Back to Journals » Cancer Management and Research » Volume 11
Protocadherin 17 is a tumor suppressor and is frequently methylated in nasopharyngeal carcinoma
Authors He Y, Wang Z, Liu C ![]() , Gong Z, Li Y, Lu T, Hu G
, Gong Z, Li Y, Lu T, Hu G ![]()
Received 16 October 2018
Accepted for publication 13 January 2019
Published 18 February 2019 Volume 2019:11 Pages 1601—1613
DOI https://doi.org/10.2147/CMAR.S191102
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Xueqiong Zhu
Ya He, 1,2 Zhihai Wang, 1 Chuan Liu, 1 Zhitao Gong, 1 Yanshi Li, 1 Tao Lu, 1 Guohua Hu 1
1Department of Otolaryngology Head and Neck Surgery, The First Affiliated Hospital of Chongqing Medical University, Chongqing, China; 2Department of Otolaryngology Head and Neck Surgery, Southwest Hospital, Third Military Medical University (Army Medical University), Chongqing, China
Purpose: Several PCDH genes were shown to be downregulated or silenced in carcinomas and act as candidate tumor suppressor genes. However, the functions of PCDH17 in nasopharyngeal carcinoma (NPC) remain unclear. Here, we investigated the PCDH17 promoter methylation status and its impact on the expression and functions of PCDH17 in NPC.
Patients and methods: To determine the mRNA levels and promoter methylation status of PCDH17 in NPC cell lines as well as 42 NPC patient specimens, we performed reverse transcription PCR, methylation-specific PCR, and bisulfite genome sequencing. The effects of ectopic PCDH17 expression in NPC cell lines were determined by colony formation, cell proliferation, wound healing, in vitro human umbilical vein endothelial cells tube formation, migration, invasion, cell cycle, and apoptosis assays and an in vivo subcutaneous tumor model.
Results: PCDH17 expression was almost absent or significantly reduced in 100% of the NPC cell lines (5/5). However, 5-aza-2′-deoxycytidine and trichostatin A treatment restored PCDH17 expression. Promoter methylation was involved in PCDH17 silencing. Ectopic expression of PCDH17 in silenced NPC cells reduced colony formation, cell migration, angiogenesis, VEGF secretion, and tumorigenicity.
Conclusion: PCDH17 plays a tumor suppressor role in NPC. PCDH17 methylation may be a tumor-specific event and can be used as an epigenetic biomarker for NPC.
Keywords: nasopharyngeal carcinoma, PCDH17, tumor suppressor gene, methylation, epigenetic inactivation
This paper has been retracted.
Introduction
Nasopharyngeal carcinoma (NPC), a rare form of head and neck cancer, is highly prevalent and severe in major parts of southern China. NPC is primarily due to infection with the EB virus, diet, environment, and genetic susceptibility;1 however, its molecular mechanisms are complex and not well understood.2 Patient death is due to tumor metastasis in ~30%–40% of the cases.3 Therefore, effective treatments are urgently needed.
Cadherins are a family of calcium-dependent cell adhesion molecules containing an extracellular cadherin repeat of ~110 amino acid residues. These proteins are further subdivided into the classical, desmosome, and protocadherin (PCDH) groups.4,5 In recent years, PCDHs (PCDH20, PCDH17, PCDH10, PCDH9, and PCDH8) have been found to act as tumor suppressor genes (TSGs).6 Epigenetic changes in TSGs often involve promoter CpG island methylation. Studies on the promoter methylation of CpG islands and the discovery of new TSGs have revealed the epigenetic mechanisms of tumorigenesis, thereby identifying early detection biomarkers for NPC.2 Several PCDH genes are downregulated or silenced in carcinomas and act as candidate TSGs: PCDH20 in non-small-cell lung cancers;7 PCDH10 in hematologic, gastric, testicular, cervical, breast, esophageal, colorectal, nasopharyngeal, lung, and hepatocellular cancers;8–15 PCDH17 in colorectal and gastric cancers, esophageal squamous cell carcinoma (ESCC),16,17 and laryngeal squamous cell carcinoma;18 PCDH9 in glioblastoma;19 and PCDH8 in breast cancer and hematologic cancers.20,21 Abnormal expression of PCDH8, 10, and 17 represses tumor cell proliferation and migration but induces apoptosis and autophagy.11,16,17,21 Recent studies have shown involvement of PCDH17 methylation in ESCC, gastric and colorectal cancers,22 and urological cancer.16,23 PCDH17 is silenced in ESCC, which is associated with a poor differentiation state, suggesting that PCDH17 is a TSG. However, the underlying mechanism is still unclear.16 These findings indicate a role of promoter CpG methylation in PCDH silencing in carcinomas, which leads to tumorigenesis. However, the role of PCDH17 and whether it is epigenetically silenced in NPC are unknown. Herein, we aimed to investigate the expression of PCDH17 and its promoter methylation status in NPC. Our results demonstrate the key involvement of promoter methylation in inhibiting PCDH17 expression in NPC. Additionally, we studied the functions of PCDH17 in tumor cell proliferation, migration, and angiogenesis and reported that PCDH17 might act as a pleiotropic tumor suppressor in NPC. However, the underlying mechanisms still need to be uncovered.
Patients and methods
Tissue samples
The Department of Otolaryngology (Chongqing, China) provided 42 primary NPC tumor biopsies. Donors were informed, and they consented to therapy. Patients were diagnosed according to the WHO classification by trained pathologists. The controls included 17 histological hyperplasia tissues obtained from symptomatically NPC-positive patients who showed negative results for tumor cells in nasopharyngeal biopsies. The biopsy tissues obtained were then cryo-frozen in liquid nitrogen and further stored at –80°C until use. All of the procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. This study was approved by the ethics committee of Chongqing Medical University. Written informed consent was obtained from all the patients for the publication of this report.
