Back to Journals » Journal of Inflammation Research » Volume 19
Protective Effects of CR6-Interacting Factor 1 Against Angiotensin II-Induced Atrial Fibrillation by Regulating the SIRT1/eNOS Signaling Pathway and Cardiomyocyte Remodeling
Received 23 January 2026
Accepted for publication 27 April 2026
Published 10 June 2026 Volume 2026:19 596132
DOI https://doi.org/10.2147/JIR.S596132
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Durga Misra
Luoning Zhu, Zhongping Ning, Yuying Gu
Department of Cardiology, Shanghai Pudong New Area Zhoupu Hospital (Shanghai Health Medical College Affiliated Zhoupu Hospital), Shanghai, 201318, People’s Republic of China
Correspondence: Zhongping Ning, Department of Cardiology, Shanghai Pudong New Area Zhoupu Hospital (Shanghai Health Medical College Affiliated Zhoupu Hospital) No.1500 Zhou Yuan Road, Pudong New District, Shanghai, 201318, People’s Republic of China, Tel +86-21-68135590, Email [email protected] Yuying Gu, Department of Cardiology, Shanghai Pudong New Area Zhoupu Hospital (Shanghai Health Medical College Affiliated Zhoupu Hospital), No. 1500 Zhou yuan Road, Pudong New District, Shanghai, 201318, People’s Republic of China, Tel +86- 18101635903, Email [email protected]
Background: Atrial fibrillation (AF) is a common arrhythmia associated with myocardial injury, oxidative stress, and inflammatory remodeling. Mitochondrial protein CR6-interacting factor 1 (CRIF1) has emerged as a potential regulator of cardiomyocyte homeostasis; however, its role in AF remains unclear.
Methods: An AF model was established in C57BL/6 mice via subcutaneous infusion of angiotensin II (Ang II, 2.0 mg/kg/day) for 28 days. CRIF1 expression levels and its functional effects were evaluated in atrial tissue and Ang II-treated HL-1 cardiomyocytes using RT-qPCR, immunohistochemistry, ELISA, TUNEL, and DHE staining. CRIF1 was overexpressed to assess its effects on the SIRT1/eNOS pathway, apoptosis, hypertrophy, inflammation, and oxidative stress.
Results: Ang II infusion promoted atrial remodeling and increased susceptibility to atrial fibrillation. During electrophysiological assessment, AF episodes were triggered by transesophageal burst pacing, revealing prolonged AF duration, increased AF inducibility, elevated creatine kinase-MB (CK-MB) and lactate dehydrogenase (LDH) levels, and enhanced atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) expression in Ang II-treated mice compared with controls. CRIF1 expression was markedly downregulated in the atrial tissue of AF mice and Ang II-treated HL-1 cells. CRIF1 overexpression activates the SIRT1/eNOS pathway, attenuates cardiomyocyte apoptosis, and reduces Ang II–induced hypertrophy. Furthermore, CRIF1 suppressed the expression of inflammatory cytokines (TNF-α, IL-1β, and IL-6) and diminished intracellular reactive oxygen species accumulation, thereby restoring antioxidant enzyme activity and nitric oxide (NO) production. The protective effects of CRIF1 on apoptosis are largely dependent on SIRT1 signaling.
Conclusion: CRIF1 plays a critical protective role in Ang II–induced atrial remodeling by activating SIRT1/eNOS signaling and mitigating oxidative stress, inflammation, hypertrophy, and apoptosis. These findings highlight CRIF1 as a potential therapeutic target for preventing AF and its associated cardiac injuries.
Keywords: atrial fibrillation, CRIF1, SIRT1/eNOS signaling, oxidative stress, cardiomyocyte remodeling
Introduction
Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia worldwide and significantly contributes to morbidity and mortality, particularly via stroke, heart failure, and thromboembolic complications.1,2 The pathogenesis of AF is multifactorial and involves electrical, structural, and molecular remodeling of the atrial myocardium. Structural remodeling is driven in part by neurohormonal activation, especially via angiotensin II (Ang II), which promotes atrial fibrosis, inflammation, and hypertrophy.3,4 Mechanistically, Ang II activates signaling via the AT1 receptor, triggering pro-fibrotic pathways (eg, MAPK and TGF-β/Smad), oxidative stress (eg, via NOX-derived ROS), and inflammatory cascades (eg, NF-κB and JAK/STAT), thereby increasing susceptibility to AF.2,5,6
Mitochondrial dysfunction is an important contributor to cardiovascular diseases, including arrhythmias and cardiomyopathy.7,8 Mitochondria are essential for cardiomyocyte energy production through oxidative phosphorylation (OXPHOS) and for maintaining cellular redox homeostasis. Mitochondrial dysfunction results in ATP depletion, excessive reactive oxygen species (ROS) generation, and activation of apoptotic and inflammatory signaling pathways that promote cardiac remodeling.9 One key regulator of mitochondrial protein synthesis is CR6-interacting factor-1 (CRIF1), a mitochondrial inner-membrane protein required for the translation and insertion of OXPHOS polypeptides into the mitochondrial membrane.10 Previous studies have shown that CRIF1 deficiency impairs mitochondrial respiration, disrupts OXPHOS complex assembly, increases oxidative stress, and contributes to cellular injury and metabolic dysfunction.10,11
Emerging evidence suggests that CRIF1 participates in cardiovascular regulation by modulating the SIRT1/endothelial nitric oxide synthase (eNOS) signaling pathway. Sirtuin-1 (SIRT1) is an NAD⁺-dependent deacetylase that regulates oxidative stress responses, mitochondrial function, and cell survival in cardiovascular tissues.12 SIRT1 directly deacetylates eNOS, thereby enhancing nitric oxide (NO) production and maintaining vascular and myocardial homeostasis.13 Early study demonstrated that endothelial-specific deletion of CRIF1 in mice resulted in reduced SIRT1 expression, increased oxidative stress, and impaired cardiac function, whereas pharmacological activation of SIRT1 restored eNOS activity and attenuated inflammatory responses.14 These findings highlight a functional link between CRIF1, mitochondrial homeostasis, and SIRT1/eNOS signaling in cardiovascular protection.
