")
Back to Journals » International Journal of Nanomedicine » Volume 13
Protein nanoparticles with ligand-binding and enzymatic activities
Authors Morozova OV , Pavlova ER , Bagrov DV , Barinov NA, Prusakov KA, Isaeva EI , Podgorsky VV, Basmanov DV , Klinov DV
Received 19 June 2018
Accepted for publication 18 August 2018
Published 18 October 2018 Volume 2018:13 Pages 6637—6646
DOI https://doi.org/10.2147/IJN.S177627
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Olga V Morozova,1,2 Elizaveta R Pavlova,1,3 Dmitry V Bagrov,1,4 Nikolay A Barinov,1 Kirill A Prusakov,1,3 Elena I Isaeva,2 Victor V Podgorsky,1 Dmitry V Basmanov,1 Dmitry V Klinov1
1Department of Biophysics, Federal Research and Clinical Center of Physical-Chemical Medicine of Federal Medical Biological Agency (FRCC PCM), Moscow, Russia; 2Ivanovsky Institute of Virology of the National Research Center of Epidemiology and Microbiology of N.F. Gamaleya of the Russian Ministry of Health, Moscow, Russia; 3Moscow Institute of Physics and Technology, Moscow, Russia; 4Department of Bioengineering, Faculty of Biology, Lomonosov Moscow State University, Moscow, Russia
Purpose: To develop a general method for NP fabrication from various proteins with maintenance of biological activity.
Methods: A novel general approach for producing protein nanoparticles (NP) by nanoprecipitation of the protein solutions in 1,1,1,3,3,3-hexafluoroisopropanol is described. Protein NP sizes and shapes were analyzed by dynamic light scattering, scanning electron and atomic force microscopy (SEM and AFM). Chemical composition of the NP was confirmed using ultraviolet (UV) spectroscopy, energy-dispersive X-ray spectroscopy (EDX) and circular dichroism (CD). Biological properties of the NP were analyzed in ELISA, immunofluorescent analysis and lysozyme activity assay.
Results: Water-insoluble NP were constructed from globular (bovine serum albumin (BSA), lysozyme, immunoglobulins), fibrillar (fibrinogen) proteins and linear polylysines by means of nanoprecipitation of protein solutions in fluoroalcohols. AFM and SEM revealed NP sizes of 20–250 nm. The NP chemical structure was confirmed by UV spectroscopy, protease digestion and EDX spectroscopy. CD spectra revealed a stable secondary structure of proteins in NP. The UV spectra, microscopy and SDS-PAA gel electrophoresis (PAGE) proved the NP stability at +4°C for 7 months. Co-precipitation of proteins with fluorophores or nanoprecipitation of pre-labeled BSA resulted in fluorescent NP that retained antigenic structures as shown by their binding with specific antibodies. Moreover, NP from monoclonal antibodies could bind with the hepatitis B virus antigen S. Besides that, lysozyme NP could digest bacterial cellular walls.
Conclusion: Thus, the water-insoluble, stable protein NP were produced by nanoprecipitation without cross-linking and retained ligand-binding and enzymatic activities.
Keywords: protein nanostructures, nanoprecipitation, atomic force and electron microscopy, energy-dispersive X-ray spectroscopy, ultraviolet and circular dichroism spectroscopy, ELISA, lysozyme activity assay
Introduction
Biocompatible biodegradable NP are used for targeted delivery and prolonged release of bioactive compounds in the pharmaceutical, food and cosmetic industries, in dentistry, hemostasis, for immunodiagnostics, as adjuvants of inactivated vaccines and in other biomedical applications.1–4 In order to ensure safe parenteral administration and efficient cellular uptake, the NP sizes must be from 10 to 1,000 nm, preferably less than 500 nm. The most commonly used materials for NP fabrication are polylactic acid and polylactic-co-glycolic acid.5,6 Their surfaces can be functionalized for targeted drug delivery.5–7 However, possible toxicological problems caused by hydrolysis of the synthetic polymers into acid or alcohol monomers constrain their implementation.8 In addition, the possible toxicity due to long-term overloading of the polymers exists.9
Bio-reactivity of different NP is commonly based on protein interactions. Various proteins are helpful in the treatment of a wide array of diseases: to compensate for the deficiency of hormones and cytokines, growth factors and enzymes as well as for antibody-mediated targeted delivery. However, because of their enzymatic degradation in gastric and intestinal fluids, poor membrane permeability and tissue penetration, proteins are not administered orally.10 Therefore, although non-invasive peroral, intranasal and pulmonary administration of non-degradable proteins is highly desirable, parenteral administration remains the standard delivery method for proteins. Evident advantages of protein NP in comparison with other polymers include their biological activity, natural biodegradation, absence of toxicity, ready-to-use functional amino, carboxy and thiol groups for chemical modifications and subsequent covalent attachment of different compounds. Protein NP exploit natural pathways to selectively deliver drugs to cells. The protein defined primary structures and developed tools of genetic engineering permit the use of their surface functional groups both for the covalent binding of drugs and the targeting of ligands.11 Moreover, NP from bioactive proteins can be used as therapeutic agents without additional modification. Proteins are known to be immunogenic and, therefore, the corresponding NP can be used for vaccine production although the risk of allergic reactions is higher compared to synthetic polymers. However, lack of a convenient, general method of protein NP preparation due to the great diversity of proteins and their low solubility in organic solvents hampers their application.