Cell culture
HK1,24 C666-1,25 CNE1,26 HONE1,27 HNE1,24 and NP6927 cell lines were kind gifts from Prof Qian Tao of the Chinese University of Hong Kong and were approved by Chongqing Medical University for use in this study. C666-1, HNE1, CNE1, HONE1, and HK1 cell lines were cultured in RPMI-1640 media containing 10% FBS, 1% GlutaMax, and 1% penicillin–streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). NP69 cells were cultured in keratinocyte serum free medium (K-SFM) medium (Thermo Fisher Scientific), as described previously.28 We treated the cells for 3 days with 10 µM of the demethylating chemical 5-aza-2′-deoxycytidine (5-Aza-C; Sigma-Aldrich Co., St Louis, MO, USA) followed by treatment with 100 ng/mL of the histone deacetylase inhibitor trichostatin A (TSA; Cayman Chemical Co., Ann Arbor, MI, USA) for another 24 hours.9,29 Thereafter, the cells were harvested for DNA and RNA extraction.
Semi-quantitative reverse transcription PCR (RT-PCR)
mRNA expression was quantified by RT-PCR, as described previously.29 In brief, RNA was isolated from tissue samples or cell pellets using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol. Subsequently, the samples were reverse transcribed and amplified using semi-quantitative RT-PCR involving 32 cycles with 55°C as the annealing temperature. The primers used for this experiment are shown in Table 1.
 | Table 1 PCR primers used in this study Abbreviations: RT-PCR, reverse transcription PCR; MSP, methylation-specific PCR; BGS, bisulfite genome sequencing. |
Methylation level analysis
Methylation of the promoter of the PCDH17 gene was determined by a technique known as methylation-specific PCR (MSP) and bisulfite genome sequencing (BGS). DNA from tissue samples was isolated using the Animal Genome extraction kit (Axygen Biosciences, Inc., Union City, CA, USA). After bisulfite-mediated modification of the sample DNA, we carried out MSP and BGS as described previously.30,31 The PCR reaction system of MSP included 2 µL of modified DNA, 12.5 µL of Premix Ex Taq DNA polymerase mix, 8.5 µL of ddH2O, and 2 µL of primers that were either methylation or non-methylation specific. We then used Platinum PCR SuperMix High Fidelity (Thermo Fisher Scientific) for bisulfite sequencing PCR of the bisulfite-modified DNAs with primers specific to the PCDH17 gene promoter. The methylation level was analyzed as described previously.32,33 Table 1 lists the primers that we used in this study for both MSP and BGS.
PCDH17-expressing plasmid and transfection
The NPC cell line CNE1 was transfected with the pCMV-PCDH17 plasmid or empty vector pCMV (gifts from Prof Qian Tao, Chinese University in Hong Kong). CNE1 cells were seeded on six-well plates at a density of 2×105 cells/well and incubated overnight for attachment. For transfection of CNE1 cells with the PCDH17 expression plasmid or the empty vector, we used Lipofectamine 2000 (Thermo Fisher Scientific). After 48 hours of transfection, we harvested the cells, replated them, and selected them with G418 (400 µg/mL). Cells that were stably transfected with PCDH17 were obtained after 12 days of selection by G418. Quantitative PCR and Western blot analyses confirmed the ectopic expression of PCDH17.
Colony formation assays
For colony formation assays, 500 transfected cells per well were seeded and cultured for ~12 days for colony formation. Colonies formed were fixed and washed with 4% paraformaldehyde (PFA) in PBS. Following Giemsa staining, the colonies were imaged, and their numbers were counted using an inverted microscope (Olympus). The experiments were carried out in triplicate with three repeats.
Cell proliferation assay
We estimated cellular proliferation by using the Cell Counting Kit-8 (CCK-8). CNE1-PCDH17 and CNE1-vector cells were selected as described above. Briefly, we plated the cells in a 96-well plate at a density of 5,000 cells per well for 24 hours, in addition to 10 µL of CCK-8 solution. The OD density was subsequently determined after a 4-hour incubation at 37°C using a microplate reader.
Wound-healing assay
We performed a scratch wound-healing assay to assess cell mobility. CNE1-PCDH17 and CNE1-vector cells were cultured until subconfluency. Then, the cell monolayer was scraped off by using sterile pipette tips. The scraped cells were subsequently rinsed with PBS (two times) and incubated for 24 hours. The cellular gap in the cell monolayer was measured using an inverted microscope at specific time points.
In vitro migration and invasion assays
Cell migration assays were performed using 24-well transwell chambers with an 8 µm pore size (Corning Incorporated, Corning, NY, USA). Briefly, 5×104 cells were plated in the upper chamber and maintained at 37°C for 48 hours. Next, we removed the cells in the upper membrane and fixed the cells in the lower membrane layer using 4% PFA. The fixed cells were then stained and enumerated using a standard light microscope. For the invasion assay, we coated the upper transwell chambers for 1 hour using Matrigel (Corning Incorporated). Then, we plated the cells in the chamber. The experiments were independently performed three times.
Cell cycle and apoptosis analysis
To carry out this analysis, we harvested the cells following dissociation by trypsin-EDTA (Thermo Fisher Scientific), centrifugation, and PBS washes according to our previous protocol.34 Prior to the cell cycle analysis, we fixed the cells with ice-cold 75% ethanol and left them overnight at 4°C. After centrifugation and a PBS rinse, we stained the cells with 1% propidium iodide supplemented with 1 mg/mL RNase A for 30 minutes at 37°C. This step was followed by two washes with PBS and analysis. An Annexin V-PE/7-AAD Apoptosis Detection Kit (BD Pharmingen, San Diego, CA, USA) was used to stain the cells. Following resuspension of 1×105 cells in 1× binding buffer, they were transferred to a FACS tube. The cells were then stained with phycoerythrin Annexin V and 5 µL of 7-amino-actinomycin D, followed by incubation in the dark at room temperature for 15 minutes. After the staining, we added 400 µL of 1X binding buffer to each sample in the fluorescence activated cell sorting (FACS) tube. Cell cycle and apoptosis analysis was then performed on a BD C6 flow cytometer and analyzed by FlowJo 7.6.1 software. The analyses were performed in three independent experiments, with at least three samples for each group.