Recent studies have begun to connect CRIF1 dysfunction with atrial remodeling and arrhythmogenesis. Experimental evidence indicates that CRIF1 deficiency disrupts mitochondrial oxidative phosphorylation, leading to mitochondrial damage, excessive reactive oxygen species production, and impaired cellular metabolism.15,16 Moreover, reduced CRIF1 expression has been shown to downregulate SIRT1 and impair eNOS activity, thereby decreasing NO bioavailability and promoting oxidative stress-related cardiac dysfunction.17 In a high-fat diet-induced atrial remodeling model, CRIF1 downregulation reduced NO production through the CRIF1/SIRT1/eNOS/P21 signaling axis, which promoted atrial neural remodeling and increased susceptibility to atrial fibrillation.18 These findings suggest that CRIF1 deficiency may contribute to AF pathogenesis by disturbing mitochondrial homeostasis, redox balance, and NO-dependent signaling pathways that are essential for maintaining atrial structural and electrical stability.
Despite these advances, the potential role of CRIF1 in Ang II–induced atrial fibrillation and its involvement in cardiomyocyte remodeling under neurohormonal stress remain poorly understood. In particular, whether CRIF1 can protect against Ang II-mediated mitochondrial dysfunction and oxidative stress in atrial cardiomyocytes has not been fully elucidated. Therefore, in the present study, we hypothesized that CRIF1 exerts a protective effect against Ang II–induced AF by preserving mitochondrial integrity and activating the SIRT1/eNOS signaling pathway, thereby attenuating apoptosis, hypertrophy, inflammation, and oxidative stress in atrial cardiomyocytes. To test this hypothesis, we employed both in vivo (Ang II–infused mice) and in vitro (Ang II-treated HL-1 cells) models to investigate the cardioprotective role and mechanistic actions of CRIF1 in AF pathogenesis.
Materials and Methods
Animals
All experimental procedures involving the sixteen male C57BL/6 mice (8–10 weeks of age) were approved by the Animal Care and Use Committee of Shanghai Pudong Zhoupu Hospital and were conducted in compliance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85–23, revised 1996). To establish the atrial fibrillation model, mice were subjected to a 28-day subcutaneous infusion of Ang II (2.0 mg/kg/day, MedChemExpress, HY-13948) delivered by osmotic mini-pumps.19,20 A control group was maintained, receiving saline via the same method. All animal procedures, including Ang II pump implantation and electrophysiological pacing experiments, were performed by trained researchers at the Experimental Animal Center of Shanghai Pudong Zhoupu Hospital under the supervision of experienced investigators.
Transesophageal Burst Pacing
On the 28th day following the initiation of Ang II infusion, mice were anesthetized by intraperitoneal injection of 1% pentobarbital sodium. An 8-electrode catheter (Japan Lifeline, Tokyo, Japan) was positioned in the esophagus dorsal to the left atrium to record a surface electrocardiogram (ECG). AF was induced by applying three 2-second burst pacing trains using an automated stimulator (MadLab-4C/501H, ZS Dichuang Co., Ltd., Beijing, China). The stimulation parameters were set at 20 V, 4 mA, with a 6 ms wave width, 5 ms pulse duration, and a 40 ms cycle length. During burst pacing stimulation, AF was the primary arrhythmia observed. An AF episode was defined as a rapid, irregular atrial rhythm accompanied by variable R-R intervals lasting for at least 1s. The duration of each AF episode was measured from the cessation of burst pacing until the appearance of the first sinus P wave.21 Although occasional brief episodes of atrial tachycardia were observed immediately after pacing, these events were transient and spontaneously reverted to sinus rhythm. No sustained ventricular arrhythmias were detected during electrophysiological recordings.
Biochemical Analysis
After completion of the experimental protocol, mouse blood was drawn via cardiac puncture under anesthesia to obtain the serum for analyzing cardiac injury markers. The blood was left to clot for 30 min at room temperature and then centrifuged at 3000 × g for 10 min. The activities of creatine kinase-MB (CK-MB) and lactate dehydrogenase (LDH) in the serum were measured using commercial colorimetric kits (Nanjing Jiancheng Bioengineering Institute; CK-MB: A032-1-1, LDH: A020-2-2) as per the manufacturer’s guidelines. Additionally, serum levels of atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) were determined using mouse-specific ELISA kits (Elabscience; ANP: E-EL-M0166c, BNP: E-EL-M0204c). All measurements were conducted in duplicate, and a microplate reader was used to record absorbance.
Histology and Immunohistochemistry
Tissue processing and staining commenced with harvesting cardiac tissues, which were subsequently rinsed in phosphate-buffered saline and fixed in 4% paraformaldehyde solution for 24 h. The specimens were then dehydrated through a graded ethanol series, embedded in paraffin blocks, and sectioned to a thickness of 4 µm. A subset of sections was subjected to hematoxylin and eosin (H&E) staining using a commercial kit (Beyotime, C0105S) to facilitate morphological evaluation. For immunohistochemical localization of CRIF1, tissue sections were deparaffinized, rehydrated, and subjected to heat-induced epitope retrieval using a citrate-based buffer (pH 6.0) in a microwave. Endogenous peroxidase activity was suppressed by incubation with 3% hydrogen peroxide, and nonspecific binding sites were blocked with 5% BSA. The sections were then incubated with a rabbit polyclonal primary antibody directed against CRIF1 (Proteintech, 16260-1-AP) at 4°C overnight. A rabbit-derived antibody was intentionally selected to minimize nonspecific background staining commonly observed in mouse-on-mouse immunohistochemistry, as the tissue samples were obtained from mice. This strategy reduces potential cross-reactivity with endogenous mouse immunoglobulins, thereby improving staining specificity and sensitivity. After incubation with an appropriate HRP-conjugated secondary antibody, immunoreactivity was detected using a DAB chromogen, and cell nuclei were counterstained with hematoxylin. Imaging was performed using a light microscope, and a quantitative assessment of staining intensity was conducted on randomly selected areas.
Cell Culture and Treatment
The mouse cardiac muscle cell line HL-1 was purchased from Procell (CL-0605, China). HL-1 cells were cultivated in complete Claycomb Medium (Sigma-Aldrich). Louis, MO, USA) supplemented with 10% FBS (Gibco, Waltham, MA, USA) and 100 U/mL penicillin/streptomycin. HL-1 cells were treated with 1 μM Ang II for 24 h to generate an in vitro AF model. To explore the role of SIRT1, the SIRT1 inhibitor EX527 (10 μM, HY-15452, MedChemExpress) was used to treat HL-1 cells.
Cell Grouping
HL-1 cells were divided into four groups: (i) control group: (ii) Model group (Ang II +Vector): cells were transfected with vector for 24 h, and then treated with Ang II (1 μM) for further 24 h; (iii) CRIF1 overexpression group (Ang II+OE-CRIF1): cells were transfected with OE-CRIF1 for 24 h, and treated with Ang II for 24 h; (iv) SIRT1 pathway inhibitor group (Ang II+OE-CRIF1+EX-527): cells were transfected with OE-CRIF1 for 24 h, and then incubated with a SIRT1 inhibitor EX527 (10 μM) and Ang II for 24 h.
