Back to Journals » Psoriasis: Targets and Therapy » Volume 6
Psoriasis and inflammatory bowel disease: links and risks
Authors Vlachos C, Gaitanis G, Katsanos K, Christodoulou D ![]() , Tsianos E, Bassukas I
, Tsianos E, Bassukas I ![]()
Received 18 December 2015
Accepted for publication 7 April 2016
Published 20 July 2016 Volume 2016:6 Pages 73—92
DOI https://doi.org/10.2147/PTT.S85194
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Uwe Wollina
Christoforos Vlachos,1 Georgios Gaitanis,1 Konstantinos H Katsanos,2 Dimitrios K Christodoulou,2 Epameinondas Tsianos,2 Ioannis D Bassukas1
1Department of Skin and Venereal Diseases, 2Division of Gastroenterology, Faculty of Medicine, School of Health Sciences, University of Ioannina, Ioannina, Greece
Abstract: Psoriasis and the spectrum of inflammatory bowel diseases (IBD) are chronic, inflammatory, organotropic conditions. The epidemiologic coexistence of these diseases is corroborated by findings at the level of disease, biogeography, and intrafamilial and intrapatient coincidence. The identification of shared susceptibility loci and DNA polymorphisms has confirmed this correlation at a genetic level. The pathogenesis of both diseases implicates the innate and adaptive segments of the immune system. Increased permeability of the epidermal barrier in skin and intestine underlies the augmented interaction of allergens and pathogens with inflammatory receptors of immune cells. The immune response between psoriasis and IBD is similar and comprises phagocytic, dendritic, and natural killer cell, along with a milieu of cytokines and antimicrobial peptides that stimulate T-cells. The interplay between dendritic cells and Th17 cells appears to be the core dysregulated immune pathway in all these conditions. The distinct similarities in the pathogenesis are also reflected in the wide overlapping of their therapeutic approaches. Small-molecule pharmacologic immunomodulators have been applied, and more recently, biologic treatments that target proinflammatory interleukins have been introduced or are currently being evaluated. However, the fact that some treatments are quite selective for either skin or gut conditions also highlights their crucial pathophysiologic differences. In the present review, a comprehensive comparison of risk factors, pathogenesis links, and therapeutic strategies for psoriasis and IBD is presented. Specific emphasis is placed on the role of the immune cell species and inflammatory mediators participating in the pathogenesis of these diseases.
Keywords: psoriasis, inflammatory bowel disease, Crohn’s disease, ulcerative colitis, immune cells, inflammation
Introduction
Psoriasis and the group of inflammatory bowel diseases (IBD) are chronic, inflammatory, organotropic conditions. The former affects the skin of 2%–3% of the population, with hyperproliferation of keratinocytes, impaired epidermal barrier function at the sites of skin lesions, and skin infiltration by activated inflammatory cells.1 Ulcerative colitis (UC) and Crohn’s disease (CD) are the two most prevalent representatives of the IBD group. UC usually affects the rectum and may extend to the colon, with restriction of the inflammatory process to the mucosa and submucosa layers, while CD can affect any site of the digestive tract, with segmentary distribution of the lesions and inflammatory infiltration of all intestinal wall layers.2
Immune dysregulation, determined by genetic predisposition and environmental assaults, underlies all three conditions. A coincidence of psoriasis and IBD has been observed in various clinical settings: they can appear as concomitant disease events in the same patient or as “paradoxical” treatment-related adverse events.3,4
Moreover, a wide spectrum of therapeutic modalities is used to treat psoriasis and IBD, a fact that further underscores pathophysiological disease similarities. There are also cases of distinct discrepancies in the effectiveness of certain modalities that highlight differences in the pathophysiology between skin and gastrointestinal (GI) diseases and even within the IBD themselves.
The objectives of this paper were to review the most relevant literature data on the role of microbiota, inflammatory cell species, and humoral inflammation mediators in psoriasis and IBD immunopathogenesis and treatment response. For this purpose, corresponding core review articles were compiled, and a focused literature search was performed for retrieving the updated information. The search was conducted periodically during progressing text editing, employing Medline (PubMed). Two search filters were applied (language “English” and field “Title/abstract”), and the identified material was explored for selecting papers with adequate content. The full text of selected articles was retrieved, each study was individually evaluated, and the resulting material was integrated in this descriptive report.
After a short comparison of the genetic collation, we will survey the role of microbiota and selected innate and adaptive immunity inflammatory cell species and humoral inflammation mediators in the pathogenesis of these diseases. Finally, we discuss similarities and discrepancies in the response of psoriasis and IBD patients to the established pharmacologic therapies of either to additionally highlight the nosologic relationships of these conditions.
Diseases coincidence
Psoriasis and IBD cluster at all genetic levels of human populations, ie, they share the same geographic/ethnic, kindred, and patient niches. These diseases are more frequent in Northern Europe and North America (psoriasis prevalence: ∼2%; UC and CD incidences: 19.2–24.3 and 12.7–20.2 per 100,000 person-years, respectively) compared to most regions of Africa, Asia, and the Middle East (psoriasis prevalence: <0.5%; UC and CD incidences: 6.3 and 5.0 per 100,000 person-years, respectively).5–7
Both psoriasis and CD present with much higher incidence among family members of patients than in the general population.8,9 Almost one-third of patients with psoriasis also have a first-degree relative affected with the disease.9 In contrast, the risk of the siblings of a CD patient to be affected is only ∼5%.8 Moreover, differences in concordance rates between monozygotic and dizygotic twins both in psoriasis (70% vs 23%, respectively) and CD (37% vs 7%) support the important role of genetic background.10,11
Psoriasis and CD also coexist in the same patient more often than expected by chance: the prevalence of psoriasis in patients with CD is 9.6% (vs 2.2% in controls).12 The relative risks of psoriasis are significantly increased in both UC (1.56-fold) and CD (1.52-fold) patients and psoriasis patients are at increased risk to develop IBD (2.49-fold for CD and 1.64-fold for UC).13,14
Genetic collation
A shared genetic susceptibility background probably underlies the pathobiological and clinical overlap of psoriasis and IBD. To date, the literature describes 13 psoriasis susceptibility loci (designated PSORS1-13), and genome-wide association studies implicate 32 loci in the pathogenesis of CD and 17 in UC (Table 1).15–17
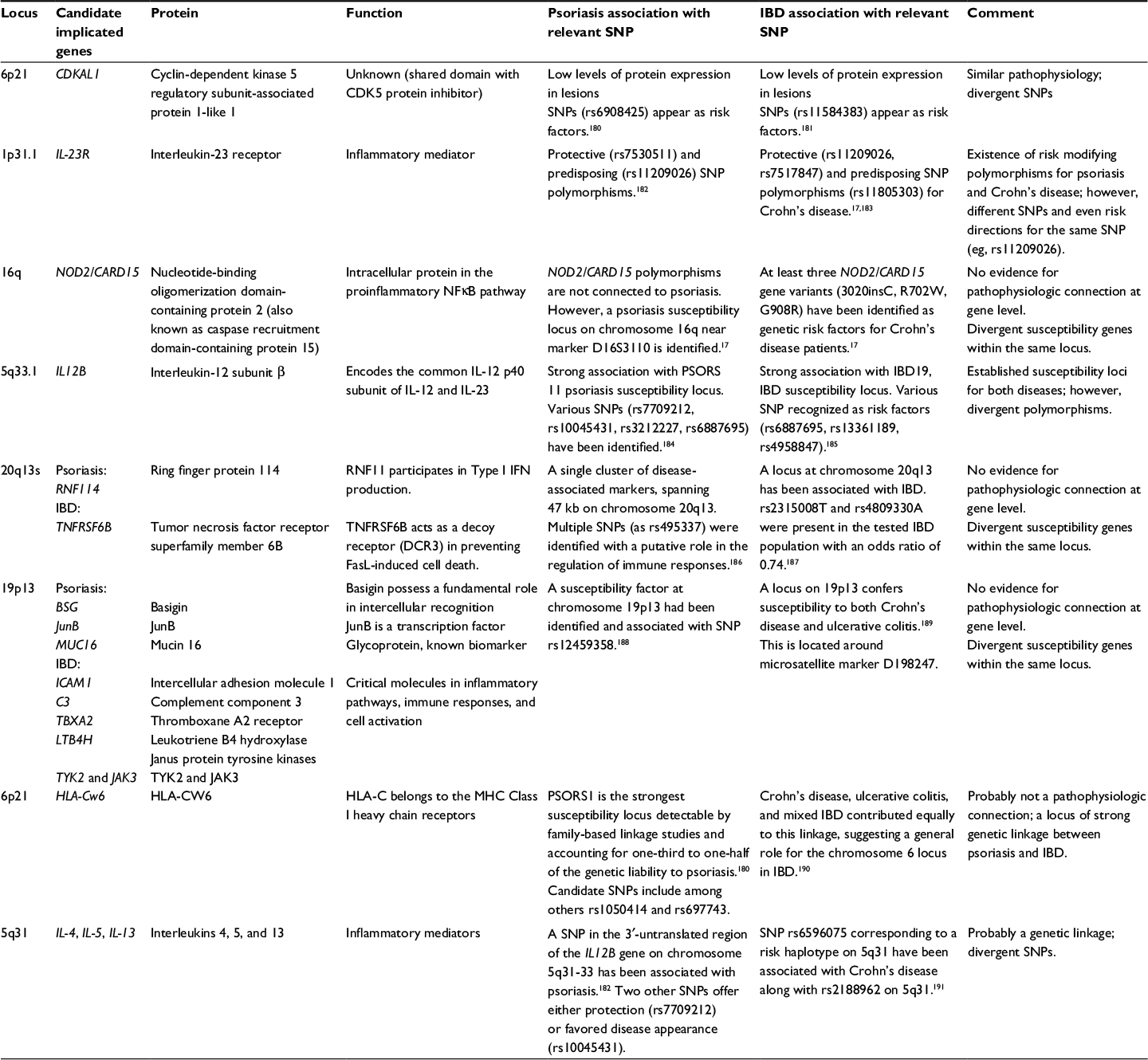 | Table 1 Genetic susceptibility loci associated with psoriasis and IBD Notes: Genetic association studies have discovered various genome areas that are associated with both diseases. Only eight loci are described with well-established associations for both diseases. However, within the same locus, the involved genes and polymorphisms may diverge between the skin and gut diseases. Abbreviations: SNP, single nucleotide polymorphism; IBD, inflammatory bowel disease; MHC, major histocompatibility complex. |
The epidemiologic coincidence of the conditions is partly explained by the sharing of disease susceptibility loci: 20q13 corresponds to PSORS12 and IBD24, 19p13 to PSORS6 and IBD6, 6p21 to PSORS1 and IBD3, and 5q31 to PSORS11 and IBD5.17 However, despite loci concordance, the involved genes are often different. For example, in the 6p21 region, psoriasis seems to be linked to the HLA-Cw*0602 allele, while IBD are mainly linked to polymorphisms in the TNF-α gene promoter.17
Of note, a recent meta-analysis of genome-wide association studies that included >4,500 patients and 10,000 controls recognized seven susceptibility loci outside the human leukocyte antigen region (9p24 near JAK2, 10q22 at ZMIZ1, 11q13 near PRDX5, 16p13 near SOCS1, 17q21 at STAT3, 19p13 near FUT2, and 22q11 at YDJC) shared by psoriasis and CD and confirmed four already established common psoriasis and CD risk loci (IL23R, IL12B, REL, and TYK2).18 Future functional studies that explore these genetic relationships will contribute to better understanding of the resonance of genetic colocalization to the pathophysiology of the diseases.
Microbiota
Microbiota play a significant role in the pathogenesis of different diseases.19 Skin and intestine are the two major niches of prokaryotic and eukaryotic symbiotic microorganism flora in humans.20,21 The inflammatory diseases evaluated herein primarily affect the structure and function of the corresponding organism–environment interface linings, resulting in distinct sustained alterations in the respective microenvironments of exposed tissue surfaces and, simultaneously, also of the species composition of the microorganism populations (dysbiosis).
