Back to Journals » Neuropsychiatric Disease and Treatment » Volume 13
Psychosis in parkinsonism: an unorthodox approach
Authors Onofrj M, Carrozzino D, D'Amico A, Di Giacomo R, Delli Pizzi S, Thomas A, Onofrj V, Taylor JP, Bonanni L
Received 30 June 2016
Accepted for publication 3 December 2016
Published 16 May 2017 Volume 2017:13 Pages 1313—1330
DOI https://doi.org/10.2147/NDT.S116116
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Marco Onofrj,1,2 Danilo Carrozzino,3,4 Aurelio D’Amico,1,2 Roberta Di Giacomo,1,2 Stefano Delli Pizzi,1 Astrid Thomas,1,2 Valeria Onofrj,5 John-Paul Taylor,6 Laura Bonanni1,2
1Department of Neuroscience Imaging and Clinical Sciences, University “G. d’Annunzio” of Chieti-Pescara, 2CE.S.I. University Foundation, 3Department of Psychological, Health, and Territorial Sciences, University “G. d’Annunzio” of Chieti-Pescara, Chieti, Italy; 4Psychiatric Research Unit, Psychiatric Centre North Zealand, Copenhagen University Hospital, Hillerød, Denmark; 5Department of Bioimaging, University Cattolica del Sacro Cuore, Rome, Italy; 6Institute of Neuroscience, Campus for Ageing and Vitality Newcastle University Newcastle upon Tyne, Newcastle upon Tyne, UK
Abstract: Psychosis in Parkinson’s disease (PD) is currently considered as the occurrence of hallucinations and delusions. The historical meaning of the term psychosis was, however, broader, encompassing a disorganization of both consciousness and personality, including behavior abnormalities, such as impulsive overactivity and catatonia, in complete definitions by the International Classification of Diseases-10 (ICD-10) and the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5). Our review is aimed at reminding that complex psychotic symptoms, including impulsive overactivity and somatoform disorders (the last being a recent controversial entity in PD), were carefully described in postencephalitic parkinsonism (PEP), many decades before dopaminergic treatment era, and are now described in other parkinsonisms than PD. Eminent neuropsychiatrists of the past century speculated that studying psychosis in PEP might highlight its mechanisms in other conditions. Yet, functional assessments were unavailable at the time. Therefore, the second part of our article reviews the studies of neural correlates of psychosis in parkinsonisms, by taking into account both theories on the narrative functions of the default mode network (DMN) and hypotheses on DMN modulation.
Keywords: postencephalitic parkinsonism, Parkinson’s disease, psychosis, default mode network, atypical parkinsonism
Introduction
Psychotic symptoms have been reported to occur in ~60% of patients with Parkinson’s disease (PD)1 and are even more common in patients with dementia with Lewy bodies (DLB); for example, visual hallucinations (VHs) and delusions occur in 60% to 70% of DLB patients, respectively.2,3
PD, PD with dementia (PDD), and DLB are facets of a disorder whose central pathology is the deposition of alpha synuclein aggregates (known as Lewy bodies) in subcortical and cortical brain structures.2
Psychosis in PD, PDD, and DLB is currently rated on the basis of different neuropsychological test scores (eg, mainly by using the Neuropsychiatric Inventory),4 which typically do not include in-depth phenomenological interviews. The result of this approach is that psychosis in PD and parkinsonisms is currently titrated, perhaps simplistically, as the occurrence of just two core symptoms, hallucinations and delusions.1 In contrast, the broader definition provided by the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) for psychotic disorder describes psychosis within the classification item schizophrenia spectrum,5 as being characterized by abnormalities in one or more of the following five domains: delusions, hallucinations, but also disorganized thinking, grossly disorganized or abnormal motor behavior (including catatonia), and negative symptoms. The International Classification of Diseases-10 (ICD-10)6 uses the term “psychotic” more widely than DSM-55 to include abnormal perceptions, thought contents, and behaviors, without a constraint to the schizophrenia spectrum.
From a historical perspective, the term psychosis has received a number of different definitions, ranging from a broad “impairment that grossly interferes with the capacity to meet ordinary demands of life”,7 “gross impairment in reality testing”8 to the narrow restriction of prominent hallucinations in any modality.1,9
Examining this in more detail, earlier French and German classifications used the term psychosis in such a broad sense that it was considered “sufficient to define any mental disorder”.10 For example, Einheit-psychosis indicated a severe disorganization of (self) consciousness or personality, while Névrose-Neurosis indicated lesser disorganizations,11–13 same as previously reported in ICD-9.6
This review article aimed at challenging the current restrictive definition of psychosis in Parkinsonisms, and we posit that a broader psychosis definition might be better suited for capturing the diversity of behavioral symptoms observed in these disorders. To show that psychotic and behavioral symptoms of parkinsonism, which are considered relatively recently identified clinical entities in the orthodox approach14–18 to these diseases, are in fact not a novelty, our review opens by reporting an analysis of mental disorders historically described in epidemic encephalitis (Von Economo disease) postencephalitic parkinsonism (PEP).10
We will then compare behavioral and psychiatric disorders observed in PD and other parkinsonisms with the categorization items of DSM-5,5 highlighting the incongruity between observed disorders and categorization items.
Finally, we review potential anatomical circuits and substrates whose derangement may explain the observed disorders.
Within the neural correlate section, we repropose the idea of prominent neuropsychiatrists of the past century10 that understanding the possible mechanisms of psychosis in parkinsonisms might shed light on the mechanisms of psychosis in other clinical conditions.
Methods
Research on PEP was performed through examination of historical reports present in our institutional and personal libraries and the library of the Catholic University, Medical School of Rome.
Medline research was performed using the following search terms: postencephalitic parkinsonism/Economo encephalopathy/dementia with Lewy bodies/Parkinson’s disease/parkinsonisms and psychosis/behavior/nonmotor symptoms.
Neural correlates of psychotic symptoms and of impulse control disorders in Parkinsonism were searched on PubMed and Scopus databases without any type of date limitations by using the following keywords acting as search terms: Parkinson, parkinsonism, nonmotor symptoms, psychosis, fMRI, MRI, imaging studies, PET, and SPECT.
The mental disorders observed in PEP
The lethargic, epidemic, Von Economo-Cruchet encephalitis appeared in Europe as a pandemia in 1916–1917 and spread to the rest of the world in 1918–1920. The infective vector, also known as Spanish flu, was lately identified as H1N1 virus.19 Neuropathological lesions due to this disease affect predominantly the mesodiencephalic regions. The accompanying characteristic neurological symptoms are diplopia, oculogyric crisis, parkinsonism and dystonia, disorders of vigilance and consciousness in the acute phase (lethargic oneiroid confusion), and complex mental disorders in the chronic phase.20–23 The chronic parkinsonism induced by lethargic encephalitis was termed PEP. A recent revision study24 suggests that PEP is a controversial entity, as the direct relationship of PEP symptoms with epidemic encephalitis appears weak in some of the described cases, and it may be argued that some of the cases were affected by PD or other parkinsonisms.24 This argument does not impinge on the purpose of our review, which is focused to evidence that mental disorders, presented in recent articles and consensus as a novelty, were in fact described in detail in PEP patients.
According to early French authors, the epidemic encephalitis showed, for the first time in history, that a viral infection could induce a range of psychoses (as outlined in the following paragraphs), which were, until then, considered as endogenous or constitutional and interpreted according to psychodynamic theories, which were innovative and prominent at the time.10,25 Observations of psychosis in PEP, including compulsive and conversion symptoms, according to the broad nosographic definition10–13 prompted some authors to define the epidemic encephalitis with a, nowadays inacceptable, term of “maladie expérimentale” (experimental disease) quoted in detail by Ey et al,10 and by other authors,20,25,26 as offering an insight into the possible mechanisms of mental diseases.
The most frequently described mental disorders observed in the chronic phase of epidemic encephalitis are listed in Box 1.
 | Box 1 Mental disorders described in the chronic phase of epidemic encephalitis |
As pointed out by ICD-106 and DSM-55 classifications, hysteria is today considered as an inappropriate or impolite term, and the use of substitutive terms, ie, somatic symptom and related disorders, is recommended. Yet, the term hysteria is still used in several studies,27–29 despite attempts to develop equivalent terms that describe symptoms not fully explained by organic anatomical and physiological changes. The area remains unclear and controversial, with “functional” or “psychogenic” definitions being the focus of a number of editorial debates.30–33
Notably in PEP, “pseudoneurological”, psychogenic symptoms, including sensory, motor, gait disorders, and dystonic postures, which should be now classified as conversion disorders according to DSM-IV-TR34 or somatic symptoms according to DSM-55 or ICD-106 definitions, were described by several authors.10,25,35–37 They also clearly reported patients who were histrionic and displayed theatrical behaviors and suggestibility, being suggestibility the epitome of pithiatism in hysteria.38 For example, Van Bogaert25 pointed out that the symptoms appearing in chronic epidemic encephalitic evidenced that “hysteroid symptoms previously attributed to pithiatism or simulation” could be determined by “undebatable organic disorders”.10,25 Thus, somatic symptoms in the context of PEP appear to have an organic basis that undermines their prior purported functional etiology.
Beyond the psychomotor automatisms consisting of stereotyped behaviors, echopraxia, and echolalia, the attention of early authors was mainly focused on the occurrence of impulse control and sexual behavior disorders. In this regard, hypersexuality, paraphilias, and changes in sexual orientation were observed, although undoubtedly some of these descriptions were driven by the social norms and morality of the time.10,20–23,25,35–37,39 Among disorder characterized by loss of impulse control, kleptomania, mostly defined grossly as “stealing”, was discussed by several authors,10,35–44 as well as erratic actions (eg, wandering) and restlessness.45
These symptoms were the focus of several clinical observations with catatonia classified, mostly with hypertonic-rigid symptoms, inside the typical negativism of psychosis.10 The chronic hallucinatory psychosis was mostly attributed to oneirism (dream enactment) or oneiroid status, and this reflected the different categorization concepts that were topical at the time.10,21,22
In contrast with the recent attitude toward VHs in PD,46,47 the description of hallucinations was mostly focused on delusional hallucinations or paraphrenia, ie, complex hallucinations with intrinsic logic and narrative, while there was little interest in simple hallucinations, which were called eidolic hallucinations or pareidolias, and attributed to disordered perception and “compatible with reason, according to ancient authors”,10 ie, Aristoteles (De Anima), as quoted by Ey et al.10
Box 1 also reports viscosity, which was considered by the different authors among the mood and impulsivity disorders.10,20,35–37,45 Many studies described this disordered behavior, and Menger45 defined it “inopportunité assidue” (assiduous inopportunity)45 by describing it as the relentless questioning, fixation on topics and clinginess. This aberrant behavior was lately considered by Waxman and Geschwind48 as one of the psychiatric manifestations of temporal lobe epilepsy.48 Viscosity has never been listed among the mental disorders in parkinsonism, although every day clinical practice seems to suggest its presence.
Psychotic symptoms in other parkinsonisms
It is quite difficult to define properly the “other parkinsonisms” as the revolution provoked by genetic studies has progressively eroded the concept of idiopathic PD and prompted a revitalization of the term parkinsonian syndrome.49
It could be relevant to know precisely what is the prevalence of the different mental disorders in the different genetically determined parkinsonisms, but data are insufficient or incongruent at present.