Western blotting
We performed Western blot analysis for proteins using previously described methods.35 Briefly, we lysed the cells using a lysis buffer containing a protease inhibitor cocktail. This step was followed by centrifugation at 13,000× g for 15 minutes in a centrifuge that had been cooled down to 4°C. The protein concentration of each sample was then determined using the bicinchoninic acid method. Subsequently, the protein samples were resolved by SDS-PAGE and transferred onto polyvinylidene fluoride membranes. Each membrane was then blocked with blocking buffer comprised of Tris-buffered saline, which had 0.1% Tween 20 and 5% fat-free milk added. The membranes were then incubated with primary antibodies at a dilution of 1:1,000 and a secondary antibody. Primary antibodies against the following proteins were used: PCDH17 (Abcam, Cambridge, UK), caspase-3 (Abcam), Beclin-1 (Novus Biologicals), LC3B (Abcam), caspase-9 (Thermo Fisher Scientific), Bax (Abcam), Bcl2 (Abcam), and GAPDH (Abcam). The bands were imaged by enhanced chemiluminescence. Image Lab was used to normalize the intensity of each band to that of GAPDH and analyze each band.
In vitro tube formation assay
First, 1×104 pretreated in vitro human umbilical vein endothelial cells (HUVECs)/well were plated onto a layer of Matrigel (BD Bioscience, San Diego, CA, USA) and incubated in a 37°C incubator with 5% CO2 for 24 hours. Then, the tubular structures were photographed and quantified under a microscope. The number of formed capillary tubes was counted manually. Each experimental condition was tested in triplicate.
ELISA
CNE1-PCDH17 and CNE1-vector cells were incubated for 48 hours to determine the VEGF level. Then, the conditioned media were harvested and assayed by sandwich ELISA kits according to the manufacturer’s protocol. The optical intensity was measured at 450 nm (Molecular Devices, Sunnyvale, CA, USA). The data are representative of three independent experiments.
In vivo tumor model
All animal experiments that were performed during our study had been granted prior approval from the animal ethics committee of Chongqing Medical University. All animal experiments were carried out according to the Guide for the Animal Welfare of Chongqing Medical University. We performed subcutaneous administration of 3.5×106 viable CNE1-PCDH17 and CNE1-vector cells into the right dorsal flank of 6-week-old female BALB/c nude mice. After every interval of 5 days, we assessed the tumor volume for 40 days. Tumor volume was calculated by the following formula: V (mm3)=(short diameter)2×(long diameter)/2.
In situ apoptosis detection
The detection involved staining the cells using an In-Situ Cell Death Detection Kit (Hoffman-La Roche Ltd., Basel, Switzerland). Paraffin-embedded sections of the tumor tissues from CNE1-PCDH17- and CNE1-vector cell-inoculated mice were stained according to the manufacturer’s protocol. For the staining of active caspase-3, the tumor sections were preincubated with 0.5% H2O2 dissolved in PBS for 30 minutes. This treatment reduced the endogenous peroxidase levels. Then, the sections were incubated with 0.5% Triton X-100 in PBS for 30 minutes and blocked with BSA to prevent nonspecific binding. The sections were treated with biotin-conjugated secondary caspase-3 antibody raised in rabbits (Abcam). The prepared sections were then visualized using microscopy. During enumeration, five randomly selected fields were used to count apoptotic cells at a magnification of 400×.
Statistical analysis
In this study, all statistical analyses were performed using SPSS 18.0 software (SPSS Inc., Chicago, IL, USA). Pearson’s chi-squared test or Fisher’s exact test were used to analyze associations between methylated samples and clinicopathological features of NPC patients. The results of the colony formation assays and other assays are presented as the mean ± SD, and a paired t-test was used to determine the significance of differences. P<0.05 was considered significant.
Results
PCDH17 is silenced and methylated in NPC cell lines
We measured the transcriptional levels of PCDH17 in normal adult human tissues, five cell lines (CNE1, HK1, C666-1, HNE1, HONE1), and in an immortalized nasopharyngeal epithelial cell line (NP69) by semi-quantitative RT-PCR. PCDH17 was expressed in normal adult human tissues (Figure 1A) and NP69 (a normal NP epithelial cell line; Figure 1B) but not in the NPC cell lines (Figure 1B).
 | Figure 1 PCDH17 is silenced and methylated in NPC cell lines. Notes: (A) PCDH17 expression in normal adult tissues was verified by semi-quantitative RT-PCR, with GAPDH as a control. (B) PCDH17 expression in NPC cell lines and the normal cell line NP69 was tested by semi-quantitative RT-PCR, with GAPDH as a control. MSP showed silencing of PCDH17 by promoter methylation. M: methylated; U: unmethylated. The results showed that PCDH17 was highly methylated in PCDH17-silenced cell lines. (C) Pharmacological demethylation with 5-Aza-C and histone acetylation with TSA. C: untreated control; A: 5-Aza-C-treated group; A+T: 5-Aza-C plus TSA-treated group. Representative results are shown. (D) Detailed BGS analyses of PCDH17 promoter methylation: vertical lines, individual CpG sites; circles, CpG sites analyzed; filled circle, methylated CpG site; open circle, unmethylated CpG site. Abbreviations: NPC, nasopharyngeal carcinoma; RT-PCR, reverse transcription PCR; MSP, methylation-specific PCR; BGS, bisulfite genome sequencing; 5-Aza-C, 5-aza-2′-deoxycytidine; TSA, trichostatin A. |
Epigenetic modification is a key player in regulating the expression of TSGs during tumorigenesis. We used MSP, 5-Aza-C treatment, and TSA for treatment to validate the important role of DNA methylation and histone deacetylation in silencing PCDH17 in NPC cell lines. The MSP results showed that PCDH17 was highly methylated in PCDH17-silenced cell lines (Figure 1B). In addition, following treatment with 5-Aza-C for demethylation and histone acetylation treatment with TSA, PCDH17 expression in NPC cell lines was significantly restored, and the unmethylated alleles increased significantly (Figure 1C).