Commensal microbes produce numerous biologically active metabolites that impact the physiology and immune response of the underlying epithelia. Notably, members of the phylum Firmicutes, the number of which are decreased in the gut lumen of CD patients, produce short-chain fatty acid metabolites (acetate, butyrate) with anti-inflammatory actions in the normal gut that may fail in IBD patients.22–24 Aryl hydrocarbon receptor (AhR), the promiscuous receptor that mediates the recognition of xenobiotics and modifies immune responses, may play a distinct role in mediating these pathophysiologic alterations. AhR-deficient mice, a mutant known to develop enhanced Th17-driven inflammatory immune responses, are characterized by a high prevalence of segmented filamentous bacteria in their gut. Interestingly, the administration of antibiotics that target these bacteria results in the reduction of intestinal IL-17 production as well.24
Microbiota act both as triggers and inhibitors of inflammation; to date, the balance of these effects have been better studied in IBD compared to psoriasis patients.19 IBD patients are characterized by a decrease of anaerobic bacteria (Bacteroides, Eubacterium, Lactobacillus).25 Particularly, CD patients present with a reduction in Firmicutes and Clostridium leptum populations, along with a simultaneous increase in Proteobacteria.26,27 These alterations have been linked to genetic determinants of the immune response (as the NOD2 gene) or genes that are associated with intracellular degradation of proteins, such as ATG16L1.28 Likewise, Paneth cell function and the FUT2 genotype (a gene involved in the expression of ABO blood group antigens in the GI mucosa) have been linked to substantial differences in the species composition of the microbial populations that colonize the gut lumen and modify the risk of developing CD.29,30 Interestingly, similarly to patients with a microbial infection, IBD are characterized by a substantial loss of microorganism species diversity in the gut and by a shift from an innocuous multispecies homeostatic flora to a narrower spectrum of proinflammatory and pathogenic microbial species.31,32 The impact of antibiotic use in this shift is a matter of debate, yet certain antibacterial agents (such as the 5-aminosalicylic acid) suppress the ability of gut bacteria to build adherent biofilms in patients with IBD, resulting in disease improvement.33
Genes affecting epidermal barrier function and adaptive immune responses with subsequent alterations in bacterial colonization have been suspected to contribute to the pathogenesis of psoriasis.34 The interrelationship between microbes and psoriasis pathogenesis is currently under intense investigation. Many studies agree that a certain degree of dysbiosis is a landmark of the skin lesions and that psoriasis may, at least in part, be associated with a substantial alteration in the composition of the cutaneous microflora.35 The genus most frequently identified in diseased skin areas is Corynebacterium as opposed to Propionibacterium in healthy controls. In addition, the representation of Propionibacterium and Actinobacteria species is lower and that of Firmicutes is higher in affected skin.36,37 Infections also play a central role in the course of psoriasis. Bacteria have been suspected to be an important source of antigens that trigger the immune system activation, which is a characteristic of this condition.38 Guttate psoriasis flares are usually preceded by streptococcal pharyngitis episodes. Moreover, the distinct differences in the prevalence of psoriasis in different populations have been attributed to the effect of natural selection for gene polymorphisms associated with more vigorous immunity against infectious agents such as invasive streptococci infections or leprosy pandemics in the past.39,40 T-cell clones primed by streptococcal tonsillitis or pharyngitis are supposed to be reactivated in the skin as a result of their exposure to cross-reactive epitopes expressed there (molecular mimicry).41
Microbes seem to further interact with the cells of the innate immune system, resulting in increased expression of antimicrobial peptides, production of cytokines, and subsequently, stimulation of T-cells.42,43 As in the gut of IBD patients, the absence of AhR enhances skin inflammation both in the skin of patients with psoriasis and in a murine model of psoriasiform skin inflammation.44 Notably, the potent AhR agonist indirubin, also produced on the surface of the skin by Malassezia yeasts, is an ingredient used in topical remedies for psoriasis in traditional Chinese medicine.45,46
In conclusion, alterations of the microbial flora in the gut and the skin may not only result from disease-associated tissue perturbations but may also exacerbate and maintain the disease state in a vicious self-perpetuating cycle.
The role of innate immune cells and the IL-23/Th17 axis
Innate and adaptive immune processes are expected to participate in the pathogenesis of both psoriasis and IBD (Figure 1, Table 2). Until recently, the prevailing opinion in the literature favored adaptive immunity as the major player in the pathogenesis of these diseases; however, current evidence highlights a more decisive role of the innate immunological mechanisms.
 | Figure 1 The epithelial barrier in both diseases appears more permeable than normal. Notes: The increased rate of epithelial cell apoptosis in IBD and the acanthosis/parakeratosis in psoriasis are accompanied by a reduction in the structural complexity of the intercellular junctions. Thus, antigen and pathogen crossing-through is increased, resulting in binding to pattern recognition receptors (PRRs) and immune system activation. The dendritic cells are the main regulators of immune reaction, armored with a wide range of PRRs, including toll-like receptors and nod-like receptors (NLRs). Under inflammation, they produce cytokines, as IL-6 or IL-23, contributing to T helper-17 (Th17) cell formation. Macrophages have a complementary role in eliminating and displaying antigens to T-lymphocytes, besides producing cytokines to recruit other immune cells to the inflammation site. Tissue macrophages are also involved in the generation of T-regulatory cells. The IL-23/Th17 axis interaction with epithelial and synovial cells is one of the main events in the initiation and maintenance of the inflammation in both diseases. Abbreviation: IBDs, inflammatory bowel diseases. |
 | Table 2 Summary of the role of innate and adaptive immunity cell species in the pathophysiology of psoriasis and inflammatory bowel diseases (IBD) Abbreviations: TLR, toll-like receptors; PRR, pattern recognition receptors; TNF, tumor necrosis factor; IFN, interferon; CD, Crohn’s disease; UC, ulcerative colitis; pDCs, plasmacytoid DCs. |
The innate immune system includes the host’s primary response mechanisms against foreign agents: protective physical barriers, inflammation-related proteins like the complement system and C-reactive protein, antimicrobial peptides, such as defensins and cathelicidins, and pattern recognition receptors (PRRs), cytokines, phagocytic cells, dendritic cells (DCs), and natural killer (NK) cells.22,43
The intestinal epithelial barrier is a physical defense line that encompasses intestinal epithelial cells, tight junctions, and a layer of mucous. The resident microbiota can activate specific PRRs in the epithelial cells,47 effecting downstream signaling that leads to proinflammatory protein synthesis and in the case of IBD to a more susceptible and permeable epithelial barrier.48,49 The anatomical and functional integrity of the barrier is further compromised by proinflammatory cytokines (such as TNF-α), which disturb the integrity of intercellular tight junctions by affecting the synthesis of occludins and other related proteins in the gut epithelium.50 In addition, mucous production is either up- or downregulated in IBD patients, further contributing to disease processes.51
Compared to healthy individuals, intestinal epithelial cells of IBD patients express an altered pattern of Toll-like receptors (TLRs): increased TLR4 expression in UC patients and decreased TLR4 and TLR3 expression in CD patients.52 An overexpression of TLR4 and TLR2 in intestinal wall macrophages during inflammation also contributes to a state of gut hyperresponsiveness to xenobiotics in these patients.53 Based on the aforementioned observations, in IBD patients, the defective epithelial barrier function facilitates intestinal antigens to reach and bind to PRRs, activate the immune system, and ultimately herald inflammation.
Likewise, defects in the structure and function of the epidermal barrier seem to play an important role in the pathogenesis of psoriasis. Similar to the aforementioned IBD findings, the expression of structural proteins such as claudin and zonula occludens-1 is reduced in the epidermis of psoriatic lesions, which results in alterations in the structure of epidermal tight junctions.54 The overall pathophysiologic consequence is a partially compromised epidermal barrier capacity that enables enhanced invasion of microorganisms, their products, and other foreign molecules, ultimately leading to the stimulation of inflammatory skin processes.55
On the other hand, the expression of TLRs is also modified in psoriasis.56 TLR 2 is constitutively expressed in the suprabasal layers of normal skin, but it is limited to the basal cell layers of the epidermis in diseased skin. There is also a characteristic tissue shift in epidermal TLR 4 expression, with an inversion of the spatial expression gradient from a basal to superficial pattern in the normal skin to a more pronounced expression in more superficial layers in psoriasis lesions. These shifts are associated with the disturbed skin barrier, and are interpreted as an adaptation of the tissue to meet the need for enhanced immune surveillance and avoidance of overstimulation due to pathophysiological inflammation cascades in diseased skin.56
Dendritic cells (DCs) are the main regulators of immunity since they are able to provoke inflammatory responses or impose immune tolerance.22 They are equipped with a wide range of PRRs, including TLRs and Nod-like receptors (NLRs), allowing them to distinguish between microbiota and pathogens. So far DCs have not been stimulated by immunological insults their state corresponds to an “immature” phenotype that maintains the T-lymphocytes inactive.
In the intestine, submucosal DCs reach the intestinal lumen by projecting their dendrites through the tight junctions boundaries between epithelial cells.57 In IBD, gut inflammation and the defective mucous membrane barrier allow an increased amount of intestinal lumen antigens to reach the DCs, resulting in their maturation and activation, thus enhancing immune responses.58 The population of mucosal DCs and the number of CD40+ DCs is significantly increased in UC and CD, when compared to controls.59 Among intestinal DCs, the CD103+ cell subpopulation seems to be essential for maintaining the homeostasis of the mucosa and for regulation of the balance between effector and regulatory T-lymphocytes.60 During an IBD disease flare, DC subsets expressing E-cadherin (a CD103 receptor) in the intestine and the draining mesenteric lymph nodes and provide proinflammatory cytokines, such as IL-6 and IL-23, which are suggested to contribute toward Th17 cell maturation and colon damage in at least a subset of IBD patients.61,62
Also in psoriasis the cellular composition of cutaneous DC subsets is modified compared to healthy skin: Langerhans cells (LCs) are markedly reduced, whereas plasmacytoid DCs (pDCs) are increased in human psoriasis lesions and in the animal skin of a murine psoriasis model (DKO* mice).63 Particularly, the depletion of LCs in the latter model aggravated the disease, while depletion of pDCs (before disease onset) resulted in an overall milder phenotype. The proposed mechanisms included IL-10 and IL-23 decrease. The modulating role of LCs in psoriasis is further highlighted by the fact that after treatment with adalimumab, the density of epidermal LCs increases rapidly (with a concomitant normalization of their spatial localization within the epidermis).64
pDCs predominate among the immune cells of the dermis of lesions and uninvolved skin of psoriasis patients.65 In healthy skin, pDCs are almost absent, but they are abundant in the skin of psoriatic plaques, particularly in early lesions, where they become activated and produce interferon-α (INF-α), an inflammatory mediator essential for the development of psoriasis lesions.65 A less distinct role for pDCs is presumed for the chronic-stationary state of psoriasis lesions;66 other dermal DC subsets appear to be more important for the stabilization of disease state and the persistence of these lesions.67
Macrophages participate in innate and adaptive immune processes at the epithelium, acting as an environmental barrier through phagocytosis and antigen presentation.68 Two major subtypes of macrophages and further cellular subsets are distinguished: inflammatory (M1) and anti-inflammatory (M2a, M2b, M2c), reflecting the vast cellular diversity and functional plasticity of this cell species in the skin and intestine.69
In healthy individuals, the main intestinal population of macrophages bears the CX3CR1high phenotype and, although, nonresponsive to inflammatory stimuli, these cells preserve phagocytic capacity. Under the proinflammatory circumstances of the gut tissues in IBD, another subpopulation of CX3CR1low/int macrophages expands there due to the emigration and differentiation of precursor monocytes.70 This M1 macrophage subset that overexpresses antigen receptors and PRRs augments the environmental signaling for the synthesis of proinflammatory cytokines (TNF, IL-1, IL-6) and contributes to local immune response exacerbations.71
The M1 macrophages subset is also closely linked to the proinflammatory situation of the psoriatic plaques.72 A variety of exogenous pathogen-associated molecular patterns and endogenous ligands, recognized through PRRs, are implicated in the molecular pathways by which macrophage activation is induced after the skin barrier is breached.73 Thepen et al74 showed that local elimination of macrophages led to resolution of focuses of cutaneous inflammation. A number of endogenous anti-inflammatory mediators produced by macrophages have also been identified, including IL-10, TGF-β, lipoxins, and prostaglandins.75 In addition, tissue macrophages have been involved in the generation of T-regulatory cells.76 Taken together, macrophage subsets in psoriatic skin may delineate both pro- and anti-inflammatory roles in the pathogenesis of psoriasis. Given the central role of macrophages in maintaining tissue inflammation, better understanding of their pathophysiological role is predicted to contribute innovative macrophage-targeting therapeutic modalities for psoriasis and IBD in the future.