In Synuclein triplication impulse control disorders, hallucinations and delusions have been reported as phenotypic features.50 Psychiatric disorders are also reported in patients affected by glucocerebrosidase A (GBA) mutations, but the description of these are not detailed51,52 although in our local cohorts, we have observed impulse control disorders, hallucinations, and delusions in 9 of 12 genetically ascertained GBA mutations (25%).53 For leucine-rich repeated kinase 2 (LRRK2) mutation, it is known that the presenting phenotype may consist of bipolar disorder, dementia, and schizotypic personality disorder rather than of parkinsonism, but we do not know clearly about the overlap of mental disorders and parkinsonism.54 A recent research report showed high prevalence of nonmotor disorders in LRRK2, as evidenced by standardized rating scales.55
Hallucinations and impulse control disorders are not described in multiple system atrophy (MSA) and progressive supranuclear palsy (PSP), according to cohort study,56 which concludes that the presence of hallucinations should be considered as an exclusion criterion for MSA diagnosis, although it is notable that a single case57 and a cohort study have also described possible hallucinations in MSA.58 Another study showed that hallucinations are reported when the apparent MSA phenotype is due to atypical–atypical variants59 arising from genetic or mitochondrial diseases.
In contrast, in frontotemporal lobar degenerations (FTLDs) with parkinsonism, the hallucinatory, delusional, and impulse control disorders symptoms are clearly described.60 In particular, hallucinations and delusions are prominent in chromosome 9 open reading frame 72 (C9ORF72) mutation, which is the most common cause of FTLD phenotype occurrence.61,62
Impulsive–compulsive disorders consisting of, for example, recycling metal and plastic, stomping ants, solitaire, crosswords and puzzles playing, weeding, collecting coins, dish washing, searching family photographs, cataloguing daily events, and skipping pavement cracks have all been reported in C9ORF72 and transactive response DNA-binding protein 43 (TDP-43) and fused in sarcoma (FUS) genetic mutations.62–66
Mental disorders in PD
Table 1 lists the mental disorders described in PD, in comparison with DSM-5-based classifications of similar disorders.
 | Table 1 Mental disorders described in PD, in comparison with DSM-5-based classifications of similar disorders |
From the comparisons of the two lists, it is evident that disorders observed in parkinsonisms do not match DSM-5 classifications.5
The incongruence is evident, for example, for impulse control disorders of PD (PD-ICD), that are not listed in DSM-5,5 despite the fact that they appeared in the previous DSM-IV-TR revision, which included only intermittent explosive disorder, kleptomania, pyromania, pathological gambling, and trichotillomania.34 The same disorders are listed in ICD-10.6
In DSM-5,5 some impulse control disorders now appear under the obsessive–compulsive disorder categorization, yet the specifier, reported in Table 1, is that the key factor is obsession, as persistent ideas, thoughts, and impulses that are experienced as intrusive and inappropriate and cause marked anxiety or distress.5 Compulsions are repetitive behaviors, the goal of which is to prevent anxiety or distress.5
The recent history of PD-ICD is initially characterized by case reports describing those disorders as secondary to dopamine agonist administration or to L-DOPA treatment.41,42,67,68 Later on, systematic studies evidenced a presence of this disorder in 5%–8% of untreated PD patients,42–44 with a prevalence only slightly higher than the prevalence obtained in control population. However, the increased prevalence and occurrence of PD-ICD due to dopamine agonist treatment appears today undebatable.41–44,67,68 What remains undescribed is whether the obsession present in PD or the impulses are irresistibly enacted upon without the conscious perception of anxiety as a driver.
The presence of obsession would frame PD-ICD in the categories of DSM-55 and ICD-10,6 but its absence would assimilate these disorders to the impulsivity described in PEP where a clouding of conscience and surrendering to the impulse drive was clearly described by the neuropsychiatrists of the time.10,20–23,25,35–37,39
Furthermore, most of the PD-ICD presentations described in parkinsonism do not appear in DSM-5 psychiatric classifications, and hypersexuality, gambling, wandering, and repetitive activities are framed by DSM-55 under different categories, than obsessive–compulsive disorder.
Hallucinations in PD have been extensively studied.46,69–75 However, it is important to remind the reader that hallucinations in PD appear in two formats, one is early, consisting of misperceptions/illusions (also termed pareidolias), or simple hallucinations or moving hallucinations, which are recognized by the patient as unreal and that disappear when the patient focuses his attention.73 The other type is instead consistent of complex visual experiences, which are often not recognized as hallucinations and are associated with dementia.46,69–80 Cohort systematic studies evidenced that the two types of hallucinations cannot be considered as separate clinical entities, in opposition to the concept of benign vs malignant hallucinations, as the occurrence of the first type is mostly a harbinger to occurrence of the second type.76 These second “complex hallucinations” match with the description of hallucinatory psychosis (or paraphrenia) described earlier in PEP.
Delusions in parkinsonism are less frequently described than PD-ICD, and the prevalence of VHs greatly varies between 2% and 16% of patients, according to different studies,1,77–84 probably due to different disease durations in the described cohorts or because the reported data were not controlled for pharmacological management of psychosis. In this regard, a recent cohort study reported that delusions of sin/guilt or grandiosity were relatively frequent (35% of delusional patient), as well as delusions of reference (30%), while delusions of jealousy were less frequent (13%).1 Other studies instead reported that delusion of jealousy was more common than the other types.81–83 Delusion of infestation was also recently reported as a novelty,77,84 although it was described in prior studies.85–87 Somatic delusions in PEP were described as hysteria–pithiatism, while in PD were discussed in comparison with somatoform disorders, with the purpose to highlight the psychotic characteristics of symptoms that did not conform to any known anatomical pathways and physiological mechanisms.85–87
In a PD cohort study, somatic delusions accounted for just a 4% of cases,1 this evidencing furthermore that different prevalences emerge from different cohort or case series.
Somatoform disorders alternatively termed as functional or psychogenic symptoms were only recently described in L-DOPA-responsive parkinsonism,85 in a comparative study including MSA, PSP, frontotemporal lobe degeneration (FTLD), Alzheimer’s disease (AD), and DLB.85 This introductory study was followed by several confirmatory reports,31,88–97 concluding that, after all, “functional overlap in Parkinson disease can commonly be seen in clinical practice”.31
A careful search of historical literature, however, showed that somatic disorder (ie, somatization) or hysteria was also described in PD at the dawn of the L-DOPA era.98–101
Neuropsychological scale-based studies have demonstrated a high incidence of somatization and alexithymia in PD, reaching a prevalence of 40%,89,102 whereas in our cohort study of somatoform disorders, the effort to classify, confidently, different motor, and nonmotor symptoms, as functional, and specifically differentiating them from motor and nonmotor symptoms in PD, resulted in a much lower incidence in PD of 7%–9% and 15%–19% in DLB.85
Somatoform disorders were not observed in FTLD, AD, and MSA and appeared in 1 of 17 patients with PSP.85 Absence of somatoform disorders in MSA is intriguing, as MSA patients do present with high anxiety (trait and state) scores, similar to PD patients without hallucinations or delusions.85,103 This finding may suggest that somatization is not an output of anxiety. The absence of VH and somatoform disorders in MSA may suggest, inversely, that a common mechanism underlying the occurrence of both VH and somatic symptom disorders could not be present in MSA.
The presence of somatoform disorders in PD and DLB is predictive of cognitive decline,85 which is correlated with hallucinations.76 In contrast, the absence of these disorders in AD, FTLD, and PSP was puzzling, and in counterdistinction to what is observed in PD and DLB, it has been suggested that in these disorders, severe cognitive decline is incompatible with the production of functional symptoms. Clearly, different patterns of neurodegeneration influence the profile of symptoms, and this needs to be taken into account.
In studies following the initial cohort analysis,86,87 the psychotic traits accompanying functional disorders in parkinsonism were underlined by evidencing that some of the bizarre symptoms needed a proper classification as somatic delusions, rather than as somatoform disorders.86,87
Finally, we must underline a relevant innovation introduced by DSM-5,5 which includes a helpful new diagnostic categorization element, that being, “somatic disorders in a coexisting medical disease”.5 This is a new and a much needed classification item, where the functional–psychogenic overlaps in PD could be usefully framed and categorized.
PD-ICD can be extremely disruptive of previous personality traits and behavioral aspects; impressive examples can be found not only in scientific literature but also in websites reporting personal experiences and results of legal actions. The disruption of personality and the resistance to reality testing constitute the reasons to introduce ICD6 in a discussion of psychosis in parkinsonisms. Similar to delusions and complex hallucinations, the disruptive effects of ICD on personality and on failures on insight and reality testing suggest that some ICDs might be more easily framed into the notion of psychosis, as used by pre-DSM psychiatrists.10,20–23
The unorthodox question derived from the first two sections would be thus summarized as: Is it acceptable to frame the complex mental and behavioral disorders of PD into categories resulting from the reduction of DSM guidelines into simplified questionnaires?
We would argue that the older, etymologically, and historically accepted meaning of psychosis could be better suited to encompass the complexity of mental disorders in parkinsonisms.
At least its use would remind us of the long and rich intellectual history of discussions that have helped frame the current study of mental disorders, when neurology and psychiatry were structuring their identities, and would underline the differences between the mental disorders observed in PD and those analyzed by DSM systems, which, useful to remind, quote Parkinson only in the Dementia chapter.5
Neural substrate of psychosis in parkinsonisms
The reward system, including orbitofrontal (OF) projection to striatothalamic circuit, was the main focus of studies on PD-ICD.104–106 We will not describe these aspects in the present report. We mentioned those disorders just to underline the altered mentation that accompanies these symptoms. In brief, studies on ICD posit the centrality of the dopaminergic reward system, of projections to striato-pallidal structures, and of the effect of long-term potentiation on the aforementioned circuits.107,108
Primary idiotypic and associative visual cortex and temporal lobes have been the target of studies on VHs. These studies however considered pareidolias, ie, illusions or misperceptions, as their main target.109,110
In “Conclusion” section we will discuss, instead, only the possible substrates of complex hallucinations, in particular, those endowed with narrative characteristics, ie, those in which there is a contextual meaning of the perceived hallucination, or secondary delusions, and of somatization symptoms of which, in previous studies,85–87 the delusional characterization was evidenced.
Even though the complex, kinematic hallucinations are, apparently, less frequent than illusions and simple hallucinations, the intrinsic narrative linked to these hallucinations is an intriguing phenomenon.46,73–75 For hallucinations associated with narratives, there appears to be an intrinsic logic that structures the hallucinatory perceptions, and one which is often overlapping with dream themes.47,69,70,111,112 For example, in several studies, funerals, fighting demons, theatrical performances of Lilliputian personages, and dancing figures are clearly described as hallucinations and with secondary delusions.47,112–114
We postulate that the structure providing this narrative is probably the default mode network (DMN). Figure 1 represents DMN regions activation during resting state.
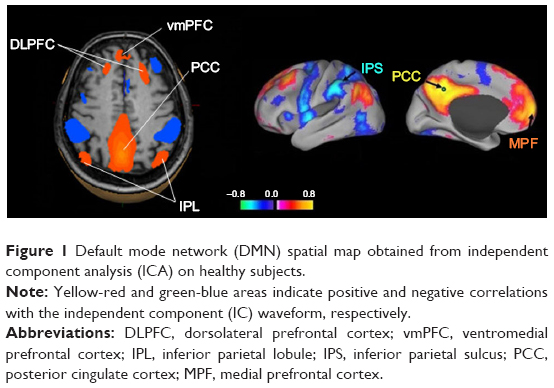 | Figure 1 Default mode network (DMN) spatial map obtained from independent component analysis (ICA) on healthy subjects. |
DMN has emerged as an evolutionary novel functional network in human brains as shown in studies comparing with lower order primates.115 It consists of a set of cortical regions including medial prefrontal, posterior cingulate, lateral inferior parietal cortex, and medial temporal lobes. As posterior cingulate cortex (PCC) is anatomically framed inside the limbic structures and because of its possible functions, DMN is considered part of the limbic system, as a system driving memory, emotions, and behavior.116 Figure 2 shows a schematic representation of the limbic areas.
