Back to Journals » Journal of Hepatocellular Carcinoma » Volume 9
Protodioscin Induces Mitochondrial Apoptosis of Human Hepatocellular Carcinoma Cells Through Eliciting ER Stress-Mediated IP3R Targeting Mfn1/Bak Expression
Authors Yu CL, Lee HL, Yang SF ![]() , Wang SW, Lin CP, Hsieh YH
, Wang SW, Lin CP, Hsieh YH ![]() , Chiou HL
, Chiou HL
Received 31 December 2021
Accepted for publication 20 April 2022
Published 24 April 2022 Volume 2022:9 Pages 327—341
DOI https://doi.org/10.2147/JHC.S355027
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Imam Waked
Chen-Lin Yu,1,2,* Hsiang-Lin Lee,3,4,* Shun-Fa Yang,2,5 Shih-Wei Wang,1,6 Ching-Pin Lin,4,7 Yi-Hsien Hsieh,2,5 Hui-Ling Chiou8,9
1Institute of Biomedical Science, Mackay Medical College, New Taipei City, Taiwan; 2Institute of Medicine, Chung Shan Medical University, Taichung, Taiwan; 3Department of Surgery, Chung Shan Medical University Hospital, Taichung, Taiwan; 4School of Medicine, Chung Shan Medical University, Taichung, Taiwan; 5Department of Medical Research, Chung Shan Medical University Hospital, Taichung, Taiwan; 6Graduate Institute of Natural Products, College of Pharmacy, Kaohsiung Medical University, Kaohsiung, Taiwan; 7Division of Hepatology and Gastroenterology, Department of Internal Medicine, Chung Shan Medical University Hospital, Taichung, Taiwan; 8School of Medical Laboratory and Biotechnology, Chung Shan Medical University, Taichung, Taiwan; 9Department of Clinical Laboratory, Chung Shan Medical University Hospital, Taichung, Taiwan
*These authors contributed equally to this work
Correspondence: Yi-Hsien Hsieh; Hui-Ling Chiou, Tel +886-4-2473-0022, Fax +886-4- 2472-3229, Email [email protected]; [email protected]
Objective: Protodioscin (PD), a steroidal saponin, has a diverse pharmacological activity including neuroprotection, male fertility improvement, and cytotoxicity against various cancers cell lines of different origins. However, the effect of PD on hepatocellular carcinoma (HCC) is still unclear.
Methods: Cell viability, colony formation and flow cytometry analysis for apoptosis profile, mitochondrial membrane potential endoplasmic reticulum (ER) expansion were employed to determine the effect of PD against HCC cells. Transient transfection of siRNA, immunofluorescent imaging and immunoprecipitation were used to elucidate the anti-cancer mechanism of PD. The in vivo toxicity and efficacy of PD were assessed by a xenograft mouse model.
Results: PD induced apoptosis, loss of mitochondrial membrane potential and ER expansion in HCC cells. Either downregulation of Mfn1 or Bak reversed PD-induced apoptosis and loss of mitochondrial membrane potential. Further analysis revealed that Mfn1 and Bak will form a complex with IP3R to facilitate the transfer of Ca2+ from ER to mitochondria and apoptosis. In addition, our tumour xenograft model further verifies the in vivo anti-tumour effect of PD.
Conclusion: Our study sheds light on the understanding of the anti-HCC effects of PD and may open new aspects for the development of novel treatment for human hepatocellular carcinoma.
Keywords: hepatocellular carcinoma, protodioscin, mitochondrial, apoptosis, endoplasmic reticulum
Graphical Abstract:

Introduction
Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer and the third leading cause of cancer-related death worldwide.1 Limitations in implementation of early detection may be responsible for the extreme high incidence-to-mortality ratio.2 Whilst sequential treatment optimization with sorafenib followed by regorafenib achieved significant survival benefit, almost all patients within the intermediate and the advanced stage inevitably died of cancer.3 Therefore, novel drugs with better efficacy and less side effects are in urgent demand.
Protodioscin (PD), a steroidal saponin found in Dioscorea nipponica and Tribulus terrestris, has been shown to possess activities of neuroprotection,4 male fertility improvement,5 and anti-proliferation.6 Moreover, protodioscin exhibits a spectrum of cytotoxicity against many cancer cell lines of different origins, including lung, cervix, and white blood cells.6–8 However, the effects of protodioscin against HCC and the underlying mechanisms remain unclear.
To maintain ER function and ensure proper protein folding, the unfolded protein response (UPR) is initiated by the ER transmembrane protein sensors: inositol-requiring enzyme 1 (IRE1), protein kinase RNA-like endoplasmic reticulum kinase (PERK) and activating transcription factor 6 (ATF6).9 Bip, a major ER chaperone, is another sensor for the UPR. Under resting condition, Bip binds and inhibits the activation of IRE1 and PERK. On ER stress, Bip will preferably bind to unfolded protein and trigger the dissociation of the sensor-Bip complex, which lead to the activation of PERK and IRE1.10 However, when UPR is unable to resolve the irreversible ER damage, cells enter apoptotic programs. Under prolonged ER stress, the pro-survival aspect of IRE1 was attenuated, whereas PERK/CHOP signaling persists.11 Activated PERK will phosphorylate the eukaryotic translation initiation factor 2 subunit-α (eIF2α), which lead to the subsequent upregulation of activating transcription factor 4 (ATF4) and its proapoptotic target protein proapoptotic factor C/EBP Homologous Protein (CHOP). Ca2+ release form the inositol 1,4,5-trisphosphate receptor (IP3R) on the ER and the uptake of Ca2+ through the voltage-dependent anion channel (VDAC) on the outer membrane of mitochondria also trigger the apoptotic response of cells.12 Recent study has found that PERK is able to facilitate the tethering of ER to mitochondria at mitochondria-associated membranes (MAMs), leading to increased ROS production and subsequent apoptotic cell death.13 Even though the ER-mitochondrial tether protein mitofusin 2 (Mfn 2) has been shown to form a complex with PERK to attenuate pro-apoptotic UPR signaling,13 the other tether protein mitofusin 1 (Mfn 1) has been shown to facilitate apoptosis by binding and activating the pro-apoptotic Bcl-2 family protein Bak.14 Therefore, targeting the interplay between ER stress and mitochondrial apoptosis is a promising antitumor therapy.15 Through this study, we aimed to clarify if PD may induce apoptosis via the activation of ER stress and the interaction between Mfn 1 and Bak in human HCC cells.
Materials and Methods
Chemicals and Reagents
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide) and DAPI were bought from Sigma (St. Louis, MO, USA). Protodioscin (CFN99517) of 99% purity was purchased from ChemFaces (Wuhan, Hubei, PRC). Antibodies (against Bak, Mcl-1, Bcl-2, β-actin and cytochrome c), siRNA (against Mfn1 or Bak), and horseradish peroxidase-conjugated secondary antibodies (anti-mouse, and -rabbit) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Antibodies against cleaved-caspase-3, cleaved-caspase-9, cleaved-PARP, Bip, PERK, ATF6, eIF2α, p-eIF2α, ATF4 and CHOP were purchased from Cell Signaling Technology (Beverly, MA, USA). TUDCA (tauroursodeoxycholic acid) was obtained from Cayman Chemical (Ann Arbor, MI, USA). Fetal bovine serum (FBS) and penicillin/streptomycin were purchased from Cytiva (Marlborough, MA, USA).