Mast cells (MCs) are another cell species with a recognizable role in the pathogenesis of IBD and psoriasis. There are two subsets of MCs, mucosal and connective tissue MCs. The human mucosal MCs are a functionally T-cell-dependent population mainly found in the lungs and the intestine. Connective tissue MCs are not as much T-cell dependent and are found in the skin and the intestinal submucosa.77
The largest number of MC progenitors are found in the intestine. These cells continue to differentiate, mature, and establish residence there.78 Increased number of MCs are found in biopsies from UC- or CD-affected gut areas.79,80 Under conditions of tissue inflammation, intestinal MCs degranulate and release substances such as tryptase-β and chymase-181 leading to increased tissue levels of histamine, prostaglandin D2, and leukotrienes.82 In the intestine, MCs also play an important homeostatic role by regulating the baseline permeability of the gut epithelium83 and by modulating epithelial–neuromuscular interactions in the gut. Thus, they participate in the pathogenesis of altered motility patterns seen in functional GI disorders, postoperative ileus, food allergy, and also IBD.84
In the human skin, connective tissue MCs play an essential role in diverse physiological and pathological processes such as the mediation of type I hypersensitivity reactions and allergic diseases. Upon activation, they produce large amounts of TNF-α, IFN-γ, IL-8, IL-33, and multiple other mediators, such as VEGF, contributing to the creation of a tissue microenvironment essential for the recruitment of neutrophils and lymphocytes during innate and T-cell-mediated inflammatory processes.85 The increased number of MC that are found in psoriasis skin, underscore the important role of these cells in the pathophysiology of this disease.85,86
In particular, a postulated interaction between stress and inflammatory processes in both the skin and the intestine is suggested to be mediated via the function of MCs.87,88 MC–neuronal interactions are thought to be involved in translating stress insults into psoriasis flares.89
The natural killer (NK) cells are a specialized subset of CD56+CD16+ cells that can recognize injured cells and promote their death through a non-MHC-dependent manner.90 They also produce cytokines, particularly IFN-γ, TNF-α, and TGF-β but also IL-2, IL-15, and IL-23, and affect the maturation of DCs.91
In IBD, NK cells are characterized by increased IL-21R expression, which amplifies the effect of IL-21 and the downstream proinflammatory cytokine cascade.92 In the intestinal mucosa of CD patients, there are alterations in the ratio of NKp44- and NKp46-expressing NK cells.93 Increased numbers of IFN-γ-producing NKp46+ cells are found in CD-affected mucosa sections, whereas IL-22-producing NKp44+ cells are significantly decreased compared to UC patients and healthy controls.
In the acute state of psoriatic plaques, ∼5%–8% of the inflammatory cellular infiltrate consists of NK cells expressing the CD56+CD3− phenotype and the activation antigen CD69. Compared to the uninvolved skin of psoriasis patients or healthy controls, NK cells in the lesional psoriasis skin produce large quantities of IFN-γ and express higher levels of perforin, a pore-forming protein found in the cytotoxic granules of NK cells and a key mediator of NK cells cytotoxicity.94,95
Finally, decreased number of circulating NK cells is found in patients with long-standing psoriasis. Decreased NK cell activity was also observed in the peripheral blood of IBD patients, but this could not be attributed to decreased NK cell number.96
Another leukocyte species involved in IBD pathogenesis is the innate lymphoid cell (ILCs).22 ILCs are abundant at barrier surfaces of the body such as the skin and the intestine.97 These cells play a key role in tissue remodeling and controlling microbe tissue load, lymphoid tissue development, and tissue homeostasis.98,99 ILCs are divided into subtypes based on the patterns of synthesized cytokines and on the expression of specific transcription factors.99,100
Besides their important role in gut homeostasis, these cells are probably also involved in the pathogenesis of IBD, where increased numbers are found compared to controls.101 In a proinflammatory environment, these cells produce IL-22; a pathophysiologic adaptation that limits apoptosis, preserves intestinal barrier, and protects the gut during inflammation.101–103
Blood and lesional skin biopsies of patients with psoriasis showed enrichment of NCR+ ILC3; however, similar frequencies of CD161+ ILC1 and CRTH2+ ILC2 compared to controls.104 ILC3 is an innate source of IL-22 and IL-17 in the psoriatic skin, and an increased skin concentration of NKp44+ ILC3 cells has been associated with disease severity.105 It is worth noting that treatment of psoriasis patients with adalimumab reversed the number of these cells in parallel with clinical disease improvement.106
Regarding the role of the adaptive immunity, the latest pathophysiologic hypotheses consider an interaction of Th17, T-regulatory lymphocytes (Tregs), and Th1 cells crucial for the pathogenesis of both psoriasis and IBD.107
Th17 cells comprise a subpopulation of T-helper cells that produce mainly IL-17 and IL-21.108 They comprise a T-lymphocyte subset evolutionarily distinct from the Th1 and Th2 lymphocyte subpopulations, with a key role in chronic organotropic inflammatory processes such as psoriasis and CD.109 Some Th17 subtypes also produce IL-22, which is associated with disease aggravations in psoriasis; however, it has a protective function in IBD. Inflammatory foci of the GI mucosa in IBD patients display a massive infiltration of Th17 cells, and Th17-related cytokines are produced in excess in both CD- and UC-affected tissues.110 Genome-wide association studies have revealed increased frequencies of certain Th17-related gene polymorphisms (such as STAT3 or IL-23R) in IBD patients, thus supporting the involvement of the Th17 pathway in IBD pathogenesis.111 Despite earlier findings, to date, targeting Th17 cells,112 either by blocking their proliferation and differentiation or by inhibiting and neutralizing their specific cytokines, is a highly disputed, rather ineffective therapeutic pathway for IBD (see “Comparison of pharmacologic treatment modalities” section).
Recent advances in understanding the psoriasis pathogenesis indicate to an interaction of the IL-23/Th17 cells axis with keratinocytes and synovial cells as one of the main pathophysiologic events in the initiation and maintenance of tissue inflammation in this disease.113 IL-17-producing Th17 lymphocytes promote the synthesis of inflammatory molecules and induce acanthosis, hyperkeratosis, and parakeratosis.114,115 As in the case of IBD, polymorphisms of genes have been observed in psoriasis that relate to development and functional polarization of Th17 lymphocytes. For example, polymorphisms in the aforementioned genes STAT3 and IL23 are strongly associated with the psoriasis phenotype.116 The key role of IL-17 in psoriasis is further underlined by the distinct effectiveness of treatment modalities (contrary to IBD) that either target the IL-17 cytokine directly (secukinumab, bimekizumab, ixelkizumab) or block the IL-17 receptor (IL-17R: brodalumab).113
Another lymphocyte subpopulation with increasing importance in IBD and psoriasis research is the Tregs. In various organs, including intestine and skin, these cells counteract the abnormal activation of the immune system, preventing excessive stimulation and contributing to self-tolerance.117
Compared to controls, Tregs are reduced in peripheral blood in the active phase of both CD and UC; however, they normalize during disease remissions. This observation suggests that during the course of IBD, Tregs migrate from peripheral blood to the inflamed intestinal mucosal sites, the lamina propria, and mesenteric lymph nodes.118,119 It is worth noting that at the level of the intestine, the tissue concentration of Tregs is higher in IBD irrespective of the activity state of the disease, when compared to healthy controls.120,121
Tregs were significantly increased in the dermis and epidermis of psoriatic skin too, as compared to normal skin.122 Functional and numerical abnormalities of Tregs have also been documented in the peripheral blood of psoriasis and IBD patients.123 However, although Foxp3 polymorphisms appear to contribute to the risk of developing psoriasis (at least in certain patient subpopulations), a similar association could not be verified in IBD patients.124,125
Comorbidities
Chronic inflammatory conditions such as psoriasis and IBD also share a series of important comorbidities. Most of these connections have been attributed to the systemic action of sustainably increased tissue levels of inflammatory mediators, shared genetic risk factors, and, in some cases, predictable adverse effects of the employed treatments.126 Patients with severe psoriasis are at increased risk of premature death from most leading mortality causes in the population, including cardiovascular (CV) disease, malignancies, chronic lower respiratory disease, diabetes, dementia, infection, and kidney disease.127 Moreover, in psoriasis, besides the characteristic arthropathy and the concordance with IBD, variable evidence for a higher prevalence has been observed for CV disease, obesity, diabetes, hypertension, dyslipidemia, metabolic syndrome, nonalcoholic fatty liver disease, cancer, and anxiety/depression.128 On the other hand, apart from psoriasis, usual comorbidities of IBD include arthritis, asthma, pericarditis, certain malignancies, and also psychiatric conditions. Notably, among IBD, patients with UC (but not with CD) are additionally at increased risk of developing chronic renal failure and multiple sclerosis.129 It is worth noting that overall mortality rates are also slightly increased in IBD patients at a rate comparable to that observed in the case of severe psoriasis.130 Table 3 compares the evidence for selected major comorbidities in patients with psoriasis and IBD. Corresponding data concerning three selected major comorbidities (cancer, obesity, and CV diseases) will be briefly discussed.
 | Table 3 Psoriasis and IBD share a series of important comorbidities. Notes: Most of these connections have been attributed to the systemic action of sustainably increased levels of inflammatory mediators, shared genetic risk factors, and employed treatments. Abbreviations: NAFLD, nonalcocholic fatty liver disease; PSC, primary sclerosing cholangitis; NMSC, nonmelanoma skin cancer; CD, Crohn’s disease; UC, ulcerative colitis; PsA, psoriatic arthritis; IBD, inflammatory bowel disease. |
Besides epidemiologic evidence, the rationale for the search for possible connections of psoriasis and IBD, on one hand, and malignancies, on the other, is the bidirectional relationship between chronic inflammation and cancer. Chronic inflammatory conditions are frequently associated with the induction CD8+ cytotoxic T-lymphocytes and increased NK cell activities. Moreover, INF-γ and TNF-α produced by the activated CD4+ Th1 cells in inflammatory conditions may cause tumor cell senescence and tumor control. In addition, Th1 immunity may induce antiangiogenic chemokines that in certain circumstances could counteract cancer growth.131 On the contrary, other studies indicate that inflammation contributes directly to tumorigenesis.132 Chronic inflammation can initiate tumors directly by causing DNA alterations or by making cells more susceptible to mutagens. In addition, inflammation can also act as a tumor promoter since inflammatory mediators such as cytokines (TNF-α, IL-1, IL-6), growth factors, chemokines, and proteases produced by tumor-associated lymphocytes, and macrophages can enhance tumor cell growth and metastasis.133,134 However, taking available data together, there is no evidence of increased cancer incidence among patients with psoriasis and IBD, with the exception of the association of UC with certain GI malignancies. Probably these opposing mechanisms generally balance themselves in the case of the present chronic inflammatory conditions, resulting in overall unaffected cancer risks. Nonmelanoma skin cancer represents an exception among malignancies for both conditions; however, their increased incidence is most likely secondary to the application of therapeutic modalities with recognized tumor-promoting risks (ie, ultraviolet radiation and azathioprine in psoriasis and IBD, respectively).135
From a pathophysiologic point of view, obesity is an interesting comorbidity that potentially highlights the shared pathogenesis mechanisms of psoriasis and IBD. Obesity is currently considered a chronic, low-grade inflammatory condition;136 proinflammatory adipose tissue macrophages stimulate the secretion of proinflammatory mediators, such as TNF-α, IL-6, and leptin, which establish and maintain an inflammatory tissue state.137 With a doubled odds ratio compared to controls, obesity is a recognized serious health risk factor in patients with psoriasis.138 In addition a role for obesity in the pathogenesis of psoriasis is speculated.139 More specific, significantly increased blood levels of leptin are found in patients with psoriasis, a mediator that might have participated to psoriasis induction by promoting Type 1 cytokines synthesis.140 On the other hand, due to the nature of IBD (with abdominal pain and diarrhea, weight loss, and some degree of malnutrition affecting the majority of the patients), obesity was not predicted to be an important comorbidity of IBD patients.141 However, it is surprising that a recent study finds an increasing prevalence of obesity in patients with IBD (15%–20%), with a further 40% of patients being overweight.142 Chronic inflammation, the type of intestinal microbiota, the medication for controlling IBD, and the lack of physical activity are all discussed as putative factors that favor overweight and obesity in the IBD patients.143–145 Future studies should also clarify to what extent the improved efficacy of IBD treatment modalities might per se have contributed to increased obesity prevalence among these patients.