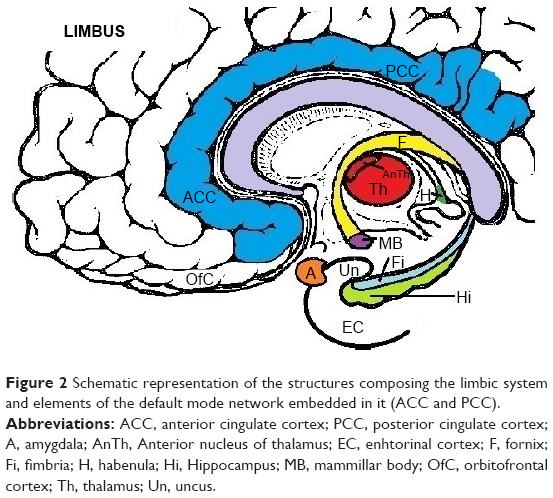 | Figure 2 Schematic representation of the structures composing the limbic system and elements of the default mode network embedded in it (ACC and PCC). |
Imaging studies found brain regions with highly correlated activity during rest and deactivation during task, which were named DMN, and showed that DMN has roles in imagery production and incorporation of self-referential information into consciousness.117,118
The original DMN role in self-referential information production has been reconsidered in theoretical studies that seek to explain the occurrence of hallucinations by suggesting that DMN could introduce self-referential information into misperceptions.109,110
From a broader network perspective, the DMN is believed to act in balance with the attentional control networks, ie, the dorsal attentional network (DAN), consisting of dorsolateral prefrontal cortex (DLPFC), posterior parietal cortex, and corpus striatum, as well as the ventral attentional network (VAN), consisting of the basolateral amygdala, lateral and inferior prefrontal cortex, temporoparietal junction, and ventral striatum.110 Cortical and subcortical structures now considered as pertinent to the DAN and VAN networks were previously named as salience network or Petersen cingulo-opercular task control network119 (Table 2).
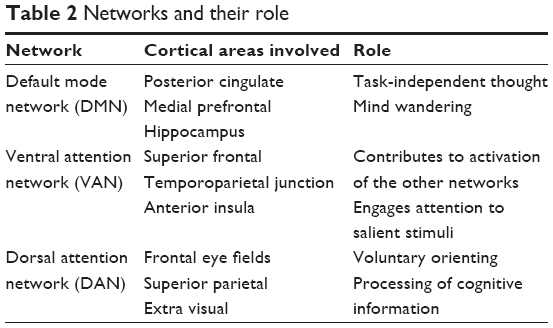 | Table 2 Networks and their role |
According to a recent hypothesis and theory paper, entry into an oneiric, consciousness clouding, by dreaming or transliminality state,120 depends on collapse of the normally highly organized activity within the DMN and decoupling between the DMN and other cerebral structures.121
This model suggests that the oneiric state is due to DMN decoupling and represents an entropic, and thus highly disordered, state of the brain as opposed to the tendency of entropy suppression, which typifies normal working consciousness with a constrained (ordered) quality and associated metacognitive factors, including reality testing and social awareness.121
In the highly entropic state, DMN functional connectivity is poised at a critical point, with connectivity motifs that form and fragment across time.121
This hypothesized model rehabilitates many Freudian metapsychology concepts122 and defines the DMN decoupling state as a primitive or primary state of consciousness, ie, the one that preceded the development of the modern, adult, linear, and normal working consciousness.121
Envisioning energy states, with representation of low entropy/high entropy states, can be assimilated to the Freudian metapsychology frames,122 depicting shifts of energy from conscious to unconscious activities; in particular, the DMN-driven entropic states may be analogous to the activities of subconscious structures, variably defined as “es” or “id” by Freudian psychoanalysts, who always depicted a preserved consciousness of the individual identity for this metapsychological structure, despite the lack of control by the “adult, linear normal waking consciousness” defined by the same authors as “ego”.121,122
Similar concepts were exposed in studies suggesting that the brain is in a metastable condition but can easily reenter into entropic states if required. In normal function, return to stable motifs can occur rapidly.123–126 The study by Carhart-Harris et al121 has the merit of pointing to DMN with clarity and of evidencing the similarities of possible DMN functions with Freudian metapsychology.122
The concluding parts of the present review focus on three hypotheses: role of DMN, possible driver of DMN, and mechanisms of somatic delusions.
Hypothesis 1
The first hypothesis of the present review suggests that DMN disinhibition is key mechanism for psychosis in parkinsonisms.
This hypothesis is also supported by data suggesting that DMN hyperactivity has been shown in other psychotic manifestations and in psychotic depression with suicidal behavior.127–132
The existence or predisposition for a DMN disinhibited entropic state in Parkinsonisms is difficult to evidence, as evidence would require a clear cut definition of what is a normal activity for DMN. Most of the studies involving DMN activity were actually exploiting the property of DMN, consisting of being active in resting state, without the requirement of performing any task, and thus investigated connectivity mostly as related to motor symptoms or motor phenotypes.133,134
Nevertheless, increased activity or connectivity of DMN, or preserved activity and connectivity compared with other nonpsychotic conditions, has been reported as C9orf72 mutation carriers62 in Lewy body dementia and135–138 in parkinsonism with hallucinations in comparison with other disorders without hallucinations.103,139,140
Several articles showed increased DMN connectivity or activity in PD,103,139,140 while the articles focused on motor symptoms reached different conclusions.133,134,141–146 It is notable that in PD without VH and in parkinsonisms without VH, similar to MSA, DMN activity and connectivity are significantly reduced.103
Increased activity is undoubtedly evidenced by the so-called “cingulate island sign”, now considered a diagnostic marker of DLB.147 The sign indicates preserved activity of the PCC, a main hub of DMN, as observed with PET,147 in the comparison between DLB and controls or other conditions characterized by cognitive decline.147 Figure 3 shows an example of intense activity of PCC in a DLB patient.
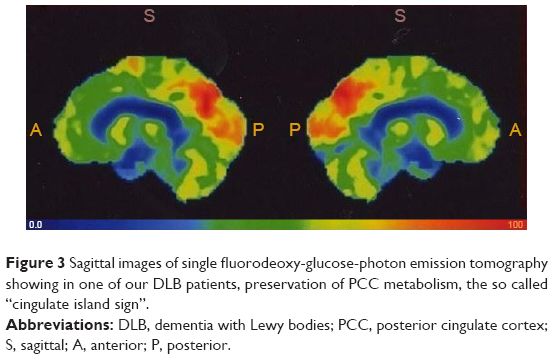 | Figure 3 Sagittal images of single fluorodeoxy-glucose-photon emission tomography showing in one of our DLB patients, preservation of PCC metabolism, the so called “cingulate island sign”. |
From task-based dynamical studies, altered DMN inhibition during a visual-attention task was described in DLB,138 and increased coupling between DMN and visual networks, during a cognitive task, has been described in PD with VH.140
In AD patients without VH, it is well established that DMN activity and connectivity are reduced.117,148–157
The reduction of DMN activity in AD has been related to a lack of compensatory mechanisms, which may depend on the greater cellular loss that occurs in this disease117 in posterior cortex, which is supposed to be one of the main targets of amyloid pathology (of note, a recognized AD variant consists specifically in prominent posterior cortical atrophy).158
Similarly, altered and reduced connectivity has been described in PSP,159 a disease without VH, also characterized by significant brain atrophy in posterior cortex.
In the study on MSA, decreased DMN activity, which was similar to reductions observed in AD, was observed, yet no cellular loss was evidenced as demonstrated by preservation of a normal cortical thickness.103 This finding suggests that functional modulatory mechanisms rather than structural differences are relevant.
Table 3A and B shows the results of different studies on the activation/connectivity of DMN in different neurodegenerative diseases. Table 3A reports findings in AD patients; Table 3B reports findings in different parkinsonisms.
 | Table 3A Findings obtained in previous studies on DMN in resting state fMRI in the comparison between patient groups (AD) and healthy control group |
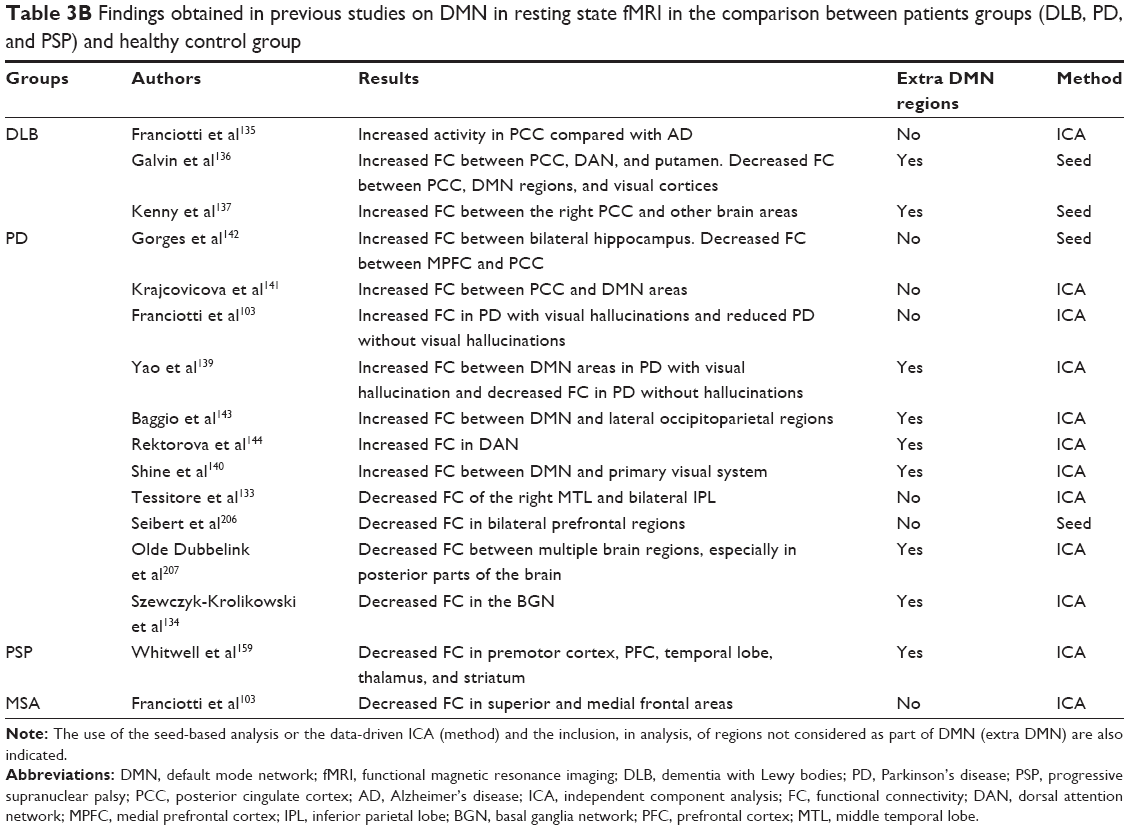 | Table 3B Findings obtained in previous studies on DMN in resting state fMRI in the comparison between patients groups (DLB, PD, and PSP) and healthy control group |
Therefore, the comparison between different diseases, with different etiologies, suggests that DMN enhancement or inhibition is not a disease-related process but rather is an aspect linked to the expression of clinical phenotypes.