We examined the methylation profile of CpG islands in the PCDH17 promoter region using high-resolution BGS analysis and further verified the results of the MSP experiment (Figure 1D). The results clearly indicated that the two NPC cell lines, CNE1 and HONE1, showed a significantly higher methylation level of the PCDH17 CpG islands than the immortalized nasopharyngeal epithelial cell line NP69 (Figure 1D). In conclusion, the re-expression of PCDH17 after treatment with 5-Aza-C and TSA and the hypermethylation of PCDH17 in NPC cell lines suggest that PCDH17 transcriptional silencing in NPCs is mediated by methylation.
PCDH17 is downregulated and methylated in primary NPCs
To detect the promoter methylation level of PCDH17 in human NPC tissues and in NP69, we used MSP. The methylation rates of PCDH17 in human NPC tissues and normal NP tissues were 83.3% (35/42) and 17.6% (3/17), respectively (P<0.05; Figure 2).
 | Figure 2 MSP analysis of PCDH17 in primary NPCs. Notes: MSP results of 42 primary NPC tissues and 17 normal NP tissues are shown. M, methylated; U, unmethylated. Abbreviations: NPC, nasopharyngeal carcinoma; MSP, methylation-specific PCR. |
We analyzed the correlations between PCDH17 clinicopathological parameters and promoter methylation levels (Table 2). No significant correlation was found in NPC patients between the methylation/unmethylation of the PCDH17 promoter and age, gender, tumor TNM stage, or lymph node metastasis (Table 2). Thus, PCDH17 methylation status could be a new tumor marker for NPC.
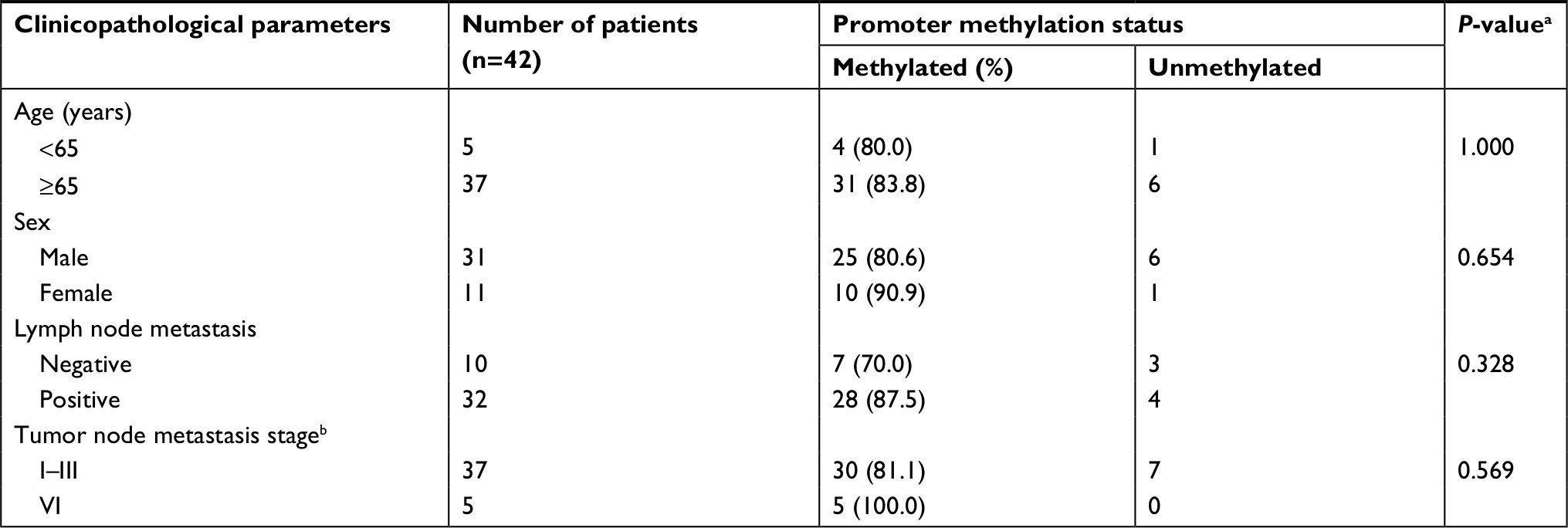 | Table 2 Correlations between PCDH17 methylation and clinicopathological parameters in NPC patients Notes: aP<0.05 was statistically significant, values are from chi-squared or Fisher’s exact test. bStaging according to the International Union Against Cancer. Abbreviation: NPC, nasopharyngeal carcinoma. |
Ectopic expression of PCDH17 inhibits tumor cell growth of NPC
DNA methylation leads to PCDH17 transcriptional silencing in NPC, indicating that PCDH17 may play a tumor suppressor role in NPC. We investigated how cancer cell growth was affected by the increase in PCDH17 expression. We stably transfected the PCDH17 gene into CNE1 cells, and real-time PCR (Figure 3A) and Western blot analyses (Figure 3B) confirmed that PCDH17 expression was increased in CNE1 cells. Colony formation assay results showed that compared with CNE1-vector cell colonies, the CNE1-PCDH17 cell colonies were significantly reduced (P<0.05; Figure 3C, D). These results suggest that PCDH17 can inhibit the colony formation of CNE1 cells. CCK-8 assays showed that the growth rate of CNE1-PCDH17 cells was significantly slower than that of CNE1-vector cells (Figure 3E), suggesting that PCDH17 also inhibited the proliferation of tumor cells.