Various methods are available for preparation of NP from natural and synthetic polymers. They can be applied to pre-formed polymers (solvent evaporation method; spontaneous emulsification/solvent diffusion method; nanoprecipitation, salting out methods, spray drying) or to their monomers (emulsion or interfacial polymerization etc).1 Anti-solvent precipitation is based on mixing of a polymer solution in its corresponding solvent with a non-solvent resulting in NP nucleation and subsequent growth. With modifications, some of the previously developed approaches including alcohol or acetone desolvation, emulsification, spray drying and thermal denaturation can be applied for a number of proteins.2,9,11 To produce protein NP ethanol,12 acetone13 and polyvalent ions14 can be used as anti-solvents or desolvation/precipitation agents.15 Unfortunately, most currently known methods of protein NP construction can be applied to certain proteins only. Native and recombinant proteins originate from various sources and exhibit a wide diversity of molecular and physicochemical characteristics, eg, molecular weight, conformation, flexibility, polarity, charge, isoelectric point, solubility, and interactions. As a result, protein NP can be assembled using numerous methods, from one or more types of proteins or from a combination of a protein and another biopolymer (usually a polysaccharide).2,4,16 Self-assembling NP, including virus-like particles, can be formed from a number of native proteins with high affinity of binding.17–19 Water-insoluble proteins in hydro-alcoholic solvents with surfactants and phospholipids at optimal pH can also be aggregated in NP14 that may be stabilized by cross-linking with glutaraldehyde.11 Thermal denaturation of tobacco mosaic virus was shown to produce NP.20
Our goal was to develop a general method for NP fabrication from various proteins with maintenance of biological activity.
Materials and methods
Materials
BSA, lysozyme, polylysines 15–30 kDa and 30–70 kDa were purchased from (Sigma-Aldrich Co., St Louis, MO, USA). Fibrinogen was obtained from Technology Standard, Barnaul, Russia. HFIP was purchased from P&M-Invest, Moscow, Russia, and Sigma-Aldrich Co., St Louis, MO, USA. Mab against HBsAg were kindly provided by L.E. Matveev (Biosan, Novosibirsk, Russia).
Protein nanoprecipitation
Protein NPs were constructed by nanoprecipitation according to the following method5 with some modifications. Proteins with concentrations up to 20 mg/mL were dissolved in HFIP but not in acetone as previously described for synthetic polymers.5,6 For higher concentrations of proteins (up to 100 mg/mL) their prolonged dissolvement in HFIP (up to a week) at room temperature with shaking was required.
Then protein solutions in HFIP were added dropwise, either manually or by syringe pump (KD Scientific Inc., Holliston, MA, USA), to 40% ethanol in water (1/10 part of total volume resulting in final protein concentration of 2 mg/mL). Permanent vigorous stirring was necessary to maintain the stability of the three-phase system including two solvents (water and HFIP) and one anti-solvent (ethanol). The mixture was immediately placed at 58.2°C (HFIP boiling point). To accelerate evaporation of both alcohols, nanoprecipitation was performed at pressures less than 25 mBar. Alcohol evaporation time depended on both pressure and volume of the mixture. Then water-insoluble particles were pelleted at 15,000 g and washed with deionized water three times to remove the residual protein molecules. An alternate way of NP purification was gel-chromatography using Sephadex G200 with subsequent centrifugation at 500 g. Microparticles were removed by additional differential centrifugation at 700–1,000 g, leaving the NP in the supernatant.
AFM
Aliquots of 10 μL of 20 μg/mL BSA in HFIP or 10 μL of freshly prepared NP suspension in water were placed on a freshly cleaved mica surface for 10 seconds and dried with a flow of argon. Protein solutions and NP were analyzed using Ntegra Prima (NT-MDT, Moscow, Russia) AFM. All the AFM observations were performed with high-resolution silicon cantilevers with resonance frequencies from 190 to 325 kHz in attraction regime of intermittent contact mode at a scan rate of 1 Hz. Free amplitude of the cantilever in the air was in the range 1–10 nm. FemtoScan Online (Advanced technologies center, Russia) was used for the basic image processing (lines fitting, slope subtraction) and presentation, SPM Image Magic (https://spm-image-magic.software.informer.com/) for the height measurements.
SEM
Immediately before sample deposition, silicon wafers were treated in plasma cleaner Electronic Diener (Plasma Surface Technology, Ebhausen, Germany). The protein NP were then deposited onto them and characterized using a Zeiss Merlin microscope equipped with GEMINI II Electron Optics (Zeiss, Oberkochen, Germany). SEM imaging was done at 1–3 kV accelerating voltage and 30–80 pA probe current.
EDX was performed by SEM via Silicon Drift Detector (SDD) X-MaxN 150 (Oxford Instruments, Abingdon, Oxfordshire, UK) and AztecEnergy EDX Software (Version 3.0).
DLS
Protein NP sizes were determined by DLS using NANO-flex 180° (Microtrac Inc., Montgomeryville, PA, USA). Refraction index (n) of protein NP was 1.4, n (water)=1.3 and n (HFIP)=1.27.
UV spectroscopy
UV absorption spectra of protein solutions in water and HFIP as well as protein NP were measured using NanoDrop 2000c UV-Vis spectrophotometer (Thermo Scientific, Waltham, MA, USA).
CD spectroscopy
To probe protein conformational changes, CD spectra of BSA, fibrinogen and lysozyme solutions in water and HFIP as well as corresponding NP suspensions were obtained by Chirascan spectrophotometer (Applied Photophysics Limited, Randalls Rd, Leatherhead, UK). For quantitative analysis of CD spectra, CDNN 2.1 (Gerald. Bohm, 1997, CD Spectra Deconvolution, Delphi, Halle) software was used.