This hypothesis expands the possible role of DMN, by considering that DMN enhancement may underlie the expression of VH (with extreme manifestations including psychosis and delusions), while DMN inhibition might be linked to a phenotype characterized by hyperarousal or increased anxiety (with extreme manifestations including, for example, akathysia).103,135 In conclusion, it might be hypothesized that a phenotype characterized by fluctuations in alertness and oniroid hallucinations and imagery might be linked to persistent DMN activity, while a phenotype characterized by increased arousal and severe anxiety (eg, Godot syndrome)160 might be linked to deficient DMN activity.135
This hypothesis is focused on early phenotypical aspects, as it is likely that in the late course of any dementia, psychosis could appear because of a collapse of the systems. In late course, psychosis may emerge because of widespread low energy states, in which modulations and malfunctions are set at lower connectivity and activity levels, with emergence of ontogenetically elementary reflexes (nurture, withdrawal). Thus, the final path for late dementia may converge to the same phenotype, but our hypothesis is that the starting point may be different.
Our hypothesis, driven by the DMN-entropia model,121 implies that more studies should be performed in different conditions, following the preliminary studies showing enhanced or disinhibited DMN activity/connectivity in parkinsonisms with hallucinations or delusions.
Relevant information should be gained from studies on DMN inhibition (task/rest comparisons) rather than from studies on DMN activation (like the simple resting state). Understanding dynamical changes during specific tasks and impact of DMN on other systems should be the target of future studies.
In a practical example, the evidence of impaired right hemisphere connections in DLB, as observed in one of our studies,135 might suggest a specific role of a right hemisphere dysfunction in the occurrence of fluctuating cognition, in accordance with several early and late hypotheses on anatomical correlates of attention. However, specific activation paradigms will need to be developed as only the evidence of abnormal DMN activities during rest, coupled with abnormal activations during task, can properly distinguish the role of different cortical areas, as shown by a recent report.138
The energetic frame in which DMN is embedded,121 with connection motifs forming and fragmenting through time, also suggests that the stability and variability through time of DMN connections need proper assessments, together with a validation of what is a normal DMN activity/connectivity and under which condition.
Further investigations should answer the following questions:
Does DMN power and connectivity represent a mere epiphenomenon or are the key drivers of the clinical phenotype?
Is DMN inhibition/lack of inhibition dependent on anatomical factors (like in AD) or on functional modulatory activities (like in MSA)?
Hypothesis 2
The role of thalamus
Could there be a driver of DMN lack of inhibition? A possible answer comes from studies in other diseases characterized by the occurrence of bizarre, rich in narrative, confabulations, or delusions.161–163 Bizarre confabulations are typically presented by patients affected by ruptures of aneurisms of the anterior communicating artery (AcoA syndrome),161,162 by patients with the Korsakoff syndrome,164–166 and by patients with ischemic lesions of the anteromedial thalamic nuclei.163,167–170
Figure 4 shows a reconstruction of thalamic nuclear lesions in patients with confabulations, and amnesia.
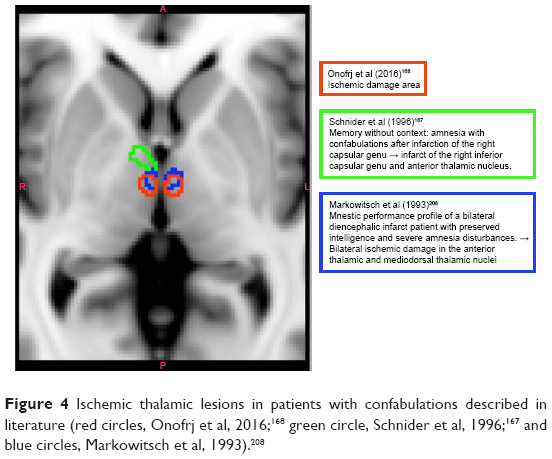 | Figure 4 Ischemic thalamic lesions in patients with confabulations described in literature (red circles, Onofrj et al, 2016;168 green circle, Schnider et al, 1996;167 and blue circles, Markowitsch et al, 1993).208 |
In patients with Korsakoff syndrome, metabolic degeneration of anteromedial thalamic nuclei induces severe inhibition of anterior cingulate cortices (ACCs) and probably OF cortex.164
In patients with ischemic lesions of anteromedial thalamic nuclei, projections to BA 32 and BA 25 areas of ACC are predominantly involved.168
In the AcoA syndrome, the OF and ACC, which are the targets of anterior thalamic radiations, are variably disrupted by aneurismal ruptures.161,162
These observations suggest that the anteromedial part of the thalamus or the cortical regions that are the target of thalamic projections from anteromedial nuclei have a relevant role in disinhibition of confabulatory narratives.
To further explain the mechanisms of bizarre confabulations in patients with ACC lesions, it has been hypothesized that ACC and OF may control reality monitoring and representation of ongoing reality.167
An alternative hypothesis could suggest that ACC inhibits PCC, which is the main hub of DMN functions and of DMN role in self-narrative production. Similarly, the ACC has phylogenetically developed in humans in parallel with frontal lobes expansion, in order to control PCC.
ACC could modulate the activity of PCC by prevalent inhibition. Lesions of anteromedial thalamic nuclei, by altering glutamatergic facilitatory projections to ACC, could inhibit ACC and consequently disinhibit PCC and DMN. The ensuing disinhibition could explain the occurrence of florid narratives.
In conclusion, in parkinsonisms, thalamic structures could indirectly drive the DMN disinhibition, as disruption of anterior and medial thalamic functions would affect excitatory projections to ACC and OF: suppression or depression of thalamic afference to ACC could inhibit ACC–OF and thus disinhibit PCC (Figure 5).
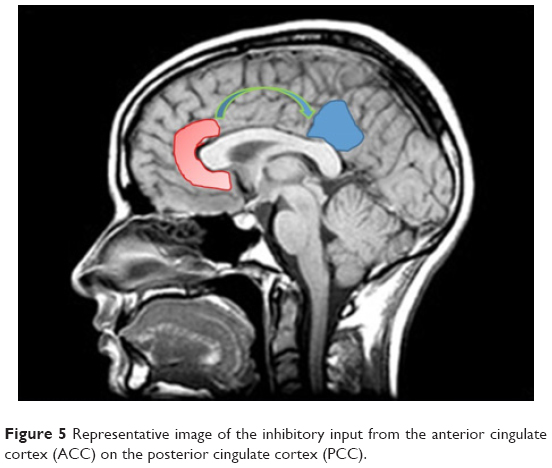 | Figure 5 Representative image of the inhibitory input from the anterior cingulate cortex (ACC) on the posterior cingulate cortex (PCC). |
Many studies have evidenced noradrenergic, dopaminergic, or cholinergic denervation in the thalamus of patients with PD and DLB.171–173 Prominent thalamic degeneration and denervation have also been observed in C9ORF72 FTD.60,63 Studies on DLB are ongoing, and although a hypothesized alteration of cholinergic thalamic interneurons has not been found,174 some recent reports have suggested a cholinergic deafferentation of the thalamus from pedunculopontine nuclei projections.175,176
DLB and PD patients present with specific symptoms characterized by clouding of consciousness (fluctuating cognition),2 sleep disturbances (mind–body dissociation), and VHs (within-mind dissociation),2 which have all being demonstrated to be associated with abnormalities of thalamic nuclei or thalamocortical connectivity.177,178
The EEG of DLB and PDD patients is characterized by appearance of fast theta at 5.6–7.8 Hz, also termed prealpha,179 activity during wakefulness, an activity substituting pseudocyclically the normal posterior alpha activity or appearing as a dominant posterior rhythm.
A possible pathophysiological explanation of the peculiar appearance of a dominant fast theta rhythm in DLB patients relies on the role of thalamic neurons, in the genesis of different oscillatory states.180 One of the most impressive network modulations in the brain is the one which generates the transition from wakefulness to sleep and vice versa. This event involves a high number of neurons and yet may take a very short time to occur as the state transition from tonic gamma band-based cognition to the sleep burst mode (characterized by global slow coherence). These two states are mutually exclusive each other under normal conditions.181,182 But there are situations, such as the state dissociation,183 or deafferentation syndromes, where a set of neurons in the thalamus displays low rhythmicity in an otherwise awake brain state.180
It has been recently demonstrated that even in normal conditions, extensive cortical territories remain activated for several minutes after the thalamic deactivation at sleep onset, a situation that may be propitious to the development of hypnagogic experiences so common during the wake–sleep transition.180
There is therefore a close relationship among specific thalamic neurons activity, their associated rhythms, and different states of consciousness.
In dissociative states, a dysrhythmic state in a portion of the thalamocortical system is trapped in spindle-like activity, whereas the rest of the system remains in the usual waking state.
A possible explanation is that such a dysrhythmic state is the consequence of a disfacilitation or deafferentation. Alternatively protracted inhibition, due to excessive inhibitory input from globus pallidus or the pedunculopontine nucleus (PPN) projection to the thalamic reticular nucleus via abnormal slow cortical oscillations may be a driver.181
The role of PPN in alertness and wakefulness has also been recently demonstrated in a PD patient who underwent deep brain stimulation of the PPN.175 When the stimulator was switched off, the patient experienced increased confusion and worsening of cognitive performances.175
Moreover, the presence of fast theta (ie, prealpha) rhythm is thought to be generated in the medial pulvinar nuclei,180,184 which are connected with the parieto-occipital cortex, which has been implicated in the genesis of specific clinical symptoms, such as VHs, and showing the presence of specific abnormal EEG rhythms.
The relevant thalamic nuclei for the genesis of VH should be medial pulvinar, the reticular relay, and anteromedial nuclei. Idiotypic proprioceptive thalamic nuclei (ventral-anterior [VA]; geniculate) could be unaffected in PD-DLB. A recent work showed that lateral geniculate nucleus (LGN), a visual relay center between the retina and visual cortex, possibly involved in the genesis of VH, is unaffected in DLB patients and therefore suggested that the early visual system is relatively spared in DLB, which implies that upstream visual structures may be largely responsible for the generation of hallucinatory percepts.185
Thalamic projections to the ACC include the anteromedial nuclei and medial pulvinar, and notably, a recent review of the pulvinar functions suggests that loss of thalamocortical connectivity via this nuclei may explain fluctuations in cognition177 and hallucinations in DLB.178
The first and the second hypotheses described so far are mostly focused on the explanation of complex hallucinations, as endowed with narrative elements, and delusions. DMN disinhibition, because of the loss of reality testing and insight, could however represent also a possible background for somatic delusions and for somatization manifestations of PD and DLB.
