")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Proteomic response to 5,6-dimethylxanthenone 4-acetic acid (DMXAA, vadimezan) in human non-small cell lung cancer A549 cells determined by the stable-isotope labeling by amino acids in cell culture (SILAC) approach
Authors Pan S, Zhou Z, He Z, Zhang X, Yang T, Yang Y, Wang D, Qiu J, Zhou S
Received 18 October 2014
Accepted for publication 28 November 2014
Published 17 February 2015 Volume 2015:9 Pages 937—968
DOI https://doi.org/10.2147/DDDT.S76021
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan

Shu-Ting Pan,1,* Zhi-Wei Zhou,2,3,* Zhi-Xu He,3 Xueji Zhang,4 Tianxin Yang,5 Yin-Xue Yang,6 Dong Wang,7 Jia-Xuan Qiu,1 Shu-Feng Zhou2
1Department of Oral and Maxillofacial Surgery, The First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi, People’s Republic of China; 2Department of Pharmaceutical Sciences, College of Pharmacy, University of South Florida, Tampa, FL, USA; 3Guizhou Provincial Key Laboratory for Regenerative Medicine, Stem Cell and Tissue Engineering Research Center and Sino-US Joint Laboratory for Medical Sciences, Guiyang Medical University, Guiyang, 4Research Center for Bioengineering and Sensing Technology, University of Science and Technology Beijing, Beijing, People’s Republic of China; 5Department of Internal Medicine, University of Utah and Salt Lake Veterans Affairs Medical Center, Salt Lake City, UT, USA; 6Department of Colorectal Surgery, General Hospital of Ningxia Medical University, Yinchuan, 7Cancer Center, Daping Hospital and Research Institute of Surgery, Third Military Medical University, Chongqing, People’s Republic of China
*These two authors contributed equally to this work
Abstract: 5,6-Dimethylxanthenone 4-acetic acid (DMXAA), also known as ASA404 and vadimezan, is a potent tumor blood vessel-disrupting agent and cytokine inducer used alone or in combination with other cytotoxic agents for the treatment of non-small cell lung cancer (NSCLC) and other cancers. However, the latest Phase III clinical trial has shown frustrating outcomes in the treatment of NSCLC, since the therapeutic targets and underlying mechanism for the anticancer effect of DMXAA are not yet fully understood. This study aimed to examine the proteomic response to DMXAA and unveil the global molecular targets and possible mechanisms for the anticancer effect of DMXAA in NSCLC A549 cells using a stable-isotope labeling by amino acids in cell culture (SILAC) approach. The proteomic data showed that treatment with DMXAA modulated the expression of 588 protein molecules in A549 cells, with 281 protein molecules being up regulated and 306 protein molecules being downregulated. Ingenuity pathway analysis (IPA) identified 256 signaling pathways and 184 cellular functional proteins that were regulated by DMXAA in A549 cells. These targeted molecules and signaling pathways were mostly involved in cell proliferation and survival, redox homeostasis, sugar, amino acid and nucleic acid metabolism, cell migration, and invasion and programed cell death. Subsequently, the effects of DMXAA on cell cycle distribution, apoptosis, autophagy, and reactive oxygen species (ROS) generation were experimentally verified. Flow cytometric analysis showed that DMXAA significantly induced G1 phase arrest in A549 cells. Western blotting assays demonstrated that DMXAA induced apoptosis via a mitochondria-dependent pathway and promoted autophagy, as indicated by the increased level of cytosolic cytochrome c, activation of caspase 3, and enhanced expression of beclin 1 and microtubule-associated protein 1A/1B-light chain 3 (LC3-II) in A549 cells. Moreover, DMXAA significantly promoted intracellular ROS generation in A549 cells. Collectively, this SILAC study quantitatively evaluates the proteomic response to treatment with DMXAA that helps to globally identify the potential molecular targets and elucidate the underlying mechanism of DMXAA in the treatment of NSCLC.
Keywords: DMXAA, non-small cell lung cancer, cell cycle, apoptosis, autophagy, SILAC
Introduction
Lung cancer is the most common cancer and the leading cause of cancer-related death in humans worldwide.1,2 There were about 1.8 million new cases diagnosed with lung cancer in 2012, accounting for 12.9% of the total cases of cancer.1,2 Small-cell lung cancer and non-small cell lung cancer (NSCLC) are the two major types of lung cancer. NSCLC is the most common type, accounting for 70%–85% of all cases of lung cancer. In the USA, there were 207,339 new cases of lung cancer and 156,953 deaths resulting from lung cancer in 2011,3 and it is estimated that there were 224,210 new cases of lung cancer and 159,260 deaths due to lung cancer in 2014.4 In the People’s Republic of China, lung cancer is the most common cancer and the leading cause of cancer-related death, with a skyrocketing increase in incidence and mortality rates.2,5 In 2009, the incidence rate for lung cancer was about 53.57/100,000, accounting for 18.74% of overall new cases of cancer; the mortality rate for lung cancer was about 45.57/100,000, accounting for 25.24% of cancer-related deaths.2,5 Current therapies for lung cancer include surgery, chemotherapy, radiotherapy, immunotherapy, and targeted therapy, which are used alone or in combination. However, the therapeutic outcome for lung cancer is often disappointing, in particular for advanced NSCLC,6,7 due to the poor response to current therapeutics, drug resistance, and severe side effects, which highlights an urgent need for discovery of efficacious and safe new agents for the treatment of NSCLC.
5,6-Dimethylxanthenone-4-acetic acid (DMXAA, Figure 1), also known as vadimezan and ASA404, is a vascular-disrupting agent that reduces the blood supply to tumoral tissue, resulting in tumor regression.8,9 However, the molecular targets and exact mechanisms of action of DMXAA are elusive so far. DMXAA shows inhibitory effects against several protein kinases, with the most potent effects being on the vascular endothelial growth factor receptor tyrosine kinase family.10,11 DMXAA is a potent inducer of tumor necrosis factor-α and activates host immune effectors that assist in killing cancer cells.10 DMXAA induces rapid vascular collapse and subsequent tumor hemorrhagic necrosis via induction of apoptosis in tumor vascular endothelial cells and indirect vascular effects induced by various cytokines, in particular, tumor necrosis factor-α, serotonin, and nitric oxide.10 The pharmacokinetics of DMXAA has also be investigated. In cancer patients, DMXAA concentration-time profiles are well described by a three-compartment model with saturable elimination.12 Body surface area and sex are significant covariates on the volume of distribution of the central compartment and the maximum elimination rate, respectively.12 DMXAA is extensively metabolized in human liver microsomes and cancer patients. There are two major metabolites of DMXAA, ie, DMXAA acyl glucuronide and 6-hydroxymethyl-5-methylxanthenone-4-acetic acid (6-OH-MXAA). Cytochrome P450 1A2 is responsible for the conversion of DMXAA to 6-OH-MXAA, with an apparent Km of 6.2 μM and a Vmax of 0.014 nmol/minute/mg.13 DMXAA is also extensively metabolized by uridine 5’-diphospho-glucuronosyltransferase 1A2 (UGT1A2) and UGT2B7, with a greater contribution from UGT2B7.14 DMXAA has been tested mainly in the treatment of NSCLC, and also in prostate cancer and human epidermal growth factor receptor 2-negative breast cancer.15–20 In these clinical studies, DMXAA is used alone and more often in combination with other cytotoxic drugs. A Phase II clinical trial showed that DMXAA in combination with carboplatin and paclitaxel had a potent anticancer effect in NSCLC patients.15 This triple combination therapy prolonged the survival of about 5 months when compared with the monotherapy.15 However, the Phase III clinical trial conducted by Lara et al showed that the triple chemotherapy of DMXAA with carboplatin and paclitaxel failed to improve the efficacy of the monotherapy.16 This may be due to the complexity of the mechanisms of action of DMXAA. DMXAA has been shown to target the stimulator of interferon gene (STING) pathway and this effect is only observed in mice but not in humans.21–23 However, this cannot provide a convincing explanation for the failure of DMXAA in the Phase III trial in NSCLC patients. Therefore, it is of great importance to globally understand and uncover the molecular targets and related signaling pathways involved in the anticancer effect of DMXAA and DMXAA-based combination therapies.
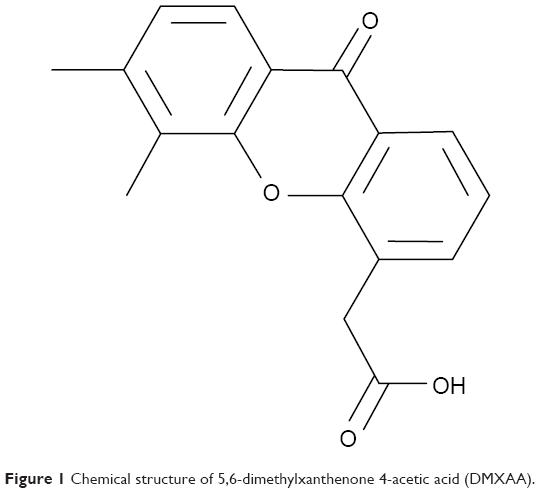 | Figure 1 Chemical structure of 5,6-dimethylxanthenone 4-acetic acid (DMXAA). |
So far, there are many studies on the mechanisms of action of DMXAA in the treatment of NSCLC, showing that DMXAA can activate STING-dependent innate immune pathways and mitogen-activated protein kinases and inhibit vascular endothelial growth factor receptor.11,21–23 However, there is a lack of evidence to depict the global molecular targets and related signaling pathways for the NSCLC cell killing effects of DMXAA, such as cell proliferation, programmed cell death, and cell migration and invasion. Notably, targeting cell cycle progression, apoptosis, autophagy, and epithelial to mesenchymal transition (EMT) has been proposed for treatment of NSCLC.24 Therefore, an approach that can evaluate cellular proteomic responses to the DMXAA is important for the optimal treatment of NSCLC. Stable-isotope labeling by amino acids in cell culture (SILAC) is a practical and powerful approach to uncovering the global proteomic response to drug treatment and other interventions.25 In particular, it can be used to systemically and quantitatively assess the target network of drugs, to evaluate drug toxicity, and to identify new biomarkers for the diagnosis and treatment of important diseases, including NSCLC.25–27 In this regard, we investigated the molecular targets of DMXAA in A549 cells using a combination of proteomic and functional approaches, with a focus on cell cycle distribution, apoptosis, autophagy, and redox homeostasis.
Materials and methods
Chemicals and reagents
DMXAA (purity ≥98%), 13C6-L-lysine, L-lysine, 13C6 15N4-L-arginine, L-arginine, RNase A, propidium iodide, Dulbecco’s phosphate-buffered saline (PBS), heat-inactivated fetal bovine serum (FBS), dialyzed FBS, and Roswell Park Memorial Institute (RPMI)-1640 medium for SILAC were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). The 5-(and 6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) was sourced from Invitrogen Inc. (Carlsbad, CA, USA). A FASP™ protein digestion kit was purchased from Protein Discovery Inc. (Knoxville, TN, USA). RPMI-1640 medium for general cultural use was obtained from Corning Cellgro Inc. (Herndon, VA, USA). The polyvinylidene difluoride membrane was purchased from EMD Millipore Inc. (Bedford, MA, USA). Proteomic quantitation kits for acidification, desalting, and digestion, ionic detergent compatibility reagent, a Pierce bicinchoninic acid protein assay kit, and Western blotting substrate were obtained from Thermo Fisher Scientific Inc. (Waltham, MA, USA). Primary antibodies against human cytochrome c, cleaved caspase 3, microtubule-associated protein 1A/1B-light chain 3-I (LC3-I), LC3-II, and beclin 1 were all purchased from Cell Signaling Technology Inc. (Beverly, MA, USA). The antibody against human β-actin was obtained from Santa Cruz Biotechnology Inc. (Dallas, TX, USA).
Cell line and cell culture
The A549 NSCLC cell line was obtained from American Type Culture Collection (Manassas, VA, USA) and cultured in RPMI-1640 medium supplemented with 10% heat-inactivated FBS. The cells were maintained at 37°C in a 5% CO2/95% air humidified incubator. DMXAA was dissolved in dimethyl sulfoxide at a stock concentration of 20 mM and stored at −20°C. It was freshly diluted to predetermined concentrations with culture medium. The final concentration of dimethyl sulfoxide was 0.05% (v/v). The control cells received the vehicle only.