 | Figure 3 PCDH17 ectopic expression inhibits tumor cell growth of NPC. Notes: (A) PCDH17 expression levels in CNE1-vector and CNE1-PCDH17 cells were confirmed by real-time PCR. Vector: vector-transfected CNE1 cells (CNE1-vector); PCDH17: PCDH17-transfected CNE1 cells (CNE1-PCDH17). *P<0.05. (B) PCDH17 expression levels in CNE1-vector and CNE1-PCDH17 cells were confirmed by Western blot analyses. (C) A colony formation assay is shown. The independent experiments were carried out in triplicate. (D) Quantitative analysis of colony formation. The colony number of CNE1-vector cells was set to 100%, and CNE1-PCDH17 is presented as the mean ± SD. *P<0.05. (E) Growth curves of CNE1-PCDH17 and CNE1-vector cells by CCK-8 assays. The values are shown as the mean from three independent experiments. *P<0.05. Abbreviations: NPC, nasopharyngeal carcinoma; CCK-8, Cell Counting Kit-8. |
Ectopic PCDH17 expression inhibits NPC cell invasion and migration
We performed wound-healing, transwell migration, and transwell invasion assays to determine whether PCDH17 plays a role in inhibiting the migration and invasion of NPC cells. The migration of CNE1-PCDH17 cells as well as the wound edge was significantly slower than that of CNE1-vector cells (Figure 4A), indicating the role of PCDH17 expression in inhibiting tumor cell migration.
 | Figure 4 PCDH17 ectopic expression inhibits the migration and invasion of NPC cells. Notes: (A) Wound-healing assays of CNE1-vector cells and CNE1-PCDH17 cells. Twenty-four hours after the cell surface was scratched, microscopic observations were recorded. All experiments were performed in triplicate, and a representative image is shown. Scale bar: 100 µm. *P<0.05. (B) The migrated CNE1-PCDH17 and CNE1-vector cells in transwell migration assays. *P<0.05. (C) Invaded CNE1-PCDH17 and CNE1-vector cells in Matrigel invasion assays. Scale bar: 50 µm. *P<0.05. Abbreviation: NPC, nasopharyngeal carcinoma. |
We observed that the number of CNE1-PCDH17 cells passing into the lower chamber through the membrane was significantly lower than that of CNE1-vector cells (P<0.05, Figure 4B, C). These results indicate a significant reduction in the migration and invasion of CNE1-PCDH17 cells, confirming that PCDH17 inhibits the migration and invasion of NPC cells.
Ectopic PCDH17 expression promotes cell cycle arrest and induces apoptosis of NPC cells
Flow cytometry was used to determine the effect of PCDH17 on the cell cycle and apoptosis of NPC cells. The cell cycle distribution of CNE1-vector cells is shown in Figure 5A; cells in S phase comprised 20.63%±2.80%, and cells in G1 phase comprised 61.67%±1.15%. For the cell cycle distribution of CNE1-PCDH17 cells, cells in S phase comprised 11.07%±0.35%, and cells in G1 phase comprised 78.99%±0.56%. A significantly higher percentage of CNE1-PCDH17 cells in G1 phase compared to that of CNE1-vector cells was observed, and the percentage of S phase CNE1-PCDH17 cells was significantly lower than that of control cells (P<0.05). These results clearly showed that PCDH17 plays a regulatory role in the cell cycle of CNE1 cells.