Hypothesis 3
Somatic delusions and the driver of psychogenic-functional abnormal motor output
In functional, psychogenic, or somatoform disorders, the motor manifestations should be accounted for. A recent model of these disorders, defined under the umbrella term of “hysteria” was a Bayesian model of hierarchical organization of perceptions-motor outputs.27
This model was developed from previous studies suggesting that the human brain acts as a Bayesian system where perceptions are integrated in a feedback/feedforward system predicting the outcome of perception. The theory of conditioned probability of Bayes was initially applied to explain memory functions, suggesting that memory may represent not just a passive collection of past events, but a demiurge of new perceptions. The active role of memory presupposes the existence of monitoring systems, the absence of which may lead to the formation of abnormal prediction of the outcome of perception and consequently to false perceptions. It was hypothesized that a complex system based on reality monitoring and confrontation of retrieved aspects of experience and actual perception may coexist as complementary aspects of the Bayesian model.27,186
Both these monitoring and confrontation systems can be conceptualized as rapid, automatic, and relatively impenetrable to reasoning, thus acting at a “preconscious” level. Both are directly represented and have immediate affinity with emotions and reward system but act with different timing. The monitoring system may, in fact, represent a system acting in a continuum, its function would be necessary and sufficient in order to place perceptions into the context of “reality”; this system may belong to the ACC.167,168 Confrontation systems may instead work as “double check” systems sufficient but not necessary for perception, and activity in regard to this may be attributed to the DLPFC.27 Both these systems provide the mechanism by which reality is perceived and disruption of both these mechanisms may result in reality break up.
The Bayesian model applied to hysteria hypothesizes, in human, the formation of an abnormal intermediate level of prediction (called prior in the model) related to a sensation or to a movement. This prior should be the induction of the hysteric motor output, which is reinforced as “attentional misdirection increases the precision of the abnormal intermediate level prior and drives perception and/or action consistent with it”.27
While attentional misdirection can be attributed to DMN decoupling, the ensuing motor output should be properly defined.
Our hypothesis is that the ensuing motor output is the complete or partial release of the archetypal “sham death” reflex (thanatosis). This hypothesis is in agreement with continuum theories linking hysteria and catatonia.28,29,85
We suggest that when the attentional misdirection, dependent on DMN decoupling and entry into a psychotic state, drives a motor output, this output should elicit the release of the thanatosis reflex, which would consist of limited manifestations (hysteria) or complete manifestations (catatonia) or intermediate conditions including the Ganser187 and the Cotard syndromes,85,188 once the psychotic state has found its narratives.
The common path driving this motor output could be located in the OF-habenular189 connections (Figure 6).
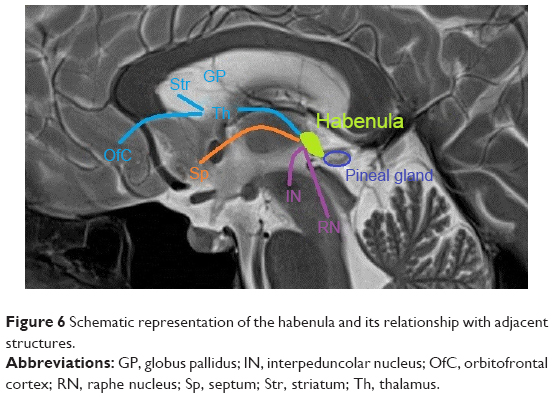 | Figure 6 Schematic representation of the habenula and its relationship with adjacent structures. |
Notably, the Habenula,189 which receives serotoninergic and dopaminergic projections, is altered in PD, in parallel with alterations of the basal OF pathway, its primary function is to suppress motor activity under stress conditions (thanatosis).190,191
Conclusion
The unorthodox approach of our review study of PD psychosis consists in the attempt to provide different perspectives than the epidemiological–statistical scale score-based dominant approach. In line with our conceptual and methodological point of view, previous research studies have highlighted the diagnostic inadequacy of many psychometric assessment instruments (eg, statistically based self-rating scales) conceived as an obstacle192–194 for detecting psychological symptoms in the clinical setting of neurology, clinical psychology,195 and psychiatry due to its lack of clinical validity196 and sensitivity to change, as well as because of its quest for homogeneous components (eg, Cronbach’s coefficient alpha).197 Our review shows that the diagnostic categories of mental disorders listed in DSM-55 and ICD-106 (both classification systems still significantly influenced by the psychometric principles)192–197 do not fully match the clinical findings in parkinsonisms.
Moreover, we criticize the clinically reductive definitions of the term psychosis in light of historical literature and conceptualization categories: altered adherence to reality checking, poor insight, and behavior monitoring is the underlying definition that emerges from our review. A comparison with descriptions of psychosis in PEP is provided to show that all the mental disorders recently described in PD as a clinical novelty are in fact not new and have been described in PEP.
PEP was considered by last century authors as a maladie expérimentale, which allowed an insight into the organicity of behaviors that were considered dependent on psychodynamics or personality structures.10,25
The psychosis appearing in PD and other parkinsonisms may provide a similar insight into mechanisms of psychosis, with the unprecedented advantage of having, today, imaging tools that, appropriately addressed, may help to reveal the underlying mechanisms.
In the discussion of psychosis, our attention is focused on complex, paraphrenic hallucination, those endowed with an intrinsic narrative, rather than to pareidolic illusions-misinterpretations.
We suggest that the probable mechanism underlying the production of complex narrative, untouched by reality checking, is disinhibition of the DMN.
We review the studies showing increased activity/connectivity of DMN in different parkinsonisms presenting with psychosis.
We review the theories of DMN decoupling effects, without hiding or dismissing the proposed conjectures aligning DMN as the set of ego (ACC) and id (PCC).121
An aberrant homunculus seated in DMN, driving the psychotic narratives, may emerge from our discussion as a Cartesian image; rather than concealing this obviously paradoxical representation, we suggest that it might represent a good start to facilitate further debates.
We speculate on the potential role of thalamic alterations and on the possible intrusion of dissociative status in driving the DMN disinhibition. Finally, the presence of alexithymia, psychogenic (functional) overlays, and catatonia in parkinsonisms may have a Bayesian basis and highlight the potential role of habenula in motor suppression.
Strengths and limitations
The major limitation of our study is that it reflects a personal opinion of the authors who were trained to attribute paramount importance to the patient interview and therefore to their thought content, rather than to rely solely on the statistical output of neuropsychological test scores. Although in neurology or psychiatry very few reports have focused their research study directly on clinical judgment analysis,197,198 in daily clinical practice, when making an initial clinical assessment of the patient, the experienced clinician is the key for performing a clinically valid medical evaluation.192–199 In the next step of the clinical assessment of the patient, a detailed analysis of the symptoms and other clinical signs needs to be integrated by using clinically valid indexes and measures.192–199 This scientifically based assessment method combining the experienced clinician as a test of clinical validity with the item response theory models200,201 is defined clinimetric approach.202–204
Thus, the review addresses peculiarly the narrative reported by patients and tries to identify the anatomical substrate for the narrative involved in hallucinations, delusions, and somatic complaints. With this approach, relevant information coming from the connectome theory-based studies103,133,139,140,205 might be overlooked, yet we impudently stated that our approach was unorthodox, leaving to the orthodox quantification the analysis of connectivity results, which we consider proper of a parallel, and complementary analytic approach.
The strength of our review, at least the one we would like to highlight, is that it addresses a functional role of DMN inside the limbic system, which we believe was not sufficiently described in the overwhelming number of connectivity studies.
By reviewing the findings of last century neuropsychiatrists, we also hope to have reached the result of reminding the reader of the concept that our current knowledge is contingent on the legacy of those we often forget.
Disclosure
The authors report no conflicts of interest in this work.
References
Factor SA, Scullin MK, Sollinger AB, et al. Cognitive correlates of hallucinations and delusions in Parkinson’s disease. J Neurol Sci. 2014;347(1–2):316–321. | ||
McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65(12):1863–1872. | ||
Aarsland D, Brønnick K, Ehrt U, et al. Neuropsychiatric symptoms in patients with Parkinson’s disease and dementia: frequency, profile and associated caregiver stress. J Neurol Neurosurg Psychiatry. 2007;78(1):36–42. | ||
Cummings JL, Mega M, Gray K, et al. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology. 1994;44(12):2308–2314. | ||
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5). 5th ed. Washington, DC: American Psychiatric Association; 2013. | ||
World Health Organization. International Statistical Classification of Disease and Related Health Problems, Tenth Revision (ICD-10). Geneva: WHO; 1992. | ||
Topor A, Andersson G, Denhov A, et al. Psychosis and poverty: coping with poverty and severe mental illness in everyday life. Psychosis. 2014;6(2):117–127. | ||
Gaebel W, Zielasek J. Future classification of psychotic disorders. Eur Arch Psychiatry Clin Neurosci. 2009;259(suppl 2):213. | ||
Flaum M, Schultz SK. The core symptoms of schizophrenia. Ann Med. 1996;28(6):525–531. | ||
Ey H, Bernard P, Brisset C. Manuel de Psychiatrie. Paris: Masson; 1960. | ||
Hoff P. Emil Kraepelin und die Psychiatrie als Klinische Wissenschaft. Ein Beitrag zum Selbstverstandnis Psychiatrischer Forschung. Berlin, Germany: Springer; 1994. | ||
Decker HS. The psychiatric works of Emil Kraepelin: a many-faceted story of modern medicine. J Hist Neurosci. 2004;13(3):248–276. | ||