Cell Culture
HCC cell lines (Huh-7, SK-Hep-1, HA22T/VGH, PLC/PRF/5 and HepG2) were purchased from the Bioresources Collection and Research Center of the Food Industry Research and Development Institute (Hsinchu, Taiwan). Huh-7, SK-Hep-1 and HA22T/VGH were maintained in Dulbecco’s Modified Eagle’s Medium (Gibco BRL, Carlsbad, CA, USA). PLC/PRF/5 and HepG2 were maintained in minimum essential medium (Gibco BRL, Carlsbad, CA, USA). All mediums were supplemented with penicillin–streptomycin, nonessential amino acids and 10% fetal bovine serum. All cells were incubated in a humidified atmosphere with 5% CO2 at 37°C.
Cell Viability Assay
MTT assay were used to determine the cell viability. HCC cells (2 × 104/well) were seeded in 24-well plates and treated with vehicle control or indicated drugs for 24 h. The mediums were then replaced with fresh medium containing 0.5 mg/mL of MTT and further incubated for 4 h at 37°C. Cell viability was directly proportional to the production of formazan by the reduction of MTT in the mitochondria. Color intensities of each well were measured after dissolution of formazan in methanol at 570 nm using a Multiskan MS ELISA reader (Labsystems, Helsinki, Finland). Each experiment was performed in independent triplicate manner.
Measurement of Apoptotic Profile
Muse® Annexin V & Dead Cell Kit (Luminex Corporate, Austin, TX, USA) was employed to analyze the apoptotic profile of PD-treated cells. HCC Cells (4 × 105/well) were seeded into 6-well plates in the presence of various concentrations of PD for 24 h. At the end of the treatment, cells were trypsinised and processed according to the manufacturer’s protocol. Finally, PD-treated cells were analyzed using the Muse® cell analyzer (Luminex Corporate, Austin, TX, USA).
Measurement of Mitochondrial Membrane Potential
Muse® MitoPotential Kit (Luminex Corporate, Austin, TX, USA) was employed to analyze the apoptotic profile of PD-treated cells. At the end of 24-hour PD treatment, cells were trypsinised and processed according to the manufacturer’s protocol and then analyzed using the Muse® cell analyzer (Luminex Corporate, Austin, TX, USA).
ATP Content Assay
StayBrite™ Highly Stable ATP Bioluminescence Assay Kit (BioVision Inc., Milpitas, CA, USA) were used to measure the intracellular level of ATP. After treatment with various concentrations of PD for 24 h, HCC cells were trypsinized and subjected to 100 µL of reaction buffer/104 cells followed by pelleting at max speed for 30 sec to remove debris. Supernatant was placed into 96-well plates (10 µL/well) and 90 µL of reaction mix was then added into each well. The luminescence intensity was measured using SpectraMax M5 (Molecular Devices, San Jose, CA, USA).
Quantification of ER Expansion
The ER-ID® Red kit (Enzo Life Science, Farmingdale, NY, USA) was used to quantify the expansion of the ER lumen. HCC Cells (4 × 105/well) were seeded into 6-well plates in the presence of various concentrations of PD for 24 h. The cells were then trypsinised and incubated in the reaction mix at 37°C for 15 min in the dark. The fluorescence intensities were measured using FACSCalibur flow cytometer and the data were analyzed with Cell Quest software (BD Biosciences, Franklin Lakes, NJ, USA).
Western Blotting
PD-treated HCC cells were trypsinised and lysed in NETN lysis buffer containing protease/phosphatase inhibitor cocktail (Roche Molecular Biochemicals, Penzberg, Upper Bavaria, Germany). Cell lysates were then subjected to sonication on ice followed by 4°C centrifugation at 12,000 ×g for 15 min to isolate total proteins. Equal amounts of total proteins (20 μg) were separated by 8%–12% SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes (PALL, Port Washington, NY, USA). Non-specific binding sites on PVDF membrane were blocked with 5% dry milks. Blocked PVDF membranes were then incubated with primary antibodies overnights at 4°C, washed and then further incubated with HRP-conjugated secondary antibodies. Immobilon HRP Substrate (Millipore, Burlington, MA, USA) was then added to the PVDF membranes to develop chemiluminescence signals and visualize with ImageQuant LAS 4000 mini (Cytiva, Marlborough, MA, USA).
siRNA Transient Transfection
SK-Hep-1 cells (5 × 105) were seeded on the day before transfection on 6-cm dishes at 37°C for 24 h. On the day of transfection, the mediums were changed to serum-free DMEM. siRNA against Mfn1 (10 nM) or Bak (10 nM) and RNAiMAX reagents (Thermo Fisher Scientific, Waltham, MA, USA) were prepared and mixed in serum-free DMEM for the formation of RNA-lipid complexes and added directly to SK-Hep-1 cells. After 24 h of incubation, siRNA transfected SK-Hep-1 cells were treated with PD for another 24 h.
Immunofluorescence Staining
SK-Hep-1 cells were seeded onto Lab-Tek 8-well chamber slides (Thermo Fisher Scientific, Waltham, MA, USA) and incubated in the absence or presence of PD (6 µM) for 24 h. PD-treated cells were fixed and permeabilized with 4% paraformaldehyde and 0.5% Triton X-100, respectively. Anti-IP3R and anti-Mfn1 primary antibodies were then added to PD-treated cells for an overnight incubation at 4°C. DAPI (4ʹ,6ʹ-diamidino-2-phenylindole) and DyLight-conjugated anti-rabbit or anti-mouse secondary antibodies were then added and incubated for 1 h. Fluorescence images were acquired by a laser scanning confocal microscope (Zeiss LSM 510 META, Jena, Germany).
Co-IP Assay
SK-Hep-1 cells were seeded onto 10 cm culture dish (2 × 106 cells/dish) and treated with 6 µM of PD for 24 h. PD-treated HCC cells were then lysed in NETN lysis buffer containing protease/phosphatase inhibitor cocktail (Roche Molecular Biochemicals, Penzberg, Upper Bavaria, Germany) and subjected to sonication on ice. Sonicated lysates were centrifuged under 4°C at 12,000 ×g for 30 min. Two micrograms of specific antibodies (IP3R or Mfn1) were added to 500 µL of cell extracts and rotated at 4°C overnight. Immunoprecipitation (IP) was performed using Protein G magnetic beads (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions. Briefly, after adding the magnetic beads, the IP solutions were rotated for 2 h at 4°C. Immunoprecipitates were collected by centrifugation under 4°C at 4000 rpm for 5 min and washed 3 times with NETN buffer. Afterward, the immunoprecipitates were resuspended and subjected to Western blot analysis as described above.
In vivo Tumor Xenograft Nude Mice Model
The animal experiment protocol was approved by the Institutional Animal Care and Use Committee of Chung Shan Medical University (IACUC number: 2311) in accordance with guidelines for the ethical review of laboratory animal welfare, Taiwan (2018). Five-week-old female athymic BALB/c nude mice (National Laboratory Animal Centre, Taipei, Taiwan) and kept in a light- and humidity-regulated environment (12 h light/12 h dark cycle) at 23°C. The mice were free to feed and drink sterilized water. SK-Hep-1 cells (5 × 106/100 µL) were subcutaneously injected into the right flank of BALB/c nude mice (National Laboratory Animal Centre, Taipei, Taiwan). The tumor size was measured with a digital caliper manually once per week and the tumor volume was estimated by the following formula: length × width2 × 0.52. Once the tumor volume exceeded 80 mm3, animals were randomly assigned to three groups to receive orally administered PD (25 or 50 mg/kg) twice per week for 3 weeks. Each mouse was weighed once per week to evaluate the toxicity of PD. After the completion of treatment, the mice were sacrificed, and the tumor masses were excised for H&E staining and immunohistochemical staining for Ki-67 (cell proliferation marker).