Cell Transfection
HL-1 cells were seeded in 6-well plates (1.0×105 cells/well) and allowed to reach 70–80% confluence. pcDNA3.1-OE-CRIF1 (OE-CRIF1) or its negative control (pcDNA3.1 vector) was introduced into HL-1 cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Transfection efficiency was verified using RT-qPCR and Western blotting 48 h after transfection.
Immunofluorescence
A series of fluorescence-based assays were performed on HL-1 cells cultured on sterilized coverslips following treatment. After washing with PBS and fixation in 4% paraformaldehyde, the cells were analyzed for apoptosis, cytoskeletal structure, and oxidative stress. The TUNEL method (Beyotime, C1086) was utilized to identify apoptotic cells. For immunofluorescence, cells were permeabilized with 0.3% Triton X-100, blocked with 5% BSA, and probed with an anti-α-actinin primary antibody (Abcam, ab108198) overnight at 4°C, followed by a fluorophore-conjugated secondary antibody and DAPI counterstaining. Concurrently, intracellular ROS production was quantified by staining with dihydroethidium (DHE; Beyotime, S0063). Imaging was performed using a fluorescence microscope at magnifications of 200 X (TUNEL, DHE) and 400 X (α-actinin), maintaining identical exposure settings within each assay for quantitative comparison.
Oxidative Stress Evaluation
To analyze oxidative stress, HL-1 cells were homogenized in ice-cold buffer to create a 10% (w/v) suspension. The homogenates were centrifuged at 3000 × g for 10 min at 4°C, and the resulting supernatants were used for subsequent assays. Lipid peroxidation was evaluated by measuring malondialdehyde (MDA) levels using a commercial kit (Beyotime, S0131S). The activities of the antioxidant enzymes superoxide dismutase (SOD; Beyotime, S0109) and catalase (CAT; Beyotime, S0051) were determined according to the manufacturer’s instructions. Additionally, NO production was quantified using a specific kit (Nanjing Jiancheng Bioengineering Institute, A012-1-2). All measurements were performed in duplicate, and a microplate reader was used to record absorbance.
Enzyme-Linked Immunosorbent Assay (ELISA)
The secretion profile of pro-inflammatory cytokines was assessed in the conditioned medium of HL-1 cells. Post-treatment, the medium samples were harvested and centrifuged at 1000 × g for 10 min at 4°C to pellet any residual cells or debris. The clarified supernatant was used in enzyme-linked immunosorbent assays (ELISAs) to determine the concentrations of tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6), employing specific commercial kits (R&D Systems; TNF-α: MTA00B, IL-1β: MLB00C, IL-6: M6000B). The assays were performed according to the manufacturer’s guidelines, with absorbance measured at 450 nm. Final cytokine concentrations were derived from their respective standard curves, and all analyses were conducted in duplicate.
Reverse Transcription Quantitative PCR (RT-qPCR)
A standard reverse transcription-quantitative PCR (RT-qPCR) protocol was employed to evaluate gene expression. Total RNA was initially purified from HL-1 cells using a Takara extraction kit (Cat. No. RR820A). The concentration and purity of the eluted RNA were determined spectrophotometrically, and samples meeting the quality threshold (A260/A280 ratio of 1.8–2.0) were selected for cDNA synthesis. This was achieved using 1 µg of total RNA and a reverse transcription kit from Yeasen Biotechnology (Cat. No. 11201ES50). The qPCR amplifications were performed in a reaction mixture containing the synthesized cDNA, gene-specific primers, SYBR Green master mix, and nuclease-free water. The thermal profile included an initial denaturation step (95 °C for 5 min) and 40 amplification cycles (95 °C for 10s, 60 °C for 30s). Post-amplification, a melting curve was generated to ensure primer specificity. The relative quantification of mRNA expression was conducted by normalizing to GAPDH and applying the 2−ΔΔCt algorithm. The sequences for all primers are detailed in Table 1.
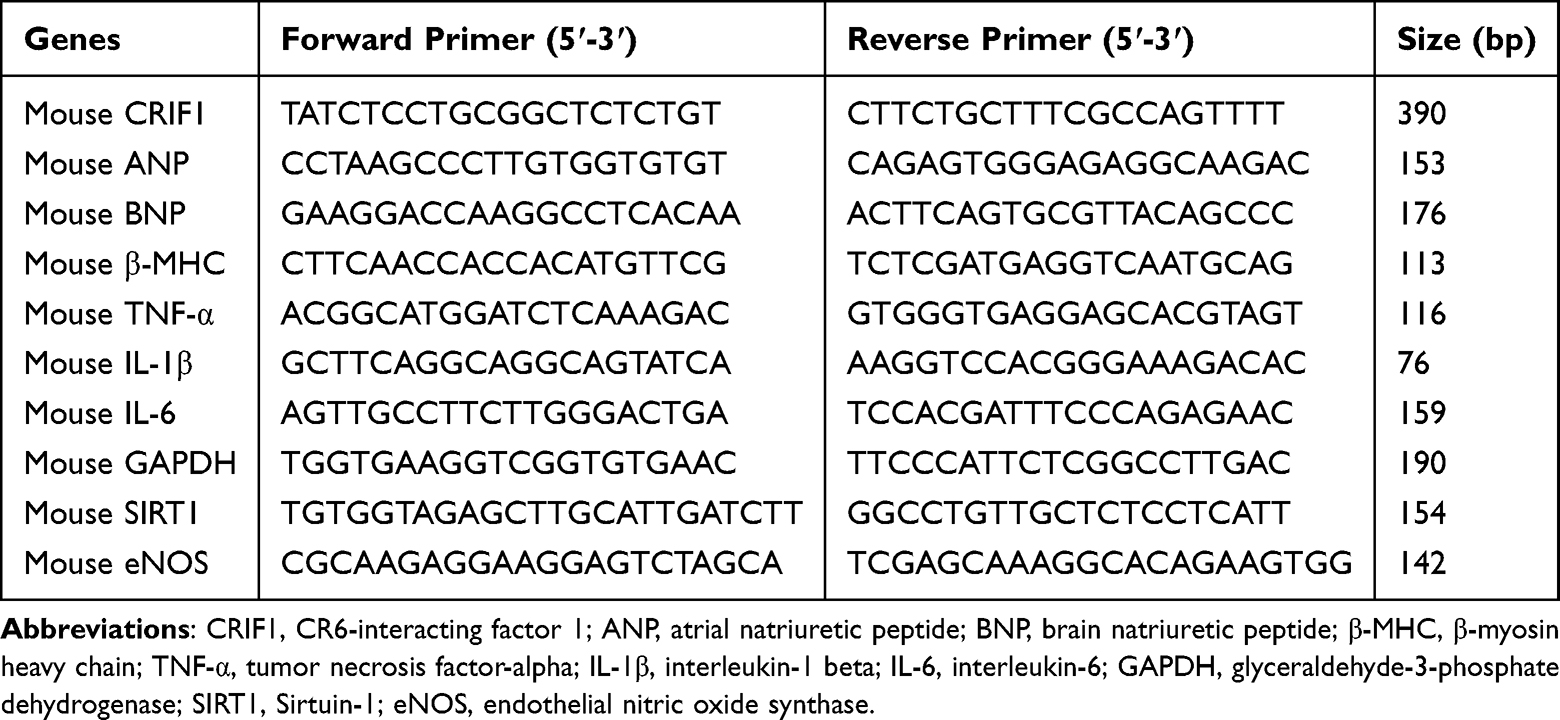 |
Table 1 Oligonucleotide Primer Sequences Used in This Study |
Statistical Analysis
SPSS 30.0 software and Prism version 9.0 (GraphPad Software) were used for statistical analysis and graph production. Data are expressed as the mean ± standard deviation (SD). A two-tailed independent t-test was used to compare the two groups. A one-way analysis of variance was used to compare multiple groups, followed by the post hoc Tukey’s test. Statistical significance was set at P < 0.05.