Quantitative proteomic study using SILAC
Quantitative proteomic experiments were performed using a SILAC-based approach as described previously.25,26,28 Briefly, A549 cells were cultured in RPMI-1640 medium (for SILAC) with (heavy) or without (light) stable isotope-labeled amino acids (13C6 L-lysine and 13C6 15N4 L-arginine) and 10% dialyzed FBS. A549 cells cultured in heavy medium were treated with 10 μM DMXAA for 24 hours after six cell doubling times. After treatment with DMXAA, A549 cell samples were harvested and lysed with hot lysis buffer (100 mM Tris base, 4% sodium dodecyl sulfate [SDS], and 100 mM dithiothreitol). The cell lysate was denatured at 95°C for 5 minutes and then sonicated for 3 seconds with six pulses. The samples were then centrifuged at 15,000× g for 20 minutes at room temperature and the supernatant was collected. The protein concentration was determined using ionic detergent compatibility reagent. Subsequently, equal amounts of heavy and light protein samples were combined to reach a total volume of 30–60 μL containing 300–600 μg protein. The combined protein sample was digested using an filter-aided sample prep (FASP™) protein digestion kit. After digestion, the resulting sample was acidified to a pH of 3 and desalted using a C18 solid-phase extraction column. The samples were then concentrated using a vacuum concentrator at 45°C for 120 minutes, and the peptide mixtures (5 μL) were subjected to the hybrid linear ion trap (LTQ Orbitrap XL™, Thermo Fisher Scientific Inc.). Liquid chromatography-tandem mass spectrometry was performed using a 10 cm long, 75 μm (inner diameter) reversed-phase column packed with 5 μm diameter C18 material having a pore size of 300 Å (New Objective Inc., Woburn, MA, USA) with a gradient mobile phase of 2%–40% acetonitrile in 0.1% formic acid at 200 μL per minute for 125 minutes. The Orbitrap full mass spectrometry scanning was performed at a mass (m/z) resolving power of 60,000, with positive polarity in profile mode (M + H+). The peptide SILAC ratio was calculated using MaxQuant version 1.2.0.13. The SILAC ratio was determined by averaging all peptide SILAC ratios from peptides identified of the same protein. The proteins were identified using Scaffold 4.3.2 (Proteome Software Inc., Portland, OR, USA) and the pathway was analyzed using ingenuity pathway analysis (IPA) from QIAGEN Inc. (Redwood City, CA, USA).
Cell cycle analysis using flow cytometry
The effect of treatment with DMXAA on the cell cycle was determined by flow cytometry as described previously.29 Briefly, A549 cells were treated with DMXAA at concentrations of 0.1, 1, and 10 μM for 24 hours. In separate experiments, A549 cells were treated with 10 μM DMXAA over a 72-hour period. The cells were suspended, fixed in 70% ethanol, washed in PBS, and resuspended in 1 mL of PBS containing 1 mg/mL RNase A and 50 μg/mL propidium iodide. The cells were incubated in the dark for 30 minutes at room temperature. Next, the cells were subject to cell cycle analysis using a flow cytometer (BD LSR II Analyzer; Becton Dickinson Immunocytometry Systems, San Jose, CA, USA). The flow cytometer collected 10,000 events for analysis.