Fluorescent protein NP fabrication and microscopy
Fluorescent NP were fabricated by nanoprecipitation of the preliminary labeled BSA or by co-precipitation of proteins with different fluorophores. BSA was labeled with rhodamine B (Rho B) in 0.1 M Na2CO3 solution at pH 9.3 for 1 hour at room temperature and was then purified by Sephadex G25 gel-chromatography followed by mild centrifugation at 700 g. Co-precipitation of proteins with Rho B solution prepared immediately before the experiment was performed in 40% ethanol. The fluorescent protein NP were imaged using the fluorescent microscope Nikon Eclipse Ti, with a Nikon Apo TIRF 100x/1.49 Oil objective, camera Andor Ixon ULTRA 512×512 (Andor Technology, Belfast, Northern Ireland, UK) and filters providing excitation for RhoB at 528–553 nm and emission at 590–650 nm.
Binding of fluorescent BSA NP with immobilized anti-BSA antibodies
Subsequent 10-fold dilutions of polyclonal antibodies from the whole antiserum against BSA (Sigma-Aldrich, USA) were immobilized onto activated polystyrene 96-well plates and, after blocking with 1% gelatin in PBS, were used for binding with fluorescent BSA-RhoB NP fabricated from BSA-RhoB. Unspecific binding of immobilized total human IgG with the same BSA-RhoB NP was considered as negative control. Specific binding of immobilized anti-BSA polyclonal antibodies with fluorescent protein BSA-RhoB was used as positive control. To detect RhoB, fluorescence emission at 580 nm was visualized and quantitated using Typhoon (GE Healthcare Life Sciences, Marlborough, MA, USA). All binding titers were determined in triplicate with subsequent averaging of values.
Detection of antibodies against HBV S antigen in NP
The NP from the Mab HV301-HBV against the HBsAg were constructed by nanoprecipitation as described above. To compare the antigen-binding properties of the original antibodies and the corresponding NP, ELISA, using kit “B-DS-IFA-anti-HBsAg” (Diagnostic systems, Moscow, Russia) was performed according to the manufacturer’s instructions. PBS was used as negative control whereas original Mab HV301-HBV as positive control. Additional negative control was binding with NP from unspecific Mab against the hepatitis C virus. Geometric mean titers of antibodies were calculated from triplicate repeats.
Lysozyme NP enzymatic assay
Lysozyme NP was constructed from lysozyme solution (50 mg/mL) in HFIP. Lysozyme activity of the NP was assayed with Micrococcus lysodeikticus cell walls (Sigma-Aldrich Co.) in PBS in dynamics and compared with the activity of a fresh solution of lysozyme in water, as described in21 using both AFM and UV-visible spectroscopy.
Statistical analysis
Continuous variables were compared using Student’s t-test. P-values <0.05 were assumed to be significant.
Results
Fabrication of protein NP
Nanoprecipitation is a simple, fast and inexpensive method of forming of NP from polymers based on their desolvation in hydroalcoholic solution and subsequent fast solvent evaporation.1
The scheme of the nanoprecipitation (Figure 1) includes dropwise addition of protein solutions in HFIP to 40% ethanol using an automatic pipette, a syringe or a syringe pump with different speeds. Ethanol concentrations may be in the range 30%–50%. The protein nanoprecipitation appeared to occur at elevated temperatures near the HFIP boiling point, both under atmospheric and reduced (25 mBar) pressure.
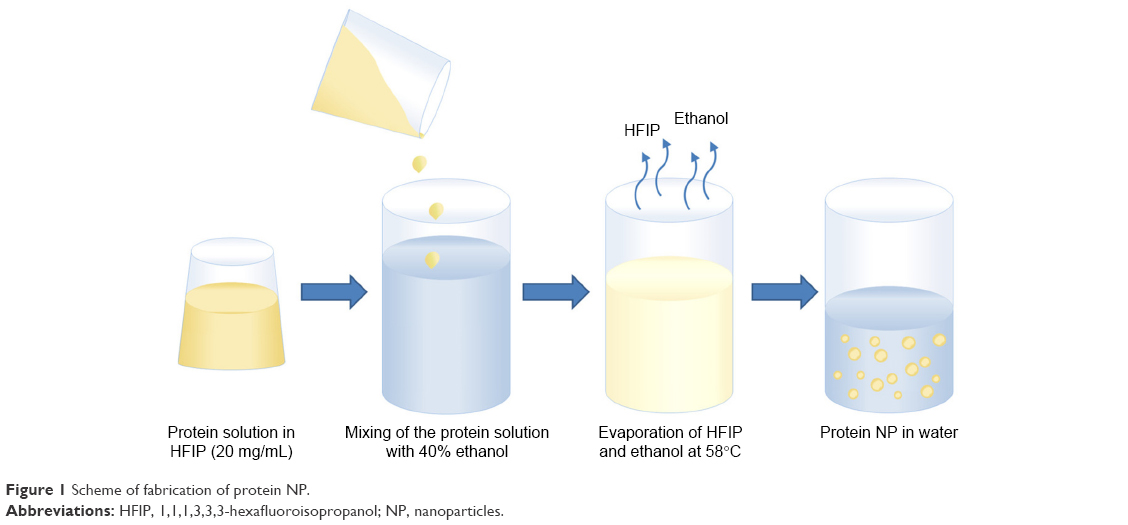 | Figure 1 Scheme of fabrication of protein NP. |
BSA NP shapes and sizes
Albumins from blood plasma have a defined structure convenient for functionalization via thiol, amino and carboxyl groups. They are poorly immunogenic and able to act as extracellular antioxidants providing competitive protection from free radicals and other harmful chemicals.9 Many drugs and endogenous molecules are known to bind with albumins. BSA was selected because of its high water solubility (up to 40% w/v) as well as stability in a wide range of pH (4–9) and temperatures (up to 60°C for 10 hours).11
Complete dissolution of BSA in HFIP to separate molecules was confirmed by AFM (Figure 2A and C). The experimentally observed spheres of 1–3 nm height (Figure 2A and C) on mica corresponded to previously described heights of single globular protein molecules adsorbed on mica from water solution.22 The nanoprecipitation resulted in spherical BSA NP of 20–250 nm height (Figure 2B and D). This is the interquartile range between 25th and 75th percentiles of the histogram (Figure 2D). Both DLS (Supporting Information Table S1), AFM and SEM revealed BSA NP of similar sizes after nanoprecipitation of BSA in a broad concentration range of 0.1 μg/mL to 2 mg/mL. Final NP sizes and their distribution did not correlate with the original protein molecular weight, shape or concentration (Table S1). But the nanoprecipitation of 20 mg/mL BSA resulted in a nano-film (Supporting Information Figure S1).