Results
Ang II Infusion Increases Susceptibility to Atrial Fibrillation and Induces Myocardial Injury in Mice
Burst pacing predominantly induced atrial fibrillation (AF) in Ang II-treated mice. Although brief episodes of atrial tachyarrhythmia were occasionally observed immediately after pacing, these events were transient and were not included in the quantitative analysis of AF inducibility or duration.
To establish an AF-susceptible model, mice were subcutaneously infused with Ang II (2.0 mg/kg/day) for 28 days. Electrophysiological evaluation using transesophageal burst pacing demonstrated that Ang II-treated mice exhibited marked electrophysiological abnormalities compared with controls (Figure 1A). Ang II infusion significantly increased AF susceptibility, as evidenced by a higher rate of AF inducibility and prolonged AF duration following burst pacing (Figure 1B and C). Consistent with AF-associated myocardial injury, serum levels of cardiac damage markers CK-MB and LDH were significantly elevated in Ang II-treated mice compared with controls (Figure 1D and E). Furthermore, ELISA analysis revealed increased circulating levels of ANP and BNP, indicating atrial remodeling and cardiac stress (Figure 1F and G). Collectively, these findings confirm that Ang II infusion promotes atrial remodeling and enhances susceptibility to AF in mice.
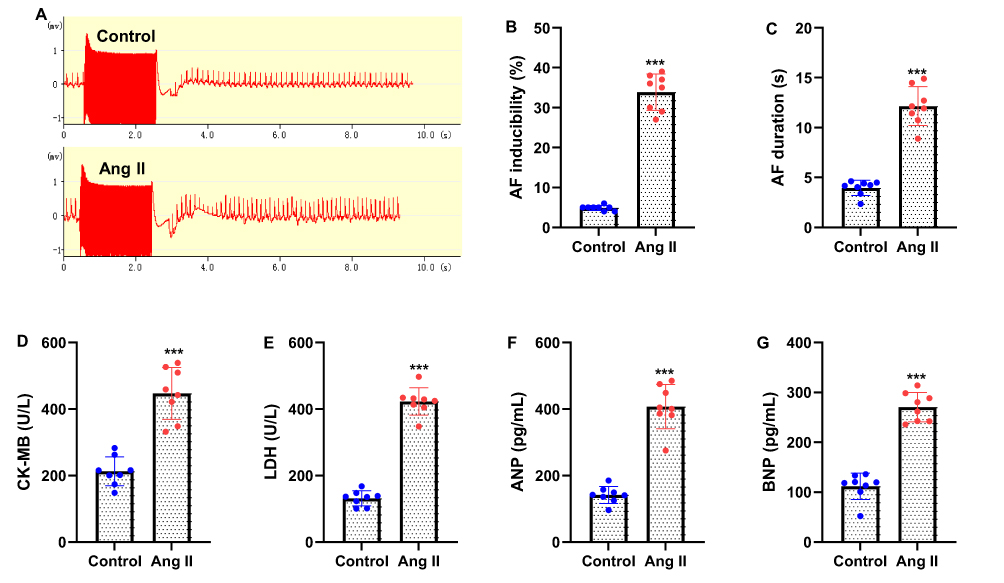 |
Figure 1 Mice is subcutaneously infused with angiotensin II (Ang II, 2.0 mg/kg/day) for 28 days to establish atrial fibrillation (AF) model. (A) Representative surface electrocardiogram (ECG) traces were recorded during the transesophageal burst pacing. The marked regions indicate typical electrophysiological abnormalities characteristic of atrial fibrillation, including the disappearance of organized P waves, rapid irregular atrial electrical activity, and variable R-R intervals in Ang II-treated mice compared to controls. (B) After burst pacing, the percentage of successful AF inducibility was recorded. (C) Average AF duration. (D and E) Serum of mice were collected to measure the concentrations of myocardial injury-related molecules, CK-MB and LDH. (F and G) ELISA was used to measure serum levels of ANP and BNP. Data are presented as mean ± SD (n=8 mice in each group). t-test was applied. ***P<0.001 vs control group. |
CRIF1 Expression Is Downregulated in Atrial Tissue of AF Mice and Ang II-Treated HL-1 Cells
To determine whether CRIF1 is involved in Ang II–induced atrial remodeling, its expression was examined in atrial tissues of AF mice. H&E staining revealed notable structural alterations in the atria of Ang II–infused mice compared to controls, indicating myocardial disorganization and remodeling (Figure 2A). Immunohistochemical analysis further demonstrated a marked reduction in CRIF1 expression in atrial tissues from mice with AF (Figure 2A, 200×). Consistent with the immunohistochemical findings, RT-qPCR analysis showed significantly decreased CRIF1 mRNA levels in atrial tissues of Ang II-treated mice (Figure 2B). To validate the regulatory effect of Ang II on CRIF1 expression at the cellular level, HL-1 cells were exposed to increasing concentrations of Ang II (0, 0.1, 0.2, 0.5, 1, and 2 μM) for 24 h. Ang II treatment induced a dose-dependent reduction in CRIF1 mRNA expression, as shown by RT-qPCR (Figure 2C). Collectively, these results indicated that CRIF1 was significantly downregulated in both Ang II–induced AF mice and Ang II-treated HL-1 cells, suggesting a potential role for CRIF1 in atrial pathological remodeling.