Measurement of intracellular reactive oxygen species (ROS) levels
Intracellular levels of ROS were measured by a fluorometer using CM-H2DCFDA according to the manufacturer’s instructions. Cell-permeant CM-H2DCFDA passively diffuses into cells and is retained in the cells after cleavage by intracellular esterases. Upon oxidation by ROS, the nonfluorescent CM-H2DCFDA is converted to highly fluorescent CM-DCF. Briefly, A549 cells were seeded into a 96-well plate at a density of 1×104 cells/well. After treatment with DMXAA at 0.1, 1, and 10 μM for 48 hours, the cells were incubated with CM-H2DCFDA at 5 μM in PBS for 30 minutes. The fluorescence intensity was detected at wavelengths of 485 nm (excitation) and 530 nm (emission). The control cells were treated with vehicle only (0.05% dimethyl sulfoxide, v/v).
Western blotting analysis
A549 cells were washed with pre-cold PBS after 24-hour treatment with DMXAA at 0.1, 1, and 10 μM, lysed with radioimmunoprecipitation (RIPA) buffer containing the protease inhibitor and phosphatase inhibitor cocktails, and centrifuged at 3,000× g for 10 minutes at 4°C. Protein concentrations were measured using a Pierce bicinchoninic acid protein assay kit. An equal amount of protein sample (30 μg) was resolved by SDS polyacrylamide gel electrophoresis (PAGE) sample loading buffer and electrophoresed on 12% SDS-PAGE minigel after thermal denaturation at 95°C for 5 minutes. The proteins were transferred onto an Immobilon polyvinylidene difluoride membrane at 400 mA for 1 hour at 4°C. Membranes were blocked with skim milk and probed with the indicated primary antibody overnight at 4°C and then blotted with appropriate horseradish peroxidase-conjugated secondary anti-mouse or anti-rabbit antibody. Visualization was performed using a ChemiDoc™ XRS system (Bio-Rad, Hercules, CA, USA) with enhanced chemiluminescence substrate, and the blots were analyzed using Image Lab 3.0 (Bio-Rad). The protein level was normalized to the matching densitometric value of the internal control β-actin.
Statistical analysis
The data are presented as the mean ± standard deviation (SD). Comparisons of multiple groups were evaluated by one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison procedure. Values of P<0.05 were considered to be statistically significant. Assays were performed at least three times independently.
Results
Overview of proteomic response to DMXAA treatment in A549 cells
To reveal the potential molecular targets of DMXAA in the treatment of NSCLC, we conducted proteomic experiments to evaluate the interactome of DMXAA in A549 cells. There were 588 protein molecules identified as potential molecular targets of DMXAA in A549 cells, with 281 protein molecules being upregulated and 306 protein molecules being downregulated (Table 1). Subsequently, these proteins were subjected to IPA. The results showed that 256 signaling pathways and 184 cellular functional proteins were regulated by DMXAA in A549 cells (Tables 2 and 3). These