Berrios GE, Hauser R. The early development of Kraepelin’s ideas on classification: a conceptual history. Psychol Med. 1988;18(4):813–821. | ||
Chang A, Fox SH. Psychosis in Parkinson’s disease: epidemiology, pathophysiology, and management. Drugs. 2016;76(11):1093. | ||
Marsh L, Williams JR, Rocco M, Grill S, Munro C, Dawson TM. Psychiatric comorbidities in patients with Parkinson disease and psychosis. Neurology. 2004;63(2):293–300. | ||
Mack J, Rabins P, Anderson K, et al. Prevalence of psychotic symptoms in a community-based Parkinson disease sample. Am J Geriatr Psychiatry. 2012;20(2):123–132. | ||
Lee AH, Weintraub D. Psychosis in Parkinson’s disease without dementia: common and comorbid with other non-motor symptoms. Mov Disord. 2012;27(7):858–863. | ||
Factor SA, Steenland NK, Higgins DS, et al. Disease-related and genetic correlates of psychotic symptoms in Parkinson’s disease. Mov Disord. 2011;26(12):2190–2195. | ||
Czermak M, Jean T. Von Economo-Cruchet lethargic encephalitis and its relation to HIV infection. Encephale. 1990;16(5):375–382. | ||
Pavan L, Torta R, Catalano M, et al. Disturbi mentali in relazione ad altre malattie. In: Cassano GB, Pancheri P, editors. Trattato Italiano di Psichiatria. Milano: Masson; 1999:1227–1287. | ||
Tanzi E, Lugaro E. Trattato delle malattie mentali. Milano: Società Editrice Libraria; 1923. | ||
Achard C. L’encéphalite léthargique. Paris: J. B. Baillière et fils; 1921. | ||
Van Bogaert L. Encéphalite léthargique. Hb. Spez. Path. Anat. Vol 13, Issue 2. Berlin: Springer; 1958:313–361. | ||
Vilensky JA, Gilman S, McCall S. Does the historical literature on encephalitis lethargica support a simple (direct) relationship with postencephalitic Parkinsonism? Mov Disord. 2010;25(9):1124–1130. | ||
Van Bogaert L. l’hystérie et les fonctions diencéphaliques (étude neurologique). Rapport de neurologie présenté lors du congress des médecins aliénistes et neurologists de France et des pays de langue française à Bruxelles. 1935. | ||
Lancereaux E. De la maladie expérimentale comparée à la maladie spontanée. Paris: Delahaye; 1872. | ||
Edwards MJ, Adams RA, Brown H, Pareés I, Friston KJ. A Bayesian account of ‘hysteria’. Brain. 2012;135:3495–3512. | ||
Shorter E. Hysteria and catatonia as motor disorders in historical context. Hist Psychiatry. 2006;17:461–468. | ||
Halligan W, Bass C, Marshall J. Contemporary Approaches to the Study of Hysteria: Clinical and Theoretical Perspectives. Oxford: Oxford University Press; 2001:171–183. | ||
Onofrj M. Functional or somatoform disorders in Parkinson’s disease? Mov Disord. 2013;28(13):1907. | ||
Parees I, Saifee TA, Edwards MJ. Functional motor or somatoform disorders in PD? Mov Disord. 2013;28:1907. | ||
Edwards MJ, Stone J, Lang AE. Functional/psychogenic movement disorders: do we know what they are? Mov Disord. 2014;29(13):1696–1697. | ||
Fahn S, Olanow CW. “Psychogenic movement disorders”: they are what they are. Mov Disord. 2014;29(7):1697–1698. | ||
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders-Text Revised (DSM-IV-TR). 4th ed. Washington, DC: American Psychiatric Association; 2000. | ||
Masquin H, Masquin L. Troubles mentaux au cours des encephalitis et des névraxites. Encyclopédie Médico-Chirurgicale. Paris: Edition Techniques; 1970:1–18. | ||
Bergouignan M, Loiseau P. Encéphalite épidémique: maladie d’Economo-Cruchet. Encyclopédie Medicale Sur Le Systéme Nerveux; 1964:1–12. | ||
Levy S. Post-encephalitic behavior disorder; a forgotten entity: a report of 100 cases. Am J Psychiatry. 1959;115(12):1062–1067. | ||
Babinski J. Hypnotism and hysteria. J Nerv Ment Dis. 1892;17(6):383–395. | ||
Robin G. Le perversions instinctives des encéphalites [Thèse de Paris]; 1923. | ||
Juqueller P, Vinchon J. Revue de psychiatrie et de psychologies experimental. L’Historie de la Kleptomanie. 1914:47–64. | ||
Calandrella D, Antonini A. Pathological gambling in Parkinson’s disease: disease related or drug related? Expert Rev Neurother. 2011;11(6):809–814. | ||
Voon V, Fernagut P-O, Wichens J, et al. Chronic dopaminergic stimulation in Parkinson’s disease: from dyskinesias to impulse control disorders. Lancet Neurol. 2009;8(12):1140–1149. | ||
Weintraub D. Impulse control disorders in Parkinson’s disease: prevalence and possible risk factors. Parkinsonism Relat Disord. 2009;15(suppl 3):S110–S113. | ||
Colosimo C, Morgante L, Antonini A, et al. Non-motor symptoms in atypical and secondary parkinsonism: the PRIAMO study. J Neurol. 2009;257(1):5–14. | ||
Menger P. Les perversions morales postencéphalitiques. Paris: Maloine; 1929. | ||
Onofrj M, Taylor JP, Monaco D, et al. Visual hallucinations in PD and Lewy body dementias: old and new hypotheses. Behav Neurol. 2013;27(4):479–493. | ||
Onofrj M, Thomas A, Martinotti G, et al. The clinical associations of visual hallucinations. In: Collerton D, Mosimann UP, Perry E, editors. The Neuroscience of Visual Hallucinations. Vol 5. Oxford: WILEY Blackwell; 2015:91–117. | ||
Waxman SG, Geschwind N. The interictal behavior syndrome in temporal lobe epilepsy. Arch Gen Psychiatry. 1975;32:1580–1586. | ||
Albin RL, Dauer WT. Parkinson syndrome. Heterogeneity of etiology; heterogeneity of pathogenesis? Neurology. 2012;79(3):202–203. | ||
Olgiati S, Thomas A, Quadri M, et al. Early-onset parkinsonism caused by alpha-synuclein gene triplication: clinical and genetic findings in a novel family. Parkinsonism Relat Disord. 2015;21(8):981–986. | ||
Mata IF, Leverenz JB, Weintraub D, et al. GBA Variants are associated with a distinct pattern of cognitive deficits in Parkinson’s disease. Mov Disord. 2016;31(1):95–102. | ||
Brockmann K, Srulijes K, Hauser AK, et al. GBA-associated PD presents with nonmotor characteristics. Neurology. 2011;77(3):276–280. | ||
Bonanni L, Onofrj M, Valente EM, et al. Recurrent and fatal akinetic crisis in genetic-mitochondrial parkinsonisms. Eur J Neurol. 2014;21(9):1242–1246. | ||
Shanker V, Groves M, Heiman G, et al. Mood and cognition in leucine-rich repeat kinase 2 G2019S Parkinson’s disease. Mov Disord. 2011;26(10):1875–1880. | ||
Marras C, Alcalay RN, Caspell-Garcia C, et al. Motor and nonmotor heterogeneity of LRRK2-related and idiopathic Parkinson’s disease. Mov Disord. 2016;31(8):1192–1202. | ||
Bertram K, Williams DR. Visual hallucinations in the differential diagnosis of parkinsonism. J Neurol Neurosurg Psychiatry. 2012;83:448–452. | ||
Spaccavento S, Del Prete M, Loverre A, Craca A, Nardulli R. Multiple system atrophy with early cognitive deficits: a case report. Neurocase. 2013;19(6):613–622. | ||
Koga S, Naoya A, Uitti RJ, et al. When DLB, PD, and PSP masquerade as MSA. An autopsy study of 134 patients. Neurology. 2015;85:404–412. | ||
Stamelou M, Quinn NP, Bhatia KP. “Atypical” atypical parkinsonism: new genetic conditions presenting with features of progressive supranuclear palsy, corticobasal degeneration, or multiple system atrophy-a diagnostic guide. Mov Disord. 2013;28(9):1184–1199. | ||
Sha SJ, Takada LT, Rankin KP, et al. Frontotemporal dementia due to C9ORF72 mutations: clinical and imaging features. Neurology. 2012;79(10):1002–1011. | ||
Le Ber I, Camuzat A, Guillot-Noel L, et al. C9ORF72 repeat expansions in the frontotemporal dementias spectrum of diseases: a flow-chart for genetic testing. J Alzheimers Dis. 2013;34(2):485–499. | ||
Lee SE, Khazenzon AM, Trujillo AJ, et al. Altered network connectivity in frontotemporal dementia with C9orf72 hexanucleotide repeat expansion. Brain. 2014;137:3047–3060. | ||
Piguet O, Hornberger M, Mioshi E, et al. Behavioral-variant frontotemporal dementia: diagnosis, clinical staging, and management. Lancet Neurol. 2011;10(2):162–172. | ||
Dormann D, Rodde R, Edbauer D, et al. ALS – associated fused in sarcoma (FUS) mutations disrupt Transportin – mediated nuclear import. EMBO J. 2010;29:2841–2857. | ||
Snowden JS, Rollinson S, Thompson JC, et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72. Brain. 2012;135:693–708. | ||
Simòn-Sànchez J, Dopper EG, Cohn-Hokke PE, et al. The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain. 2012;135(pt 3):723–735. | ||
Beaulieu-Boire I, Lang AE. Behavioral effects of levodopa. Mov Disord. 2015;30(1):90–102. | ||
Samuel M, Rodriguez-Oroz M, Antonini A, et al. Management of impulse control disorders in Parkinson’s disease: controversies and future approaches. Mov Disord. 2015;30(2):150–159. | ||
Fénelon G, Goetz CG, Karenberg A. Hallucinations in Parkinson disease in the prelevodopa era. Neurology. 2006;66(1):93–98. | ||
Pappert EJ, Goetz CG, Niederman FG, et al. Hallucinations, sleep fragmentation, and altered dream phenomena in Parkinson’s disease. Mov Disord. 1999;14(1):117–121. | ||
Arnulf I, Bonnet AM, Damier P, et al. Hallucinations, REM sleep, and Parkinson’s disease: a medical hypothesis. Neurology. 2000;55(2):281–288. | ||
Comella CL, Tanner CM, Ristanovic RK. Polysomnographic sleep measures in Parkinson’s disease patients with treatment induced hallucinations. Ann Neurol. 1993;34:710–714. | ||
Diederich NJ, Goetz CG, Stebbins GT. Repeated visual hallucinations in Parkinson’s disease as disturbed external/internal perceptions: focused review and a new integrative model. Mov Disord. 2005;20(2):130–140. | ||
Onofrj M, Thomas A, Bonanni L. New approaches to understanding hallucinations in Parkinson’s disease: phenomenology and possible origins. Expert Rev Neurother. 2007;7(12):1731–1750. | ||
Onofrj M, Bonanni L, Albani G, Mauro A, Bulla D, Thomas A. Visual hallucinations in Parkinson’s disease: clues to separate origins. J Neurol Sci. 2006;248(1–2):143–150. | ||
Goetz CG, Fan W, Leurgans S, et al. The malignant course of ‘benign hallucinations’ in Parkinson disease. Arch Neurol. 2006;63(5):713–716. | ||
Weintraub D. Progress regarding Parkinson’s disease psychosis: it’s no illusion. Mov Disord. 2016;3(5):431–434. | ||
Fenelon G, Soulas T, Zenasni F, Cleret de Langavant L. The changing face of Parkinson’s disease-associated psychosis: a cross-sectional study based on the new NINDS-NIMH criteria. Mov Disord. 2010;25(6):763–766. | ||
De la Riva P, Smith K, Xie SX, Weintraub D. Course of psychiatric symptoms and global cognition in early Parkinson disease. Neurology. 2014;83(12):1096–1103. | ||
Kurita A, Murakami M, Takagi S, Matsushima M, Suzuki M. Visual hallucinations and altered visual information processing in Parkinson disease and dementia with Lewy bodies. Mov Disord. 2010;25(2):167–171. | ||
Poletti M, Perugi G, Logi C, et al. Dopamine agonists and delusional jealousy in Parkinson’s disease: a cross-sectional prevalence study. Mov Disord. 2012;27(13):1679–1682. | ||