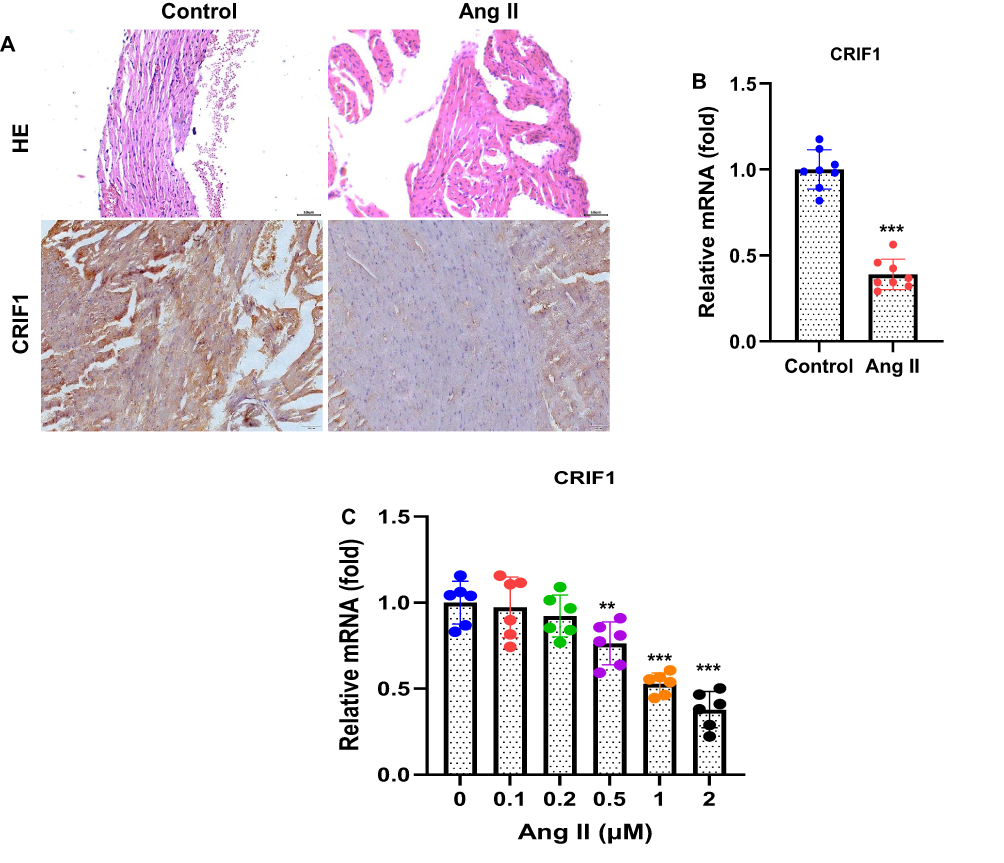 |
Figure 2 CRIF1 was downregulated in the AF mice. (A) Atrial tissue of mice was stained with hematoxylin and eosin (H&E) to display the morphological structure. CRIF1 expression in the atrial tissue of mice was detected using immunohistochemistry (200×). (B) The CRIF1 mRNA levels in the atrial tissue were detected by RT-qPCR. The mice cardiac muscle cell line HL-1 was treated with Ang II (0, 0.1, 0.2, 0.5, 1 and 2 μM) for 24 h. (C) The CRIF1 mRNA expression was determined by RT-qPCR. Data are presented as mean ± SD (n=8 mice in each group). t-test was applied. ***P<0.001 vs control group. |
CRIF1 Overexpression Activates the SIRT1/eNOS Pathway and Attenuates Ang II-Induced Apoptosis in HL-1 Cells
To investigate whether CRIF1 regulates the SIRT1/eNOS signaling pathway in cardiomyocytes exposed to Ang II, HL-1 cells were transfected with pcDNA3.1-CRIF1 (OE-CRIF1) or an empty vector and subsequently treated with Ang II. RT-qPCR analysis confirmed successful CRIF1 overexpression and demonstrated that CRIF1 upregulation markedly enhanced SIRT1 protein levels compared to vector-transfected Ang II-treated cells (Figure 3A and B). Moreover, CRIF1 overexpression significantly increased eNOS, indicating the activation of the eNOS signaling pathway (Figure 3C). To determine whether the protective effects of CRIF1 depend on SIRT1 activity, HL-1 cells overexpressing CRIF1 were co-treated with the SIRT1 inhibitor, EX527. TUNEL staining revealed that CRIF1 overexpression markedly reduced Ang II–induced apoptosis (Figure 3D). However, the inhibition of SIRT1 with EX527 significantly reversed the anti-apoptotic effect of CRIF1, resulting in a higher number of TUNEL-positive cells. Quantitative analysis confirmed that apoptosis levels were significantly lower in the OE-CRIF1 group but were restored upon SIRT1 inhibition (Figure 3E). Collectively, these findings demonstrate that CRIF1 overexpression activates the SIRT1/eNOS pathway and protects cardiomyocytes against Ang II–induced apoptosis, and that these effects are mediated largely through SIRT1 signaling.