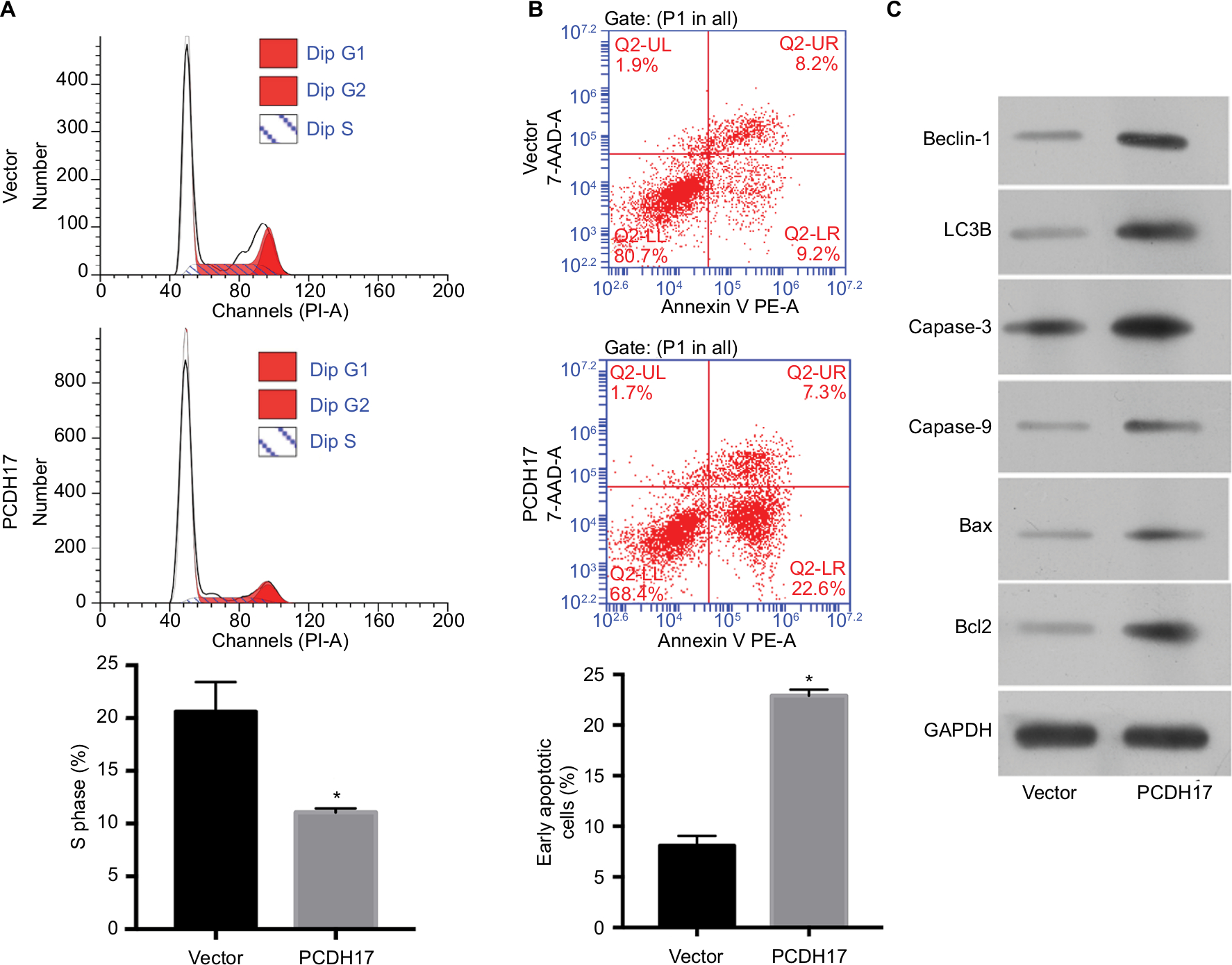 | Figure 5 PCDH17 ectopic expression promotes cell cycle arrest and induces apoptosis in NPC cells. Notes: (A) Flow cytometry was used to determine the cell cycle distribution of CNE1-vector cells and CNE1-PCDH17 cells. The percentage of S phase cells is shown. *P<0.05. (B) The Annexin-V kit was used to detect the apoptosis of CNE1-PCDH17 cells and CNE1-vector cells. The percentage of early apoptotic cell numbers is shown. *P<0.05. (C) Western blot was used to examine the expression of several key apoptosis and autophagic regulators. Abbreviations: NPC, nasopharyngeal carcinoma; PE, phycoerythrin; PI, propidium iodide; 7-AAD, 7-amino-actinomycin D. |
We used the Annexin-V kit to detect the apoptosis of NPC cells. The population of early apoptotic cells increased significantly in CNE1-PCDH17 cells (22.9%±0.6%) compared with CNE1-vector cells (8.1%±1.0%) (Figure 5B). Upregulation of several key apoptosis regulators by ectopic PCDH17 expression is shown through Western blot analysis (Figure 5C). Thus, PCDH17 regulates the cell cycle and apoptosis of NPC cells.
PCDH17 ectopic expression inhibits angiogenesis and VEGF secretion
We examined the role of PCDH17 in modulating the angiogenic capacity of NPC cells by HUVEC tube formation assays. Herein, we showed that the HUVEC tube formation ability of CNE1-PCDH17 cells was significantly attenuated compared to that of CNE1-vector cells (P<0.05; Figure 6A). VEGF is considered a key growth factor in angiogenesis. We used VEGF ELISAs to examine whether the inhibition of HUVEC tube formation by PCDH17 was related to the inhibition of VEGF secretion. The results showed that the secretion of VEGF in CNE1-PCDH17 cells was significantly lower than that in CNE1-vector cells (P<0.05; Figure 6B). The percent inhibition of VEGF secretion in CNE1-PCDH17 cells was 35.37%±0.32%. Thus, we concluded that the anti-angiogenic activity of PCDH17 in NPC is associated with decreased secretion of VEGF.
 | Figure 6 PCDH17 ectopic expression inhibits angiogenesis and VEGF secretion. Notes: (A) Representative images of HUVEC tube formation assays. Scale bar: 50 µm. (B) VEGF ELISA results are shown. *P<0.05. Abbreviation: HUVEC, in vitro human umbilical vein endothelial cell. |
PCDH17 inhibits tumor growth in nude mice
The gold standard for evaluating candidate TSGs is the ability to inhibit tumor growth in vivo. We injected CNE1-PCDH17 and CNE1-vector cells subcutaneously into nude mice to assess the effect of PCDH17 on NPC cell growth in vivo and to determine its potential for inhibiting tumor growth. The growth curves showed that after injection into nude mice for 40 days, the size of the tumors formed by CNE1-PCDH17 cells was significantly smaller than that of CNE1-vector cells (P<0.05; Figure 7A), suggesting that PCDH17 can significantly reduce the tumorigenicity of NPC cells.
 | Figure 7 PCDH17 inhibits tumor growth in nude mice. Notes: (A) CNE1-PCDH17 and CNE1-vector cells were injected subcutaneously into nude mice, and the growth curves are shown. *P<0.05. (B) Tumor sections from nude mice were stained for apoptotic cells using active caspase-3 staining and TUNEL assays. *P<0.05. |
We prepared tumor tissue sections from nude mice and studied the role of PCDH17 in tumor cell apoptosis. Our results show that apoptotic cells were positive for TUNEL and that active caspase-3 staining was significantly increased in CNE1-PCDH17 tumor tissue sections compared with CNE1-vector tumor tissue sections (Figure 7B). These results indicated that PCDH17 plays a tumor suppressor role in NPC by promoting tumor cell apoptosis.