functional proteins were involved in a number of important cellular processes, including cell proliferation, redox homeostasis, cell metabolism, cell migration and invasion, cell survival, and cell death. The signaling pathways included the G1 and G2 checkpoint regulation pathways, the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR)signaling pathway, the 5′-AMP-activated protein kinase (AMPK) signaling pathway, the nuclear factor erythroid 2-related factor 2 (Nrf2)-mediated oxidative stress response pathway, the epithelial adherens junction signaling pathway, regulation of the epithelial-mesenchymal transition signaling pathway, the nuclear factor-κB signaling pathway, and the apoptosis signaling pathway. The IPA results showed that the top ten targeted signaling pathways were the eukaryotic initiation factor (eIF) 2 signaling pathway, mTOR signaling pathway, eIF4 and p70S6K signaling pathway, epithelial adherens junction signaling pathway, remodeling of epithelial adherens junctions pathway, Nrf2-mediated oxidative stress response signaling pathway, RhoA signaling pathway, integrin signaling pathway, Rho-mediated regulation of actin-based motility signaling pathway, and Fcγ receptor-mediated phagocytosis signaling pathway (Table 2).
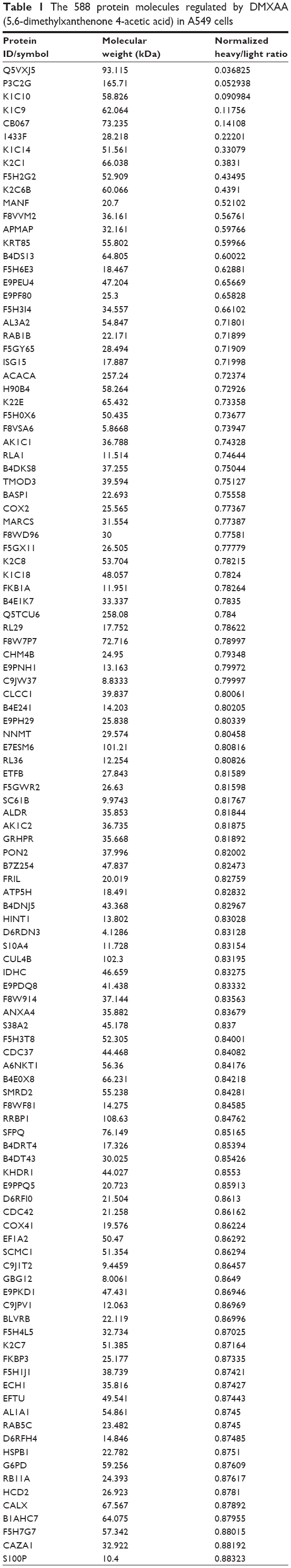 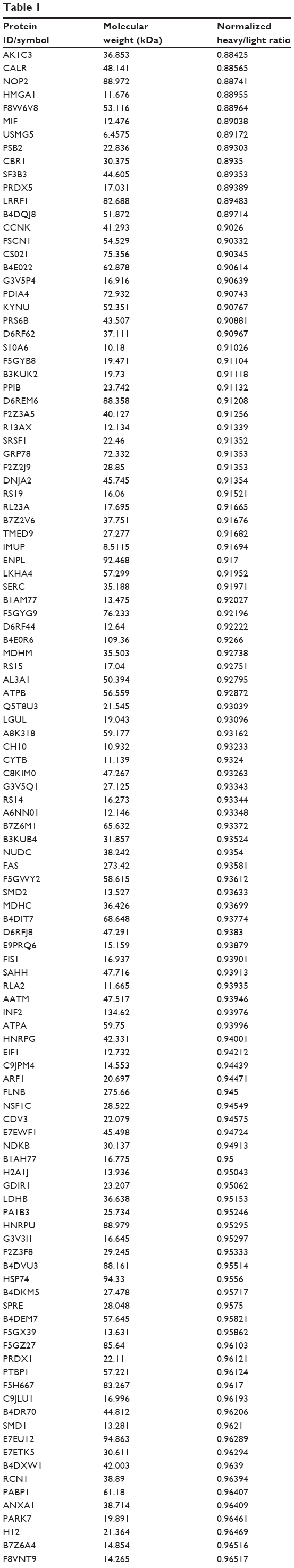 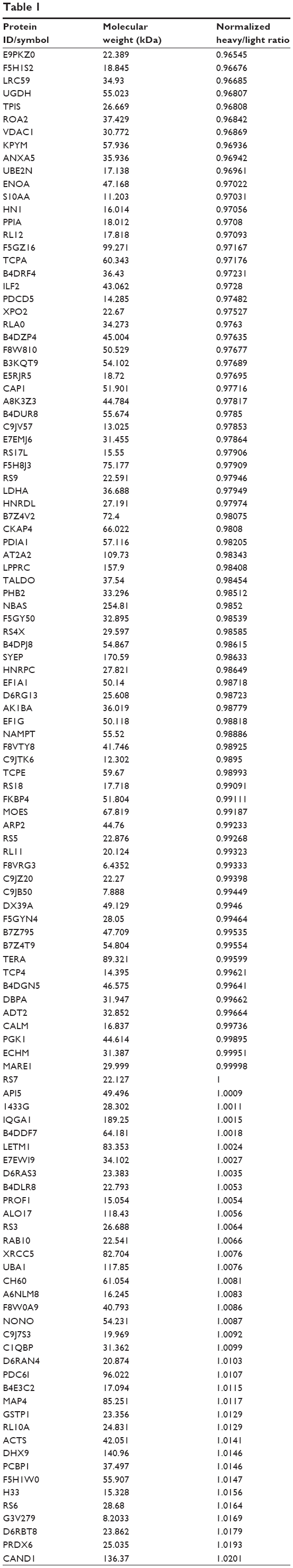 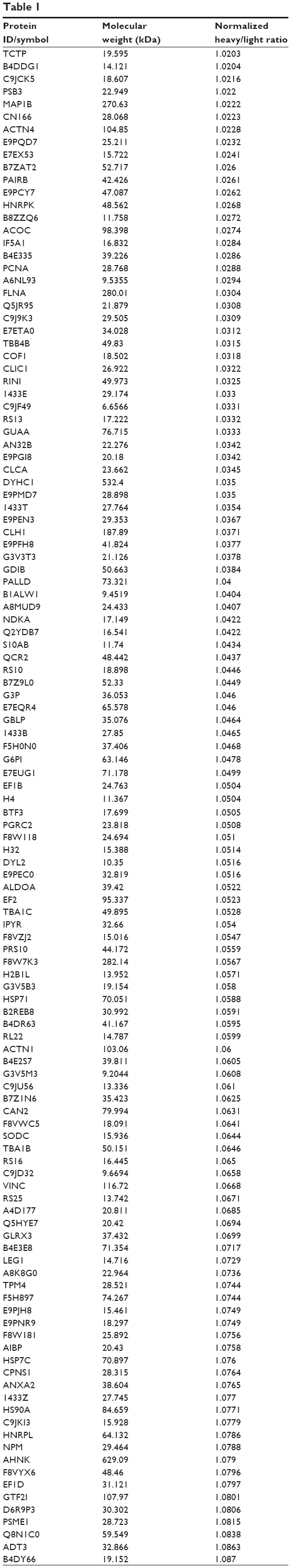 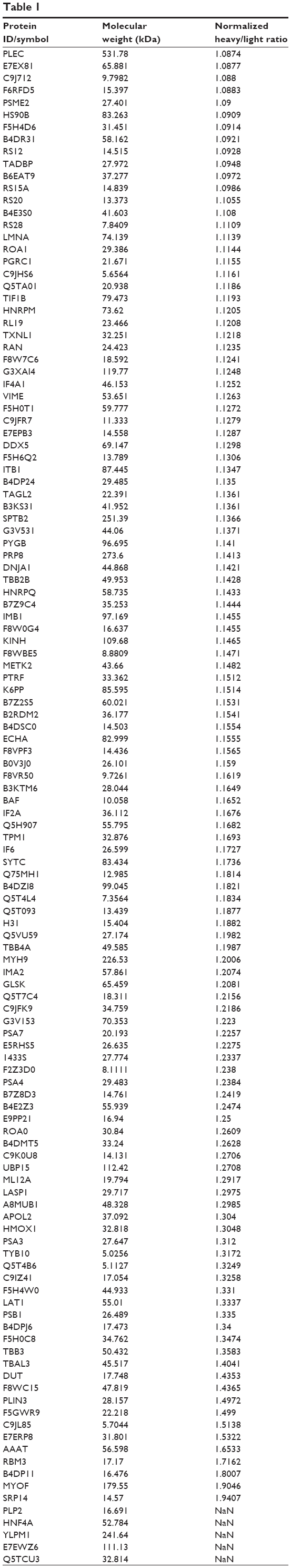 |
Table 1 The 588 protein molecules regulated by DMXAA (5,6-dimethylxanthenone 4-acetic acid) in A549 cells |
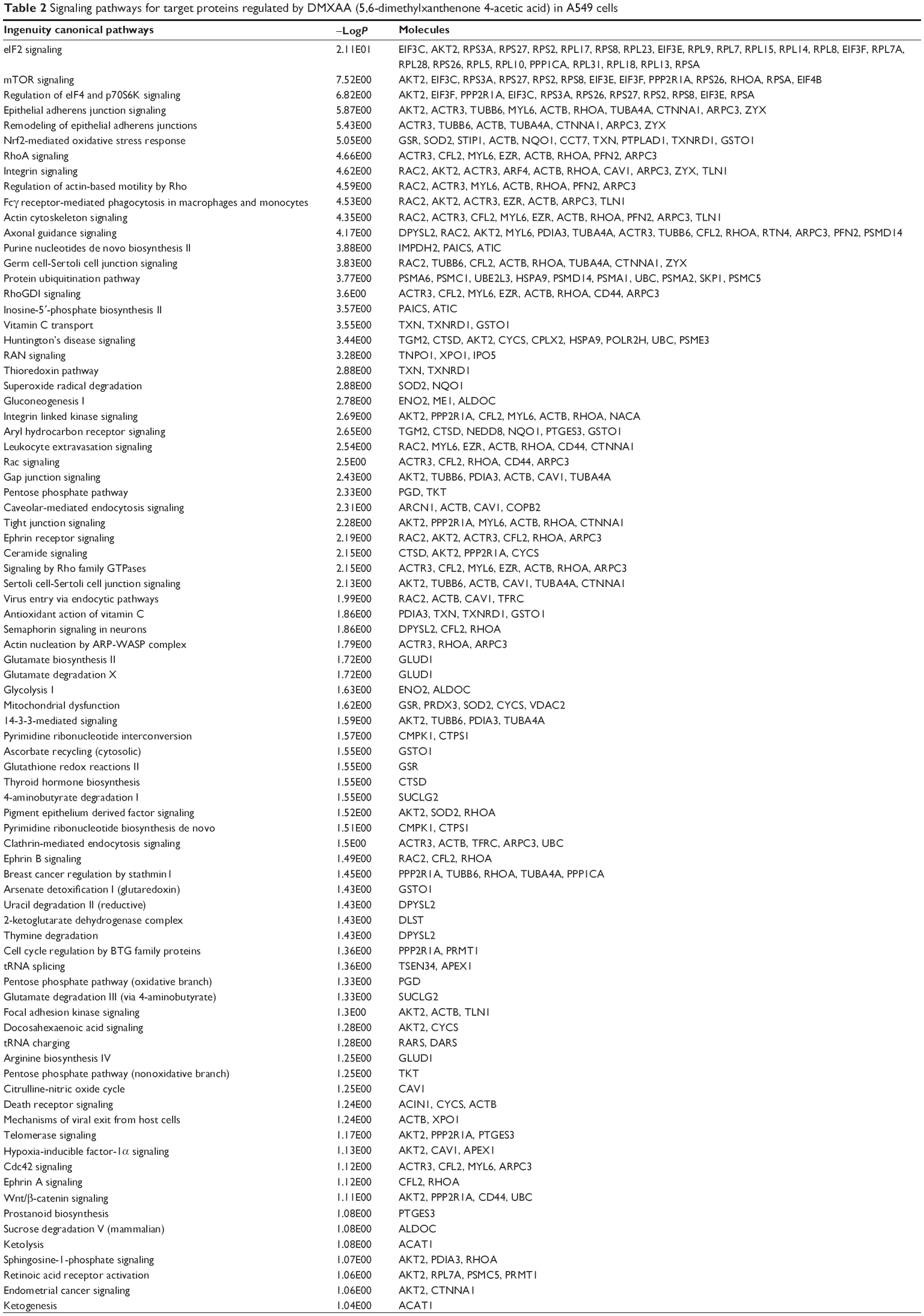 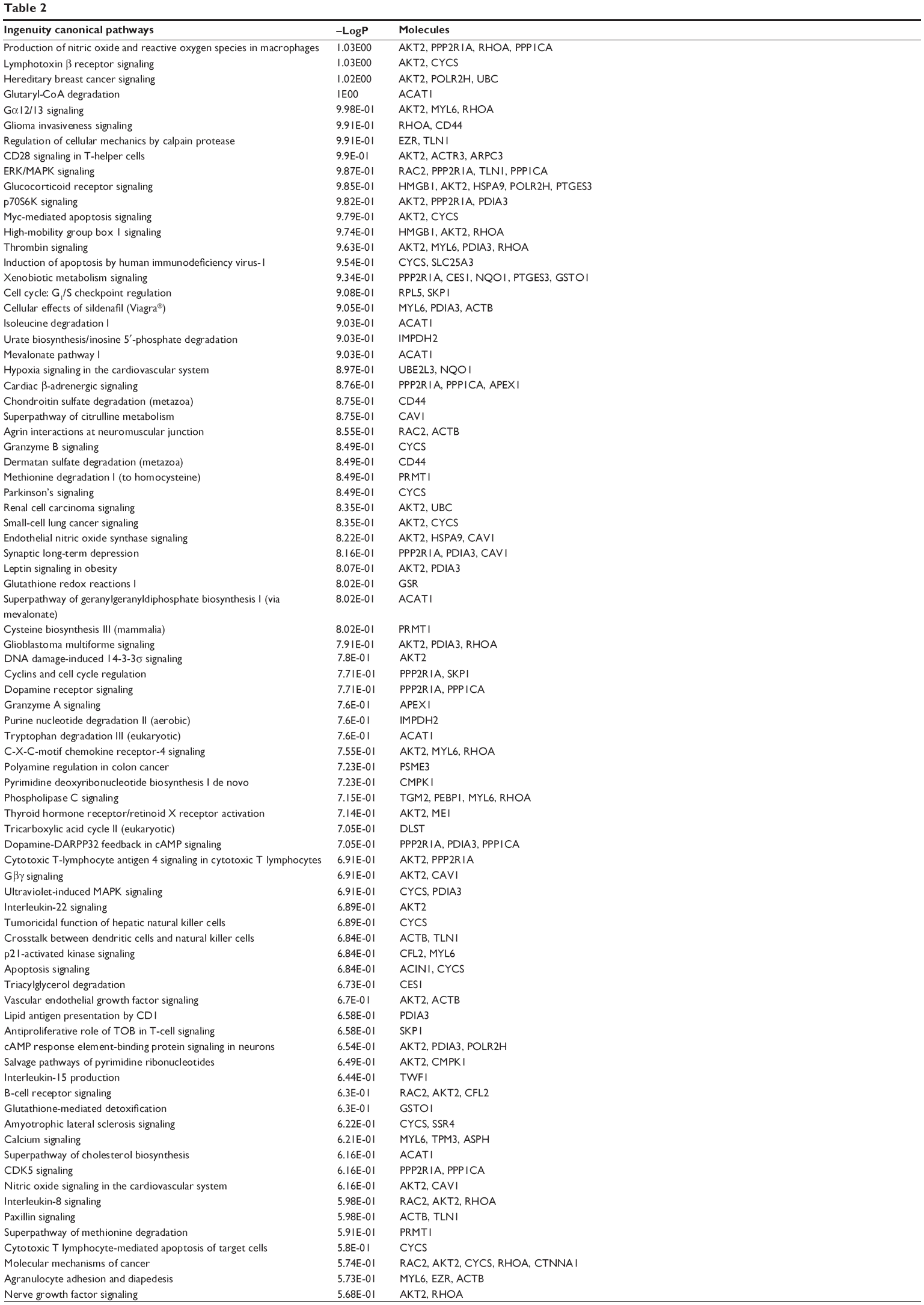 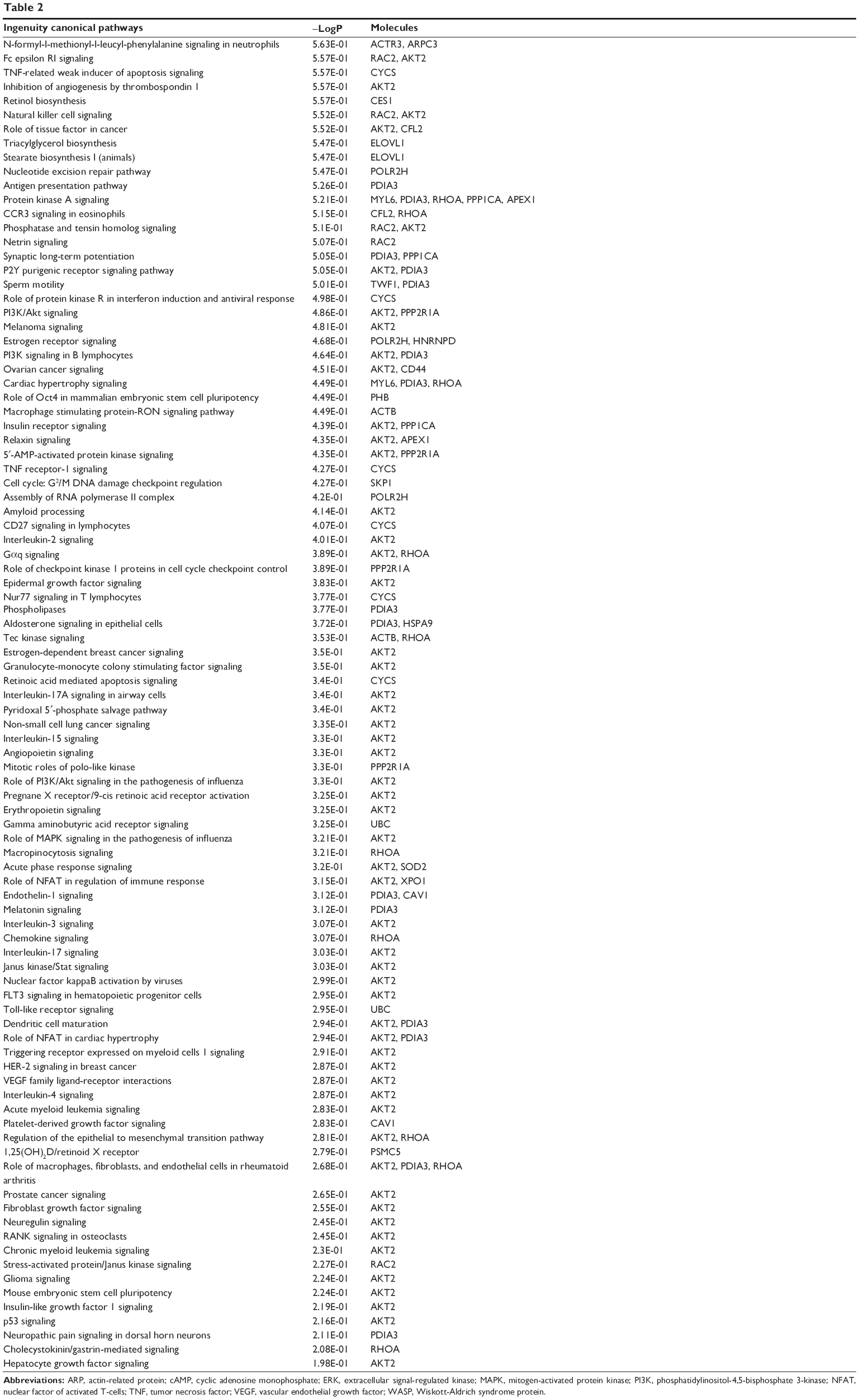 | Table 2 Signaling pathways for target proteins regulated by DMXAA (5,6-dimethylxanthenone 4-acetic acid) in A549 cells |
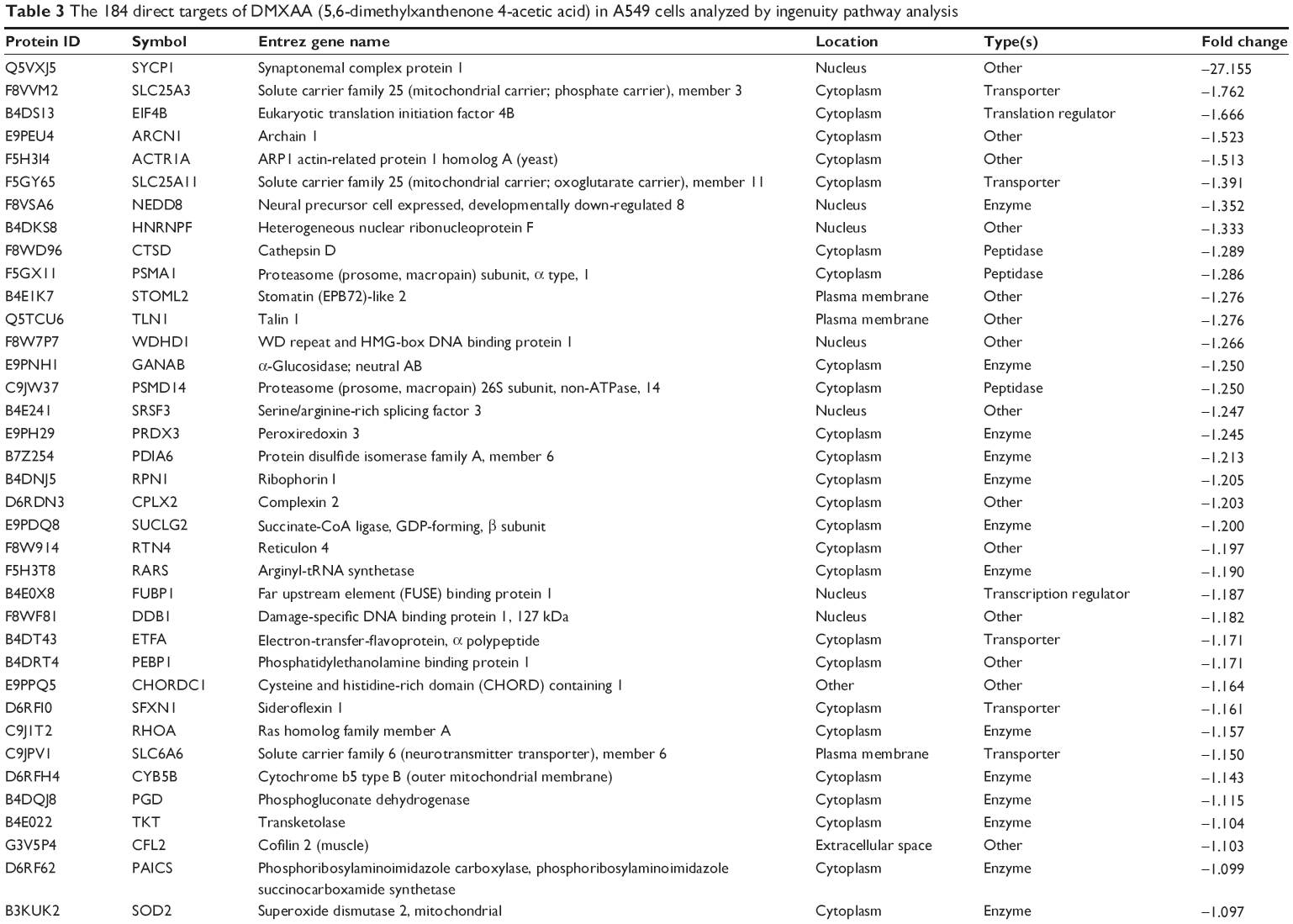 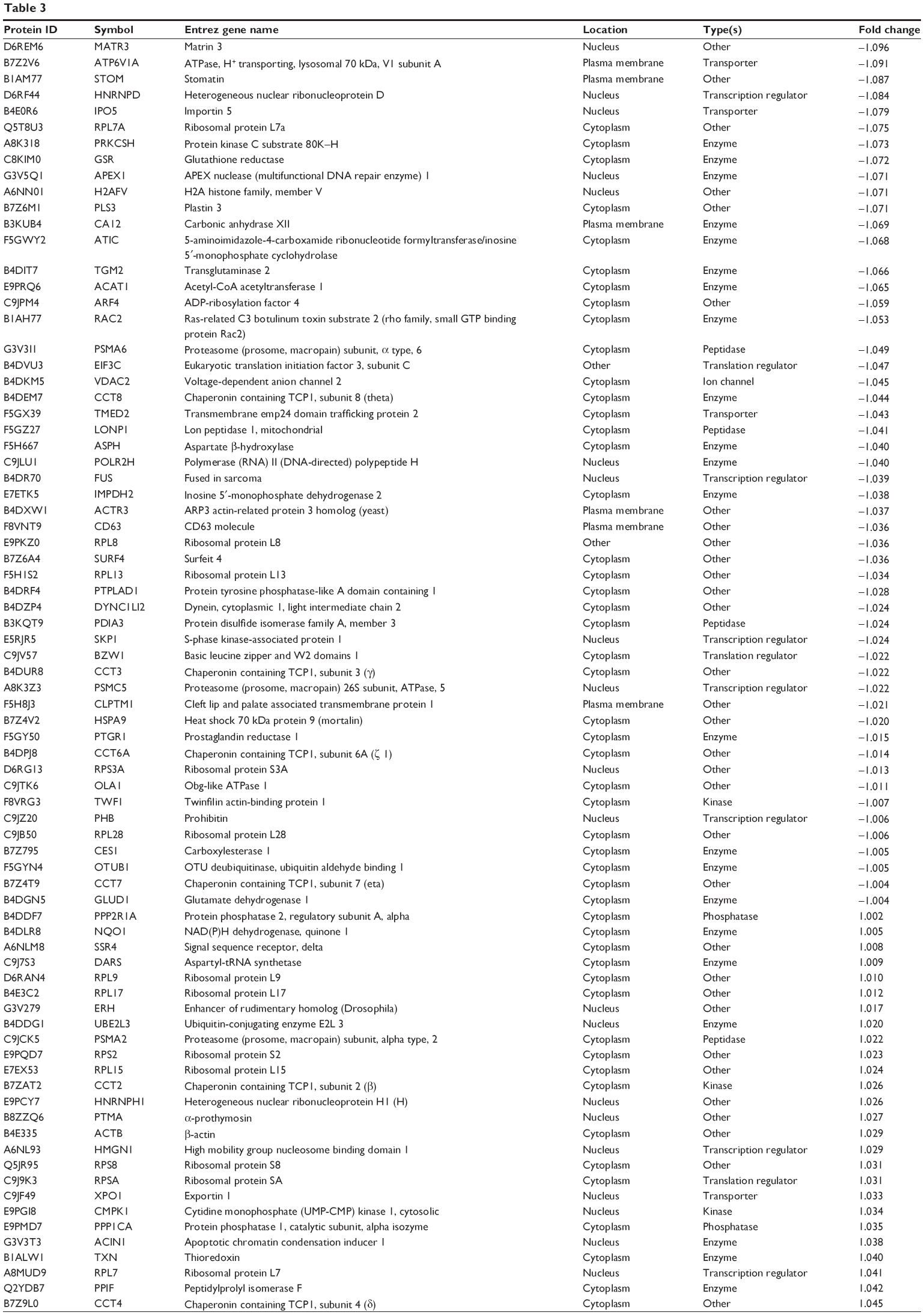 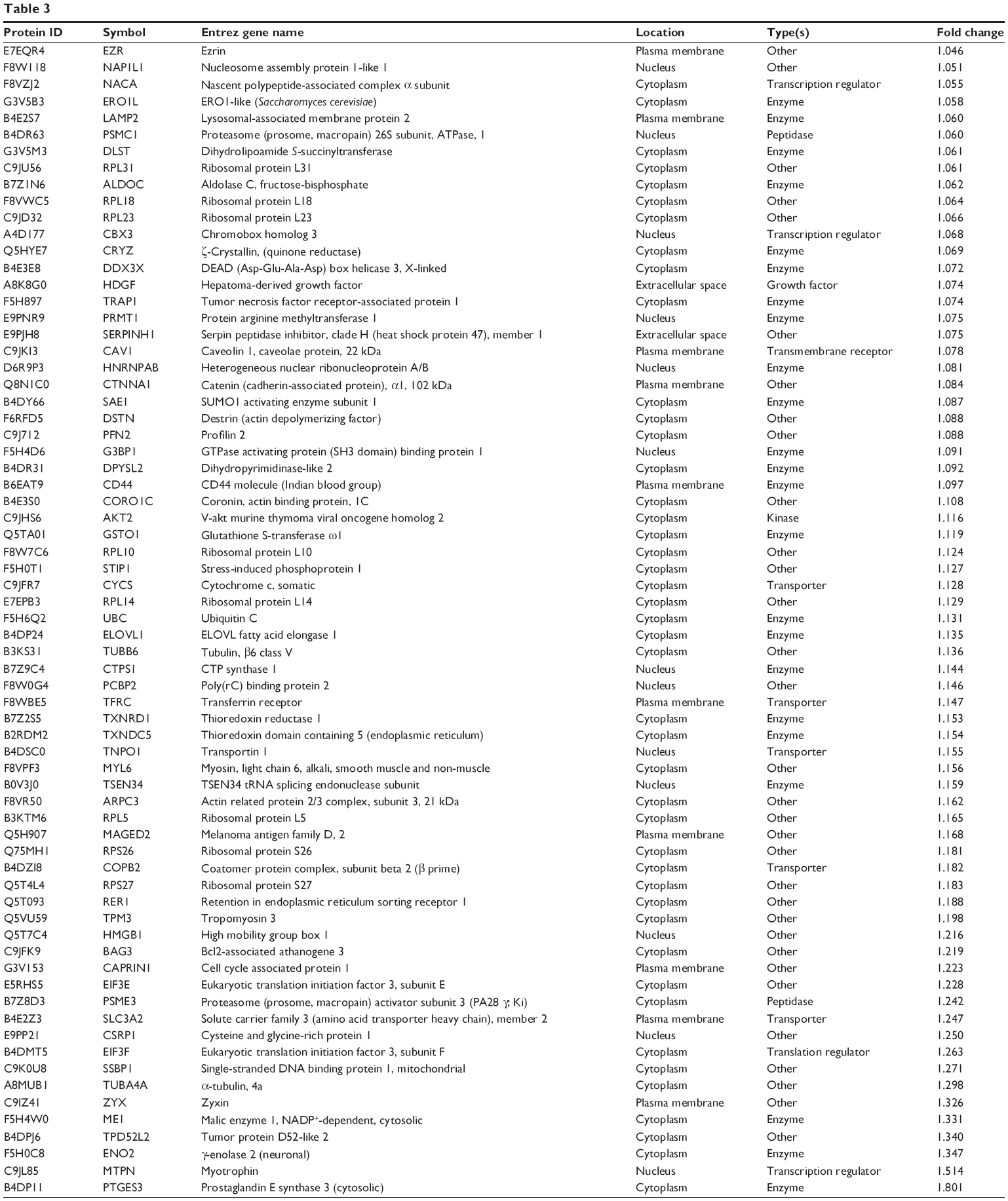 | Table 3 The 184 direct targets of DMXAA (5,6-dimethylxanthenone 4-acetic acid) in A549 cells analyzed by ingenuity pathway analysis |
DMXAA modulates networked signaling pathways in A549 cells
As seen in Figures 2 and 3, DMXAA showed an ability to regulate a number of networked signaling pathways that have critical roles in the regulation of cellular processes. IPA classified the top ten networks of signaling pathways responding to DMXAA in A549 cells (Table 4). These signaling networks have important roles in pathophysiological functions and the development of many important diseases. They included gene expression, DNA replication, recombination and repair, protein synthesis, small molecule biochemistry, carbohydrate metabolism, lipid metabolism, energy production, cellular response to therapeutics, connective tissue development and function, cellular assembly and organization, cellular compromise, cell morphology, free radical scavenging, cell death and survival, neurological disease, skeletal and muscular disorders, cardiac damage, cardiac fibrosis, development and function of cardiovascular system, and development of cancer.