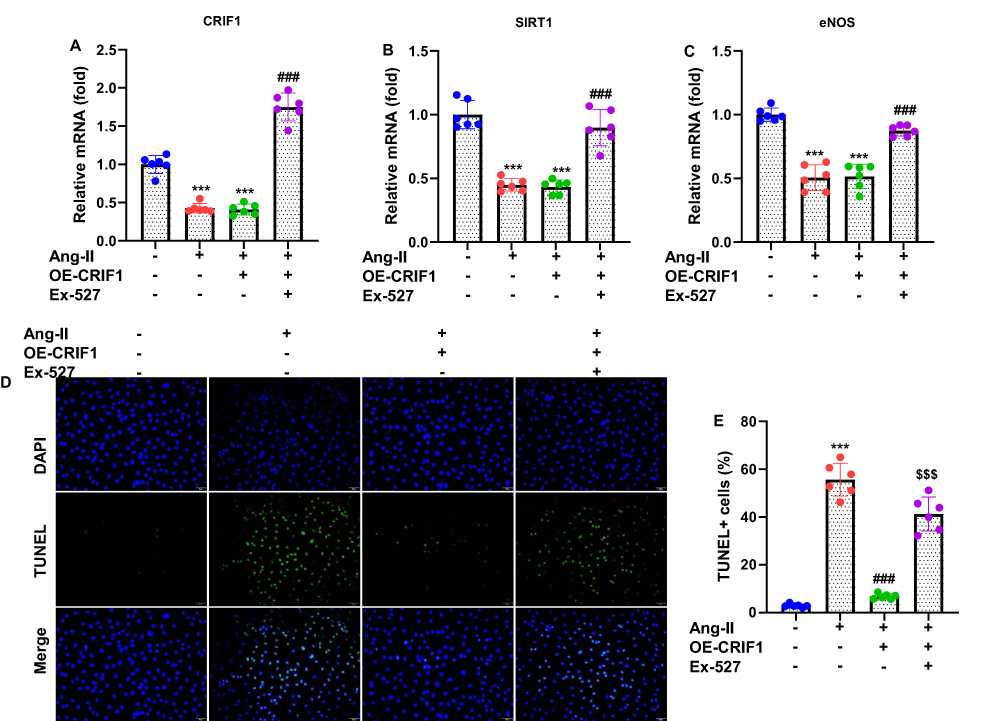 |
Figure 3 Overexpression of CRIF1 activates the SIRT1/eNOS pathway in cardiomyocytes treated with Ang II. HL-1 cells were transfected with pcDNA3.1-CRIF1 (OE-CRIF1) or vector for 24 h, and then treated with Ang II (1 μM) for an additional 24 h. The mRNA expression levels of (A) CRIF1, (B) SIRT1 and (C) eNOS were evaluated using RT-qPCR. HL-1 cells were transfected with pcDNA3.1-CRIF1 (OE-CRIF1) or vector for 24 h, and were then cotreated with a SIRT1 inhibitor EX527 (10 μM) and Ang II (1 μM) for further 24 h. (D) The cell apoptosis was assessed by staining with TUNEL and DAPI, and observed under a fluorescence microscope (200 X). (E) Apoptosis was quantified by calculating TUNEL positive cells (normalized to DAPI-stained cells). Data are presented as mean ± SD in triplicates. ***P<0.001 vs control group; ###P<0.001 vs Ang II+Vector group; $$$P<0.001 vs OE-CRIF1 group. |
CRIF1 Overexpression Attenuates Ang II-Induced Cardiomyocyte Hypertrophy
To determine whether CRIF1 plays a protective role against Ang II–induced hypertrophic remodeling, HL-1 cells were transfected with pcDNA3.1-CRIF1 (OE-CRIF1) or a vector and subsequently exposed to Ang II. Immunofluorescence staining for α-actinin revealed that Ang II treatment markedly increased cell size, which was consistent with hypertrophic enlargement (Figure 4A). In contrast, CRIF1 overexpression substantially reduced Ang II–induced increases in cardiomyocyte surface area, indicating the suppression of hypertrophic growth. To further validate these observations, the expression of hypertrophy-associated genes was examined using RT-qPCR. Ang II stimulation significantly elevated mRNA levels of ANP, BNP, and β-myosin heavy chain (β-MHC) (Figure 4B–D). Notably, CRIF1 overexpression markedly attenuated the Ang II–induced upregulation of all three markers, demonstrating a robust antihypertrophic effect. Collectively, these results indicate that CRIF1 overexpression effectively inhibits Ang II–induced hypertrophic remodeling in HL-1 cardiomyocytes.
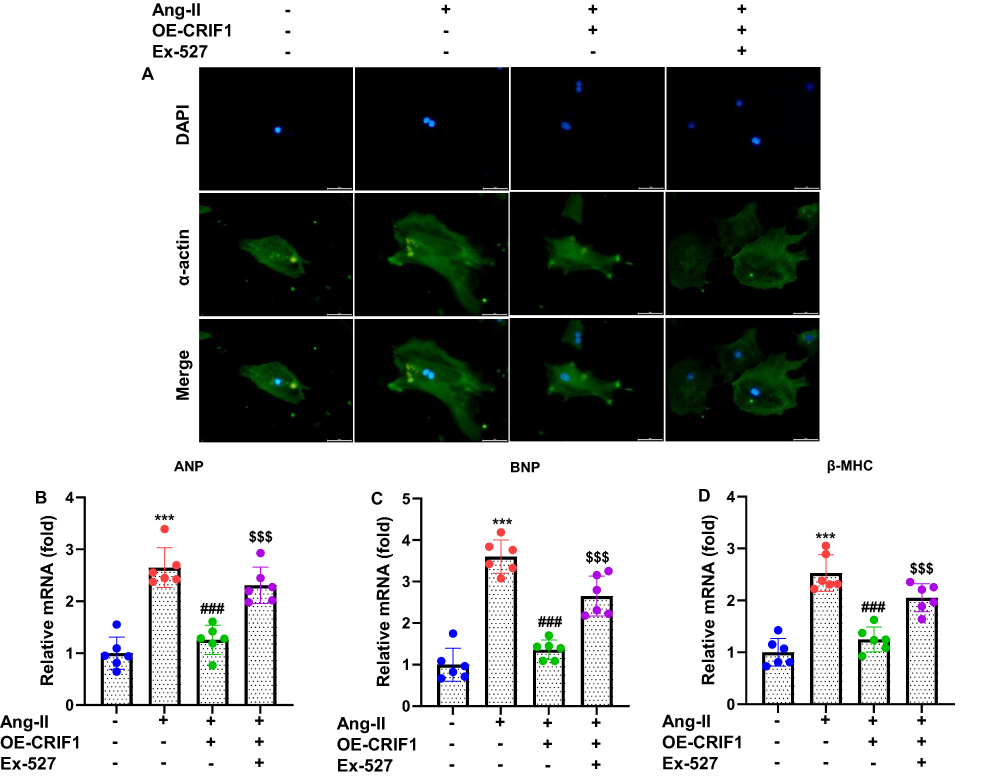 |
Figure 4 CRIF1 overexpression inhibits Ang II–induced hypertrophy of HL-1 cells. (A) Cells were stained with α-actinin antibody and observed under a fluorescence microscope. Representative images are shown (200×). RT-qPCR was used to assess the mRNA expression levels of hypertrophy-related genes, including (B) ANP, (C) BNP, and (D) β-MHC. Data are presented as mean ± SD in triplicate. ***P<0.001 vs control group; ###P<0.001 vs Ang II+Vector group; $$$P<0.001 vs OE-CRIF1 group. |
CRIF1 Overexpression Suppresses Ang II-Induced Inflammatory Responses in Cardiomyocytes
To assess whether CRIF1 modulates the inflammatory response triggered by Ang II, the expression of key pro-inflammatory cytokines was examined in HL-1 cells overexpressing CRIF1. RT-qPCR analysis revealed that Ang II stimulation markedly elevated the mRNA levels of TNF-α, IL-1β, and IL-6 compared those in control cells (Figure 5A–C). In contrast, CRIF1 overexpression significantly reduced Ang II–induced upregulation of all three cytokines, indicating a strong anti-inflammatory regulatory effect at the transcriptional level. Consistent with the mRNA findings, ELISA results showed that Ang II treatment substantially increased the secretion of TNF-α, IL-1β, and IL-6 into the culture medium (Figure 5D–F). CRIF1 overexpression effectively suppressed the elevation of these cytokines, demonstrating reduced release of inflammatory mediators. Together, these findings indicate that CRIF1 overexpression robustly attenuates Ang II–induced inflammatory signaling in HL-1 cardiomyocytes.