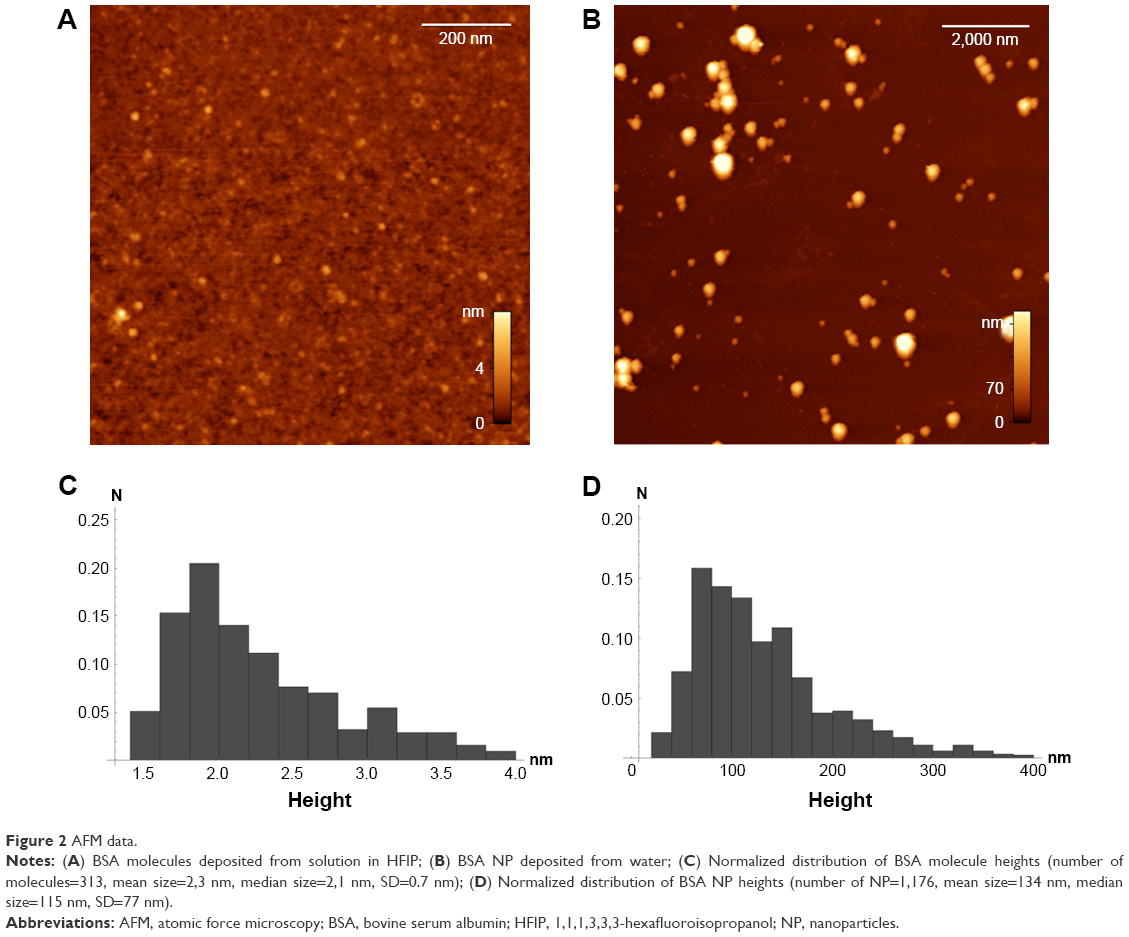 | Figure 2 AFM data. |
Chemical composition of the NP
The protein NP chemical structure was assayed by UV absorption spectroscopy and EDX analysis. UV absorption spectra of the NP suspensions in water with two peaks at 230 nm and 280 nm, typical for proteins (Supporting Information Figure S2), were similar to those for protein solutions in water and HFIP. At least half of the total proteins became water-insoluble after the nanoprecipitation and the first differential centrifugation. After subsequent washes with water, the trace amounts of the remaining free protein molecules were below the known UV spectroscopy sensitivity limit (0.1 mg/mL). Taken together, the available data allow us to estimate the reaction yield of the nanoprecipitation as more than 50%.
In order to confirm the protein nature, the NP were treated with proteases (trypsin and pronase) for 1 hour at +37°C. SEM indicated NP absence after exhaustive proteolytic digestion.
EDX spectrum of the BSA NP (Figure 3) revealed the presence only of oxygen (O), carbon (C) and nitrogen (N) atoms and might serve as additional evidence of the protein NP.