Statistical Analysis
All experiments were conducted with at least three independent biological replicates. Data were analyzed using GraphPad Prism4 software (San Diego, CA, USA) and presented as the mean ± standard error (SE). Student’s t-test or one-way analysis of variance (ANOVA) with a post-hoc analysis using Tukey’s multiple-comparison test was used to perform statistical comparisons with p < 0.05 or p < 0.01 being considered as statistically significant.
Results
PD Inhibited Cell Viability and Induced Apoptosis in HCC Cells
To evaluate the effect of PD (structure depicted in Figure 1A) on cell viability, several HCC cell lines (Huh-7, HepG2, PLC/PRF/5, SK-Hep-1, and HA22T/VGH) were treated with an increment of concentration of PD (0, 2, 4, 6, 8, and 10 µM) for 24 h, and analyzed for cell viability using the MTT assay. Our results showed that PD inhibited the cell viability in Huh-7 cells (17%, 38%, 43%, 62% and 82%), SK-Hep-1 cells (8%, 35%, 43%, 64% and 81%), PLC/PRF/5 (3%, 25%, 51%, 61% and 75%), HA22T/VGH cells (6%, 44%, 68%, 87% and 88%) and HepG2 cells (8%, 35%, 58%, 56% and 70%) for 24 h (Figure 1B). The distribution of apoptotic cells was then evaluated with Muse cell analyzer using Annexin V and dead cell assay. Figure 1C shows that the proportion of apoptotic cells increased in accordance with the concentration of PD in Huh-7 cells from 7.4% (control) to 56.3% (6 µM, p<0.01) and 65.3% (8 µM, p<0.01); in SK-Hep-1 cells from 6.2% (control) to 32.5% (6 µM, p<0.01) and 45.6% (8 µM, p<0.01); in PLC/PRF/5 cells from 10.2% (control) to 42.5% (6 µM, p<0.01) and 49.8% (8 µM, p<0.01) were observed, respectively. In addition, we also demonstrated that PD induced the protein expression of cleaved-caspase-9 (c-caspase-9) in HCC cells, compared with control cells (Figure 1D). Therefore, these results demonstrated that PD inhibited the proliferation of HCC cells through inducing apoptosis.
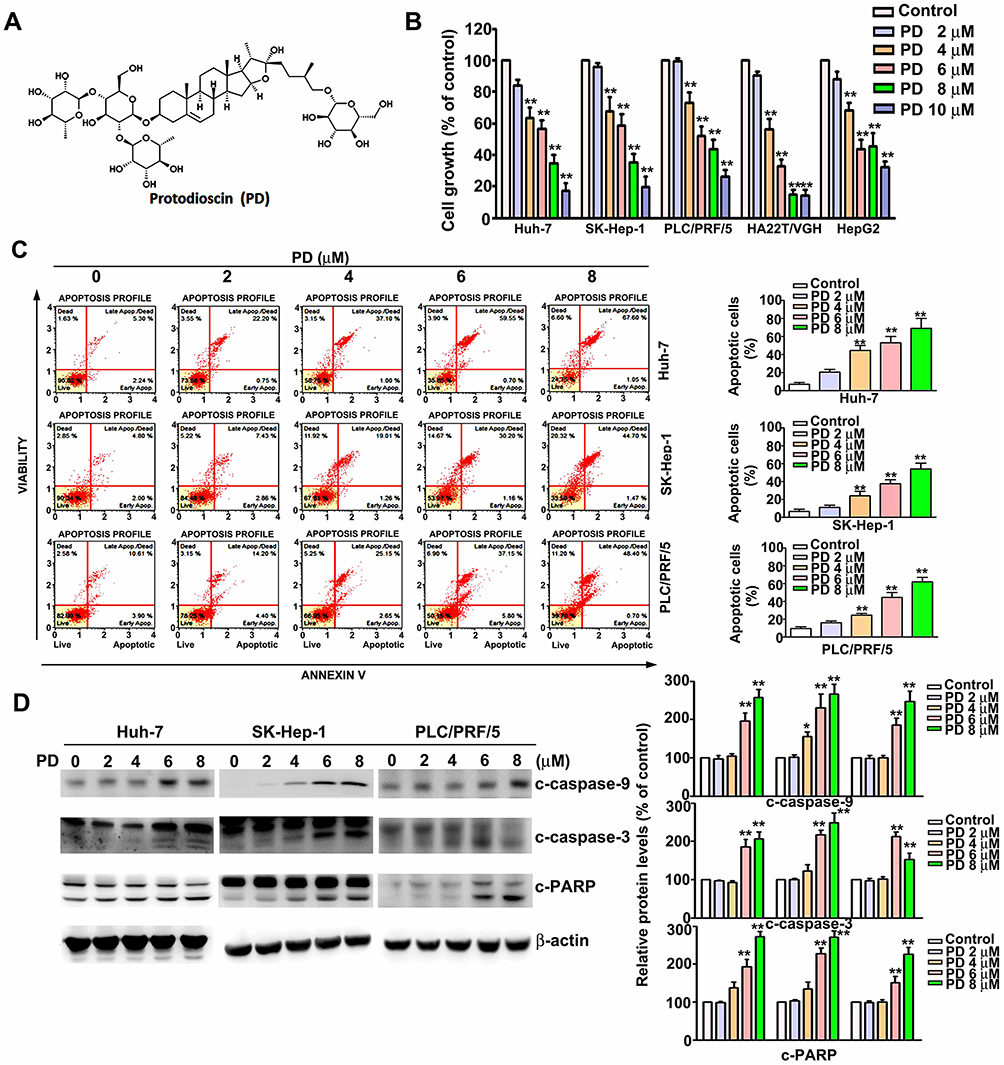 |
Figure 1 Protodioscin (PD) inhibits the proliferation of hepatocellular carcinoma (HCC) cell and induces apoptosis. (A) Structure of protodioscin (PD). (B) Human HCC cell lines (Huh-7, HepG2, PLC/PRF/5, SK-Hep-1, and HA22T/VGH) were treated with a series of concentration of PD (0, 2, 4, 6, 8, 10 µM) for 24 h. The cell viability was determined by MTT assay. (C) HCC cells treated with PD were subjected to Annexin V and dead cell assay to determine the distribution of the apoptotic cells using the Muse cell analyzer. (D) The expression of c-caspase-3, c-caspase-9, c-PARP and β-actin were staining indicated antibodies by Western blotting. *p<0.05, **p<0.01, versus control. Data are presented as the mean ± SE of at least three independent experiments. |
PD Induced Mitochondrial Apoptosis in HCC Cells
Disruption of mitochondrial membrane potential (MMP) is a key feature of the intrinsic apoptosis pathway.16 Therefore, we analyzed the changes in MMP in PD-treated HCC cells with Muse cell analyzer using MitoPotential assay. Loss of membrane potential were observed in all PD-treated HCC cells and the portion of depolarized cells increased in Huh-7 cells from 1.7% (control) to 24.9% (6 µM, p<0.01) and 34.4% (8 µM, p<0.01); in SK-Hep-1 cells from 3.1% (control) to 37.8% (6 µM, p<0.01) and 53.4% (8 µM, p<0.01); in PLC/PRF/5 cells from 7.2% (control) to 32.9% (6 µM, p<0.01) and 49.1% (8 µM, p<0.01) were observed, respectively (Figure 2A). Another hallmark of intrinsic apoptosis is the arrest of mitochondrial ATP synthesis.17 Here, we demonstrated that PD-treatment also decreased the intracellular ATP levels in Huh-7 cells decrease to 38.9% and 15.3% (6 and 8 µM, p<0.01), in SK-Hep-1 cells decrease to 33.1% and 19.2% (6 and 8 µM, p<0.01), in PLC/PRF/5 cells decrease to 36.2% and 17.1% (6 and 8 µM, p<0.01) were observed, respectively, compared with control (Figure 2B). Decreased level of ATP has also been implicated to cause excessive mitochondrial Ca2+ uptake through the MAMs and trigger intrinsic apoptosis.18 In this regard, we also evaluated the levels of MAMs-related mitochondrial fusion proteins in PD-treated HCC cells. As shown in Figure 2C, the expression of Mfn1 and Mfn2 increased in response to PD treatments. Finally, we further examined whether PD alters the levels of Bcl-2-related proteins, key regulator of MMP,19 in HCC cells. Our results showed that PD treatment increased the expression of Smac, Bak and cytochrome c, while downregulated the expression of Mcl-1 (Figure 2D). Taken together, these results revealed that PD-induced apoptosis is mediated by the mitochondrial pathways in HCC cells.