Cannas A, Solla P, Floris G, et al. Hypersexual behavior, frotteurism and delusional jealousy in a young parkinsonian patient during dopaminergic therapy with pergolide: a rare case of iatrogenic paraphilia. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(8):1539–1541. | ||
Georgiev D, Danieli A, Ocepek L, et al. Othello syndrome in patients with Parkinson’s disease. Psychiatr Danub. 2010;22(1):94–98. | ||
Davis JL, Kurek JA, Sethi KD, Morgan JC. Delusional infestation in Parkinson’s disease. Mov Disord. 2016;86(16):324. | ||
Onofrj M, Bonanni L, Manzoli L, Thomas A. Cohort study on somatoform disorders in Parkinson disease and dementia with Lewy bodies. Neurology. 2010;74(20):1598–1606. | ||
Onofrj M, Thomas A, Bonanni L, et al. Somatoform disorders in Parkinson’s disease and dementia with lewy bodies evidence underlying psychotic traits. In: Ebmeier KP, O’Brien JT, Taylor JP, editors. Psychiatry of Parkinson’s Disease. Vol 27. Basel: Karger; 2012:125–132. Adv Biol Psychiatry. | ||
Onofrj M, Thomas A, Tiraboschi P, et al. Updates on somatoform disorders (SFMD) in Parkinson’s disease and dementia with Lewy bodies and discussion of phenomenology. J Neurol Sci. 2011;310(1–2):166–171. | ||
Stacy MA, Murck H, Kroenke K. Responsiveness of motor and nonmotor symptoms of Parkinson disease to dopaminergic therapy. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(1):57–61. | ||
Siri C, Cilia R, De Gaspari D, et al. Psychiatric symptoms in Parkinson’s disease assessed with the SCL-90R self-reported questionnaire. Neurol Sci. 2010;31(1):35–40. | ||
Stone J. Functional neurological disorders: the neurological assessment as treatment. Pract Neurol. 2016;16(1):7–17. | ||
Stone J, Pal S, Blackburn D, Reuber M, Thekkumpurath P, Carson A. Functional (Psychogenic) cognitive disorders: a perspective from the neurology clinic. J Alzheimers Dis. 2015;48(suppl 1):S5–S17. | ||
Aprahamian I, Yassuda MS, Martinelli JE. Somatoform and conversion disorder preceding Lewy body dementia: a newly described phenomenological manifestation of the disease. J Am Geriatr Soc. 2015;63(9):1967–1969. | ||
Minami A, Nakanishi A, Matsuda S, et al. Function of α-synuclein and PINK1 in Lewy body dementia. Int J Mol Med. 2015;35(1):3–9. | ||
Fujishiro H, Nakamura S, Sato K, Iseki E. Prodromal dementia with Lewy bodies. Geriatr Gerontol Int. 2015;15(7):817–826. | ||
Pareés I, Saifee TA, Kojovic M, et al. Functional (psychogenic) symptoms in Parkinson’s disease. Mov Disord. 2013;28(12):1622–1627. | ||
Mobley W, Rosenberg RN. The evolution of academic neurology: new information will bring new meaning. Arch Neurol. 2012;69(3):308–314. | ||
Edwards MJ, Bhatia KP. Functional (psychogenic) movement disorders: merging mind and brain. Lancet Neurol. 2012;11(3):250–260. | ||
Marsh GG, Markham CH. Does levodopa alter depression and psychopathology in Parkinsonism patients? J Neurol Neurosurg Psychiatry. 1973;36(6):925–935. | ||
Martin WE, Loewenson RB, Resh JA, Baker AB. Parkinson’s disease: clinical analysis of 100 patients. Neurology. 1973;23(8):783. | ||
Morel-Maroger A. Maladie de Parkinson et syndrome parkinsonien. Encyclopédie Médico-Chirurgicale sur Le Systeme Nerveux. EMC; 1975. | ||
Morel-Maroger A. Effects of levodopa on “frontal” signs in Parkinsonism. BMJ. 1977;2:1543–1544. | ||
Costa A, Peppe A, Carlesimo GA, Salamone G, Caltagirone C. Neuropsychological correlates of alexithymia in Parkinson’s disease. J Int Neuropsychol Soc. 2007;13:980–992. | ||
Franciotti R, Delli Pizzi S, Perfetti B, et al. Default mode network links to visual hallucinations: a comparison between Parkinson’s disease and multiple system atrophy. Mov Disord. 2015;30(9):1237–1247. | ||
Aracil-Bolaños I, Strafella AP. Molecular imaging and neural networks in impulse control disorders in Parkinson’s disease. Parkinsonism Relat Disord. 2016;22(suppl 1):S101–S105. | ||
Politis M, Loane C, Wu K, et al. Neural response to visual sexual cues in dopamine treatment-linked hypersexuality in Parkinson’s disease. Brain. 2013;136(pt 2):400–411. | ||
Ushe M, Perlmutter JS. Sex, drugs and Parkinson’s disease. Brain. 2013;136(pt 2):371–373. | ||
Linazasoro G. Dopamine dysregulation syndrome and levodopa-induced dyskinesias in Parkinson disease: common consequences of anomalous forms of neural plasticity. Clin Neuropharmacol. 2009;32(1):22–27. | ||
Calabresi P, Giacomini P, Centonze D, Bernardi G. Levodopa-induced dyskinesia: a pathological form of striatal synaptic plasticity? Ann Neurol. 2000;47(suppl 1):S60–S68. | ||
Uchiyama M, Nishio Y, Yokoi K, Hosokai Y, Takeda A, Mori E. Pareidolia in Parkinson’s disease without dementia: a positron emission tomography study. Parkinsonism Relat Disord. 2015;21(6):603–609. | ||
Shine JM, Halliday GM, Gilat M, et al. The role of dysfunctional attentional control networks in visual misperceptions in Parkinson’s disease. Hum Brain Mapp. 2014;35(5):2206–2219. | ||
Sharf B, Moskovitz C, Lupton MD, Klawans HL. Dream phenomena induced by chronic levodopa therapy. J Neural Transm. 1978;43(2):143–151. | ||
Collerton D, Mosimann UP, Perry E. The Neuroscience of Visual Hallucinations. Oxford: WILEY Blackwell; 2015. | ||
Lewis DJ. Lilliputian hallucinations in the functional psychoses. Can J Psychiatry. 1961;6:177–201. | ||
Kanazawa A, Hata T. Coexistence of the Ekbom syndrome and lilliputian hallucination. Psychopathology. 1992;25(4):209–211. | ||
Mantini D, Corbetta M, Romani GL, Orban GA, Vanduffel W. Evolutionarily novel functional networks in the human brain? J Neurosci. 2013;33(8):3259–3275. | ||
Catani M, Dell’acqua F, Thiebaut de Schotten M. A revised limbic system model for memory, emotion and behaviour. Neurosci Biobehav Rev. 2013;37(8):1724–1737. | ||
Greicius MD, Srivastava G, Reiss AL, Menon V. Default-mode network activity distinguishes Alzheimer’s disease from healthy aging: evidence from functional MRI. Proc Natl Acad Sci U S A. 2004;101(13):4637–4642. | ||
Northoff G. How does the ‘rest-self overlap’ mediate the qualitative and automatic features of self-reference? Cogn Neurosci. 2016;7(1–4):18–20. | ||
Neta M, Schlaggar BL, Petersen SE. Separable responses to error, ambiguity, and reaction time in cingulo-opercular task control regions. Neuroimage. 2014;99:59–68. | ||
Thalbourne MA, Bartemucci L, Delin PS, et al. Transliminality: its nature and correlates. J Am Soc Psych Res. 1997;91:305–332. | ||
Carhart-Harris RL, Leech R, Hellyer PJ, et al. The entropic brain: a theory of conscious states informed by neuroimaging research with psychedelic drugs. Front Hum Neurosci. 2014;8:20. | ||
Freud S. The Ego and the Id. London: L. and Virginia Woolfat the Hogarth Press, The Institute of Psycho-Analysis; 1927. | ||
Hellyer PJ, Scott G, Shanahan M, Sharp DJ, Leech R. Cognitive flexibility through metastable neural dynamics is disrupted by damage to the structural connectome. J Neurosci. 2015;35(24):9050–9063. | ||
Friston KJ. Transients, metastability, and neuronal dynamics. Neuroimage. 1997;5(2):164–171. | ||
Shanahan M. Metastable chimera states in community-structured oscillator networks. Chaos. 2010;20(1):013108. | ||
Tognoli E, Kelso JA. The metastable brain. Neuron. 2014;81(1):35–48. | ||
Serafini G, Pardini M, Pompili M, et al. Understanding suicidal behavior: the contribution of recent resting-state fMRI techniques. Front Psychiatry. 2016;7:69. | ||
Verrocchio MC, Carrozzino D, Marchetti D, Andreasson K, Fulcheri M, Bech P. Mental pain and suicide: a systematic review of the literature. Front Psychiatry. 2016;7:108. | ||
Whitfield-Gabrieli S, Ford JM. Default mode network activity and connectivity in psychopathology. Annu Rev Clin Psychol. 2012;8:49–76. | ||
Zhu X, Wang X, Xiao J, et al. Evidence of a dissociation pattern in resting-state default mode network connectivity in first-episode, treatment-naïve major depression patients. Biol Psychiatry. 2012;71(7):611–617. | ||
McEwen SCJ, Connolly CG, Kelly AMC, et al. Resting-state connectivity deficits associated with impaired inhibitory control in non-treatment-seeking adolescents with psychotic symptoms. Acta Psychiatr Scand. 2014;129(2):134–142. | ||
Chang X, Shen H, Wang L, et al. Altered default mode and fronto-parietal network subsystems in patients with schizophrenia and their unaffected siblings. Brain Res. 2014;1562:87–99. | ||
Tessitore A, Esposito F, Vitale C, et al. Default-mode network connectivity in cognitively unimpaired patients with Parkinson disease. Neurology. 2012;79(23):2226–2232. | ||
Szewczyk-Krolikowski K, Menke RA, Rolinski M, et al. Functional connectivity in the basal ganglia network differentiates PD patients from controls. Neurology. 2014;83(3):208–214. | ||
Franciotti R, Falasca NW, Bonanni L, et al. Default network is not hypoactive in dementia with fluctuating cognition: an Alzheimer disease/dementia with Lewy bodies comparison. Neurobiol Aging. 2013;34(4):1148–1158. | ||
Galvin JE, Price JL, Yan Z, Morris JC, Sheline YI. Resting bold fMRI differentiates dementia with Lewy bodies vs Alzheimer disease. Neurology. 2011;76(21):1797–1803. | ||
Kenny ER, Blamire AM, Firbank MJ, O’Brien JT. Functional connectivity in cortical regions in dementia with Lewy bodies and Alzheimer’s disease. Brain. 2012;135(pt 2):569–581. | ||
Firbank M, Kobeleva X, Cherry G, et al. Neural correlates of attention-executive dysfunction in Lewy body dementia and Alzheimer’s disease. Hum Brain Mapp. 2016;37(3):1254–1270. | ||
Yao N, Shek-Kwan Chang R, Cheung C, et al. The default mode network is disrupted in parkinson’s disease with visual hallucinations. Hum Brain Mapp. 2014;35(11):5658–5666. | ||
Shine JM, Muller AJ, O’Callagan C, et al. Abnormal connectivity between the default mode and the visual system underlies the manifestation of visual hallucinations in Parkinson’s disease: a task-based fMRI study. NPJ Park Dis. 2015;1:15003. | ||
Krajcovicova L, Mikl M, Marecek R, Rektorova I. The default mode network integrity in patients with Parkinson’s disease is levodopa equivalent dose-dependent. J Neural Transm. 2012;119(4):443–454. | ||
Gorges M, Müller HP, Lulé D, Ludolph AC, Pinkhardt EH, Kassubek J. Functional connectivity within the default mode network is associated with saccadic accuracy in Parkinson’s disease: a resting-state FMRI and videooculographic study. Brain Connect. 2013;3(3):265–272. | ||
Baggio HC, Segura B, Sala-Llonch R, et al. Cognitive impairment and resting-state network connectivity in Parkinson’s disease. Hum Brain Mapp. 2015;36(1):199–212. | ||
Rektorova I, Krajcovicova L, Marecek R, Novakova M, Mikl M. Default mode network connectivity patterns associated with visual processing at different stages of Parkinson’s disease. J Alzheimers Dis. 2014;42(suppl 3):S217–S228. | ||