Finally, there is currently increasing interest in the literature concerning the interrelationship between psoriasis and CV morbidity, with particular emphasis in the role of the antipsoriatic therapeutic interventions. Most studies indicate an increased CV risk of patients with psoriasis that is mediated by endothelial dysfunction and is manifested by higher incidences of stroke, atherosclerosis, coronary artery disease, and myocardial infarction.146 Interestingly, psoriasis and atherosclerosis share a common pattern of Th1 and Th17 cytokine upregulation, T-cell activation, local and systemic expression of adhesion molecules, and endothelins.147 Moreover, increased amounts of vascular endothelial growth factor produced by proliferating keratinocytes and the hyperhomocysteinemia (a common finding in patients with psoriasis) provide additional possible mechanisms for increased atherosclerosis risk in these patients.148,149 On the other hand, CV is still a common cause of mortality among IBD patients although they have no different CV risk compared to the general population.150–152 And this is in spite of the increased prevalence of a series of risk factors in patients with IBD, known to promote CV risk (prothrombotic state, subclinical atherosclerosis).153–156 This controversy is still increasing when the role of therapeutic interventions is considered. Besides reports of decreased frequency and severity of CV events both in psoriasis and IBD patients under treatment with methotrexate and probably also TNF-α inhibitors, other studies found exactly the opposite, ie, increased myocardial infarction risks, with the same modalities.157–159
Taken together, probably with the exception of psychiatric comorbidities and increased nonmelanoma skin cancer risk, there is no conclusive evidence definitively linking both psoriasis and IBD with a certain pattern of comorbid conditions.
Comparison of pharmacologic treatment modalities
The wide overlap of approved modalities employed to treat psoriasis and IBD, particularly the more targeted newer biologics, underscore the pathophysiologic proximity of the mechanisms that underlie the pathogenesis of these conditions. However, distinct differences also exist that obviate core differences in the pathophysiology of these conditions, and the knowledge of these differences is significant for the understanding of core pathomechanistic peculiarities of each condition. Table 4 depicts and compares the use of currently approved pharmacologic modalities for the systematic treatment of psoriasis and IBD.
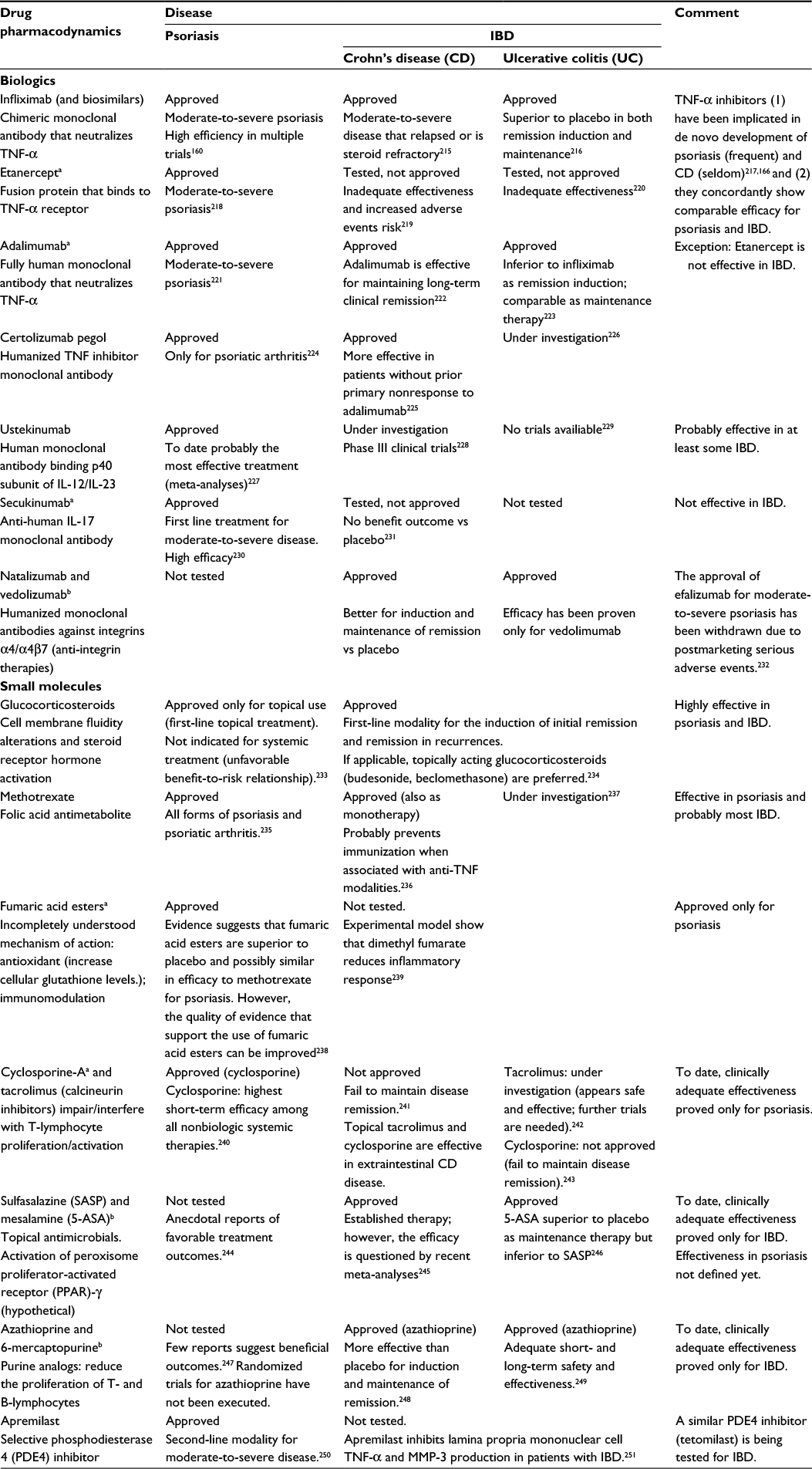 | Table 4 Comparison of the use of pharmacological treatment modalities for psoriasis and inflammatory bowel diseases (IBD) Notes: amodality approved only for psoriasis; bmodality approved only for IBD. Abbreviations: IBD, inflammatory bowel disease; TNF, tumor necrosis factor; MMP, matrix metalloproteinase. |
Although a number of different small molecules are used to treat both psoriasis and IBD, we focus on the comparison of the effectiveness of the mechanistically more predictable targeted biologic modalities.
Generally, TNF-α-targeting biologics are effective in both psoriasis and IBD with a vast differentiation: etanercept, which is approved for the treatment of psoriasis, is not effective in IBD.160,161 Paradoxically, treatment with anti-TNF-α modalities has been implicated in de novo development of psoriasis and, less frequently, of IBD.162–165 The incidence of anti-TNF-α-associated psoriasis-like manifestations has been estimated at one case per 1,000 patient-years or 3%–10% of the treated patients.107,166,167
More recently, monoclonal antibodies against IL-17 (sekukinumab, brodalimumab, ixekizumab) have been developed for the treatment of moderate-to-severe psoriasis, exhibiting exceptional efficacy.168 Secukinumab and brodalimumab have also been evaluated in randomized trials for moderate-to-severe CD, but neither reached significant effectiveness compared to placebo.169 The conflicting results provided by IL-17 inhibition verify the complex biology of this immune mechanism in CD as well as indicate the crucial differences with the role of Th-17 cells in the pathogenesis of psoriasis and IBD.170,171
Discussion
The concurrent existence of psoriasis and IBD in an increasing number of patients poses a new diagnostic and therapeutic challenge. Health professionals should be aware of the similarities and discrepancies in the pathogenesis and epidemiology of these diseases to successfully distinguish between their random coexistence, their causative concurrence, or their occurrence as a “paradoxical” side effect. IBD patients should be examined for skin lesions, as extraintestinal manifestations markedly increase the overall morbidity of these diseases.172–174
In this report, we also compared the role of immune cells in the pathophysiology of psoriasis and IBD. The anatomical and functional integrity of the tissue–environmental barriers is compromised in the skin and intestinal lumen of patients with psoriasis and IBD, respectively, allowing the invasion of allergens and immunomodulatory metabolites from an altered microflora. An increased number of DCs infiltrate the skin in psoriasis and the intestine in IBD. These cells are exposed there to a proinflammatory milieu that mainly favors a Th1 response. Through the interaction of M1 subset of macrophages with tissue MCs in both conditions, increased amounts of substances are released in the tissue environment that promotes the recruitment of neutrophils and lymphocytes. Recently, interest has focused on the role of IL-23/Th17 effector axis, as its interaction with keratinocytes and intestinal mucosa cells appears to be a central pathway for the pathogenesis of psoriasis and IBD and a promising target for treatment modalities.
The aforementioned immune similarities also form the basis for the wide overlap of treatment regimens that are used to treat psoriasis and IBD. However, there are also remarkable differences in the effectiveness of certain agents that highlight discrepancies in the pathophysiology of these conditions. Etanercept, for example, was not effective in IBD, and this has been attributed to a diminished ability of etanercept to induce T-cell apoptosis in the intestinal mucosa.175 Correspondingly, the ineffectiveness of secukinumab in the treatment of CD has been related to the importance of mycobiome in the pathogenesis of this disease.176 Given the high concordance of psoriasis and CD, it is legitimate to take into consideration the significant risk of CD flare inductions in patients receiving secukinumab, particularly as first-line treatment, for moderate-to-severe psoriasis.
It is similarly worth noting that the use of anti-TNF-α biologics to treat IBD, especially CD, has probably increased the prevalence of noninfectious dermatoses in these patients.177 Next to xerosis and atopic eczema exacerbations, psoriasis is the third most common skin manifestation provoked by the treatment with anti-TNF-α agents in these patients.177 The vast majority of the cases (76%) developed psoriasis while on infliximab, and the rest (24%) after switching to adalimumab or certolizumab, indicating that this phenomenon is not drug specific, but rather a pharmacological group effect.107 Cases of psoriasis induced by etanercept have been also reported.178 Regarding the management of paradoxical psoriasis, no specific guidelines are currently available. It has been hypothesized that individuals with newly developed psoriasis under treatment, whose cutaneous disease persists or recurs despite removal of the suspected agents, probably have an underlying previously unrecognized predisposition to psoriasis. However, those whose skin findings resolve completely after treatment modification are more likely to have suffered a true drug-induced psoriasis episode against the background of low genetic psoriasis preponderance.179
Conclusion
Available data support a high degree of overlapping epidemiologic profiles, genetic substrate, disease risk factors, inflammatory pathogenesis cascades, and also therapeutic procedures between psoriasis and IBD, particularly CD. Further studies may unravel the common mechanism that underlies these common chronic inflammatory diseases of skin and gut and may provide valuable insights for the development of innovative interventions for both conditions.
Disclosure
The authors report no conflicts of interest in this work.