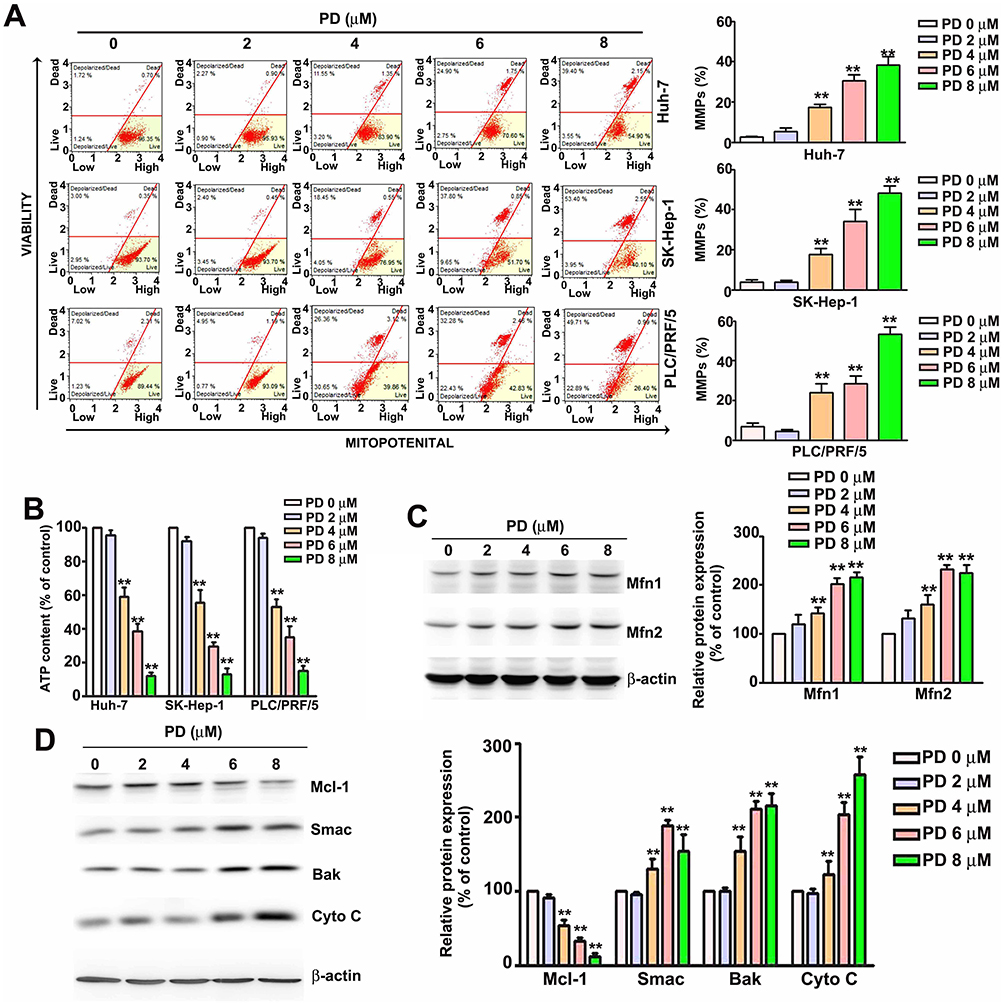 |
Figure 2 PD induces mitochondrial apoptosis in HCC cells. (A) Human HCC cell lines (Huh-7, SK-Hep-1, and PLC/PRF/5) treated with PD were subjected to MitoPotential assay and analyzed with the Muse cell analyzer. (B) HCC cells treated with PD for 24 h were subjected to ATP assay to determine the intracellular ATP levels. (C) Total cell lysates of SK-Hep-1 treated with PD for 24 h were subjected to Western blot to evaluate the expression of mitochondrial fusion-related proteins. (D) SK-Hep-1 cells treated with PD for 24 h. The total lysates were subjected to Western blot to evaluate the expression of mitochondrial apoptosis-related proteins. β-Actin was used as an internal loading control. **p<0.01, versus control. Data are presented as the mean ± SE of at least three independent experiments. |
PD Induced ER Stress in HCC Cells
The ER is the main storage site of Ca2+ in the cell and ER stress has been shown to induce apoptosis through the release and accumulation of Ca2+ in the mitochondria.20 To evaluate whether PD induce ER stress in HCC cells, the expansion of the ER lumen, one of the hallmarks of ER stress,21 was firstly assessed in PD-treated SK-Hep-1 cells. As shown in Figure 3A, PD treatment increased the ER lumen in SK-Hep-1 cells in up to 37.5% (6 µM, p<0.01) and 43.2% (8 µM, p<0.01), respectively, compared with control. Next, we examined the effect of PD on the expression of ER stress-related proteins. The expression of Bip, PERK, phosphorylated-eIF2α, and CHOP were significantly upregulated by PD treatment (Figure 3B). These results indicated that PD induced ER stress in SK-Hep-1 cells.