 | Figure 2 Proteomic analysis revealed the molecular interactome regulated by DMXAA in A549 cells. |
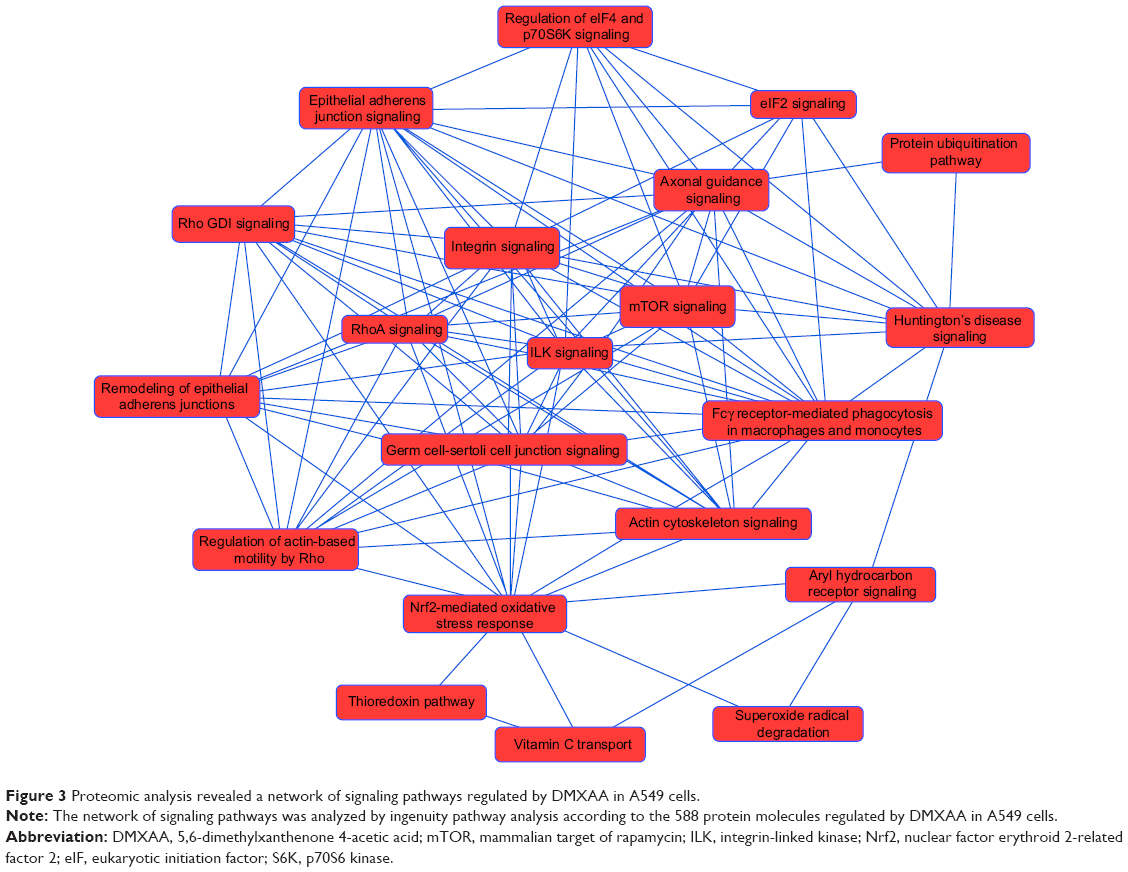 | Figure 3 Proteomic analysis revealed a network of signaling pathways regulated by DMXAA in A549 cells. |
 | Table 4 Networks of potential molecular targets regulated by DMXAA (5,6-dimethylxanthenone 4-acetic acid) in A549 cells |
DMXAA modulates important regulators involved in cell cycle distribution in A549 cells
It has been reported that regulation of cell cycle distribution is an effective approach in the treatment of lung cancer,24 and that vascular-disrupting agents exhibit modulating effects on cell cycle distribution. However, it has not been fully uncovered the molecular targets and underlying mechanisms of DMXAA. Therefore, in order to explore the effect and potential molecular targets of DMXAA on cell cycle distribution in A549 cells, we treated A549 cells with 10 μM DMXAA for 24 hours and then subjected samples of the cells to quantitative proteomic analysis. The proteomic results showed that DMXAA had an effect on the regulation of cyclins and the cell cycle distribution at G1/S and G2/M DNA damage checkpoints in A549 cells with the involvement of a number of functional proteins, such as PPP2R1A, RPL5, and SKP1 (Table 2). These findings suggest that DMXAA may modulate cell cycle distribution, contributing to its anticancer effect.
DMXAA regulates apoptosis and autophagy in A549 cells
Apoptosis and autophagy are two predominant programmed cell death pathways.30 Manipulating apoptosis and autophagy has been considered to be a promising strategy in the treatment of cancer via the regulation of mitochondria-dependent/-independent pathways.31–35 As shown in Table 2, DMXAA regulated the apoptotic signaling pathway, mitochondrial function, and death receptor signaling pathway, involving a number of functional proteins. These included ACIN1, CYCS, ACTB, AKT2, GSR, PRDX3, SOD2, and VDAC2 (Table 2). Further, the mTOR signaling pathway plays a pivotal role in the regulation of autophagy, and has been proposed to be a promising target in the treatment of NSCLC.36 Vascular disruption combined with mTOR inhibition showed an enhanced anticancer effect when compared with monotherapy.37 As shown in Table 2 and Figure 4, DMXAA showed an ability to modulate the mTOR signaling pathway in A549 cells. The results showed that DMXAA decreased the expression of EIF3C, EIF4B, RHOA, and RPS3A, but increased the expression of AKT2, EIF3E, EIF3F, PPP2R1A, RPS2, RPS8, RPS26, RPS27, and RPSA in A549 cells (Table 2), suggesting that modulation of mTOR signaling may play an important role in the cancer cell killing effect of DMXAA in A549 cells. Taken together, the results suggest that the regulatory effects of DMXAA on apoptosis, mitochondrial function, and mTOR signaling pathway contribute, at least in part, to the anticancer effect of this drug in the treatment of NSCLC.
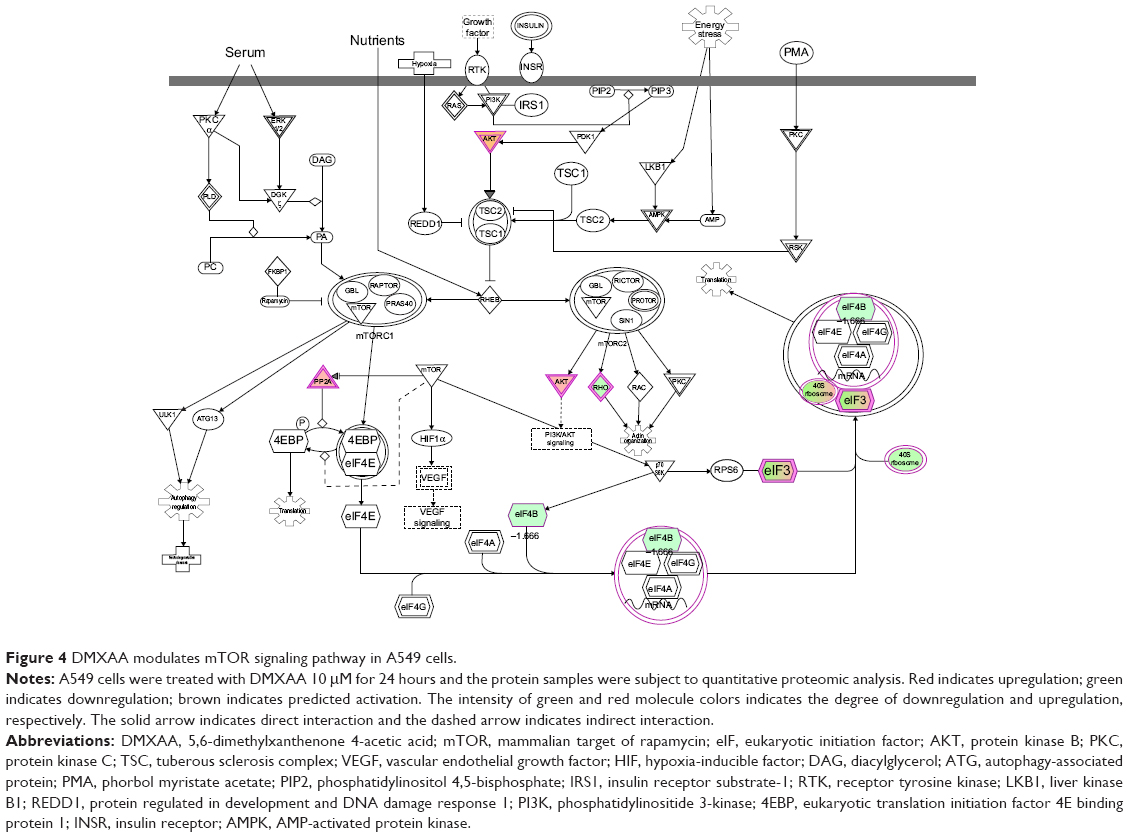 | Figure 4 DMXAA modulates mTOR signaling pathway in A549 cells. |
DMXAA regulates redox homeostasis involving ROS-mediated and Nrf2-mediated signaling pathways in A549 cells
Induction of ROS plays a critical role in the production of cytokines, contributing to the cancer cell killing effect of DMXAA.38 However, the regulatory effect of DMXAA on ROS generation-related molecules and signaling pathways is not fully understood. In this study, we observed that DMXAA regulated several critical signaling pathways related to ROS generation and redox homeostasis in A549 cells. Our quantitative proteomic study showed that treatment with DMXAA regulated oxidative phosphorylation, Nrf2-mediated oxidative stress response, and superoxide radical degradation in A549 cells (Table 2 and Figure 5). A number of functional proteins were found to be involved in these pathways, including ACTB, CCT7, GSR, GSTO1, NQO1, PTPLAD1, SOD2, STIP1, TXN, and TXNRD1 (Table 2). Of note, Nrf2-mediated signaling pathways have a critical role in the maintenance of intracellular redox homeostasis in response to various stimuli via regulating antioxidant responsive elements.39,40 The quantitative proteomic data suggest that modulation of the expression of functional proteins involved in Nrf2-mediated signaling pathways may contribute to the anticancer effect of DMXAA in the treatment of NSCLC.