Lenka A, Naduthota RM, Jha M, et al. Freezing of gait in Parkinson’s disease is associated with altered functional brain connectivity. Parkinsonism Relat Disord. 2016;24:100–106. | ||
Mathys C, Caspers J, Langner R, et al. Functional connectivity differences of the subthalamic nucleus related to Parkinson’s disease. Hum Brain Mapp. 2016;37(3):1235–1253. | ||
Graff-Radford J, Murray ME, Lowe VJ, et al. Dementia with Lewy bodies: basis of cingulate island sign. Neurology. 2014;83(9):801–809. | ||
Pievani M, de Haan W, Wu T, et al. Functional network disruption in the degenerative dementias. Lancet Neurol. 2011;10(9):829–843. | ||
Agosta F, Pievani M, Geroldi C, Copetti M, Frisoni GB, Filippi M. Resting state fMRI in Alzheimer’s disease: beyond the default mode network. Neurobiol Aging. 2012;33(8):1564–1578. | ||
Binnewijzend MA, Schoonheim MM, Sanz-Arigita E, et al. Resting-state fMRI changes in Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging. 2012;33(9):2018–2028. | ||
Chhatwal JP, Schultz AP, Johnson K, et al. Impaired default network functional connectivity in autosomal dominant Alzheimer disease. Neurology. 2013;81(8):736–744. | ||
Hahn K, Myers N, Prigarin S, et al. Selectively and progressively disrupted structural connectivity of functional brain networks in Alzheimer’s disease – revealed by a novel framework to analyze edge distributions of networks detecting disruptions with strong statistical evidence. Neuroimage. 2013;81:96–109. | ||
Jones DT, Machulda MM, Vemuri P, et al. Age-related changes in the default mode network are more advanced in Alzheimer disease. Neurology. 2011;77(16):1524–1531. | ||
Li X, Li TQ, Andreasen N, Wiberg MK, Westman E, Wahlund LO. Ratio of Aβ42/P-tau181p in CSF is associated with aberrant default mode network in AD. Sci Rep. 2013;3:1339. | ||
Zhang HY, Wang SJ, Liu B, et al. Resting brain connectivity: changes during the progress of Alzheimer disease. Radiology. 2010;256(2):598–606. | ||
Zhou J, Greicius MD, Gennatas ED, et al. Divergent network connectivity changes in behavioural variant frontotemporal dementia and Alzheimer’s disease. Brain. 2010;133(pt 5):1352–1367. | ||
Zhou B, Liu Y, Zhang Z, et al. Impaired functional connectivity of the thalamus in Alzheimer’s disease and mild cognitive impairment: a resting-state fMRI study. Curr Alzheimer Res. 2013;10(7):754–766. | ||
Tang-Wai DF, Graff-Radford NR, Boeve BF, et al. Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology. 2004;63(7):1168–1174. | ||
Whitwell JL, Avula R, Master A, et al. Disrupted thalamocortical connectivity in PSP: a resting-state fMRI, DTI, and VBM study. Parkinsonism Relat Disord. 2011;17(8):599–605. | ||
Kloszewska I. Incidence and relationship between behavioural and psychological symptoms in Alzheimer’s disease. Int J Geriatr Psychiatry. 1998;13(11):785–792. | ||
Turner MS, Cipolotti L, Yousry TA, Shallice T. Confabulation: damage to a specific inferior medial prefrontal system. Cortex. 2008;44(6):637–648. | ||
De Luca J, Cicerone KD. Confabulations following aneurism of the anterior communicating artery. Cortex. 1991;27:417–423. | ||
Markowitsch HJ. Memory and amnesia. In: Mesulam MM, editor. Principles of Behavioral and Cognitive Neurology. New York: Oxford University Press; 2000:257–293. | ||
Benson DF, Djenderedjian A, Miller BL, et al. Neural basis of confabulation. Neurology. 1996;46(5):1239–1243. | ||
Harding A, Halliday G, Caine D, Kril J. Degeneration of anterior thalamic nuclei differentiates alcoholics with amnesia. Brain. 2000;123(pt 1):141–154. | ||
Mair WG, Warrington EK, Weiskrantz L. Memory disorder in Korsakoff’s psychosis: a neuropathological and neuropsychological investigation of two cases. Brain. 1979;102(4):749–783. | ||
Schnider A, von Däniken C, Gutbrod K. The mechanism of spontaneous and provoked confabulations. Brain. 1996;119(pt 4):1365–1375. | ||
Onofrj V, Delli Pizzi S, Franciotti R, et al. Medio-dorsal thalamus and confabulations: evidence from a clinical case and combined MRI/DTI study. Neuroimage Clin. 2016;12:776–784. | ||
Ptak R, Schnider A. Confabulations after orbitofrontal damage: the role of temporal context confusion and self monitoring. Neurocase. 1999;5:243–250. | ||
Ptak R, Birtoli B, Imboden H, Hauser C, Weis J, Schnider A. Hypothalamic amnesia with spontaneous confabulations: a clinicopathologic study. Neurology. 2001;56(11):1597–1600. | ||
Kenny ER, O’Brien JT, Firbank MJ, et al. Subcortical connectivity in dementia with Lewy bodies and Alzheimer’s disease. Br J Psychiatry. 2013;203(3):209–214. | ||
Child ND, Benarroch EE. Anterior nucleus of the thalamus: functional organization and clinical implications. Neurology. 2013;81(21):1869–1876. | ||
Xuereb JH, Perry RH, Candy JM, et al. Nerve cell loss in the thalamus in Alzheimer’s disease and Parkinson’s disease. Brain. 1991;114(3):1363–1379. | ||
Perry EK, Perry RH. Acetylcholine and hallucinations: disease-related compared to drug-induced alterations in human consciousness. Brain Cogn. 1995;28(3):240–258. | ||
Ricciardi L, Piano C, Rita Bentivoglio A, et al. Pedunculopontine nucleus stimulation in Parkinson’s disease dementia. Biol Psychiatry. 2015;77(8):35–40. | ||
Kotagal V, Müller ML, Kaufer DI, Koeppe RA, Bohnen NI. Thalamic cholinergic innervation is spared in Alzheimer disease compared to parkinsonian disorders. Neurosci Lett. 2012;514(2):169–172. | ||
Delli Pizzi S, Franciotti R, Taylor JP, et al. Thalamic involvement in fluctuating cognition in dementia with Lewy bodies: magnetic resonance evidences. Cereb Cortex. 2015;25(10):3682–3689. | ||
Delli Pizzi S, Franciotti R, Taylor JP, et al. Structural connectivity is differently altered in dementia with Lewy body and Alzheimer’s disease. Front Aging Neurosci. 2015;7:208. | ||
Bonanni L, Thomas A, Tiraboschi P, Perfetti B, Varanese S, Onofrj M. EEG comparisons in early Alzheimer’s disease, dementia with Lewy bodies and Parkinson’s disease with dementia patients with a 2-year follow-up. Brain. 2008;131(pt 3):690–705. | ||
Magnin M, Rey M, Bastuji H, Guillemant P, Mauguière F, Garcia-Larrea L. Thalamic deactivation at sleep onset precedes that of the cerebral cortex in humans. Proc Natl Acad Sci U S A. 2010;107(8):3829–3833. | ||
Llinás RR, Steriade M. Bursting of thalamic neurons and states of vigilance. J Neurophysiol. 2006;95(6):3297–3308. | ||
Contreras D, Timofeev I, Steriade M. Mechanisms of long-lasting hyperpolarizations underlying slow sleep oscillations in cat corticothalamic networks. J Physiol. 1996;494(pt 1):251–264. | ||
Antelmi E, Ferri R, Iranzo A, et al. From state dissociation to status dissociatus. Sleep Med Rev. 2016;28:5–17. | ||
Benarroch EE. Pulvinar: associative role in cortical function and clinical correlations. Neurology. 2015;84(7):738–747. | ||
Erskine D, Taylor JP, Firbank MJ, et al. Changes to the lateral geniculate nucleus in Alzheimer’s disease but not dementia with Lewy bodies. Neuropathol Appl Neurobiol. 2016;42(4):366–376. | ||
Friston K. The free-energy principle: a unified brain theory? Nat Rev Neurosci. 2010;11(2):127–138. | ||
Ganser SJM. Über einen eigenartigen hysterischen Dämmerzustand. Archiv für Psychiatrie und Nervenkrankheiten. 1898;30:633–640. | ||
Cotard J. Du delire des negations. Arch Neurol. 1882;4:152–170. | ||
Benarroch EE. Habenula: recently recognized functions and potential clinical relevance. Neurology. 2015;85(11):992–1000. | ||
Hikosaka O. The habenula: from stress evasion to value-based decision-making. Nat Rev Neurosci. 2010;11(7):503–513. | ||
Sutherland RJ. The dorsal diencephalic conduction system: a review of the anatomy and functions of the habenular complex. Neurosci Biobehav Rev. 1982;6(1):1–13. | ||
Fava GA, Ruini C, Rafanelli C. Psychometric theory is an obstacle to the progress of clinical research. Psychother Psychosom. 2004;73(3):145–148. | ||
Fava GA, Tomba E, Sonino N. Clinimetrics: the science of clinical measurements. Int J Clin Pract. 2012;66(1):11–15. | ||
Fava GA, Belaise C. A discussion on the role of clinimetrics and the misleading effects of psychometric theory. J Clin Epidemiol. 2005;58(8):753–756. | ||
Bertini M, Freda MF, Fulcheri M, et al. Commenti all’articolo «Pratiche di salute, pratiche di psicologia: per una professionalizzazione della Psicologia della Salute in Italia» (Family of rules and family of affects. Which are the consequences on the well-being of adolescent offsprings?). Psicologia della Salute. 2014;2:26–55. | ||
Tomba E, Bech P. Clinimetrics and clinical psychometrics: macro- and micro-analysis. Psychother Psychosom. 2012;81(6):333–343. | ||
Carrozzino D, Marchetti D, Laino D, et al. Anxiety in adolescent epilepsy. A clinimetric analysis. Nord J Psychiatry. 2016;70(6):424–429. | ||
Bech P, Haber A, Joyce CR. Experiments on clinical observation and judgment in the assessment of depression: profiled videotapes and Judgment Analysis. Psychol Med. 1986;16(4):873–883. | ||
Bech P. Clinical Psychometrics. Oxford: Wiley Blackwell; 2012. | ||
Rash G. Probabilistic Models for Some Intelligence and Attainment Tests. Expanded ed. Chicago: Chicago University Press; 1980. | ||
Mokena RJ. Theory and Practice of Scale Analysis. Berlin: Mouton; 1971. | ||
Carrozzino D, Vass end O, Bjørndal F, Pignolo C, Olsen LR, Bech P. A clinimetric analysis of the Hopkins Symptom Checklist (SCL-90-R) in general population studies (Denmark, Norway, and Italy). Nord J Psychiatry. 2016;70(5):374–379. | ||
Feinstein AR. T. Duckett Jones Memorial Lecture-the Jones criteria and the challenges of clinimetrics. Circulation. 1982;66(1):1–5. | ||
Wright JG, Feinstein AR. A comparative contrast of clinimetric and psychometric methods for constructing indexes and rating scales. J Clin Epidemiol. 1992;45(11):1201–1218. | ||
Van Essen DC, Smith SM, Barch DM, et al. The WU-Minn Human Connectome Project: an overview. Neuroimage. 2013;80:62–79. | ||
Seibert TM, Murphy EA, Kaestner EJ, Brewer JB. Interregional correlations in Parkinson disease and Parkinson-related dementia with resting functional MR imaging. Radiology. 2012;263(1):226–234. | ||
Olde Dubbelink KT, Schoonheim MM, Deijen JB, Twisk JW, Barkhof F, Berendse HW. Functional connectivity and cognitive decline over 3 years in Parkinson disease. Neurology. 2014;83(22):2046–2053. | ||
Markowitsch HJ, Cramon DY, Schuri U. Mnestic performance profile of a bilateral diencephalic infarct patient with preserved intelligence and severe amnestic disturbance. J Clin Exp Neuropsychol. 1993;15:627–657. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.