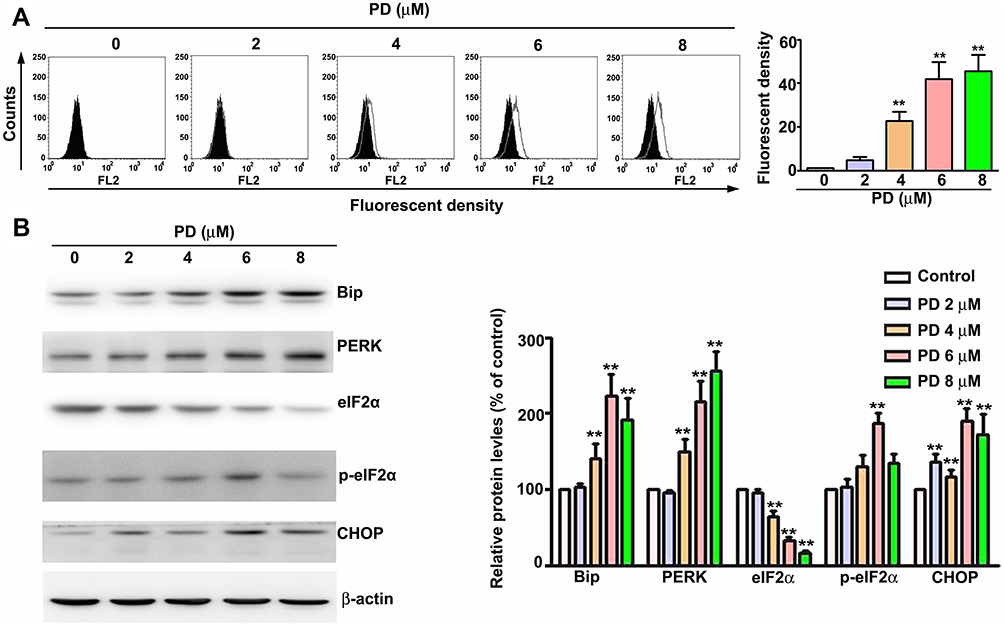 |
Figure 3 PD induces ER stress in HCC cells. SK-Hep-1 cells were treated with indicated concentrations of PD for 24 h. (A) The ER were stained with ER-ID red assay kit and further analyzed with flow cytometer to evaluate the expansion of ER. (B) The total lysates were subjected to Western blot to evaluate the expression of ER stress-related proteins. β-Actin was used as an internal loading control. **p < 0.01, versus control. Data are presented as the mean ± SE of at least three independent experiments. |
ER Stress is Essential for PD-Induced Apoptosis
To further understand the role of ER stress in PD-induced apoptosis, an ER stress inhibitor tauroursodeoxycholic acid (TUDCA)22 were employed. SK-Hep-1 cells were pre-treated with TUDCA (100 µM) prior to 24 h treatment of PD. As shown in Figure 4A and B, we found that TUDCA treatment significantly reduced to PD-induced apoptosis (17.5%; p<0.05) and loss of MMP (18.2%; p<0.05). In addition, TUDCA treatment mitigated the PD-upregulated Bip, c-PARP, Mfn1 and Bak, independent on Mfn2 (Figure 4C), suggesting that PD induced ER stress-dependent apoptosis in SK-Hep-1 cells.
 |
Figure 4 PD induces mitochondrial apoptosis through ER stress dependent pathway. SK-Hep-1 were treated with PD (6 µM) for 24 h with or without a 2 h pre-treatment of TUDCA (100 µM). (A) The apoptotic cells and (B) the mitochondrial membrane potential was determined by Muse cell analyzer with Annexin V and dead cell assay and MitoPotential assay, respectively. (C) Total lysates were subjected to Western blot with β-actin acting as an internal loading control. **p<0.01, versus control. #p<0.05, versus PD treatment alone. Data are presented as the mean ± SE of at least three independent experiments. |
PD Induced Apoptotic Cell Death Through the Mfn1 and Bak
To further determine the role of Mfn1 and Bak in PD-induced apoptosis, the expression of Mfn1 and Bak was knocked down by transfecting siRNAs against these targets in SK-Hep-1 cells. Knocking down Mfn1 attenuated the PD-induced apoptosis (19.1%; p<0.05, Figure 5A) and loss of MMP (13.7%; p<0.05, Figure 5C). Upregulations of c-PARP and Bak by PD treatment were also reversed by knocking down Mfn1 (Figure 5E). Furthermore, knocking down Bak also hindered PD-induced apoptosis (23.4%; p<0.05, Figure 5B) and loss of MMP (12.1%; p<0.05, Figure 5D). In coincident, the upregulations of cleaved PARP and Mfn1 by PD treatment were also inhibited by knocking down Bak (Figure 5F). These results indicated that upregulations of Mfn1 and Bak are mutually important in PD-induced apoptosis.
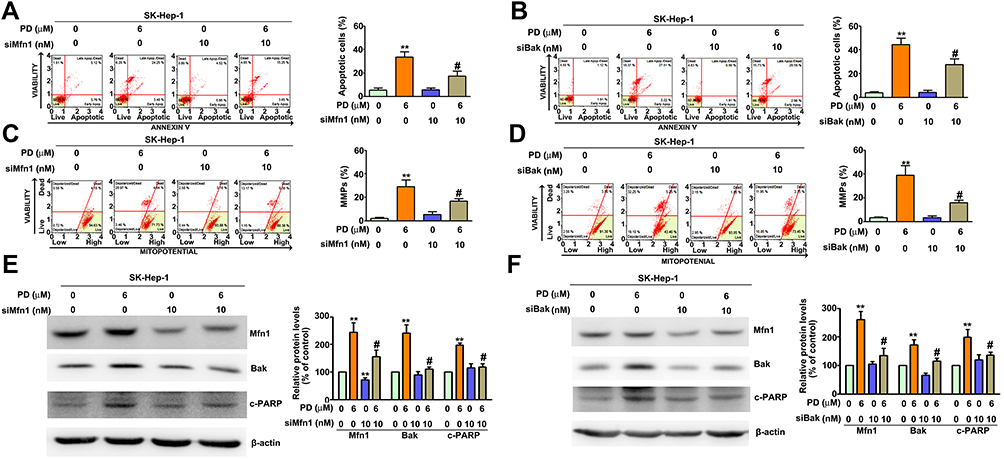 |
Figure 5 Mfn1 and Bak are both essential for PD induces mitochondrial apoptosis. SK-Hep-1 were treated with PD (6 µM) for 24 h with or without a transient transfection of si-Mfn1 (10 nM) or si-Bak (10 nM). The apoptotic cells (A, B) and the mitochondrial membrane potential (C, D) were determined by Muse cell analyzer with Annexin V and dead cell assay and MitoPotential assay, respectively. (E, F) Total lysates were subjected to Western blot with β-actin acting as an internal loading control. **p<0.01, versus control. #p<0.05, versus PD treatment alone. Data are presented as the mean ± SE of at least three independent experiments. |
PD Facilitates the Formation of Mfn1/Bak/IP3R Complex in Human HCC Cells
IP3R is the major ion channel responsible for the pro-apoptotic ER-mitochondrial Ca2+ transfers.23 To verify the role of IP3R in PD-induced apoptosis, Western blot analysis was performed. Our results indicated that PD treatment induced the upregulation of IP3R in a dose dependent manner in SK-Hep-1 cells (Figure 6A). Similar results were also observed in immunofluorescence microscopy. Additionally, Mfn1 and IP3R were colocalized in PD-treated SK-Hep-1 cells (Figure 6B). Next, to verify the role of Ca2+ in PD-induced apoptosis, mitochondria of PD-treated SK-Hep-1 were isolated and stained with Fura-2 AM. Intra-mitochondrial Ca2+ were then analyzed with a flow cytometer. Our data showed that PD increased the concentration of intra-mitochondrial Ca2+ (23.8%; p<0.05, Figure 6C) and reducing ER-Stress with TUDCA significantly diminished the concentration of intra-mitochondrial Ca2+ (10.9%; p<0.05, Figure 6C). Previous study has shown that Mfn1 will form a complex with Bak to facilitate the activation of Bak during apoptosis.14 Given the fact that both Mfn1 and IP3R are involved in the formation of MAM, we next investigated whether Mfn1/Bak/IP3R will form a complex to mediate the Ca2+ transfer from ER to mitochondria. Co-IP experiments using Mfn1 and IP3R antibodies were performed on PD-treated SK-Hep-1 cells and results revealed the presence of Mfn1, Bak and IP3R in the immunocomplex with non-specific IgG acting as control (Figure 6D and E). These findings indicated that the interaction between Mfn1, Bak, and IP3R is essential for PD-induced mitochondrial apoptosis in SK-Hep-1 cells.