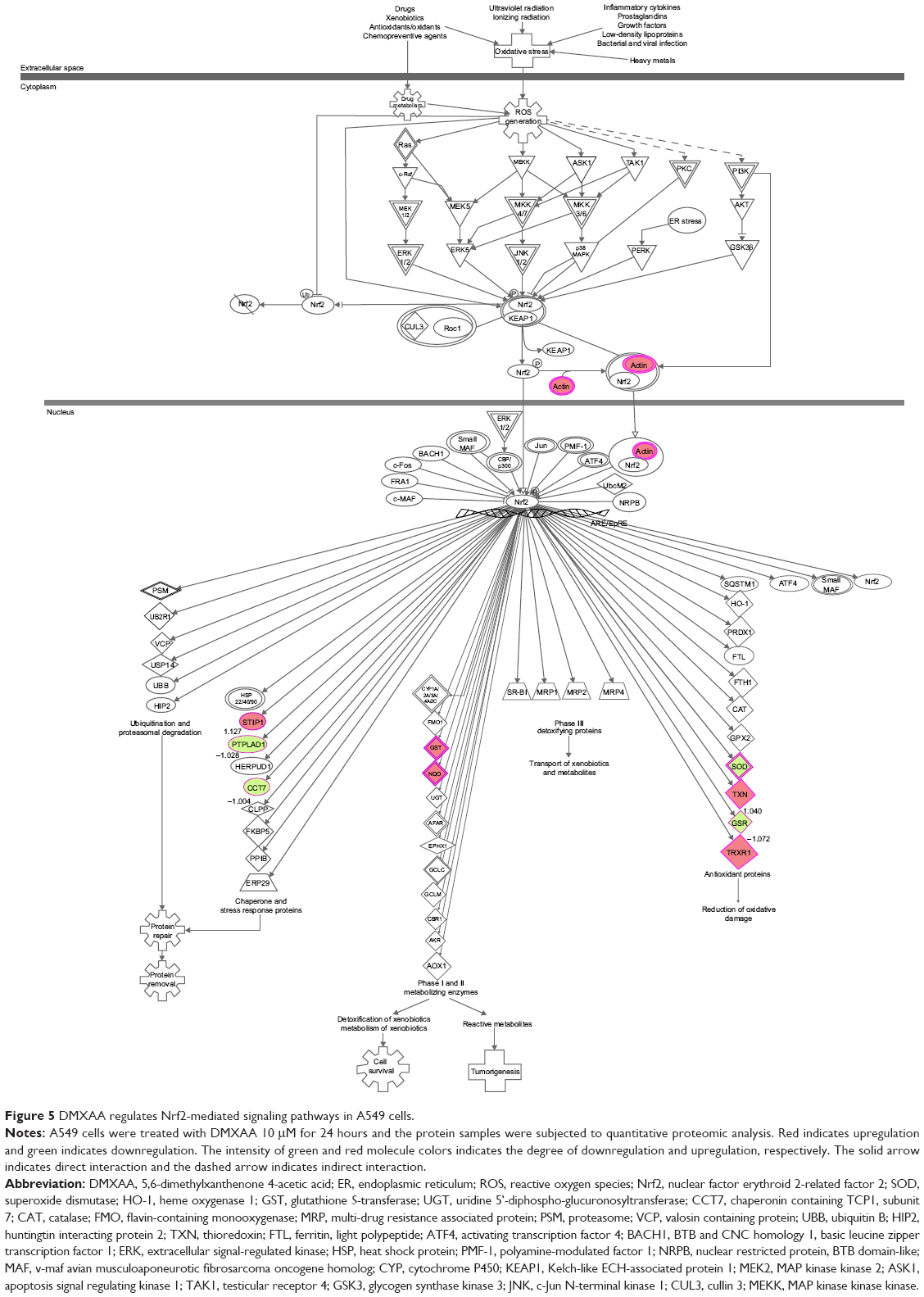 | Figure 5 DMXAA regulates Nrf2-mediated signaling pathways in A549 cells. |
Taken together, our quantitative proteomic study revealed a number of important functional proteins and associated signaling pathways that are regulated in A549 cells in response to treatment with DMXAA. These cellular signaling pathways play a pivotal role in the regulation of the cell cycle, apoptosis, autophagy, and oxidative stress. In our subsequent validation experiments, we confirmed the effect of DMXAA on cell cycle distribution, apoptosis, autophagy, and ROS generation in A549 cells.
Verification of molecular targets of DMXAA in A549 cells
The quantitative proteomic studies described above showed that DMXAA can modulate a number of functional protein molecules and related signaling pathways involved in cell proliferation, invasion and migration, death, and survival. In order to verify the quantitative proteomic data further, we investigated how DMXAA affected cell cycle distribution, apoptosis, autophagy, and redox homeostasis in A549 cells.
DMXAA induces G1 arrest in A549 cells
To validate the effect of DMXAA on cell growth, the cell cycle distribution was determined in A549 cells using flow cytometric analysis. As shown in Figure 6, a concentration-dependent increase in the cell number in G1 phase was observed after incubation of A549 cells with DMXAA at 0.1, 1, and 10 μM for 24 hours, with a 1.1-fold, 1.1-fold, and 1.4-fold increase in the number of cells arrested in G1 phase, respectively, compared with control cells treated with vehicle only (P<0.001 by one-way ANOVA, Figure 6A and B). In contrast, there was a marked decrease in the number of cells in S and G2/M phases in A549 cells treated with DMXAA at 0.1, 1, and 10 μM for 24 hours (P<0.01 or P<0.001 by one-way ANOVA, Figure 6A and B). Taken together, the results show that DMXAA can regulate the cell cycle distribution, contributing to its anticancer effect in A549 cells. Moreover, the inducing effect of DMXAA on cell cycle arrest further verifies the regulatory action of DMXAA on G1 and G2 checkpoints as determined by our proteomic study.
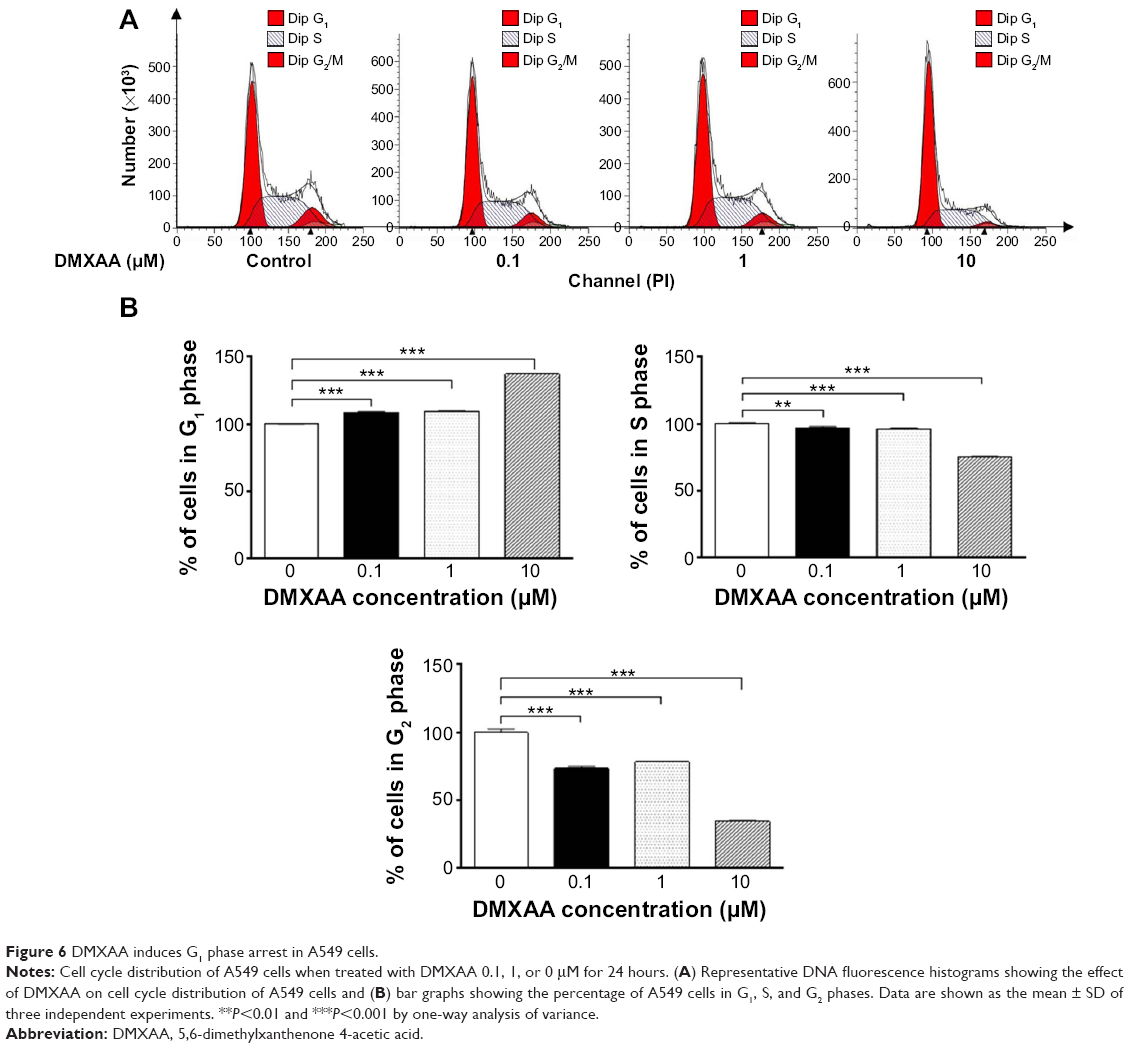 | Figure 6 DMXAA induces G1 phase arrest in A549 cells. |
DMXAA induces apoptosis and autophagy in A549 cells
As stated in our proteomic results, treatment with DMXAA induced apoptotic and autophagic responses in A549 cells involving several important signaling pathways. In the mitochondria/cytochrome c-mediated apoptotic pathway, release of cytochrome c from the mitochondria to the cytosol and resultant activation of the caspase cascade are key steps in the apoptosis process.41,42 Beclin 1 and LC3-I/II are two important markers in the initiation and progression of autophagy and are critical for formation of autophagosomes.43,44 During the autophagy process, LC3/Atg8 is cleaved at its C-terminus by Atg4 to generate cytosolic LC3-I.45 LC3-I is subsequently conjugated to phosphatidylethanolamine, then proteolytically cleaved and lipidated by Atg3 and Atg7 to form LC3-II, which attaches to the membrane of the autophagosome.
To verify the proteomic response to DMXAA with regard to cell death, we performed Western blotting assays to examine the expression of cytochrome c, caspase 3, beclin 1, and LC3-I/II in A549 cells treated with DMXAA. Incubation of A549 cells with DMXAA at 0.1, 1, and 10 μM markedly increased the cytosolic level of cytochrome c by 1.2-, 1.6-, and 1.6-fold, respectively, compared with the control cells (P<0.05 or P<0.01 by one-way ANOVA, Figure 7A and B). Cleaved caspase 3 was increased by DMXAA in A549 cells in a concentration-dependent manner. Incubation of A549 cells with DMXAA at 0.1, 1, and 10 μM significantly increased the level of cleaved caspase 3 by 1.3-, 1.4-, and 2.0-fold, respectively, compared with control cells (P<0.05 by one-way ANOVA, Figure 7A and B). These results indicate that DMXAA induces a marked increase in the cytosolic level of cytochrome c and activation of caspase 3, eventually leading to apoptotic death in A549 cells.