References
Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361(5):496–509. | ||
Baumgart DC, Sandborn WJ. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet. 2007;369:1641–1657. | ||
Lolli E, Saraceno R, Calabrese E, et al. Psoriasis phenotype in inflammatory bowel disease: a case-control prospective study. J Crohns Colitis. 2015;9(9):699–707. | ||
Zippi M, Corrado C, Pica R, et al. Extraintestinal manifestations in a large series of Italian inflammatory bowel disease patients. World J Gastroenterol. 2014;20(46):17463–17467. | ||
Molodecky NA, Soon IS, Rabi DM, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142:46–54. | ||
Loftus EV Jr. Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology. 2004;126(6):1504–1517. | ||
Parisi R, Symmons DP, Griffiths CE, et al. Global epidemiology of psoriasis: a systematic review of incidence and prevalence. J Invest Dermatol. 2013;133(2):377–385. | ||
Yang H, Plevy SE, Taylor K. Linkage of Crohn’s disease to the major histocompatibility complex region is detected by multiple non-parametric analyses. Gut. 1999;44(4):519–526. | ||
Oestreicher J, Walters I, Kikuchi T, et al. Molecular classification of psoriasis disease-associated genes through pharmacogenomic expression profiling. Pharmacogenomics J. 2001;1(4):272–287. | ||
Farber EM, Nall ML, Watson W. Natural history of psoriasis in 61 twin pairs. Arch Dermatol. 1974;109(2):207–211. | ||
Watts DA, Satsangi J. The genetic jigsaw of inflammatory bowel disease. Gut. 2002;50(Suppl 3):III31–III36. | ||
Lee FI, Bellary SV, Francis C. Increased occurrence of psoriasis in patients with Crohn’s disease and their relatives. Am J Gastroenterol. 1990;85(8):962–963. | ||
Bernstein CN, Wajda A, Blanchard JF. The clustering of other chronic inflammatory diseases in inflammatory bowel disease: a population-based study. Gastroenterology. 2005;129(3):827–836. | ||
Cohen AD, Dreiher J, Birkenfeld S. Psoriasis associated with ulcerative colitis and Crohn’s disease. J Eur Acad Dermatol Venereol. 2009;23(5):561–565. | ||
Chandra A, Ray A, Senapati S, Chatterjee R. Genetic and epigenetic basis of psoriasis pathogenesis. Mol Immunol. 2015;64(2):313–323. | ||
Imielinski M, Baldassano RN, Griffiths A, et al. Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat Genet. 2009;41(12):1335–1340. | ||
Skroza N, Proietti I, Pampena R, et al. Correlations between psoriasis and inflammatory bowel diseases. Biomed Res Int. 2013;2013:983902. | ||
Ellinghaus D, Ellinghaus E, Nair RP, et al. Combined analysis of genome-wide association studies for Crohn disease and psoriasis identifies seven shared susceptibility loci. Am J Hum Genet. 2012;90(4):636–647. | ||
Dzutsev A, Goldszmid RS, Viaud S, Zitvogel L, Trinchieri G. The role of the microbiota in inflammation, carcinogenesis, and cancer therapy. Eur J Immunol. 2015;45(1):17–31. | ||
Sekirov I, Russell L, Antunes C, Finlay B. Gut microbiota in health and disease. Physiol Rev. 2010;90( 3):859–904. | ||
Capone KA, Dowd SE, Stamatas GN, Nikolovski J. Diversity of the human skin microbiome early in life. J Invest Dermatol. 2011;131(10):2026–2032. | ||
Basso PJ, Fonseca MT, Bonfa G, et al. Association among genetic predisposition, gut microbiota, and host immune response in the etiopathogenesis of inflammatory bowel disease. Braz J Med Biol Res. 2014;47(9):727–737. | ||
Furusawa Y, Obata Y, Fukuda S, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–450. | ||
Qiu J, Guo X, Chen ZM, et al. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity. 2013;39:386–399. | ||
Ott SJ, Musfeldt M, Timmis KN, Hampe J, Wenderoth DF, Schreiber S. In vitro alterations of intestinal bacterial microbiota in fecal samples during storage. Diagn Microbiol Infect Dis. 2004;50(4):237–245. | ||
Walker AW, Sanderson JD, Churcher C, et al. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011;11:7. | ||
Willing B, Halfvarson J, Dicksved J. Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohn’s disease. Inflamm Bowel Dis. 2009;15:653–660. | ||
Frank DN, Robertson CE, Hamm CM, et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis. 2011;17:179–184. | ||
Wehkamp J, Salzman NH, Porter E, et al. Reduced Paneth cell α-defensins in ileal Crohn’s disease. Proc Natl Acad Sci U S A. 2005;102:18129–18134. | ||
Rausch P, Rehman A, Künzel S. Colonic mucosa-associated microbiota is influenced by an interaction of Crohn disease and FUT2 (Secretor) genotype. Proc Natl Acad Sci U S A. 2011;108(47):19030–19035. | ||
Tamboli CP, Neut C, Desreumaux P, Colompel JF. Dysbiosis as a prerequisite for IBD. Gut. 2004;53(7):1057. | ||
Rook GA, Raison CL, Lowry CA, Hale LP, Lochs H. Microbiota, immunoregulatory old friends and psychiatric disorders. Adv Exp Med Biol. 2014;817:319–356. | ||
Swidsinski A, Weber J, Loening-Baucke V, Hale LP, Lochs H. Spatial organization and composition of the mucosal florain patients with inflammatory bowel disease. J Clin Microbiol. 2005;43(7):3380–3389. | ||
Oka A, Mabuchi T, Ozawa A, Inoko H. Current understanding of human genetics and genetic analysis of psoriasis. J Dermatol. 2012;39(3):231–241. | ||
Gallo RL, Nakatsuji T. Microbial symbiosis with the innate immune defense system of the skin. J Invest Dermatol. 2011;131(10):1974–1980. | ||
Gao Z, Tseng CH, Strober BE, Pei Z, Blaser MJ. Substantial alterations of the cutaneous bacterial biota in psoriatic lesions. PLoS One. 2008;3(7):e2719. | ||
Alekseyenko AV, Perez-Perez GI, De Souza A. Community differentiation of the cutaneous microbiota in psoriasis. Microbiome. 2013;1(1):31. | ||
Fry L, Baker BS, Powles AV, Fahlen A, Engstrand L. Is chronic plaque psoriasis triggered by microbiota in the skin? Br J Dermatol. 2013;169(1):47–52. | ||
Bassukas ID, Gaitanis G, Hundeiker M. Leprosy and the natural selection for psoriasis. Med Hypotheses. 2012;78(1):183–190. | ||
McFadden JP, Baker BS, Powles AV, Fry L. Psoriasis and streptococci: the natural selection of psoriasis revisited. Br J Dermatol. 2009;160(5):929–937. | ||
Mak RK, Hundhausen C, Nestle FO. Progress in understanding the immunopathogenesis of psoriasis. Actas Dermosifiliogr. 2009;100(Suppl 2):2–13. | ||
Morizane S, Gallo RL. Antimicrobial peptides in the pathogenesis of psoriasis. J Dermatol. 2012;39(3):22. | ||
Sweeney CM, Tobin AM, Kirby B. Innate immunity in the pathogenesis of psoriasis. Arch Dermatol Res. 2011;303(10):691–705. | ||
Di Meglio P, Duarte JH, Ahlfors H. Activation of the aryl hydrocarbon receptor dampens the severity of inflammatory skin conditions. Immunity. 2014;40(6):989–1001. | ||
Magiatis P, Pappas P, Gaitanis G, et al. Malassezia yeasts produce a collection of exceptionally potent activators of the Ah (dioxin) receptor detected in diseased human skin. J Invest Dermatol. 2013;133(8):2023–2030. | ||
Lin YK, Leu YL, Yang SH, Chen HW, Wang CT, Pang JH. Anti-psoriatic effects of indigo naturalis on the proliferation and differentiation of keratinocytes within dirubin as the active component. J Dermatol Sci. 2009;54(3):168–174. | ||
Salzman NH. Microbiota-immune system interaction: an uneasy alliance. Curr Opin Microbiol. 2011;14(1):99–105. | ||
Gerova VA, Stoynov SG, Katsarov DS, Svinarov DA. Increased intestinal permeability in inflammatory bowel diseases assessed by iohexol test. World J Gastroenterol. 2011;17(17):2211–2215. | ||
Kiesslich R, Duckworth CA, Moussata D, et al. Local barrier dysfunction identified by confocal laser endomicroscopy predicts relapse in inflammatory bowel disease. Gut. 2012;61(8):1146–1153. | ||
Mankertz J, Tavalali S, Schmitz H, et al. Expression from the human occludin promoter is affected by tumor necrosis factor α and interferon gamma. J Cell Sci. 2000;113(Pt 11):2085–2090. | ||
Buisine MP, Desreumaux P, Leteurtre E, et al. Mucin gene expression in intestinal epithelial cells in Crohn’s disease. Gut. 2001;49(4):544–551. | ||
Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun. 2000;68(12):7010–7017. | ||
Hausmann M, Kiessling S, Mestermann S, et al. Toll-like receptors 2 and 4 are up-regulated during intestinal inflammation. Gastroenterology. 2002;122(7):1987–2000. | ||
Visconti B, Paolino G, Carotti S, et al. Immunohistochemical expression of VDR is associated with reduced integrity of tight junction complex in psoriatic skin. J Eur Acad Dermatol Venereol. 2015;29(10):2038–2042. | ||
Roberson ED, Bowcock AM. Psoriasis genetics: breaking the barrier. Trends Genet. 2010;26(9):415–423. | ||
Panzer R, Blobel C, Fölster-Holst R, Proksch E. TLR2 and TLR4 expression in atopic dermatitis, contact dermatitis and psoriasis. Exp Dermatol. 2014;23(5):364–366. | ||
Niess JH, Brand S, Gu X, et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science. 2005;307:254–258. | ||
Varol C, Zigmond E, Jung S. Securing the immune tightrope: mononuclear phagocytes in the intestinal lamina propria. Nat Rev Immunol. 2010;10(6):415–426. | ||
Baumgart DC, Thomas S, Przesdzing I, et al. Exaggerated inflammatory response of primary human myeloid dendritic cells to lipopolysaccharide in patients with inflammatory bowel disease. Clin Exp Immunol. 2009;157(3):423–436. | ||
Annacker O, Coombes JL, Malmstrom V, et al. Essential role for CD103 in the T cell-mediated regulation of experimental colitis. J Exp Med. 2005;202(8):1051–1061. | ||
Siddiqui KR, Laffont S, Powrie F. E-cadherin marks a subset of inflammatory dendritic cells that promote T cell-mediated colitis. Immunity. 2010;32(4):557–567. | ||
Leal MC, Däbritz J. Immunoregulatory role of myeloid-derived cells in inflammatory bowel disease. Inflamm Bowel Dis. 2015;21(12):2936–2947. | ||
Glitzner E, Korosec A, Brunner PM, et al. Specific roles for dendritic cell subsets during initiation and progression of psoriasis. EMBO Mol Med. 2014;6(10):1312–1327. | ||
Gordon KB, Bonish BK, Patel T, Leonardi CL, Nickoloff BJ. The tumour necrosis factor-α inhibitor adalimumab rapidly reverses the decrease in epidermal Langerhans cell density in psoriatic plaques. Br J Dermatol. 2005;153(5):945–953. | ||
Nestle FO, Conrad C, Tun-Kyi A, et al. Plasmacytoid predendritic cells initiate psoriasis through interferon-α production. J Exp Med. 2005;202(1):135–143. | ||
Albanesi C, Scarponi C, Pallotta S, et al. Chemerin expression marks early psoriatic skin lesions and correlates with plasmacytoid dendritic cell recruitment. J Exp Med. 2009;206(1):249–258. | ||
Cerio R, Griffiths CE, Cooper KD, Nickoloff BJ, Headington JT. Characterization of factor XIIIa positive dermal dendritic cells in normal and inflamed skin. Br J Dermatol. 1989;121(4):421–431. | ||