 |
Figure 6 PD induces Ca2+ transfer from the ER to mitochondria through the formation of an IP3R/Mfn1/Bak complex. (A) SK-Hep-1 cells were treated with indicated concentrations of PD for 24 h. The total lysates were subjected to Western blot to evaluate the expression of IP3R. β-Actin was used as an internal loading control. (B) Immunofluorescent detection of IP3R and Mfn1. (C) SK-Hep-1 were treated with PD (6 µM) for 24 h in the absence or presence of 2 h pre-treatment of TUDCA (100 µM). Mitochondria of PD-treated cells were isolated and stained with 10 µM of Fura-2 AM and transferred to Ca2+-free medium. Intra-mitochondrial Ca2+were then analyzed with flow cytometer. (D, E) SK-Hep-1 cells were treated with PD (6 µM) for 24 h. Total lysates were subjected to immunoprecipitation (IP) with anti-IP3R, anti-Mfn1, or control rabbit IgG antibodies. Total lysates and immunoprecipitated proteins were then resolved by SDS-PAGE and the presence of IP3R, Mfn1, and Bak were determined by immunoblotting (IB). β-Actin was used as an internal loading control. **p<0.01, versus control. #p<0.05, versus PD treatment alone. Data are presented as the mean ± SE of at least three independent experiments. |
PD Inhibits the Growth of Human HCC Cells in vivo
Finally, we assessed the effect of PD on in vivo tumor growth using SK-Hep-1 xenograft BALB/c nude mice model. After 21 days of treatment, the anti-tumor capability of PD showed significantly inhibition, and nude mice with 25, 50 mg/kg PD-treated group administration had smaller tumor than 25 mg/kg PD-treated mice group. Tumor volumes in PD-treated mice were significantly lower (25 mg/kg group, 214.32 mm3, p<0.01; 50 mg/kg group, 132.2 mm3, p<0.01) than control-treated mice group (330.2 mm3), and without difference in total body weight (Figure 7A). HE staining was observed no significant difference between control and treatment groups (Figure 7B, upper). Immunohistochemistry analysis revealed that PD (25 and 50 mg/kg) significantly inhibited the Ki-67 expression (cells proliferation index) of xenografted HCC tumor (Figure 7B, down). Furthermore, the indifference in the body weight of tumor bearing mice, suggesting that PD-treatment did not induce in vivo toxicity (Figure 7C). These finding indicated that PD inhibit the in vivo tumor growth without inducing meaningful toxic effects. Taken together, our results demonstrated that PD induce ER-stress mediated mitochondrial-apoptosis through the induction of IP3R/Mfn1/Bak complex (Figure 7D).
 |
Figure 7 In vivo anti-tumour effect of PD in SK-Hep-1 xenograft model. SK-Hep-1 cells were subcutaneously inoculated into BALB/c nu/nu mice. PD (25, 50 mg/kg) were orally administrated twice per 1 week once the xenograft tumour were established. (A) Average tumour volume. (B) Tumor tissue were subjected to H&E staining and immunohistochemical staining (Ki-67 expression). Scale bar=50 µm (C) Average body weight of mice. **p<0.01, versus control. Data are presented as the mean ± SE of at least three independent experiments. (D) Illustration of our proposed mechanism of PD induced ER-stress mediated mitochondrial apoptosis via the targeting IP3R/Mfn1/Bak axis. |
Discussion
The potential of hindering early hepatocarcinogenesis mechanisms, such as anti-viral, anti-inflammation, anti-angiogenesis, and anti-metastasis have made natural products a promising source for future therapeutic regimen for HCC. In recent years, a wide range of natural products are undergoing clinical evaluation against HCC and several liver diseases.24 Previous study showed that PD inhibits the proliferation of HL-60 leukemic cell line by inducing apoptosis.25 In cervical cancer, PD was shown to inhibit proliferation by inducing ER stress, mitochondrial dysfunction, and ROS accumulation via activation of p38 and JNK pathways and Bip/eIF2α/ATF4/CHOP axis.6 In the present study, we demonstrated that PD could inhibit the proliferation of HCC cells. Flow cytometry analysis showed PD increased the apoptosis fraction and disrupted the mitochondria membrane potential of HCC cells. Luminescent analysis further revealed that PD inhibited the production of ATP in HCC cells. The balance of fusion/fission plays a key role in the maintenance of mitochondrial integrity against cellular stress. Under mild stress, the fusion of mitochondria increases the production of ATP to mitigate cellular stress. Conversely, under severe stress, the fission of mitochondria will either lead to the elimination of damaged mitochondria via mitophagy or induced apoptosis via the release of cytochrome.26,27 Previous studies also shown that mitochondrial fusion/fission is associated with activation of the intrinsic apoptosis through the interaction with Bcl-2 family member proteins.28 Therefore, we used Western blot to analysis the expression of mitochondrial fusion/fission related proteins and Bcl-2 family proteins. Our results showed that PD increased the expression of fusion protein Mfn1 and Mfn2 in SK-Hep-1 cells. The expression of bak and cytochrome c were also increased by PD treatment in SK-Hep-1 cells. Taken together, our data demonstrated that PD inhibited the proliferation and induced mitochondrial apoptosis in HCC cells.
The endoplasmic reticulum (ER) is an essential organelle of eukaryotic cells involved in the maintenance of intracellular homeostasis including protein synthesis, Ca2+ storage, lipids synthesis, and glucose metabolism.29 Imbalances in protein secretion or disrupted ER protein caused by various internal or external insults will lead to the accumulation of misfolded or unfolded proteins in the ER- a condition referred to as ‘ER stress’.30 In the tumor microenvironment, hypoxia impairs protein folding in the ER and activates the UPR pathways. Activated PERK will phosphorylate eIF2α and inhibit the global protein synthesis. However, during prolonged ER stress, selective expression of ATF4 by p-eIF2α will increase the expression of pro-apoptotic gene CHOP.31 Recent study also showed that the activation of ATF6, another ER stress sensor protein, is critical for the upregulation of CHOP.32 Increased expression of CHOP will further evoke apoptosis by downregulating the expression of the anti-apoptotic gene Bcl-2, upregulating several pro-apoptotic BHC domain-only genes, and disrupting redox homeostasis.33 In addition, accumulation of unfolded protein will titrate away the Bip from caspase 12, leading to the cleavage of caspase 12 and the activation apoptosis.34 ER stress-mediated apoptosis inducing effects of natural products have been studied intensively through many studies on HCC cell lines.35 For instance, Licochalcone A was shown to induce ROS mediated elevated expression of Bip, ATF6, IRE1α and CHOP in HepG2 cells, leading to the cleavage of caspase 3 and 9.36 In Hep 3B cells, genistein induced calpain-mediated caspase 12 activation and induced apoptosis in a CHOP-dependent manner.37 In our experiments, PD was shown to increase the expansion of ER lumen and increase the expression of Bip, PERK, p-eIF2α and CHOP. To further elucidate the relationship between PD-induced ER stress and mitochondrial apoptosis. We attenuated PD-induced ER stress by TUDCA and discovered that TUDCA treatment also reversed the loss of mitochondrial membrane potential and the increased in mitochondrial-apoptosis related proteins caused by PD treatment. Taken together, our data suggested that ER stress is the upstream effector of PD induced mitochondrial apoptosis.