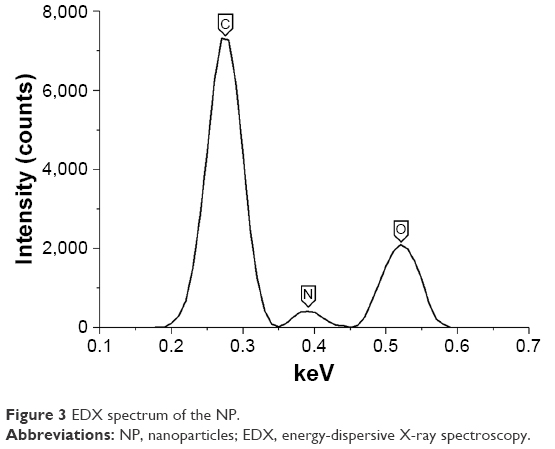 | Figure 3 EDX spectrum of the NP. |
Both UV spectroscopy and microscopy confirmed the stability of the water-insoluble NP during their storage in water suspensions at 4°C for several months (Figure S2) and in PBS at 37°C for several hours of immunoassays. Stability and biological activity of protein NP at different temperatures, pH, and ionic strength can be determined by the stability of the original proteins.10,11
Conformational analysis of protein NP
CD spectra of BSA solutions in water and HFIP as well as BSA NP (Figure 4) were similar, suggesting partial stability of the protein secondary structure in the process of nanoprecipitation (Figure 1). Protein α-helixes are known to remain stable in HFIP solution.22
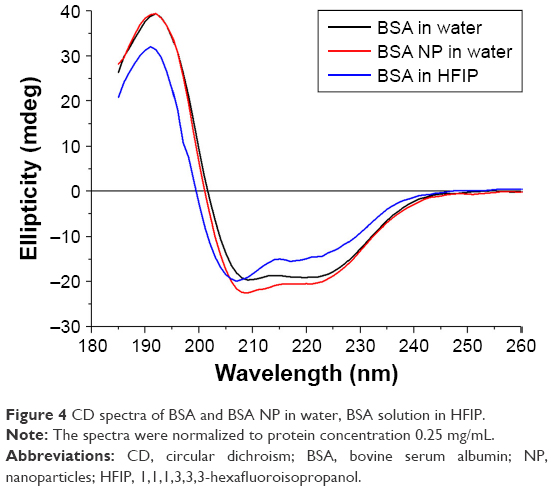 | Figure 4 CD spectra of BSA and BSA NP in water, BSA solution in HFIP. |
Conformational analysis of globular (lysozyme) and fibrillar (fibrinogen) proteins (Figure S3) also determined only slight changes of ellipticity (Table S2), suggesting relative stability of protein secondary structures in the process of the nanoprecipitation.
Production of NP from globular, fibrillar proteins and linear polypeptides
NP were prepared from both globular (BSA ~60 kDa, immunoglobulins IgG ~150 kDa, hen egg lysozyme 14.7 kDa), fibrillar (fibrinogen ~340 kDa) proteins and linear polypeptides (polylysines 15–30 kDa and 30–70 kDa). Part of available data is shown (Figure 5 and Table S1). NP diameters did not appear to depend on the molecular weight of proteins (Figure 5 and Table S1). Contamination of commercially available proteins with trace amounts of lipids or sugars did not hinder the NP formation. Moreover, protein NP can be fabricated from complex mixtures and even biological fluids.
 | Figure 5 SEM images of NP from BSA, fibrinogen, polylysine 15–30 kDa. |
DLS revealed heterogeneity of NP fabricated from globular, fibrillar proteins and linear polypeptides (Table S1). Correlation between protein NP size and original protein molecular weight was not observed (Figure 5 and Table S1).
Binding of the fluorescent BSA NP with polyclonal antibodies
Co-precipitation of all studied proteins with Rho B, FITC and Cy5 fluorescent dyes permitted fabrication of fluorescent NP. The approach permits loading of protein NP with drugs for targeted delivery, pharmacological stability and biocompatibility. However, due to permeability of the fluorescent NP, a partial leakage of the fluorescent dyes was observed after their storage for several days. Another way to produce more stable fluorescent NP is to precipitate preliminary labeled fluorescent proteins (Figure S4).
Fluorescent protein NP can be used in cellular research and immunodiagnostics. Thus, the BSA-RhoB NP after the nanoprecipitation of BSA-RhoB could bind with the immobilized specific polyclonal antibodies (Figure 6A and B). The levels of specific binding of antibodies with the BSA-RhoB protein and the corresponding fluorescent NP were comparable with the wide range of anti-BSA antibodies dilution from 1:10 to 1:10,000 and exceeded those of nonspecific binding with human IgG with the same dilution (Figure 6B). The specific binding of the RhoB-BSA NP with antibodies proved the partial stability of at least the linear protein antigenic determinants in HFIP solution and in final NP. Moreover, addition of unlabeled BSA competitively inhibited the binding of BSA-RhoB NP with the immobilized anti-BSA antibodies (Figure S5). Complete inhibition of binding of BSA-RhoB NP with the immobilized anti-BSA antibodies was observed in the presence of unlabeled BSA protein at concentrations higher 1 mg/mL (Figure S5) that coincided with BSA concentrations in NP and might serve as additional evidence of partial epitope stability. However, the immunofluorescence sensitivity limit is known to exceed those of UV spectroscopy, therefore, the trace amounts of BSA-RhoB molecules detached from the NP surface could take part in the specific binding with the immobilized specific antibodies.