Willenborg S, Eming SA. Macrophages – sensors and effectors coordinating skin damage and repair. J Dtsch Dermatol Ges. 2014;12(3):214–223. | ||
Mantovani A, Allavena P, Sica A. Tumour-associated macrophages as a prototypic type II polarised phagocyte population: role in tumour progression. Eur J Cancer. 2004;40(11):1660–1667. | ||
Bain CC, Scott CL, Uronen-Hansson H, et al. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 2013;6:498–510. | ||
Schenk M, Bouchon A, Seibold F, Mueller C. TREM-1 – expressing intestinal macrophages crucially amplify chronic inflammation in experimental colitis and inflammatory bowel diseases. J Clin Invest. 2007;117(10):3097–3106. | ||
Nickoloff BJ, Bonish BK, Marble DJ, et al. Lessons learned from psoriatic plaques concerning mechanisms of tissue repair, remodeling, and inflammation. J Investig Dermatol Symp Proc. 2006;11(1):16–29. | ||
Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823–835. | ||
Thepen T, van Vuuren AJ, Kiekens RC, Damen CA, Vooijs WC, van De Winkel JG. Resolution of cutaneous inflammation after local elimination of macrophages. Nat Biotechnol. 2000;18(1):48–51. | ||
Gilroy DW, Newson J, Sawmynaden P, Willoughby DA, Croxtall JD. A novel role for phospholipase A2 isoforms in the checkpoint control of acute inflammation. FASEB J. 2004;18(3):489–498. | ||
Lin HH, Faunce DE, Stacey M, et al. The macrophage F4/80 receptor is required for the induction of antigen-specific efferent regulatory T cells in peripheral tolerance. J Exp Med. 2005;201(10):1615–1625. | ||
Kritas SK, Saggini A, Varvara G, et al. Impact of mast cells on the skin. Int J Immunopathol Pharmacol. 2013;26(4):855–859. | ||
Hamilton MJ, Frei SM, Stevens RL. The multifaceted mast cell in inflammatory bowel disease. Inflamm Bowel Dis. 2014;20(12):2364–2378. | ||
Nishida Y, Murase K, Isomoto H, et al. Different distribution of mast cells and macrophages in colonic mucosa of patients with collagenous colitis and inflammatory bowel disease. Hepatogastroenterology. 2002;49:678–682. | ||
Nolte H, Spjeldnaes N, Kruse A, Windelborg B. Histamine release from gut mast cells from patients with inflammatory bowel diseases. Gut. 1990;31(7):791–794. | ||
Bischoff SC, Wedemeyer J, Herrmann A, et al. Quantitative assessment of intestinal eosinophils and mast cells in inflammatory bowel disease. Histopathology. 1996;28(1):1–13. | ||
Fox CC, Lazenby AJ, Moore WC, Yardley JH, Bayless TM, Lichtenstein LM. Enhancement of human intestinal mast cell mediator release in active ulcerative colitis. Gastroenterology. 1990;99:119–124. | ||
Groschwitz KR, Ahrens R, Osterfeld H, et al. Mast cells regulate homeostatic intestinal epithelial migration and barrier function by a chymase/Mcpt4-dependent mechanism. Proc Natl Acad Sci U S A. 2009;106(52):22381–22386. | ||
Wouters MM, Vicario M, Santos J. The role of mast cells in functional GI disorders. Gut. 2016;65(1):155–168. | ||
Harvima IT, Nilsson G, Suttle MM, Naukkarinen A. Is there a role for mast cells in psoriasis? Arch Dermatol Res. 2008;300(9):461–478. | ||
Cox AJ. Mast cells in psoriasis. In: Cox AJ, Farber EM, editors. Psoriasis, Proceedings of the Second International Symposium, Stanford University. New York, NY: Yorke Medical Books; 1977:36–43. | ||
Arck PC, Slominski A, Theoharides TC, Peters EM, Paus R. Neuroimmunology of stress: skin takes center stage. J Invest Dermatol. 2006;126(8):1697–1704. | ||
Rijnierse A, Nijkamp FP, Kraneveld AD. Mast cells and nerves tickle in the tummy: implications for inflammatory bowel disease and irritable bowel syndrome. Pharmacol Ther. 2007;116(2):207–235. | ||
Theoharides TC. The mast cell: a neuroimmunoendocrine master player. Int J Tissue React. 1996;18(1):1–21 | ||
Dunphy S, Gardiner CM. NK cells and psoriasis. J Biomed Biotechnol. 2011;2011:248317. | ||
Yadav PK, Chen C, Liu Z. Potential role of NK cells in the pathogenesis of inflammatory bowel disease. J Biomed Biotechnol. 2011;2011:348530. | ||
Liu Z, Yang L, Cui Y, et al. IL-21 enhances NK cell activation and cytolytic activity and induces Th17 cell differentiation in inflammatory bowel disease. Inflamm Bowel Dis. 2009;15(8):1133–1144. | ||
Takayama T, Kamada N, Chinen H, et al. Imbalance of NKp44(+)NKp46(–) and NKp44(NKp46+) natural killer cells in the intestinal mucosa of patients with Crohn’s disease. Gastroenterology. 2010;139(3):882–892, 892.e1–3. | ||
Ottaviani C, Nasorri F, Bedini C, de Pità O, Girolomoni G, Cavani A. CD56 bright CD16(–) NK cells accumulate in psoriatic skin in response to CXCL10 and CCL5 and exacerbate skin inflammation. Eur J Immunol. 2006;36(1):118–128. | ||
Yawalkar N, Schmid S, Braathen LR, Pichler WJ. Perforin and granzyme B may contribute to skin inflammation in atopic dermatitis and psoriasis. Br J Dermatol. 2001;144(6):1133–1139. | ||
Giacomelli R, Passacantando A, Frieri G, et al. Circulating soluble factor-inhibiting natural killer (NK) activity of fresh peripheral blood mononuclear cells (PBMC) from inflammatory bowel disease (IBD) patients. Clin Exp Immunol. 1999;115(1):72–77. | ||
Sonnenberg GF, Mjösberg J, Spits H, Artis D. SnapShot: innate lymphoid cells. Immunity. 2013;39(3):622–622.e1. | ||
Cupedo T, Crellin NK, Papazian N, et al. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat Immunol. 2009;10(1):66–74. | ||
Buonocore S, Ahern PP, Uhlig HH, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. 2010;464(7293):1371–1375. | ||
Lee JS, Cella M, McDonald KG, et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol. 2012;13:144–151. | ||
Longman RS, Diehl GE, Victorio DA, et al. CX3CR1+ mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Med. 2014;211(8):1571–1583. | ||
Hanash AM, Dudakov JA, Hua G, et al. Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity. 2012;24:339–350. | ||
Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity. 2008;19:947–957. | ||
Villanova F, Flutter B, Tosi I, et al. Characterization of innate lymphoid cells in human skin and blood demonstrates increase of NKp44+ ILC3 in psoriasis. J Invest Dermatol. 2014;134(4):984–991. | ||
Teunissen MB, Munneke JM, Bernink JH, et al. Composition of innate lymphoid cell subsets in the human skin: enrichment of NCR(+) ILC3 in lesional skin and blood of psoriasis patients. J Invest Dermatol. 2014;134(9):2351–2360. | ||
Salimi M, Ogg G. Innate lymphoid cells and the skin. BMC Dermatol. 2014;14:18. | ||
Fiorino G, Omodei PD. Psoriasis and inflammatory bowel disease: two sides of the same coin? J Crohns Colitis. 2015;9(9):697–698. | ||
Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28(4):454–467. | ||
Steinman L. A brief history of TH17, the first major revision in the TH1/TH2 hypothesis of T cell-mediated tissue damage. Nat Med. 2007;13(2):139–145. | ||
Zenewicz LA, Antov A, Flavell RA. CD4 T-cell differentiation and inflammatory bowel disease. Trends Mol Med. 2009;15(5):199–207. | ||
Thompson I, Lees CW. Genetics of ulcerative colitis. Inflamm Bowel Dis. 2011;17(3):831–848. | ||
Galvez J. Role of Th17 cells in the pathogenesis of human IBD. ISRN Inflamm. 2014;25:928461. | ||
Marinoni B, Ceribelli A, Massarotti M, Selmi C. The Th17 axis in psoriatic disease: pathogenetic and therapeutic implications. Auto Immun Highlights. 2014;5(1):9–19. | ||
Fitch E, Harper E, Skorcheva I, Kurtz SE, Blauvelt A. Pathophysiology of psoriasis: recent advances on IL-23 and TH17 cytokines. Curr Rheumatol Rep. 2007;9(6):461–467. | ||
Asarch A, Barak O, Loo DS, Gottlieb AB. Th17 cells: a new paradigm for cutaneous inflammation. J Dermatolog Treat. 2008;19(5):259–266. | ||
Harden JL, Krueger JG, Bowcock AM. The immunogenetics of psoriasis: a comprehensive review. J Autoimmun. 2015;64:66–73. | ||
Sakaguchi S, Wing K, Miyara M, et al. Regulatory T cells – a brief history and perspective. Eur J Immunol. 2007;37(Suppl 1):S116–S123. | ||
Saruta M, Yu QT, Fleshner PR, et al. Characterization of FOXP3+CD4+ regulatory T cells in Crohn’s disease. Clin Immunol. 2007;125(3):281–290. | ||
Yu QT, Saruta M, Avanesyan A, Fleshner PR, Banham AH, Papadakis KA. Expression and functional characterization of FOXP3+CD4+ regulatory T cells in ulcerative colitis. Inflamm Bowel Dis. 2007;13(2):191–199. | ||
Maul J, Loddenkemper C, Mundt P, et al. Peripheral and intestinal regulatory CD4+ CD25(high) T cells in inflammatory bowel disease. Gastroenterology. 2005;128(7):1868–1878. | ||
Wang Y, Liu XP, Zhao ZB, et al. Expression of CD4+ forkhead box P3 (FOXP3)+ regulatory T cells in inflammatory bowel disease. J Dig Dis. 2011;12(4):286–294. | ||
Hirahara K, Liu L, Clark RA, Yamanaka K, Fuhlbrigge RC, Kupper TS. The majority of human peripheral blood CD4+CD25highFoxp3+ regulatory T cells bear functional skin-homing receptors. J Immunol. 2006;177(7):4488–4494. | ||
Bovenschen HJ, van Vlijmen-Willems IM, van de Kerkhof PC, van Erp PE. Identification of lesional CD4+ CD25+ Foxp3+ regulatory T cells in Psoriasis. Dermatology. 2006;213(2):111–117. | ||
Gao L, Li K, Li F, et al. Polymorphisms in the FOXP3 gene in Han Chinese psoriasis patients. J Dermatol Sci. 2010;57(1):51–56. | ||
Park O, Grishina I, Leung PS, Gershwin ME, Prindiville T. Analysis of the Foxp3/scurfin gene in Crohn’s disease. Ann N Y Acad Sci. 2005;1051:218–228. | ||
Binus AM, Han J, Qamar AA, Mody EA, Holt EW, Qureshi AA. Associated comorbidities in psoriasis and inflammatory bowel disease. J Eur Acad Dermatol Venereol. 2012;26(5):644–650. | ||
Abuabara K, Azfar RS, Shin DB, Neimann AL, Troxel AB, Gelfand JM. Cause-specific mortality in patients with severe psoriasis: a population-based cohort study in the UK. Br J Dermatol. 2010;163: 586–592. | ||
Onumah N, Kircik LH. Psoriasis and its comorbidities. J Drugs Dermatol. 2012;11(5 Suppl):S5–S10. | ||
Casella G, Tontini GE, Bassotti G, et al. Neurological disorders and inflammatory bowel diseases. World J Gastroenterol. 2014;20(27):8764–8782. | ||
Card T, Hubbard R, Logan RF. Mortality in inflammatory bowel disease: a population-based cohort study. Gastroenterology. 2003;125(6):1583–1590. | ||
Müller-Hermelink N, Braumüller H, Pichler B, et al. TNFR1 signaling and IFN-gamma signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell. 2008;13(6):507–518. | ||
Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. | ||
Zumsteg A, Christofori G. Corrupt policemen: inflammatory cells promote tumor angiogenesis. Curr Opin Oncol. 2009;21:60–70. | ||