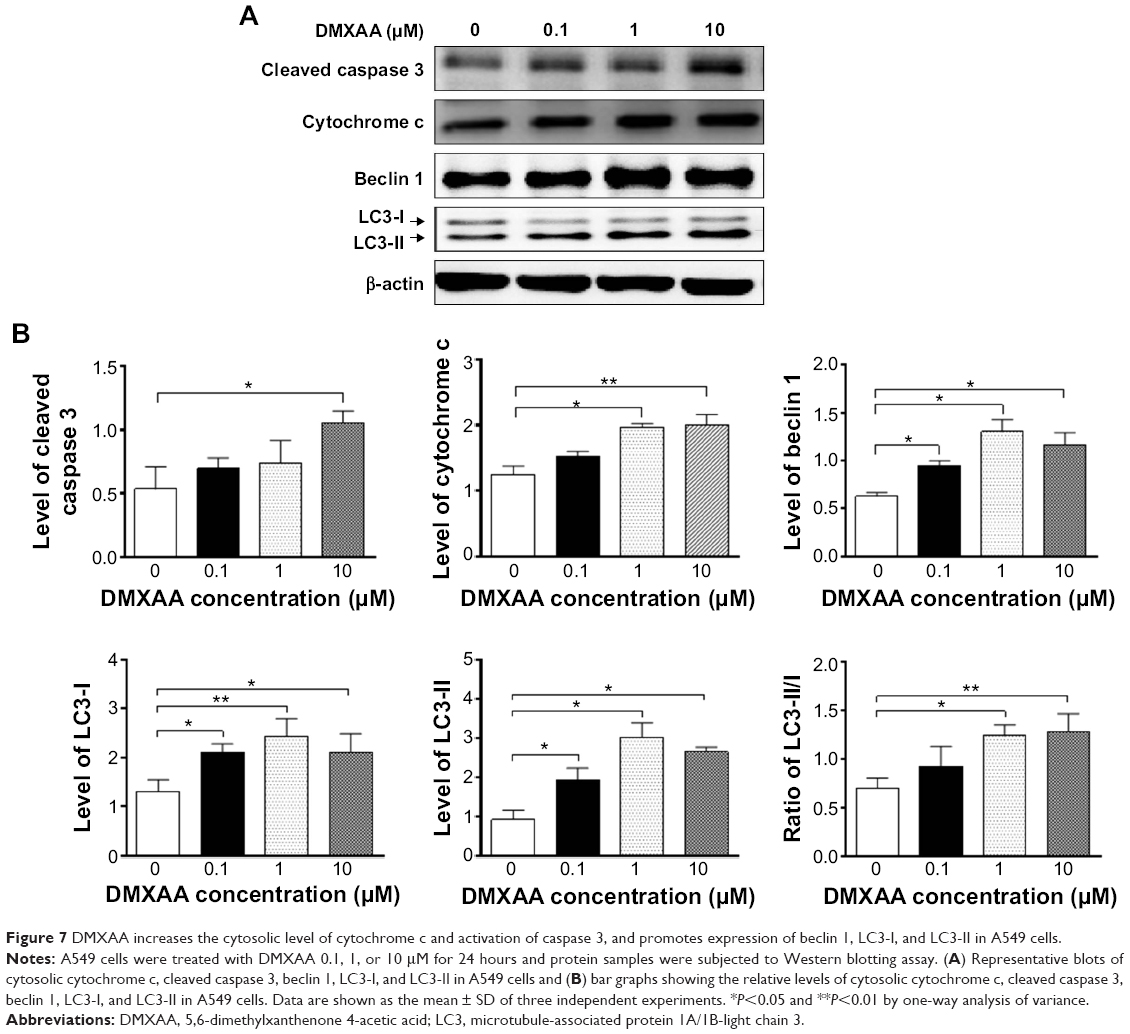 | Figure 7 DMXAA increases the cytosolic level of cytochrome c and activation of caspase 3, and promotes expression of beclin 1, LC3-I, and LC3-II in A549 cells. |
We further examined the effect of DMXAA on beclin 1 and LC3-I/II expression levels. Treatment of A549 cells with DMXAA for 24 hours significantly increased the expression of beclin 1. There was a 1.5-, 2.1-, and 1.9-fold increase in beclin 1 in A549 cells treated with DMXAA 0.1, 1, and 10 μM, respectively, for 24 hours (P<0.05 by one-way ANOVA, Figure 7A and B). Upon activation of LC3-I/II, our Western blotting analysis revealed two clear bands of LC3-I and II in A549 cells after 24 hour treatment with DMXAA (Figure 7A). Incubation of DMXAA at 0.1, 1, and 10 μM markedly increased the expression of LC3-I and LC3-II (Figure 7A and B). In comparison with the control cells, there was a 1.6-, 1.9-, and 1.6-fold increase in the level of LC3-I, and a 2.1-, 3.3-, and 2.9-fold increase in the level of LC3-II in A549 cells treated with DMXAA 0.1, 1, and 10 μM, respectively, for 24 hours. In addition, the ratio of LC3-II to LC3-I was markedly increased by 1.3-, 1.8-, and 1.8-fold in A549 cells treated with DMXAA 0.1, 1, and 5 μM, respectively (P<0.05 or P<0.01 by one-way ANOVA, Figure 7A and B). Taken together, the proteomic and Western blotting results show that DMXAA induces apoptosis and autophagy in A549 cells, contributing to the anticancer effects of DMXAA in the treatment of NSCLC.
DMXAA induces generation of intracellular ROS in A549 cells
As shown in the proteomic results, treatment with DMXAA can regulate intracellular redox homeostasis in A549 cells, which may contribute to the apoptosis-inducing and autophagy-inducing effects of DMXAA. Thus, we determined the effect of DMXAA on intracellular ROS levels in A549 cells. The intracellular levels of ROS were increased 1.1-, 1.1-, and 1.3-fold in a concentration-dependent manner when A549 cells were treated with DMXAA 0.1, 1, and 10 μM for 48 hours (P<0.001 by one-way ANOVA, Figure 8). The ROS-inducing effect of DMXAA further confirms its regulatory effect on intracellular redox homeostasis in A549 cells.
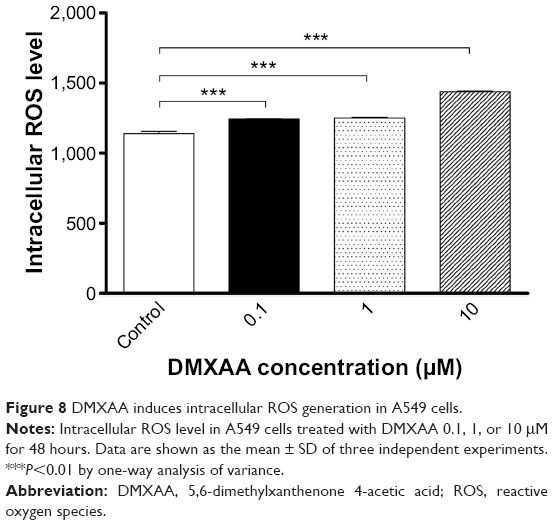 | Figure 8 DMXAA induces intracellular ROS generation in A549 cells. |
Discussion
NSCLC remains a devastating cancer, with the highest incidence and mortality rate, and treatment of the disease remains a major challenge due to the poor efficacy and severe side effects of both standard and new chemotherapeutic agents. There is an increasing interest in new agents and therapies for the treatment of lung cancer. DMXAA, a flavonoid tumor vascular-disrupting agent, has been found to have anticancer activity in vitro and in vivo in the treatment of NSCLC when used alone or in combination. It targets the established tumor blood vessels and inhibits tumor blood flow, resulting in necrosis of solid tumors. It has also been reported that DMXAA can regulate multiple signaling pathways involved in cell cycle progression, apoptosis, autophagy, and ROS generation.11,46–51 However, the global potential molecular targets and the possible mechanisms involved are not fully identified as yet. In the present study, we showed a comprehensive network of signaling pathways responding to treatment with DMXAA in A549 cells using a quantitative SILAC-based proteomic approach. The network of signaling pathways was mainly involved in cell cycle distribution, cell invasion and migration, redox homeostasis, and cell death. We verified that DMXAA arrested A549 cells in G1 phase, promoted apoptosis, induced marked autophagy, and triggered ROS generation.
The SILAC-based proteomic approach can quantitatively and comprehensively evaluate the effect of a given compound and identify its potential molecular targets and related signaling pathways.52–54 Previous studies have used this approach in A549 cells and tried to explore the potential molecular targets and possible mechanism for NSCLC therapy.55–63 In our study, we used a quantitative SILAC-based proteomic approach to evaluate the responses of A549 cells to treatment with DMXAA. This approach showed that DMXAA regulates a number of functional proteins and molecular signaling pathways involved in cell cycle progression, apoptosis, autophagy, and redox homeostasis in A549 cells, such as PPP2R1A, RPL5, SKP1, ACIN1, CYCS, ACTB, AKT2, GSR, PRDX3, SOD2, VDAC2, EIF3C, EIF4B, RHOA, RPS3A, AKT2, EIF3E, EIF3F, PPP2R1A, RPS2, RPS8, RPS26, RPS27, RPSA, ACTB, CCT7, GSR, GSTO1, NQO1, PTPLAD1, STIP1, TXN, and TXNRD1. The proteomic results suggest that DMXAA may target these molecules to elicit its anticancer effects in the treatment of NSCLC. Notably, we went on to validate the proteomic responses to DMXAA in A549 cells.
We found that DMXAA arrested A549 cells in G1 phase in a concentration-dependent manner, and speculated that the possible mechanism of DMXAA with regard to G1 arrest in A549 cells might involve a number of key regulators, including p21 Waf1/Cip1, p53, cyclins and cyclin-dependent kinases. p21 is a cyclin-dependent kinase inhibitor regulated by p53, and can bind to the Cdc2-cyclin B1 complex, thereby inducing cell cycle arrest.64 Further, cell cycle progression is tightly regulated by cyclins and cyclin-dependent kinases.65 Cyclins have no catalytic activity and are inactive in the absence of a partner cyclin. The complex formed by the association of Cdc2 and cyclin B1 plays a major role in the entry of cells into mitosis. Phosphorylation of Cdc2 at Thr161 by cyclin-dependent kinase–activating kinases is essential for the activity of Cdc2 kinase. Phosphorylation of Cdc2 at Thr14 and Tyr15 is catalyzed by Wee1 and Myt1 protein kinases, resulting in inhibition of Cdc2.65 During G2/M transition, Cdc2 is rapidly converted into the active form by dephosphorylation of Tyr14 and Tyr15, catalyzed by Cdc25 phosphatase. Thus, taking the proteomic and flow cytometric results into consideration, DMXAA-induced cell cycle arrest may occur via regulation of key modulators controlling the G1 and G2 checkpoints in A549 cells.
The present proteomic study also shows that DMXAA regulated mitochondrial function and cell death. Mitochondrial disruption and subsequent release of cytochrome c initiates the process of apoptosis, with the latter being initiated by proapoptotic members of the Bcl-2 family but antagonized by antiapoptotic members of this family.66,67 Antiapoptotic members of Bcl-2 can be inhibited by post-translational modification and/or by increased expression of PUMA, which is an essential regulator of p53-mediated cell apoptosis.68 In addition, cytochrome c released from the mitochondria can activate caspase 9, which then activates caspase 3 and caspase 7.69 In our study, we observed that the cytosolic level of cytochrome c was significantly increased and that caspase 3 was markedly activated after treatment with DMXAA. The activated caspase 3 ultimately induced apoptosis, with a decrease in the Bcl-2 level.
Further, the proteomic results show that DMXAA has a modulating effect on the mTOR signaling pathway. Under optimal growth conditions, activated mTORC1 inhibits autophagy by direct phosphorylation of Atg13 and ULK1 at Ser757.70–72 This phosphorylation inhibits ULK1 kinase activity and subsequent autophagosome formation. When the kinase activity of mTORC1 is suppressed, the autophagic machinery is initiated. In the present study, DMXAA induced autophagy in A549 cells as indicated by the increased expression of beclin 1 and the ratio of LC3-II over LC3-I. The amount of LC3-II or the ratio between LC3-II and LC3-I correlates well with the number of autophagosomes. Taken together, the autophagy-inducing effect of DMXAA may contribute to its anticancer activity via regulation of the mTOR signaling pathway.
In addition, our proteomic study showed that DMXAA regulates the Nrf2-mediated signaling pathway, which controls the basal and induced expression of a wide array of antioxidant response element-dependent genes to regulate the physiological and pathophysiological outcomes of exposure to oxidants.40,73,74 We found a significant inducing effect of DMXAA on ROS generation in A549 cells. However, the mechanism of how DMXAA induces ROS generation is unclear. Nrf2 is a nuclear transcription factor that plays a pivotal role in regulation of oxidative stress by modulating the transcription of antioxidant response elements.40 It indicates that DMXAA may induce oxidative stress via the Nrf2-mediated signaling pathway. Our results suggest that ROS may have an important role in DMXAA-induced apoptosis and autophagy in A549 cells. However, further studies are needed to elucidate how DMXAA induces generation of ROS and modulates redox homeostasis.
In summary, the quantitative SILAC-based proteomic approach used in this study showed that DMXAA inhibited cell proliferation, predominantly activated the mitochondria-dependent apoptotic pathway and induced autophagy, and increased intracellular levels of ROS in human A549 cells involving a number of key functional proteins and related molecular signaling pathways. This study may provide a clue enabling full identification of the molecular targets and elucidate the underlying mechanisms of DMXAA in the treatment of NSCLC, resulting in an improved therapeutic effect and fewer side effects in the clinical setting.