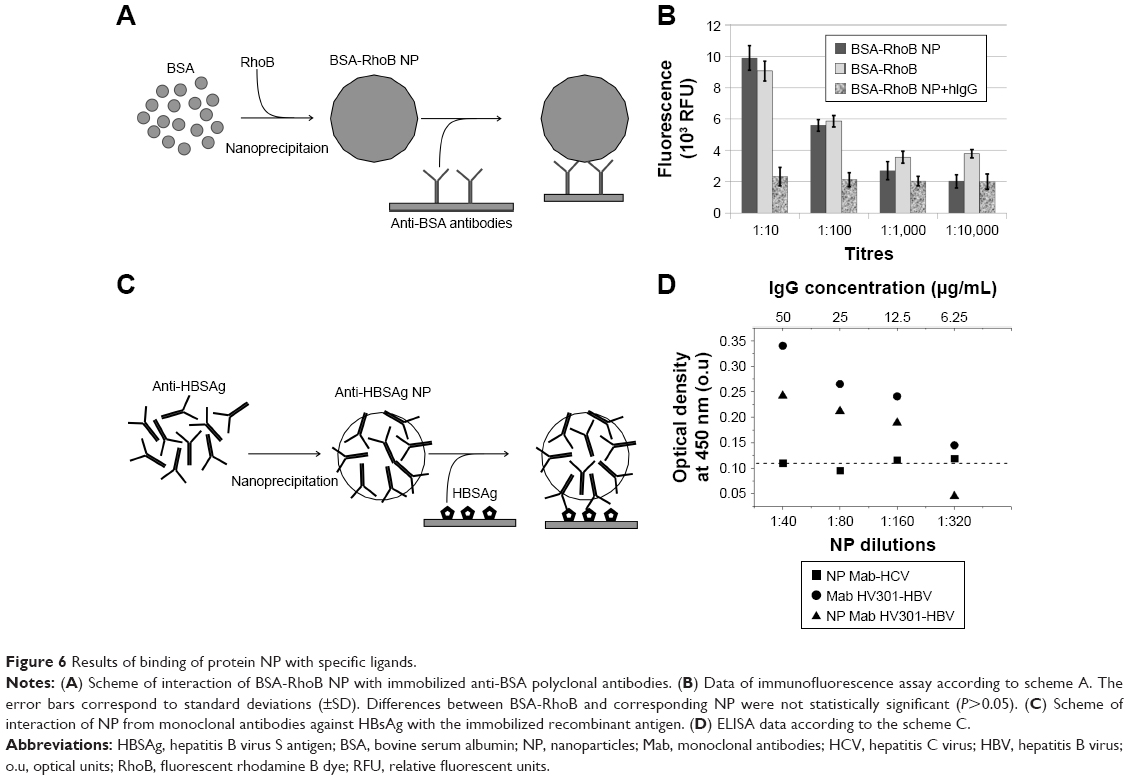 | Figure 6 Results of binding of protein NP with specific ligands. |
Additional evidence of conformational stability of proteins in NP includes binding of NP consisting of antibodies with their specific ligand. Thus, the NP from Mab against HBsAg also retained their ability to bind with the specific antigen as shown by ELISA (Figure 6C and D). Results of one of three ELISA experiments are shown in Figure 6D and the statistical analysis in Table S3. Possible separation of the immunoglobulin molecules from the NP surfaces might contribute to the ELISA results (Figure 6D) with the sensitivity limit (2 pg/mL) significantly surpassing UV spectroscopy sensitivity. However, the protein NP appeared to be stable for several months whereas ELISA usually takes a few hours. Taking into account similar OD values after ELISA of Mab HV-301 HBV and the corresponding NP with similar concentrations, one can conclude that the possible presence of trace amounts of protein molecules detached from the NP was not essential.
Lysozyme NP enzymatic activity
Lysozyme NP sizes were mainly in the range of 10 to 40 nm (Figure S6 and Table S1). Despite their relatively small sizes, the NP exceed the sizes of separate molecules by more than ten times. One should note that the lysozyme retained its enzymatic activity in the NP as shown by AFM (Figure 7) and OD measurements at 450 nm in dynamics (Figure S7).
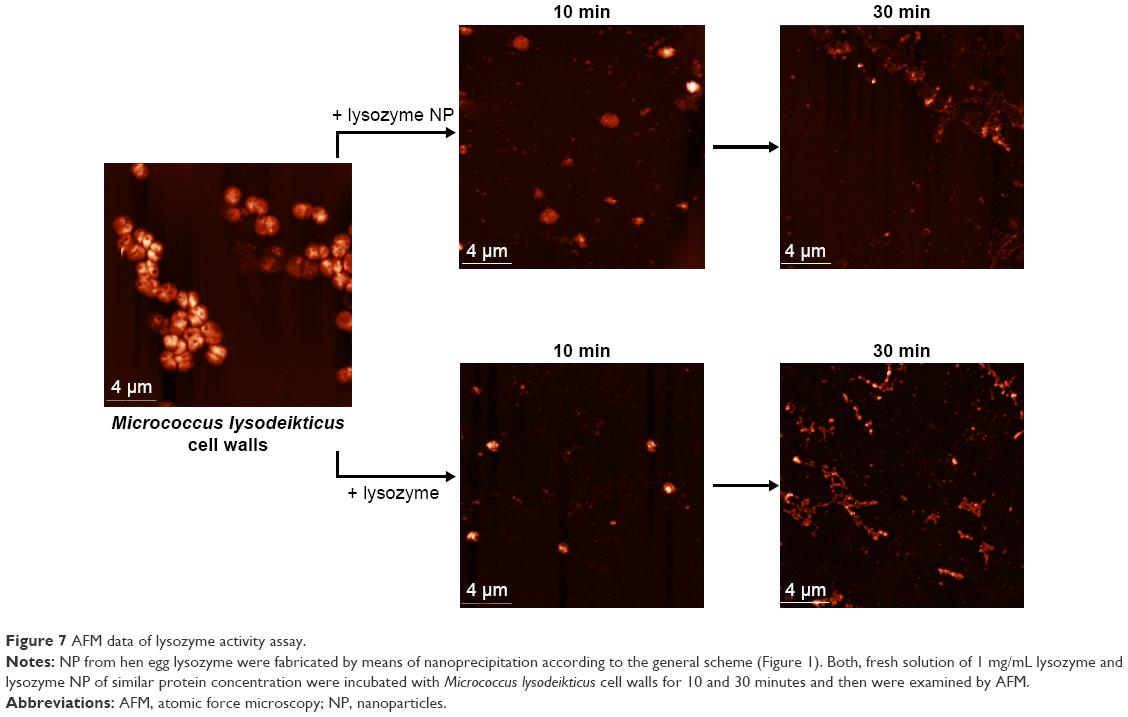 | Figure 7 AFM data of lysozyme activity assay. |
Taken together, the data demonstrated both structural and functional stability of the protein NP.