Zamarron BF, Chen W. Dual roles of immune cells and their factors in cancer development and progression. Int J Biol Sci. 2011;7:651–658. | ||
Bassukas ID, Gaitanis G, Tsianos E. Azathioprine: an explanation for the risk of skin SCC among patients with Crohn’s disease? Ann Rheum Dis. 2009;68, eLetter June 15, 2009; Available from: http://ard.bmj.com/content/68/12/1863.full/reply#annrheumdis_el_5801. Accessed May 3, 2016. | ||
Monteiro R, Azevedo I. Chronic inflammation in obesity and the metabolic syndrome. Mediators Inflamm. 2010;2010:289645. | ||
Hamminga EA, van der Lely AJ, Neumann HA, Thio HB. Chronic inflammation in psoriasis and obesity: implications for therapy. Med Hypotheses. 2006;67(4):768–773. | ||
Armstrong AW, Harskamp CT, Armstrong EJ,. The association between psoriasis and obesity: a systematic review and meta-analysis of observational studies. Nutr Diabetes. 2012;2:e54. | ||
Mattioli B, Straface E, Quaranta MG, Giordani L, Viora M. Leptin promotes differentiation and survival of human dendritic cells and licenses them for Th1 priming. J Immunol. 2005;174(11):6820–6828. | ||
Wang Y, Chen J, Zhao Y, Geng L, Song F, Chen HD. Psoriasis is associated with increased levels of serum leptin. Br J Dermatol. 2008;158(5):1134–1135. | ||
Lomer MC, Cook WB, Jan-Mohamed HJ, et al. Iron requirements based upon iron absorption tests are poorly predicted by haematological indices in patients with inactive inflammatory bowel disease. Br J Nutr. 2012;107(12):1806–11. | ||
Steed H, Walsh S, Reynolds N. A brief report of the epidemiology of obesity in the inflammatory bowel disease population of Tayside, Scotland. Obes Facts. 2009;2(6):370–372. | ||
Andrade M, Maio R, Dourado KF, Macêdo PF, Barreto Neto AC. Excessive weight muscle – depletion paradox and cardiovascular risk factors in outpatients with inflammatory bowel disease. Arg Gastroenterol. 2015;52(1):37–45. | ||
Bertin B, Desreumaux P, Dubuquoy L. Obesity, visceral fat and Crohn’s disease. Curr Opin Clin Nutr Metab Care. 2010;13(5):574–580. | ||
Chapman-Kiddell CA, Davies PS, Gillen L, Radford-Smith GL. Role of diet in the development of inflammatory bowel disease. Inflamm Bowel Dis. 2010;16(1):137–151. | ||
Mehta NN, Yu Y, Pinnelas R, et al. Attributable risk estimate of severe psoriasis on major cardiovascular events. Am J Med. 2011;124(8):775.e1–e6. | ||
Balta I, Balta S, Demirkol S, et al. Aortic arterial stiffness is a moderate predictor of cardiovascular disease in patients with psoriasis vulgaris. Angiology. 2014;65(1):74–78. | ||
Nofal A, Al-Makhzangy I, Attwa E, Nassar A, Abdalmoati A. Vascular endothelial growth factor in psoriasis: an indicator of disease severity and control. J Eur Acad Dermatol Venereol. 2009;23(7):803–806. | ||
McDonald I, Connolly M, Tobin AM. A review of psoriasis, a known risk factor for cardiovascular disease and its impact on folate and homocysteine metabolism. J Nutr Metab. 2012;2012:965385. | ||
Dorn SD, Sandler RS. Inflammatory bowel disease is not a risk factor for cardiovascular disease mortality: results from a systematic review and meta-analysis. Am J Gastroenterol. 2007;102(3):662–667. | ||
Graef V, Baggenstoss AH, Sauer WG, Spittell JA Jr. Venous thrombosis occurring in nonspecific ulcerative colitis. A necropsy study. Arch Intern Med. 1966;117:377–382. | ||
Ruisi P, Makaryus JN, Ruisi M, Makaryus AN. Inflammatory bowel disease as a risk factor for premature coronary artery disease. J Clin Med Res. 2015;7(4):257–261. | ||
Gisondi P, Cotena C, Tessari G, Girolomoni G. Anti-tumour necrosis factor-α therapy increases body weight in patients with chronic plaque psoriasis: a retrospective cohort study. J Eur Acad Dermatol Venereol. 2008;22(3):341–344. | ||
Rho YH, Chung CP, Oeser A, et al. Inflammatory mediators and premature coronary atherosclerosis in rheumatoid arthritis. Arthritis Rheum. 2009;61(11):1580–1585. | ||
Dagli N, Poyrazoglu OK, Dagli AF, et al. Is inflammatory bowel disease a risk factor for early atherosclerosis? Angiology. 2010;61:198–204. | ||
Mitchell GF. Arterial stiffness and wave reflection: biomarkers of cardiovascular risk. Artery Res. 2009;3:56–64. | ||
Micha R, Imamura F, Wyler von Ballmoos M, et al. Systematic review and meta-analysis of methotrexate use and risk of cardiovascular disease. Am J Cardiol. 2011;108(9):1362–1370. | ||
Famenini S, Sako EY, Wu JJ. Effect of treating psoriasis on cardiovascular co-morbidities: focus on TNF inhibitors. Am J Clin Dermatol. 2014;15(1):45–50. | ||
Abuabara K, Lee H, Kimball AB. The effect of systemic psoriasis therapies on the incidence of myocardial infarction: a cohort study. Br J Dermatol. 2011;165: 1066–1073. | ||
Saraceno R, Saggini A, Pietroleonardo L, Chimenti S. Infliximab in the treatment of plaque type psoriasis. Clin Cosmet Investig Dermatol. 2009;2:27–37. | ||
Akobeng AK, Zachos M. Tumor necrosis factor-α antibody for induction of remission in Crohn’s disease. Cochrane Database Syst Rev. 2004;(1):CD003574. | ||
Fiorino G, Allez M, Malesci A, Danese S. Review article: anti TNF-α induced psoriasis in patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2009;29(9):921–927. | ||
Mocciaro F, Renna S, Orlando A, Cottone M. Severe cutaneous psoriasis after certolizumab pegol treatment: report of a case. Am J Gastroenterol. 2009;104:2867–2868. | ||
Harris MD, Richards R. First case report of adalimumab-induced psoriasis in Crohn’s disease. Am J Gastroenterol. 2009;104:792–793. | ||
Verea MM, Del Pozo J, Yebra-Pimentel MT, Porta A, Fonseca E. Psoriasiform eruption induced by infliximab. Ann Pharmacother. 2004;38:54–57. | ||
Tichy M, Hercogova J. Manifestation of Crohn’s disease in a young woman during the course of treatment for severe form of chronic plaque psoriasis with etanercept. Dermatol Ther. 2014;27(4):211–214. | ||
Harrison MJ, Dixon WG, Watson KD, et al. Rates of new-onset psoriasis in patients with rheumatoid arthritis receiving anti-TNF-α therapy: results from the British Society for Rheumatology Biologics Register. Ann Rheum Dis. 2009;68:209–215. | ||
Yiu ZZ, Griffiths CE. Interleukin 17-A inhibition in the treatment of psoriasis. Expert Rev Clin Immunol. 2016;12(1):1–4. | ||
Targan S, Feagan B, Vermeire S, et al. Mo2083 a randomized, double-blind, placebo-controlled study to evaluate the safety, tolerability, and efficacy of AMG 827 in subjects with moderate to severe Crohn’s disease. Gastroenterology. 2012;143(3):e26. | ||
Ogawa A, Andoh A, Araki Y, Bamba T, Fujiyama Y. Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin. Immunol. 2004;110:55–62. | ||
Yen D, Cheung J, Scheerens H, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–1316. | ||
Monsen U, Sorstad J, Hellers G, Johansson C. Extracolonic diagnoses in ulcerative colitis: an epidemiological study. Am J Gastroenterol. 1990;85:711–716. | ||
Das KM. Relationship of extraintestinal involvements in inflammatory bowel disease: new insights into autoimmune pathogenesis. Dig Dis Sci. 1999;44(1):1–13. | ||
Vavricka SR, Brun L, Ballabeni P, et al. Frequency and risk factors for extraintestinal manifestations in the Swiss inflammatory bowel disease cohort. Am J Gastroenterol. 2011;106(1):110–119. | ||
Atreya R, Zimmer M, Bartsch B, et al. Antibodies against tumor necrosis factor (TNF) induce T-cell apoptosis in patients with inflammatory bowel diseases via TNF receptor 2 and intestinal CD14+ macrophages. Gastroenterology. 2011;141(6):2026–2038. | ||
Colombel JF, Sendid B, Jouault T, Poulain D. Secukinumab failure in Crohn’s disease: the yeast connection? Gut. 2013;62:800–801. | ||
Eligius Hellström A, Färkkilä M, Kolho KL. Infliximab-induced skin manifestations in patients with inflammatory bowel disease. Scand J Gastroenterol. 2016;51(5):563–571. | ||
Denadai R, Teixeira FV, Saad-Hossne R. Management of psoriatic lesions associated with anti-TNF therapy in patients with IBD. Nat Rev Gastroenterol Hepatol. 2012;9(12):744. | ||
Cullen G, Kroshinsky D, Cheifetz AS, Korzenik JR. Psoriasis associated with anti-tumour necrosis factor therapy in inflammatory bowel disease: a new series and a review of 120 cases from the literature. Aliment Pharmacol Ther. 2011;34(11–12):1318–1327. | ||
Gudjonsson JE, Karason A, Runarsdottir EH, et al. Distinct clinical differences between HLA-Cw*0602 positive and negative psoriasis patients – an analysis of 1019 HLA-C- and HLAB-typed patients. J Invest Dermatol. 2006;126(4):740–745. | ||
Anderson CA, Massey DC, Barrett JC, et al. Investigation of Crohn’s disease risk loci in ulcerative colitis further defines their molecular relationship. Gastroenterology. 2009;136(2):523–529.e3. | ||
Cargill M, Schrodi SJ, Chang M, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. 2007;80(2):273–290. | ||
Safrany E, Szabo M, Szell M, et al. Difference of interleukin-23 receptor gene haplotype variants in ulcerative colitis compared to Crohn’s disease and psoriasis. Inflamm Res. 2013;62(2):195–200. | ||
Capon F, Di Meglio P, Szaub J, et al. Sequence variants in the genes for the interleukin-23 receptor (IL23R) and its ligand (IL12B) confer protection against psoriasis. Hum Genet. 2007;122(2):201–206. | ||
Wolf N, Quaranta M, Prescott NJ, et al. Psoriasis is associated with pleiotropic susceptibility loci identified in type II diabetes and Crohn disease. J Med Genet. 2008;45(2):114–116. | ||
Capon F, Bijlmakers MJ, Wolf N, et al. Identification of ZNF313/RNF114 as a novel psoriasis susceptibility gene. Hum Mol Genet. 2008;17(13):1938–1945. | ||
Kugathasan S, Baldassano RN, Bradfield JP, et al. Loci on 20q13 and 21q22 are associated with pediatric-onset inflammatory bowel disease. Nature Genet. 2008;40:1211–1215. | ||
Hüffmeier U, Lascorz J, Becker T, et al. Characterisation of psoriasis susceptibility locus 6 (PSORS6) in patients with early onset psoriasis and evidence for interaction with PSORS1. J Med Genet. 2009;46(11):736–744. | ||
Rioux JD, Silverberg MS, Daly MJ, et al. Genomewide search in Canadian families with inflammatory bowel disease reveals two novel susceptibility loci. Am J Hum Genet. 2000;66(6):1863–1870. | ||
Hampe J, Shaw SH, Saiz R, et al. Linkage of inflammatory bowel disease to human chromosome 6p. Am J Hum Genet. 1999;65(6):1647–1655. | ||
Rioux JD, Daly MJ, Silverberg MS, et al. Genetic variation in the 5q31 cytokine gene cluster confers susceptibility to Crohn disease. Nature Genet. 2001;29:223–228. | ||