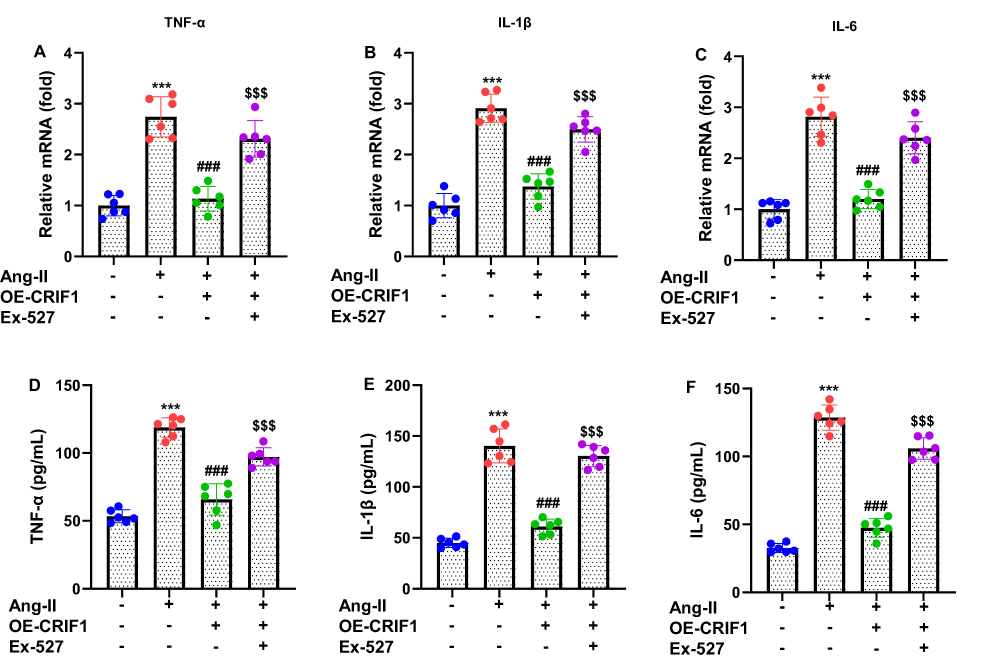 |
Figure 5 CRIF1 overexpression suppresses Ang II–induced inflammation in cardiomyocytes. (A–C) RT-qPCR was used to determine the mRNA levels of the pro-inflammatory cytokines TNF-α, IL-1β, and IL-6. (D–F) ELISA was used to measure the levels of TNF-α, IL-1β, and IL-6 in the culture medium of HL-1 cells. Data are presented as mean ± SD in triplicate. ***P<0.001 vs control group; ###P<0.001 vs Ang II+Vector group; $$$P<0.001 vs OE-CRIF1 group. |
CRIF1 Overexpression Reduces Intracellular ROS Generation and Alleviates Oxidative Stress in Ang II-Treated Cardiomyocytes
To determine whether CRIF1 modulates oxidative stress in Ang II-stimulated cardiomyocytes, intracellular ROS production was assessed using DHE staining. As expected, Ang II treatment led to a pronounced increase in ROS generation in HL-1 cells, as indicated by strong DHE fluorescence (Figure 6A). In contrast, CRIF1 overexpression markedly diminished DHE fluorescence intensity, demonstrating a substantial reduction in intracellular ROS levels. Quantitative analysis confirmed that the proportion of DHE-positive cells was significantly lower in the CRIF1-overexpressing group than in the Ang II-treated group (Figure 6B). To further evaluate oxidative stress, cell lysates were subjected to biochemical assays. Ang II stimulation significantly increased MDA levels, a marker of lipid peroxidation, whereas CRIF1 overexpression effectively suppressed this elevation (Figure 6C). Conversely, the antioxidant enzymes SOD and CAT were markedly reduced by Ang II but were significantly restored in CRIF1-overexpressing cells (Figure 6D and E). Additionally, NO production, which was reduced by Ang II exposure, was partially restored following CRIF1 overexpression (Figure 6F). Collectively, these findings indicate that CRIF1 overexpression attenuates ROS accumulation and mitigates oxidative stress in Ang II-treated cardiomyocytes, thereby highlighting its protective antioxidative role.