Discussion
The simple, reproducible, fast and cost efficient method of nanoprecipitation was found to be suitable for various globular, fibrillar proteins and linear polypeptides and provided a high yield of water-insoluble protein NP. Whereas common protein precipitation in organic solvents is known to yield water-soluble shapeless pellets, nanoprecipitation permits forming of water-insoluble and stable NP without cross-linking. Nanoprecipitation of proteins from their solutions in HFIP is based on their desolvation in hydroalcoholic solution and subsequent fast solvent evaporation at temperatures near the boiling points of alcohols.1 Acetone is the most widely used solvent in nanoprecipitation of polymers5,6 but is not suitable for proteins since the polar organic solvent causes their sedimentation. Alcohols often induce the aggregation and sedimentation of proteins at neutral pH.10 Protein precipitation in organic solvents results in water-soluble shapeless pellet. Alcohols, as moderately hydrophobic solvents, destabilize the protein native structures, often producing “open” α-helical conformations stabilized by strengthened intra molecular hydrogen bonds without strong interactions between helices. The effectiveness of alcohols depends predominantly on their hydrophobicity. Highly fluorinated alcohols such as 2,2,2-trifluoroethanol and 1,1,1,3,3,3-HFIP show marked effects due to their dynamic cluster formation.24 Cooperative formation of micelle-like clusters of HFIP in mixture with water25 may be important for induction of protein NP nucleation and maturation, as well as to maintain stable α-helixes. Therefore, HFIP with boiling point 58.2°C close to one of acetone (56.5°C) was suggested to dissolve proteins.24–26
The proposed NP production method was successfully applied to a broad range of protein concentrations up to 2 mg/mL, whereas higher concentrations resulted in nano films. NP shapes and sizes did not correlate with protein molecular weights (Figure 5 and Table S1) and concentrations. One should note that the broad range of protein NP sizes does not exclude their natural endocytosis-mediated cellular uptake.17,23
Fluoroalcohol HFIP involved in the nanoprecipitation of proteins was removed at high temperatures near boiling point and subsequent washes with water as was shown by the absence of fluorine in EDX data (Figure 3). Consequently, the final protein NP, without even trace amounts of HFIP, are biocompatible and non-toxic.
The stable protein NP do not require additional stabilization. Relatively stable secondary structure of proteins in NP was revealed by means of CD (Figure 4, Figure S3 and Table S2). Perhaps, the remarkable stability of the protein secondary structure was due to α-helixes remaining stable in HFIP.24–26 Ligand-binding properties of the protein NP were demonstrated for both antigen (BSA) and antibodies (Mab against HBsAg). The fluorescent BSA NP bound to anti-BSA polyclonal antibodies. NP from Mab against HBsAg reacted in ELISA with the specific antigen (Figure 6). Enzymatic activity was shown for lysozyme NP, capable of destroying bacterial cell walls (Figures 7 and S5). Our data correspond to previously described meta-stability of alcohol-denaturated lysozyme.10 One of possible reasons is stable α-helixes of lysozyme active center in HFIP solution.10 Lysozyme is known to be thermally stable and active in a large range of pH (6–9). The structure of lysozyme is consistent under a variety of conditions. The active site of lysozyme consists of a deep crevice, which divides the protein into two domains linked by an alpha helix.10
Taken together, the available data suggest the general protocol of fabrication of protein and fluorescent NP that are able to bind to specific ligands and to retain enzymatic activity.
Conclusion
The simple, reproducible, fast, and cost efficient method of nanoprecipitation was found to be suitable for various globular, fibrillar proteins and linear polypeptides and provided a high yield of water-insoluble protein NP. The proposed NP production method was successfully applied to a broad range of protein concentrations up to 2 mg/mL, where higher concentrations resulted in nanofilms. NP shapes and sizes did not correlate with protein molecular weights and concentrations. The stable protein NP do not require additional stabilization. Relatively stable secondary structure of proteins in NP was revealed by means of CD. The fluorescent BSA NP bound to anti-BSA polyclonal antibodies. NP from Mab against HBsAg reacted in ELISA with the specific antigen. Lysozyme NP could destroy bacterial cell walls. Taken together, the available data suggest the general protocol of fabrication of protein and fluorescent NP able to bind to specific ligands and to retain enzymatic activity.
Abbreviations
NP, nanoparticles; AFM, atomic force microscopy; DLS, dynamic light scattering; SEM, scanning electron microscopy; EDX, energy-dispersive X-ray spectroscopy; CD, circular dichroism; BSA, bovine serum albumin; HFIP, 1,1,1,3,3,3 hexafluoroisopropanol; HBsAg, hepatitis B virus S antigen; Mab, monoclonal antibodies; OD, optical density; HCV, hepatitis C virus; HBV, hepatitis B virus.