Stalder JF, Tennstedt D, Deleuran M, et al. Fragility of epidermis and its consequence in dermatology. J Eur Acad Dermatol Venereol. 2014;28(Suppl 4):1–18. | ||
Cameron AL, Kirby B, Griffiths CE. Circulating natural killer cells in psoriasis. Br J Dermatol. 2003;149(1):160–164. | ||
Hueber W, Sands BE, Lewitzky S, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61(12):1693–1700. | ||
Gladman DD. Psoriatic arthritis. Rheum Dis Clin North Am. 1998;24:829–844. | ||
Atzeni F, Defendenti C, Ditto MC. Rheumatic manifestations in inflammatory bowel disease. Autoimmun Rev. 2014;13(1):20–23. | ||
Lemos LL, de Oliveira Costa J, Almeida AM. Treatment of psoriatic arthritis with anti-TNF agents: a systematic review and meta-analysis of efficacy, effectiveness and safety. Rheumatol Int. 2014;34(10):1345–1360. | ||
Trikudanathan G, Venkatesh PG, Navaneethan U. Diagnosis and therapeutic management of extra-intestinal manifestations of inflammatory bowel disease. Drugs. 2012;72(18):2333–2349. | ||
Thapa S, Hadid H, Schairer J, Imam W, Jafri SM. Effect of inflammatory bowel disease-related characteristics and treatment interventions on cardiovascular disease incidence. Am J Med Sci. 2015;350(3):175–180. | ||
Hugh J, Van Voorhees AS, Nijhawan RI, et al. From the Medical Board of the National Psoriasis Foundation: the risk of cardiovascular disease in individuals with psoriasis and the potential impact of current therapies. J Am Acad Dermatol. 2014;70(1):168–177. | ||
Basavaraj KH, Ashok NM, Rashmi R, Praveen TK. The role of drugs in the induction and/or exacerbation of psoriasis. Int J Dermatol. 2010;49(12):1351–1361. | ||
Dubeau MF, Iacucci M, Beck PL, et al. Drug-induced inflammatory bowel disease and IBD-like conditions. Inflamm Bowel Dis. 2013;19(2):445–456. | ||
Miele L, Vallone S, Cefalo C, et al. Prevalence, characteristics and severity of non-alcoholic fatty liver disease in patients with chronic plaque psoriasis. J Hepatol. 2009;51(4):778–786. | ||
Wenk KS, Arrington KC, Ehrlich A. Psoriasis and non-alcoholic fatty liver disease. J Eur Acad Dermatol Venereol. 2011;25(4):383–391. | ||
Tsaitas C, Semertzidou A, Sinakos E. Update on inflammatory bowel disease in patients with primary sclerosing cholangitis. World J Hepatol. 2014;6(4):178–187. | ||
Gisbert JP, González-Lama Y, Maté J. Thiopurine-induced liver injury in patients with inflammatory bowel disease: a systematic review. Am J Gastroenterol. 2007;102:1518–1527. | ||
Bangemann K, Schulz W, Wohlleben J, et al. Depression and anxiety disorders among psoriasis patients: protective and exacerbating factors. Hautarzt. 2014;65(12):1056–1061. | ||
Walker JR, Graff LA, Dutz JP, Bernstein CN. Psychiatric disorders in patients with immune-mediated inflammatory diseases: prevalence, association with disease activity, and overall patient well-being. J Rheumatol Suppl. 2011;88:31–35. | ||
Lerebours E, Gower-Rousseau C, Merle V, et al. Stressful life events as a risk factor for inflammatory bowel disease onset: a population based case-control study. Am J Gastroenterol. 2007;102:122–131. | ||
Bernstein CN, Blanchard JF, Kliewer E, Wajda A. Cancer risk in patients with inflammatory bowel disease: a population-based study. Cancer. 2001;91:854–862. | ||
Beyaert R, Beaugerie L, Van Assche G, et al. Cancer risk in immune-mediated inflammatory diseases (IMID). Mol Cancer. 2013;12(1):98. | ||
Pedersen N, Duricova D, Elkjaer M, Gamborg M, Munkholm P, Jess T. Risk of extra-intestinal cancer in inflammatory bowel disease: meta-analysis of population-based cohort studies. Am J Gastroenterol. 2010;105:1480–1487. | ||
Laukoetter MG, Mennigen R, Hannig CM, et al. Intestinal cancer risk in Crohn’s disease: a meta-analysis. J Gastrointest Surg. 2011;15(4):576–583. | ||
Canavan C, Abrams KR, Mayberry J. Meta-analysis: colorectal and small bowel cancer risk in patients with Crohn’s disease. Aliment Pharmacol Ther. 2006;23:1097–1110. | ||
Dignass A, Van Assche G, Lindsay JO, et al. The second European evidence-based Consensus on the diagnosis and management of Crohn’s disease: current management. J Crohns Colitis. 2010;4(1):28–62. | ||
Akiho H, Yokoyama A, Abe S, et al. Promising biological therapies for ulcerative colitis: A review of the literature. World J Gastrointest Pathophysiol. 2015;6(4):219–227. | ||
Olteanu R, Zota A. Paradoxical reactions induced by tumor necrosis factor-α antagonists: a literature review based on 46 cases. Indian J Dermatol Venereol Leprol. 2016;82(1):7–12. | ||
Kivelevitch D, Mansouri B, Menter A. Long term efficacy and safety of etanercept in the treatment of psoriasis and psoriatic arthritis. Biologics. 2014;8:169–182. | ||
Kaser A. Not all monoclonals are created equal – lessons from failed drug trials in Crohn’s disease. Best Pract Res Clin Gastroenterol. 2014;28(3):437–449. | ||
Mozaffari S, Nikfar S, Abdolghaffari AH, Abdollahi M. New biologic therapeutics for ulcerative colitis and Crohn’s disease. Expert Opin Biol Ther. 2014;14(5):583–600. | ||
Puig L, López A, Vilarrasa E, García I. Efficacy of biologics in the treatment of moderate-to-severe plaque psoriasis: a systematic review and meta-analysis of randomized controlled trials with different time points. J Eur Acad Dermatol Venereol. 2014;28(12):1633–1653. | ||
Watanabe M, Hibi T, Mostafa NM, et al. Long-term safety and efficacy of adalimumab in Japanese patients with moderate to severe Crohn’s disease. J Crohns Colitis. 2014;8(11):1407–1416. | ||
Thorlund K, Druyts E, Mills EJ, Fedorak RN, Marshall JK. Adalimumab versus infliximab for the treatment of moderate to severe ulcerative colitis in adult patients naïve to anti-TNF therapy: an indirect treatment comparison meta-analysis. J Crohns Colitis. 2014;8(7):571–581. | ||
Cauli A, Piga M, Lubrano E, Marchesoni A, Floris A, Mathieu A. New approaches in tumor necrosis factor antagonism for the treatment of psoriatic arthritis: certolizumab pegol. J Rheumatol Suppl. 2015;93:70–72. | ||
Moon W, Pestana L, Becker B. Efficacy and safety of certolizumab pegol for Crohn’s disease in clinical practice. Aliment Pharmacol Ther. 2015;42(4):428–440. | ||
Arora Z, Shen B. Biological therapy for ulcerative colitis. Gastroenterol Rep. 2015;3(2):103–109. | ||
Campanati A, Ganzetti G, Giuliodori K, Molinelli E, Offidani A. Biologic therapy in psoriasis: safety profile. Curr Drug Saf. 2016;11(1):4–11. | ||
Simon EG, Ghosh S, Iacucci M, Moran GW. Ustekinumab for the treatment of Crohn’s disease: can it find its niche? Therap Adv Gastroenterol. 2016;9(1):26–36. | ||
Cote-Daigneault J, Bouin M, Lahaie R, Colombel JF, Poitras P. Biologics in inflammatory bowel disease: what are the data? United European Gastroenterol J. 2015;3(5):419–428. | ||
Jaleel T, Elmets C, Weinkle A, Kassira S, Elewski B. Secukinumab (AIN-457) for the treatment of psoriasis. Expert Rev Clin Pharmacol. 2016;9(2):187–202. | ||
Hueber W, Sands BE, Lewitzky S, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61(12):1693–1700. | ||
Genentech. Voluntary U.S. Market Withdrawal of Raptiva® (Efalizumab). 2009. Available from: www.fda.gov/downloads/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/UCM149681.pdf. Accessed May 2, 2016. | ||
Mrowietz U, Domm S. Systemic steroids in the treatment of psoriasis: what is fact, what is fiction? J Eur Acad Dermatol Venereol. 2013;27(8):1022–1025. | ||
Nunes T, Barreiro-de Acosta M, Marin-Jimenez I, Nos P, Sans M. Oral locally active steroids in inflammatory bowel disease. J Crohns Colitis. 2013;7(3):183–191. | ||
Czarnecka-Operacz M, Sadowska-Przytocka A. The possibilities and principles of methotrexate treatment of psoriasis – the updated knowledge. Postepy Dermatol Alergol. 2014;31(6):392–400. | ||
Vaysse T, Carbonnel F. Methotrexate in IBD: the return of the prodigal son. J Crohns Colitis. 2015;9(4):303–304. | ||
Kanwar AJ, Yadav S, Dogra S. Psoriasis: what is new in nonbiologic systemic therapy in the era of biologics? Indian J Dermatol Venereol Leprol. 2010;76(6):622–633. | ||
Atwan A, Ingram JR, Abbott R, et al. Oral fumaric acid esters for psoriasis. Cochrane Database Syst Rev. 2015;8:CD010497. | ||
Casili G, Cordaro M, Impellizzeri D, et al. Dimethyl fumarate reduces inflammatory responses in experimental colitis. J Crohns Colitis. 2016;10(4):472–483. | ||
Soleymani T, Vassantachart JM, Wu JJ. Comparison of guidelines for the use of cyclosporine for psoriasis: a critical appraisal and comprehensive review. J Drugs Dermatol. 2016;15(3):293–301. | ||
Thin LW, Murray K, Lawrance IC. Oral tacrolimus for the treatment of refractory inflammatory bowel disease in the biologic era. Inflamm Bowel Dis. 2013;19(7):1490–1498. | ||
Yamammoto T, Shimoyama T, Umegae S, Matsumoto K. Tacrolimus vs antitumor necrosis factor agents for moderately to severely active ulcerative colitis: a retrospective observational study. Aliment Pharmacol Ther. 2016;43(6):705–716. | ||
Hoentjen F, Sakuraba A, Hanauer S. Update on the management of ulcerative colitis. Curr Gastroenterol Rep. 2011;13(5):475–485. | ||
Bharti R. Mesalazine in treatment of psoriasis. Indian J Dermatol Venereol Leprol. 1996;62(4):231–232. | ||
Moja L, Danese S, Fiorino G, Del Giovane C, Bonovas S. Systematic review with network meta-analysis: comparative efficacy and safety of budesnide and mesalazine for Crohns disease. Aliment Pahrmacol Ther. 2015;41(11):1055–1065. | ||
Feagan BG, Macdonald JK. Oral 5-aminosalicylic acid for maintenance of remission in ulcerative colitis. Cochrane Database Syst Rev. 2012;10:CD000544. | ||
Du Vivier A, Munro DD, Verbov J. Treatment of psoriasis with azathioprine. Br Med J. 1974;1:49–51. | ||
Chande N, Patton PH, Tsoulis DJ, Thomas BS, MacDonald JK. Azathioprine or 6-mercaptopurine for maintenance of remission in Crohn’s disease. Cochrane Database Syst Rev. 2015;10:CD000067. | ||
Sood R, Ansari S, Clark T, Hamlin PJ, Ford AC. Long-term efficacy and safety of azathioprine in ulcerative colitis. J Crohns Colitis. 2015;9(2):191–197. | ||
Fala L. Otezla (Apremilast), an oral PDE-4 inhibitor, receives FDA approval for the treatment of patients with active psoriatic arthritis and plaque psoriasis. Am Health Drug Benefits. 2015;8(Spec Feature):105–110. | ||
Gordon JN, Prothero JD, Thornton CA. CC-10004 but not thalidomide or lenalidomide inhibits lamina propria mononuclear cell TNF-α and MMP-3 production in patients with inflammatory bowel disease. J Crohns Colitis. 2009;3(3):175–182. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.