Acknowledgments
The authors appreciate the financial support of the Startup Fund of the College of Pharmacy, University of South Florida, Tampa, FL, USA. Dr Zhi-Wei Zhou, PhD, holds a postdoctoral scholarship from the College of Pharmacy, University of South Florida.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Desantis C, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | ||
Ferlay J, Soerjomataram I, Ervik M, et al. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11. Lyon, France: International Agency for Research on Cancer; 2013. Available from: http://globocan.iarc.fr. Accessed December 10, 2014. | ||
US Cancer Statistics Working Group. United States Cancer Statistics: 1999–2011 Incidence and Mortality Web-based Report. Atlanta, GA, USA: Department of Health and Human Services, Centers for Disease Control and Prevention, National Cancer Institute; 2014. Available from: http://www.cdc.gov/cancer/npcr/pdf/USCS_FactSheet.pdf. Accessed December 10, 2014. | ||
American Cancer Society. Cancer Facts and Figures 2014. Atlanta, GA, USA: American Cancer Society; 2014. Available from: http://www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures2014/. Accessed December 10, 2014. | ||
Wang YC, Wei LJ, Liu JT, Li SX, Wang QS. Comparison of Cancer Incidence between China and the USA. Cancer Biol Med. 2012;9(2):128–32. | ||
Keith RL, Miller YE. Lung cancer chemoprevention: current status and future prospects. Nat Rev Clin Oncol. 2013;10(6):334–343. | ||
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62(1):10–29. | ||
Brown SL, Kolozsvary A, Kim JH. Vascular targeting therapies for treatment of malignant disease. Cancer. 2005;104(1):216–217. | ||
Amir E, Mandoky L, Blackhall F, et al. Antivascular agents for non-small-cell lung cancer: current status and future directions. Expert Opin Investig Drugs. 2009;18(11):1667–1686. | ||
Zhou S, Kestell P, Baguley BC, Paxton JW. 5,6-Dimethylxanthenone-4-acetic acid (DMXAA): a new biological response modifier for cancer therapy. Invest New Drugs. 2002;20(3):281–295. | ||
Buchanan CM, Shih JH, Astin JW, et al. DMXAA (vadimezan, ASA404) is a multi-kinase inhibitor targeting VEGFR2 in particular. Clin Sci (Lond). 2012;122(10):449–457. | ||
Li J, Jameson MB, Baguley BC, Pili R, Baker SD. Population pharmacokinetic-pharmacodynamic model of the vascular-disrupting agent 5,6-dimethylxanthenone-4-acetic acid in cancer patients. Clin Cancer Res. 2008;14(7):2102–2110. | ||
Zhou S, Paxton JW, Tingle MD, Kestell P. Identification of the human liver cytochrome P450 isoenzyme responsible for the 6-methylhydroxylation of the novel anticancer drug 5,6-dimethylxanthenone-4-acetic acid. Drug Metab Dispos. 2000;28(12):1449–1456. | ||
Miners JO, Valente L, Lillywhite KJ, et al. Preclinical prediction of factors influencing the elimination of 5,6-dimethylxanthenone-4-acetic acid, a new anticancer drug. Cancer Res. 1997;57(2):284–289. | ||
McKeage MJ, Von Pawel J, Reck M, et al. Randomised phase II study of ASA404 combined with carboplatin and paclitaxel in previously untreated advanced non-small cell lung cancer. Br J Cancer. 2008;99(12):2006–2012. | ||
Lara PN Jr, Douillard JY, Nakagawa K, et al. Randomized phase III placebo-controlled trial of carboplatin and paclitaxel with or without the vascular disrupting agent vadimezan (ASA404) in advanced non-small-cell lung cancer. J Clin Oncol. 2011;29(22):2965–2971. | ||
Pili R, Rosenthal MA, Mainwaring PN, et al. Phase II study on the addition of ASA404 (vadimezan; 5,6-dimethylxanthenone-4-acetic acid) to docetaxel in CRMPC. Clin Cancer Res. 2010;16(10):2906–2914. | ||
Fruh M, Cathomas R, Siano M, et al. Carboplatin and paclitaxel plus ASA404 as first-line chemotherapy for extensive-stage small-cell lung cancer: a multicenter single arm phase II trial (SAKK 15/08). Clin Lung Cancer. 2013;14(1):34–39. | ||
Hida T, Tamiya M, Nishio M, et al. Phase I study of intravenous ASA404 (vadimezan) administered in combination with paclitaxel and carboplatin in Japanese patients with non-small cell lung cancer. Cancer Sci. 2011;102(4):845–851. | ||
McKeage MJ, Reck M, Jameson MB, et al. Phase II study of ASA404 (vadimezan, 5,6-dimethylxanthenone-4-acetic acid/DMXAA) 1800 mg/m(2) combined with carboplatin and paclitaxel in previously untreated advanced non-small cell lung cancer. Lung Cancer. 2009;65(2):192–197. | ||
Prantner D, Perkins DJ, Lai W, et al. 5,6-Dimethylxanthenone-4-acetic acid (DMXAA) activates stimulator of interferon gene (STING)-dependent innate immune pathways and is regulated by mitochondrial membrane potential. J Biol Chem. 2012;287(47):39776–39788. | ||
Conlon J, Burdette DL, Sharma S, et al. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J Immunol. 2013;190(10):5216–5225. | ||
Gao P, Zillinger T, Wang W, et al. Binding-pocket and lid-region substitutions render human STING sensitive to the species-specific drug DMXAA. Cell Rep. 2014;8(6):1668–1676. | ||
Diaz-Moralli S, Tarrado-Castellarnau M, Miranda A, Cascante M. Targeting cell cycle regulation in cancer therapy. Pharmacol Ther. 2013;138(2):255–271. | ||
Ong SE, Mann M. Stable isotope labeling by amino acids in cell culture for quantitative proteomics. Methods Mol Biol. 2007;359:37–52. | ||
Mann M. Functional and quantitative proteomics using SILAC. Nat Rev Mol Cell Biol. 2006;7(12):952–958. | ||
Ong SE. The expanding field of SILAC. Anal Bioanal Chem. 2012;404(4):967–976. | ||
Ong SE, Mann M. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC). Nat Protoc. 2006;1(6):2650–2660. | ||
Farazi TA, Hoell JI, Morozov P, Tuschl T. MicroRNAs in human cancer. Adv Exp Med Biol. 2013;774:1–20. | ||
Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15(2):81–94. | ||
Shanware NP, Bray K, Abraham RT. The PI3K, metabolic, and autophagy networks: interactive partners in cellular health and disease. Annu Rev Pharmacol Toxicol. 2013;53:89–106. | ||
Morgensztern D, McLeod HL. PI3K/Akt/mTOR pathway as a target for cancer therapy. Anticancer Drugs. 2005;16(8):797–803. | ||
Wu WK, Coffelt SB, Cho CH. The autophagic paradox in cancer therapy. Oncogene. 2012;31(8):939–953. | ||
Ferreira CG, Epping M, Kruyt FA, Giaccone G. Apoptosis: target of cancer therapy. Clin Cancer Res. 2002;8(7):2024–2034. | ||
Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov. 2010;9(6):447–464. | ||
Heavey S, O’Byrne KJ, Gately K. Strategies for co-targeting the PI3K/Akt/mTOR pathway in NSCLC. Cancer Treat Rev. 2014;40(3):445–456. | ||
Ellis L, Shah P, Hammers H, et al. Vascular disruption in combination with mTOR inhibition in renal cell carcinoma. Mol Cancer Ther. 2012;11(2):383–392. | ||
Brauer R, Wang LC, Woon ST, et al. Labeling of oxidizable proteins with a photoactivatable analog of the antitumor agent DMXAA: evidence for redox signaling in its mode of action. Neoplasia. 2010;12(9):755–765. | ||
Keum YS, Choi BY. Molecular and chemical regulation of the Keap1-Nrf2 signaling pathway. Molecules. 2014;19(7):10074–10089. | ||
Ma Q. Role of Nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–426. | ||
Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9(3):231–241. | ||
Estaquier J, Vallette F, Vayssiere JL, Mignotte B. The mitochondrial pathways of apoptosis. Adv Exp Med Biol. 2012;942:157–183. | ||
Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18(4):571–580. | ||
Maiuri MC, Criollo A, Kroemer G. Crosstalk between apoptosis and autophagy within the Beclin 1 interactome. EMBO J. 2010;29(3):515–516. | ||
Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19(21):5720–5728. | ||
Downey CM, Aghaei M, Schwendener RA, Jirik FR. DMXAA causes tumor site-specific vascular disruption in murine non-small cell lung cancer, and like the endogenous non-canonical cyclic dinucleotide STING agonist, 2′3′-cGAMP, induces M2 macrophage repolarization. PLoS One. 2014;9(6):e99988. | ||
Zhang SH, Zhang Y, Shen J, et al. Tumor vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid inhibits platelet activation and thrombosis via inhibition of thromboxane A2 signaling and phosphodiesterase. J Thromb Haemost. 2013;11(10):1855–1866. | ||
Kim S, Peshkin L, Mitchison TJ. Vascular disrupting agent drug classes differ in effects on the cytoskeleton. PLoS One. 2012;7(7):e40177. | ||
Shirey KA, Nhu QM, Yim KC, et al. The anti-tumor agent, 5,6-dimethylxanthenone-4-acetic acid (DMXAA), induces IFN-β-mediated antiviral activity in vitro and in vivo. J Leukoc Biol. 2011;89(3):351–357. | ||
Woon ST, Hung SS, Wu DC, et al. NF-κB-independent induction of endothelial cell apoptosis by the vascular disrupting agent DMXAA. Anticancer Res. 2007;27(1A):327–334. | ||
Siemann DW, Chaplin DJ, Horsman MR. Vascular-targeting therapies for treatment of malignant disease. Cancer. 2004;100(12):2491–2499. | ||
Dolai S, Xu Q, Liu F, Molloy MP. Quantitative chemical proteomics in small-scale culture of phorbol ester stimulated basal breast cancer cells. Proteomics. 2011;11(13):2683–2692. | ||
Geiger T, Cox J, Ostasiewicz P, Wisniewski JR, Mann M. Super-SILAC mix for quantitative proteomics of human tumor tissue. Nat Methods. 2010;7(5):383–385. | ||
Everley PA, Krijgsveld J, Zetter BR, Gygi SP. Quantitative cancer proteomics: stable isotope labeling with amino acids in cell culture (SILAC) as a tool for prostate cancer research. Mol Cell Proteomics. 2004;3(7):729–735. | ||
Doherty MK, Hammond DE, Clague MJ, Gaskell SJ, Beynon RJ. Turnover of the human proteome: determination of protein intracellular stability by dynamic SILAC. J Proteome Res. 2009;8(1):104–112. | ||
Hammond DE, Hyde R, Kratchmarova I, Beynon RJ, Blagoev B, Clague MJ. Quantitative analysis of HGF and EGF-dependent phosphotyrosine signaling networks. J Proteome Res. 2010;9(5):2734–2742. | ||
Duan X, Kelsen SG, Clarkson AB Jr, Ji R, Merali S. SILAC analysis of oxidative stress-mediated proteins in human pneumocytes: new role for treacle. Proteomics. 2010;10(11):2165–2174. | ||
Coombs KM, Berard A, Xu W, et al. Quantitative proteomic analyses of influenza virus-infected cultured human lung cells. J Virol. 2010;84(20):10888–10906. | ||
Munday DC, Hiscox JA, Barr JN. Quantitative proteomic analysis of A549 cells infected with human respiratory syncytial virus subgroup B using SILAC coupled to LC-MS/MS. Proteomics. 2010;10(23):4320–4334. | ||
Foster MW, Thompson JW, Forrester MT, et al. Proteomic analysis of the NOS2 interactome in human airway epithelial cells. Nitric Oxide. 2013;34:37–46. | ||
Wu Q, Xu W, Cao L, et al. SAHA treatment reveals the link between histone lysine acetylation and proteome in nonsmall cell lung cancer A549 cells. J Proteome Res. 2013;12(9):4064–4073. | ||
Chiu HC, Hannemann H, Heesom KJ, Matthews DA, Davidson AD. High-throughput quantitative proteomic analysis of dengue virus type 2 infected A549 cells. PLoS One. 2014;9(3):e93305. | ||
Gray TA, Alsamman K, Murray E, Sims AH, Hupp TR. Engineering a synthetic cell panel to identify signalling components reprogrammed by the cell growth regulator anterior gradient-2. Mol Biosyst. 2014;10(6):1409–1425. | ||
Bunz F, Dutriaux A, Lengauer C, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282(5393):1497–1501. | ||
Hunter T. Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell. 1995;80(2):225–236. | ||
Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T, Snyder SH. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol. 2003;5(12):1051–1061. | ||
Yang J, Liu X, Bhalla K, et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275(5303):1129–1132. | ||
Jeffers JR, Parganas E, Lee Y, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4(4):321–328. | ||
Fesik SW, Shi Y. Structural biology. Controlling the caspases. Science. 2001;294(5546):1477–1478. | ||
Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717–1721. | ||
Chen Y, Yu L. Autophagic lysosome reformation. Exp Cell Res. 2013;319(2):142–146. | ||
Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. 2012;19(1):87–95. | ||
Suzuki T, Motohashi H, Yamamoto M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol Sci. 2013;34(6):340–346. | ||
Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.