Acknowledgments
Our research was supported by the Russian science foundation (grant no 17-75-30064). We thank Stephen J Seligman (The Rockefeller University, USA) for reviewing our manuscript and Anna Varizhuk for support with CD spectra analysis.
Disclosure
The authors report no conflicts of interest in this work.
References
Mahapatro A, Singh DK. Biodegradable nanoparticles are excellent vehicle for site directed in-vivo delivery of drugs and vaccines. J Nanobiotechnology. 2011;9(1):55. | ||
Davidov-Pardo G, Joye IJ, Mcclements DJ. Food-Grade Protein-Based Nanoparticles and Microparticles for Bioactive Delivery: Fabrication, Characterization, and Utilization. Adv Protein Chem Struct Biol. 2015;98:293–325. | ||
Silva AL, Rosalia RA, Varypataki E, Sibuea S, Ossendorp F, Jiskoot W. poly-W. Poly-(lactic-co-glycolic-acid)-based particulate vaccines: particle uptake by dendritic cells is a key parameter for immune activation. Vaccine. 2015;33(7):847–854. | ||
Hainline KM, Fries CN, Collier JH. Progress Toward the Clinical Translation of Bioinspired Peptide and Protein Assemblies. Adv Healthc Mater. 2018;7(5). | ||
Pavot V, Rochereau N, Primard C, et al. Encapsulation of Nod1 and Nod2 receptor ligands into poly(lactic acid) nanoparticles potentiates their immune properties. J Control Release. 2013;167(1):60–67. | ||
Pavot V, Climent N, Rochereau N, et al. Directing vaccine immune responses to mucosa by nanosized particulate carriers encapsulating NOD ligands. Biomaterials. 2016;75:327–339. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. | ||
Abdollahi S, Lotfipour F. PLGA- and PLA-Based Polymeric Nanoparticles for Antimicrobial Drug Delivery. Biomed Int. 2012;3(1):1–11. | ||
Sundar S, Kundu J, Kundu SC. Biopolymeric nanoparticles. Sci Technol Adv Mater. 2010;11(1):14104. | ||
Lin Y, Lee YH, Yoshimura Y, Yagi H, Goto Y. Solubility and supersaturation-dependent protein misfolding revealed by ultrasonication. Langmuir. 2014;30(7):1845–1854. | ||
Lohcharoenkal W, Wang L, Chen YC, Rojanasakul Y. Protein Nanoparticles as Drug Delivery Carriers for Cancer Therapy. Biomed Res Int. 2014;2014(6679):4–12. | ||
Wacker MG, Altinok M, Urfels S, Bauer J. Nanoencapsulation of ultra-small superparamagnetic particles of iron oxide into human serum albumin nanoparticles. Beilstein J Nanotechnol. 2014;5:2259–2266. | ||
Krishna Sailaja A, Amareshwar P. Preparation of alginate nanoparticles by desolvation technique using acetone as desolvating agent. Asian J Pharm Clin Res. 2012;5(2):132–134. | ||
Perumal O, Podaralla S, Kaushik R; South Dakota State University assignee. Method of forming non-immunogenic hydrophobic protein nanoparticles and uses therefor. United States patent US 9616021. 2014 May 11. | ||
Sadeghi R, Kalbasi A, Moosavi-Movahedi AA, Karimi M, Kokini J. Preparation of BSA Nanoparticles by desolvation method as a delivery system for nutraceuticals. Technical Proceedings of the 2013 NSTI Nanotechnology Conference and Expo, NSTI-Nanotech 2013. 2013;3 225–228. | ||
Lee SH, Heng D, Ng WK, Chan H-K, Tan RBH. Nano spray drying: A novel method for preparing protein nanoparticles for protein therapy. Int J Pharm. 2011;403(1–2):192–200. | ||
Lizotte PH, Wen AM, Sheen MR, et al. In situ vaccination with cowpea mosaic virus nanoparticles suppresses metastatic cancer. Nat Nanotechnol. 2016;11(3):295–303. | ||
Pesarrodona M, Fernández Y, Foradada L, et al. Conformational and functional variants of CD44-targeted protein nanoparticles bio-produced in bacteria. Biofabrication. 2016;8(2):25001. | ||
Sun H, Luo Q, Hou C, Liu J. Nanostructures based on protein self-assembly: From hierarchical construction to bioinspired materials. Nano Today. 2017;14:16–41. | ||
Atabekov I. The method of immune response enhancement; 2010 Russian [with English abstract]. | ||
Nash JA, Ballard TN, Weaver TE, Akinbi HT. The peptidoglycan-degrading property of lysozyme is not required for bactericidal activity in vivo. J Immunol. 2006;177(1):519–526. | ||
Schneider SW, Lärmer J, Henderson RM, Oberleithner H. Molecular weights of individual proteins correlate with molecular volumes measured by atomic force microscopy. Pflugers Arch. 1998;435(3):362–367. | ||
Zanganeh S, Hutter G, Spitler R, et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol. 2016;11(11):986–994. | ||
Yoshida K, Yamaguchi T, Adachi T, et al. Structure and dynamics of hexafluoroisopropanol-water mixtures by x-ray diffraction, small-angle neutron scattering, NMR spectroscopy, and mass spectrometry. J Chem Phys. 2003;119(12):6132–6142. | ||
Hirota N, Mizuno K, Goto Y. Cooperative alpha-helix formation of beta-lactoglobulin and melittin induced by hexafluoroisopropanol. Protein Sci. 1997;6(2):416–421. | ||
Narita M, Honda S, Umeyama H, Obana S. The Solubility of Peptide Intermediates in Organic Solvents. Solubilizing Potential of Hexafluoro-2-propanol. Bull Chem Soc Jpn. 1988;61(1):281–284. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.