.7827
81. Zhang Y, Ren S, Ji Y, Liang Y. Pterostilbene ameliorates nephropathy injury in streptozotocin-induced diabetic rats. Pharmacology. 2019;104(1–2):71–80. doi:10.1159/000500293
82. Ding RR, Huang GY, Zhang YJ, et al. Pterostilbene ameliorates renal damage in diabetic rats by suppressing hyperglycemia with inhibition of inflammatory and fibrotic responses. Biomed Environ Sci. 2021;34(12):1015–1019. doi:10.3967/bes2021.137
83. Hayes JD, Dinkova-Kostova AT, Tew KD. Oxidative stress in cancer. Cancer Cell. 2020;38(2):167–197. doi:10.1016/j.ccell.2020.06.001
84. de la Vega M R, Chapman E, Zhang DD. NRF2 and the hallmarks of cancer. Cancer Cell. 2018;34(1):21–43. doi:10.1016/j.ccell.2018.03.022
85. Bhardwaj V, He J. Reactive oxygen species, metabolic plasticity, and drug resistance in cancer. Int J Mol Sci. 2020;21(10). doi:10.3390/ijms21103412
86. Huang X, Yang W, Hu X, Peng L, Dong J, Li L. Pterostilbene induces pyroptosis in mouse breast cancer cells through the caspase-3/Gasdermin E signaling pathway and reshapes the tumor immune microenvironment. Transl Oncol. 2025;59:102454. doi:10.1016/j.tranon.2025.102454
87. Cassiano C, Eletto D, Tosco A, Riccio R, Monti MC, Casapullo A. Determining the effect of pterostilbene on insulin secretion using chemoproteomics. Molecules. 2020;25(12). doi:10.3390/molecules25122885
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Loss of Potassium and Chloride Transport Changes PM-Induced Epithelial Dysfunction
Jaworowska S, Maliszewska-Olejniczak K, Łukasiak A, Hoser J, Zając M, Bednarczyk P
Journal of Inflammation Research 2026, 19:564139
Published Date: 4 March 2026
Protective Effect of Idebenone Against UVB-Induced Photoaging in HaCaT Cells
Feng L, Tian L, Guo J, Zhang L, Wang P, Bu X
Drug Design, Development and Therapy 2026, 20:575342
Published Date: 19 March 2026