Discussion
Recent studies have revealed that many PCDHs, such as PCDH8, PCDH10, and PCDH20, often undergo gene inactivation through promoter methylation in tumors and act as tumor suppressors. These studies demonstrated that PCDH promoter methylation and genetic inactivation are closely related to tumor development.7,8,11,21,36 Herein, we report that PCDH17 frequently undergoes gene silencing or is downregulated because of promoter methylation in NPC cell lines and human NPC tissues. PCDH17 is expressed in normal tissues, and demethylation drugs can lead to the demethylation of the PCDH17 promoter and the recovery of PCDH17 expression. Based on these results, we report that the key mechanism of PCDH17 inactivation in NPC is methylation of the promoter region. Primary tumor tissue is different from cancer cell lines and generally contains infiltrating non-malignant cells; therefore, unmethylated alleles are always detectable in tumor tissues. Unmethylated alleles were also found to coexist with gene silencing in certain cell lines, suggesting that epigenetic mechanisms other than methylation, such as histone modifications, may also be involved in gene regulation. All these studies have shown that PCDH17 methylation is a common cancer-specific event, and this conclusion is consistent with other findings.16,18,23
The cadherin family mediates Ca2+-dependent binding and is a family of cell adhesion membrane proteins.37 The structure and function of PCDHs differs from that of the classical cadherins, and thus, the PCDHs may not have strong cell–cell adhesion activity. The important physiological functions of PCDHs may be cell–cell interactions and signal transduction.38,39 At present, many PCDHs have been reported to exert various tumor-suppressing effects: PCDH10 inhibits tumor cell proliferation and induces tumor cell apoptosis through the regulation of pro-apoptosis genes, anti-proliferation genes, and anti-invasion genes.11 PCDH24 plays a role in the induction of contact inhibition by inhibiting the expression of downstream target proteins of β-catenin, including cyclin D1.40 PCDH17 can induce apoptosis and autophagy, inhibit migration/invasion, and induce G1/S cell cycle arrest in tumor cells.16,17 PCDH8 and PCDH10 have also been identified as TSGs for NPC, inhibiting tumor cell growth, colony formation, migration, and invasion, which suggests that PCDH family members also play a key role in NPC.40 A recent study showed that PCDH17 expression was significantly downregulated in NPCs and that PCDH17 overexpression could significantly inhibit proliferation and promote the apoptosis of NPC cell lines.41 In our study, we used RT-PCR to detect PCDH17 expression in normal adult human tissues and the nasopharyngeal epithelial cell line NP69, but the expression was silenced in all five NPC cell lines. MSP and high-resolution BGS analysis showed that PCDH17 was highly methylated in NPCs. These results are consistent with those of a recent study. After demethylation treatment with 5-Aza-C and histone acetylation treatment with TSA, the expression of PCDH17 in NPC cell lines was significantly restored, and the unmethylated alleles increased significantly, which suggests that methylation mediates PCDH17 transcriptional silencing in NPCs. We stably transfected the PCDH17 gene into CNE1 cells and found that ectopic expression of PCDH17 inhibited the growth, migration, and invasion of NPC cells and was involved in the cell cycle, apoptosis, angiogenesis, VEGF secretion, and in vivo tumor formation. These findings suggested that PCDH17 is a pleiotropic tumor suppressor of NPC.
Conclusion
Our study found that PCDH17 frequently undergoes methylation of the promoter in NPC, resulting in gene downregulation or silencing. PCDH17 is a pleiotropic tumor suppressor of NPC, and the methylation of PCDH17 can be used as an epigenetic marker of NPC.
Acknowledgments
The authors would like to thank Professor Qian Tao (Hong Kong University, Hong Kong, China) for generously providing the NPC cell lines, plasmids, and primers, designing experiments and his technical support. Additionally, the authors appreciate the technical assistance from the Molecular Oncology and Epigenetics Laboratory from the First Affiliated Hospital of Chongqing Medical University. This work was supported by funds from the Basic Research Project of Southwest Hospital, Army Medical University (SWH2017YBXM-18, Ya He), and the Post-Doctoral Research Project of Chongqing (Xm2015100, Ya He).
Disclosure
The authors report no conflicts of interest in this work.
References
Chou J, Lin YC, Kim J, et al. Nasopharyngeal carcinoma – review of the molecular mechanisms of tumorigenesis. Head Neck. 2008;30(7):946–963. | ||
Tao Q, Chan AT. Nasopharyngeal carcinoma: molecular pathogenesis and therapeutic developments. Expert Rev Mol Med. 2007;9(12):1–24. | ||
Le QT, Tate D, Koong A, et al. Improved local control with stereotactic radiosurgical boost in patients with nasopharyngeal carcinoma. Int J Radiat Oncol Biol Phys. 2003;56(4):1046–1054. | ||
Halbleib JM, Nelson WJ. Cadherins in development: cell adhesion, sorting, and tissue morphogenesis. Genes Dev. 2006;20(23):3199–3214. | ||
Nollet F, Kools P, van Roy F. Phylogenetic analysis of the cadherin superfamily allows identification of six major subfamilies besides several solitary members. J Mol Biol. 2000;299(3):551–572. | ||
Kim SY, Yasuda S, Tanaka H, Yamagata K, Kim H. Non-clustered protocadherin. Cell Adh Migr. 2011;5(2):97–105. | ||
Imoto I, Izumi H, Yokoi S, et al. Frequent silencing of the candidate tumor suppressor PCDH20 by epigenetic mechanism in non-small-cell lung cancers. Cancer Res. 2006;66(9):4617–4626. | ||
Ying J, Li H, Seng TJ, et al. Functional epigenetics identifies a protocadherin PCDH10 as a candidate tumor suppressor for nasopharyngeal, esophageal and multiple other carcinomas with frequent methylation. Oncogene. 2006;25(7):1070–1080. | ||