The increased flow of Ca2+ from the ER to the mitochondria is another important aspect of ER-induced apoptosis.38 Mfn1/Mfn2 complex is one of the bridging complexes that govern the formation of the mitochondrial-associated membrane (MAM) between the ER and the mitochondria and Ca2+ uptake.39 Localized in both ER and mitochondria, Mfn2 forms a homo- of heterodimer with mitochondria specific Mfn1 and tethers the distance between ER and mitochondria.40 In addition, during apoptosis, pro-apoptotic protein Bak dissociates form Mfn2 and enhances the association with Mfn1.41 The association between Bak and Mfn1 will lead to the oligomerization of Bak and the subsequent increase of mitochondrial outer membrane permeabilization (MOMP), cytochrome c releases and finally apoptosis.14 Pervious study also showed that Mfn1 induced apoptosis in osteosarcoma cells and is targeted by microRNA-19b.42
In contrary, Mfn1 plays an anti-apoptosis role in cardiomyocyte, and is regulated by microRNA 140.43 Furthermore, in human prostate cancer cells, treatment with a mitochondrial calcium efflux inhibitor CGP37157 induced apoptosis through the degradation of Mfn1.44 Finally, in human HCC, low expression of Mfn1 is related to poor prognosis and vascular invasion, and it was demonstrated that Mfn1 regulates HCC metastasis through altering glucose metabolism and the epithelial to mesenchymal transition (EMT).39 These findings suggested a cell specific function of Mfn1, and further experiments are needed to define the precise mechanism of Mfn1 in human cancer. To further verify the importance of Bak and Mfn1 in the action of PD against HCC cells, we employed transient transfection of siRNA against Bak and Mfn1. Our experiments demonstrated that downregulation of either Mfn1 or Bak hindered the PD-induced apoptosis and depolarization of MMP, showing that Mfn1 and Bak are both indispensable for the anti-HCC effect of PD.
Following the line of thought that Ca2+ flux from ER to mitochondria is an important aspect in of the ER stress induced mitochondrial apoptosis, we sought to verify whether Mfn1 and Bak participate in the regulation of Ca2+ flux. Apart from the Mfn1/Mfn2 complex present in the MAM of ER, the inositol 1,4,5-trisphosphate receptors (IP3R) are the main Ca2+-releasing channels in the ER.45 In non-small cell lung cancer cells, curcumin was shown to induced apoptosis via up-regulating the expression of IP3R and the subsequent release of Ca2+.46 In accordance, our data demonstrated that PD-treatment increased the expression of IP3R and the intra-mitochondrial Ca2+ level of SK-Hep-1 cells. Furthermore, immunofluorescence confocal microscopy analysis revealed that PD-treatment induced the co-localization of Mfn1 and IP3R in SK-Hep-1 cells. Previous studies have pointed out the pro-apoptotic functions of Bak is associated with the regulation of the ER Ca2+ store47 and Mfn1 is able to activate Bak via direct interaction during apoptosis.14 In this regard, we further investigated and confirmed that Mfn1/Bak/IP3R will form a complex in PD-treated SK-Hep-1 cells. However, further analysis is needed to verify whether this complex is responsible for the Ca2+ transfer from ER to mitochondria.
Conclusions
We proposed that PD induced ER stress-dependent mitochondrial apoptosis via the formation of Mfn1/Bak/IP3R complex and the subsequent Ca2+ transfer from ER to mitochondria. Furthermore, our in vivo study showed promising anti-HCC effect of PD with no obvious sign of toxicity. The potential beneficial effect of PD may open a new era for the development of novel treatment for human hepatocellular carcinoma.
Abbreviations
ATF4, activating transcription factor 4 ATF4; CHOP, C/EBP homology protein; ER, endoplasmic reticulum; eIF2α, eukaryotic translation initiation factor 2 subunit-α; FBS, fetal bovine serum; HCC, hepatocellular carcinoma; IP3R, inositol 1,4,5-trisphosphate receptor; IP, immunoprecipitation; Mfn1, mitofusin 1; Mfn2, mitofusin 2; MMP, mitochondrial membrane potential; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; PD, protodioscin; PERK, PRKR-like endoplasmic reticulum kinase; PI, propidium iodide; siRNA, small-interfering RNA; TUDAC, tauroursodeoxycholic acid.
Ethics Statement
The animal study was approved by the Animal Ethics Committee of the Chung Shan Medical University (IACUC: 2311).
Acknowledgments
This work was supported by grants from the Ministry of Science and Technology (110-2320-B-040-014).
Disclosure
The authors declare no competing interests.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Akinyemiju T, Abera S, et al.; Global Burden of Disease Liver Cancer C. The burden of primary liver cancer and underlying etiologies from 1990 to 2015 at the global, regional, and national level: results from the Global Burden of Disease Study 2015. JAMA Oncol. 2017;3(12):1683–1691. doi:10.1001/jamaoncol.2017.3055.
3. Pinato DJ, Guerra N, Fessas P, et al. Immune-based therapies for hepatocellular carcinoma. Oncogene. 2020;39(18):3620–3637. doi:10.1038/s41388-020-1249-9
4. Zhang X, Xue X, Xian L, Guo Z, Ito Y, Sun W. Potential neuroprotection of protodioscin against cerebral ischemia-reperfusion injury in rats through intervening inflammation and apoptosis. Steroids. 2016;113:52–63. doi:10.1016/j.steroids.2016.06.008
5. Abarikwu SO, Onuah CL, Singh SK. Plants in the management of male infertility. Andrologia. 2020;52(3):e13509. doi:10.1111/and.13509
6. Lin CL, Lee CH, Chen CM, et al. Protodioscin induces apoptosis through ROS-mediated endoplasmic reticulum stress via the JNK/p38 activation pathways in human cervical cancer cells. Cell Physiol Biochem. 2018;46(1):322–334. doi:10.1159/000488433
7. Hu K, Yao X. Protodioscin (NSC-698 796): its spectrum of cytotoxicity against sixty human cancer cell lines in an anticancer drug screen panel. Planta Med. 2002;68(4):297–301. doi:10.1055/s-2002-26743
8. Oyama M, Tokiwano T, Kawaii S, et al. Protodioscin, isolated from the rhizome of dioscorea tokoro collected in Northern Japan is the major antiproliferative compound to HL-60 leukemic cells. Curr Bioact Compd. 2017;13(2):170–174. doi:10.2174/1573407213666170113123428
9. Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 2020;21(8):421–438. doi:10.1038/s41580-020-0250-z
10. Kopp MC, Larburu N, Durairaj V, Adams CJ, Ali MMU. UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nat Struct Mol Biol. 2019;26(11):1053–1062. doi:10.1038/s41594-019-0324-9
11. Lin JH, Li H, Yasumura D, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318(5852):944–949. doi:10.1126/science.1146361
12. Wang N, Wang C, Zhao H, et al. The MAMs structure and its role in cell death. Cells. 2021;10(3):45. doi:10.3390/cells10030657