Miyamoto K, Fukutomi T, Akashi-Tanaka S, et al. Identification of 20 genes aberrantly methylated in human breast cancers. Int J Cancer. 2005;116(3):407–414. | ||
Yu B, Yang H, Zhang C, et al. High-resolution melting analysis of PCDH10 methylation levels in gastric, colorectal and pancreatic cancers. Neoplasma. 2010;57(3):247–252. | ||
Yu J, Cheng YY, Tao Q, et al. Methylation of protocadherin 10, a novel tumor suppressor, is associated with poor prognosis in patients with gastric cancer. Gastroenterology. 2009;136(2):e641:640–651. | ||
Narayan G, Scotto L, Neelakantan V, et al. Protocadherin PCDH10, involved in tumor progression, is a frequent and early target of promoter hypermethylation in cervical cancer. Genes Chromosomes Cancer. 2009;48(11):983–992. | ||
Wang KH, Liu HW, Lin SR, Ding DC, Chu TY. Field methylation silencing of the protocadherin 10 gene in cervical carcinogenesis as a potential specific diagnostic test from cervical scrapings. Cancer Sci. 2009;100(11):2175–2180. | ||
Cheung HH, Lee TL, Davis AJ, Taft DH, Rennert OM, Chan WY. Genome-wide DNA methylation profiling reveals novel epigenetically regulated genes and non-coding RNAs in human testicular cancer. Br J Cancer. 2010;102(2):419–427. | ||
Ying J, Gao Z, Li H, et al. Frequent epigenetic silencing of protocadherin 10 by methylation in multiple haematologic malignancies. Br J Haematol. 2007;136(6):829–832. | ||
Haruki S, Imoto I, Kozaki K, et al. Frequent silencing of protocadherin 17, a candidate tumour suppressor for esophageal squamous cell carcinoma. Carcinogenesis. 2010;31(6):1027–1036. | ||
Hu X, Sui X, Li L, et al. Protocadherin 17 acts as a tumour suppressor inducing tumour cell apoptosis and autophagy, and is frequently methylated in gastric and colorectal cancers. J Pathol. 2013;229(1):62–73. | ||
Giefing M, Zemke N, Brauze D, et al. High resolution ArrayCGH and expression profiling identifies PTPRD and PCDH17/PCH68 as tumor suppressor gene candidates in laryngeal squamous cell carcinoma. Genes Chromosomes Cancer. 2011;50(3):154–166. | ||
de Tayrac M, Etcheverry A, Aubry M, et al. Integrative genome-wide analysis reveals a robust genomic glioblastoma signature associated with copy number driving changes in gene expression. Genes Chromosomes Cancer. 2009;48(1):55–68. | ||
Leshchenko VV, Kuo PY, Shaknovich R, et al. Genomewide DNA methylation analysis reveals novel targets for drug development in mantle cell lymphoma. Blood. 2010;116(7):1025–1034. | ||
Yu JS, Koujak S, Nagase S, et al. PCDH8, the human homolog of PapC, is a candidate tumor suppressor of breast cancer. Oncogene. 2008;27(34):4657–4665. | ||
Ying J, Li H, Yu J, et al. Wnt5a exhibits tumor-suppressive activity through antagonizing the Wnt/beta-catenin signaling, and is frequently methylated in colorectal cancer. Clin Cancer Res. 2008;14(1):55–61. | ||
Costa VL, Henrique R, Danielsen SA, et al. TCF21 and PCDH17 methylation: an innovative panel of biomarkers for a simultaneous detection of urological cancers. Epigenetics. 2011;6(9):1120–1130. | ||
Huang DP, Ho JH, Poon YF, et al. Establishment of a cell line (NPC/HK1) from a differentiated squamous carcinoma of the nasopharynx. Int J Cancer. 1980;26(2):127–132. | ||
Cheung ST, Huang DP, Hui AB, et al. Nasopharyngeal carcinoma cell line (C666-1) consistently harbouring Epstein-Barr virus. Int J Cancer. 1999;83(1):121–126. | ||
Teng ZP, Ooka T, Huang DP, Zeng Y. Detection of Epstein-Barr virus DNA in well and poorly differentiated nasopharyngeal carcinoma cell lines. Virus Genes. 1996;13(1):53–60. | ||
Glaser R, Zhang HY, Yao KT, et al. Two epithelial tumor cell lines (HNE-1 and HONE-1) latently infected with Epstein-Barr virus that were derived from nasopharyngeal carcinomas. Proc Natl Acad Sci U S A. 1989;86(23):9524–9528. | ||
Li J, Tu Z, Shen Z, et al. Quantitative measurement of optical attenuation coefficients of cell lines CNE1, CNE2, and NP69 using optical coherence tomography. Lasers Med Sci. 2013;28(2):621–625. | ||
He D, Zeng Q, Ren G, et al. Protocadherin8 is a functional tumor suppressor frequently inactivated by promoter methylation in nasopharyngeal carcinoma. Eur J Cancer Prev. 2012;21(6):569–575. | ||
Tao Q, Huang H, Geiman TM, et al. Defective de novo methylation of viral and cellular DNA sequences in ICF syndrome cells. Hum Mol Genet. 2002;11(18):2091–2102. | ||
Tao Q, Swinnen LJ, Yang J, et al. Methylation status of the Epstein-Barr virus major latent promoter C in iatrogenic B cell lymphoproliferative disease. Am J Pathol. 1999;155(2):619–625. | ||
Wang Y, Li Z, Li W, Liu S, Han B. Methylation of Cdx2 gene promoter in the prediction of treatment efficacy in colorectal cancer. Oncol Lett. 2018;16(1):195–198. | ||
Liu ZZ, Zhao XZ, Zhang XS, Zhang M. Promoter DNA demethylation of Keap1 gene in diabetic cardiomyopathy. Int J Clin Exp Pathol. 2014;7(12):8756–8762. | ||
Huang J, Jing S, Chen X, et al. Propofol administration during early postnatal life suppresses hippocampal neurogenesis. Mol Neurobiol. 2016;53(2):1031–1044. | ||
Xu P, Xu H, Tang X, et al. Liver X receptor β is essential for the differentiation of radial glial cells to oligodendrocytes in the dorsal cortex. Mol Psychiatry. 2014;19(8):947–957. | ||
Sui X, Wang D, Geng S, Zhou G, He C, Hu X. Methylated promoters of genes encoding protocadherins as a new cancer biomarker family. Mol Biol Rep. 2012;39(2):1105–1111. | ||
Suzuki ST. Protocadherins and diversity of the cadherin superfamily. J Cell Sci. 1996;109(Pt 11):2609–2611. | ||
Suzuki ST. Recent progress in protocadherin research. Exp Cell Res. 2000;261(1):13–18. | ||
Frank M, Kemler R. Protocadherins. Curr Opin Cell Biol. 2002;14(5):557–562. | ||
Ose R, Yanagawa T, Ikeda S, Ohara O, Koga H. PCDH24-induced contact inhibition involves downregulation of beta-catenin signaling. Mol Oncol. 2009;3(1):54–66. | ||
Huang D, Yang S, Dai Z, Li W, Wang R. Aberrant promoter methylation reduced the expression of protocadherin 17 in nasopharyngeal cancer. Biochem. Cell Biol. 2018;22. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.