13. Verfaillie T, Rubio N, Garg AD, et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012;19(11):1880–1891. doi:10.1038/cdd.2012.74
14. Pyakurel A, Savoia C, Hess D, Scorrano L. Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol Cell. 2015;58(2):244–254. doi:10.1016/j.molcel.2015.02.021
15. Martucciello S, Masullo M, Cerulli A, Piacente S. Natural products targeting ER stress, and the functional link to mitochondria. Int J Mol Sci. 2020;21(6):we45. doi:10.3390/ijms21061905
16. Gottlieb E, Armour SM, Harris MH, Thompson CB. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ. 2003;10(6):709–717. doi:10.1038/sj.cdd.4401231
17. Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87(1):99–163. doi:10.1152/physrev.00013.2006
18. Madreiter-Sokolowski CT, Gottschalk B, Parichatikanond W, et al. Resveratrol specifically kills cancer cells by a devastating increase in the Ca2+ coupling between the greatly tethered endoplasmic reticulum and mitochondria. Cell Physiol Biochem. 2016;39(4):1404–1420. doi:10.1159/000447844
19. Harris MH, Thompson CB. The role of the Bcl-2 family in the regulation of outer mitochondrial membrane permeability. Cell Death Differ. 2000;7(12):1182–1191. doi:10.1038/sj.cdd.4400781
20. Deniaud A, Sharaf O, Maillier E, et al. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene. 2008;27(3):285–299. doi:10.1038/sj.onc.1210638
21. Lin Y, Jiang M, Chen W, Zhao T, Wei Y. Cancer and ER stress: mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed Pharmacother. 2019;118:109249. doi:10.1016/j.biopha.2019.109249
22. Yoon YM, Lee JH, Yun SP, et al. Tauroursodeoxycholic acid reduces ER stress by regulating of Akt-dependent cellular prion protein. Sci Rep. 2016;6:39838. doi:10.1038/srep39838
23. Rosa N, Sneyers F, Parys JB, Bultynck G. Type 3 IP3 receptors: the chameleon in cancer. Int Rev Cell Mol Biol. 2020;351:101–148. doi:10.1016/bs.ircmb.2020.02.003
24. Man S, Luo C, Yan M, Zhao G, Ma L, Gao W. Treatment for liver cancer: from sorafenib to natural products. Eur J Med Chem. 2021;224:113690. doi:10.1016/j.ejmech.2021.113690
25. Hibasami H, Moteki H, Ishikawa K, et al. Protodioscin isolated from fenugreek (Trigonella foenumgraecum L.) induces cell death and morphological change indicative of apoptosis in leukemic cell line H-60, but not in gastric cancer cell line KATO III. Int J Mol Med. 2003;11(1):23–26.
26. Westermann B. Bioenergetic role of mitochondrial fusion and fission. Biochim Biophys Acta. 2012;1817(10):1833–1838. doi:10.1016/j.bbabio.2012.02.033
27. Zemirli N, Morel E, Molino D. Mitochondrial dynamics in basal and stressful conditions. Int J Mol Sci. 2018;19(2):543. doi:10.3390/ijms19020564
28. Martinou JC, Youle RJ. Mitochondria in apoptosis: bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21(1):92–101. doi:10.1016/j.devcel.2011.06.017
29. Almanza A, Carlesso A, Chintha C, et al. Endoplasmic reticulum stress signalling - from basic mechanisms to clinical applications. FEBS J. 2019;286(2):241–278. doi:10.1111/febs.14608
30. Sicari D, Delaunay-Moisan A, Combettes L, Chevet E, Igbaria A. A guide to assessing endoplasmic reticulum homeostasis and stress in mammalian systems. FEBS J. 2020;287(1):27–42. doi:10.1111/febs.15107
31. Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, Majsterek I. The role of the PERK/eIF2alpha/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Curr Mol Med. 2016;16(6):533–544. doi:10.2174/1566524016666160523143937
32. Yang H, Niemeijer M, van de Water B, Beltman JB. ATF6 is a critical determinant of CHOP dynamics during the unfolded protein response. iScience. 2020;23(2):100860. doi:10.1016/j.isci.2020.100860
33. McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21(4):1249–1259. doi:10.1128/MCB.21.4.1249-1259.2001
34. Rao RV, Peel A, Logvinova A, et al. Coupling endoplasmic reticulum stress to the cell death program: role of the ER chaperone GRP78. FEBS Lett. 2002;514(2–3):122–128. doi:10.1016/s0014-5793(02)02289-5
35. Kim C, Kim B. Anti-cancer natural products and their bioactive compounds inducing ER stress-mediated apoptosis: a review. Nutrients. 2018;10(8):543. doi:10.3390/nu10081021
36. Choi AY, Choi JH, Hwang KY, et al. Licochalcone A induces apoptosis through endoplasmic reticulum stress via a phospholipase Cgamma1-, Ca(2+)-, and reactive oxygen species-dependent pathway in HepG2 human hepatocellular carcinoma cells. Apoptosis. 2014;19(4):682–697. doi:10.1007/s10495-013-0955-y
37. Yeh TC, Chiang PC, Li TK, et al. Genistein induces apoptosis in human hepatocellular carcinomas via interaction of endoplasmic reticulum stress and mitochondrial insult. Biochem Pharmacol. 2007;73(6):782–792. doi:10.1016/j.bcp.2006.11.027
38. Marchi S, Patergnani S, Missiroli S, et al. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium. 2018;69:62–72. doi:10.1016/j.ceca.2017.05.003
39. Yang S, Zhou R, Zhang C, He S, Su Z. Mitochondria-Associated endoplasmic reticulum membranes in the pathogenesis of type 2 diabetes mellitus. Front Cell Dev Biol. 2020;8:571554. doi:10.3389/fcell.2020.571554
40. de Brito OM, Scorrano L. Mitofusin-2 regulates mitochondrial and endoplasmic reticulum morphology and tethering: the role of Ras. Mitochondrion. 2009;9(3):222–226. doi:10.1016/j.mito.2009.02.005
41. Brooks C, Wei Q, Feng L, et al. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci U S A. 2007;104(28):11649–11654. doi:10.1073/pnas.0703976104
42. Li X, Wang FS, Wu ZY, Lin JL, Lan WB, Lin JH. MicroRNA-19b targets Mfn1 to inhibit Mfn1-induced apoptosis in osteosarcoma cells. Neoplasma. 2014;61(3):265–273. doi:10.4149/neo_2014_034
43. Li J, Li Y, Jiao J, et al. Mitofusin 1 is negatively regulated by microRNA 140 in cardiomyocyte apoptosis. Mol Cell Biol. 2014;34(10):1788–1799. doi:10.1128/MCB.00774-13
44. Choudhary V, Kaddour-Djebbar I, Alaisami R, Kumar MV, Bollag WB. Mitofusin 1 degradation is induced by a disruptor of mitochondrial calcium homeostasis, CGP37157: a role in apoptosis in prostate cancer cells. Int J Oncol. 2014;44(5):1767–1773. doi:10.3892/ijo.2014.2343
45. Kania E, Roest G, Vervliet T, Parys JB, Bultynck G. IP3 Receptor-mediated calcium signaling and its role in autophagy in cancer. Front Oncol. 2017;7:140. doi:10.3389/fonc.2017.00140
46. Xu X, Chen D, Ye B, Zhong F, Chen G. Curcumin induces the apoptosis of non-small cell lung cancer cells through a calcium signaling pathway. Int J Mol Med. 2015;35(6):1610–1616. doi:10.3892/ijmm.2015.2167
47. Vervliet T, Parys JB, Bultynck G. Bcl-2 proteins and calcium signaling: complexity beneath the surface. Oncogene. 2016;35(39):5079–5092. doi:10.1038/onc.2016.31
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.