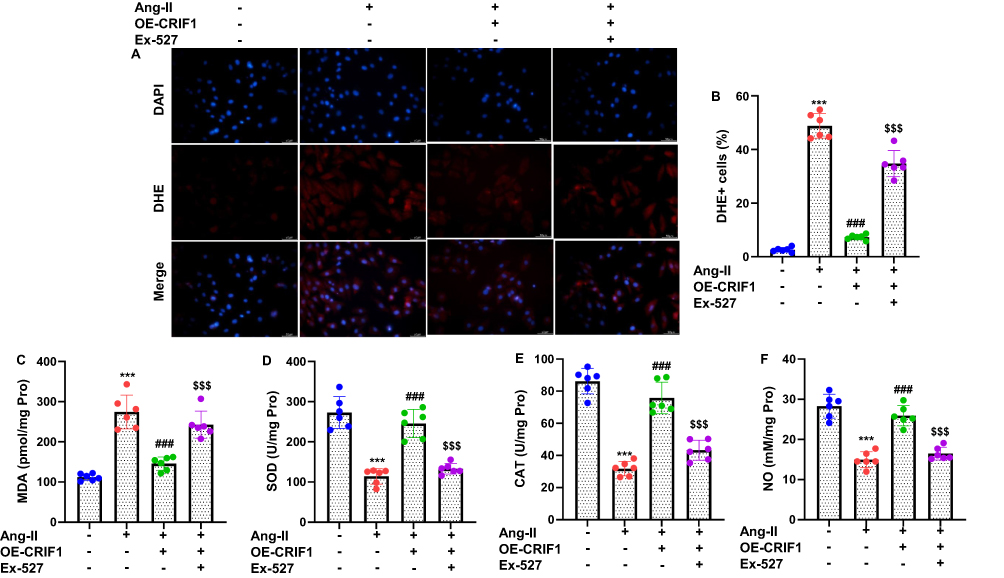 |
Figure 6 Overexpression of CRIF1 inhibits intracellular ROS generation and oxidative stress in cardiomyocytes treated with Ang II. (A) Cells were stained with DHE, and representative images of intracellular ROS are shown (200×). (B) The extent of intracellular ROS was quantified by counting DHE-positive cells (normalized to DAPI-stained cells). Cell lysates of HL-1 cells were used to detect the oxidative stress markers: (C) MDA, (D) SOD, (E) CAT, and (F) NO. Data are presented as mean ± SD in triplicate. ***P<0.001 vs control group; ###P<0.001 vs Ang II+Vector group; $$$P<0.001 vs OE-CRIF1 group. |
Discussion
In this study, we demonstrated for the first time that CRIF1 may play a protective role in Ang II–induced AF by preserving mitochondrial function, activating the SIRT1/eNOS signaling axis, and mitigating cardiomyocyte apoptosis, hypertrophy, inflammation, and oxidative stress. Our findings suggest that CRIF1 may act as a key regulator of atrial remodeling under neurohormonal stress, raising the possibility that modulating CRIF1 could contribute to AF prevention.
We found that CRIF1 expression was significantly reduced in the atrial tissue of Ang II–infused mice and in HL-1 cells treated with Ang II in a dose-dependent manner. This downregulation, in the context of pathological stress, is consistent with previous studies implicating CRIF1 in mitochondrial dysfunction and cardiovascular pathology. For example, endothelial-specific CRIF1 deletion impairs oxidative phosphorylation, increases ROS, and leads to endothelial dysfunction and cardiac injury, in part, via SIRT1 suppression.17 More broadly, CRIF1 deficiency has been linked to reduced SIRT1 expression, enhanced inflammation, and endothelial dysfunction.14 A recent study also highlighted a mechanistic axis in cardiac cells, in which CRIF1/SIRT1/eNOS signaling is disrupted, leading to reduce NO production, mitochondrial dysfunction, and increased ROS, which in turn may promote atrial neural remodeling and AF susceptibility.18 Our data extend these observations by showing that similar molecular derangements may occur in the setting of Ang II-mediated atrial stress, a clinically relevant model given the role of renin-angiotensin signaling in AF pathogenesis.
Mechanistically, our results showed that CRIF1 overexpression in HL-1 cells enhanced SIRT1 protein levels and increased eNOS phosphorylation at Ser1177, hallmarks of eNOS activation. The anti-apoptotic effect of CRIF1 on SIRT1 was supported by the reversal of protection when SIRT1 was inhibited pharmacologically with EX527. These findings align with prior data; in endothelial cells, reduced CRIF1 leads to lower SIRT1, increased ROS, and inflammation, whereas pharmacological activation of SIRT1 rescues eNOS activity and improves cellular function.18 The relevance of SIRT1 to AF is underscored by data showing that Sirt1 deficiency in atrial tissue may contribute to age-related AF via necroptosis through enhanced acetylation of RIPK1 and activation of MLKL.22 Thus, by maintaining SIRT1 levels, CRIF1 may help suppress pathological cell death and preserve atrial structural integrity.
In addition to potentially preventing apoptosis, CRIF1 overexpression reduced Ang II–induced cardiomyocyte hypertrophy, as evidenced by smaller cell size and downregulation of hypertrophic markers (ANP, BNP, and β-MHC). This anti-hypertrophic effect may be linked to the preservation of mitochondrial function and downstream SIRT1 signaling, both of which have been implicated in limiting pathological remodeling. Inflammation is a well-established driver of AF. We observed that CRIF1 overexpression attenuated Ang II–induced upregulation and secretion of proinflammatory cytokines (TNF-α, IL-1β, and IL-6). These data are consistent with vascular studies showing that CRIF1 deficiency activates nuclear factor-κB and increases the production of inflammatory mediators, whereas SIRT1 restoration suppresses these responses.23 Moreover, by reducing ROS accumulation (measured by DHE), decreasing MDA levels, and restoring antioxidant enzyme activity (SOD and catalase), CRIF1 overexpression may preserve redox homeostasis in cardiomyocytes. Restoration of NO production further supports functional recovery of eNOS signaling. Taken together, these combined anti-apoptotic, anti-hypertrophic, anti-inflammatory, and antioxidative effects suggest a central integrative role of CRIF1 in mitigating Ang II-mediated atrial pathology.16,17,24
AF remains a major unmet clinical challenge, particularly when driven by activation of the renin-angiotensin system. Current therapies, including antiarrhythmic drugs and ablation, do not directly target the molecular and mitochondrial underpinnings of atrial remodeling. Our findings indicate that CRIF1 may act as an upstream regulator of pathways critical to mitochondrial health, cell survival, and atrial structural integrity. Interventions that enhance CRIF1 expression or preserve its function may therefore provide a preventive strategy to protect against Ang II–induced atrial remodeling and reduce AF susceptibility. Furthermore, given the centrality of SIRT1 in mediating CRIF1’s protective effects, these data are in line with the interest in SIRT1 activators (eg, resveratrol) as potential cardioprotective agents. SIRT1 activation has shown benefits in various cardiac disease models, including diabetic cardiomyopathy, hypertrophy, and ischemia-reperfusion injury, by improving mitochondrial function, reducing ROS levels, and inhibiting fibrosis.25,26
Collectively, our data suggest that CRIF1 may act as an integrative regulator in the context of Ang II–induced atrial remodeling. By enhancing SIRT1/eNOS signaling, preserving mitochondrial function, and potentially attenuating apoptosis, hypertrophy, inflammation, and oxidative stress, CRIF1 appears to contribute to maintaining atrial structural and functional integrity under neurohormonal stress. These findings support the hypothesis that targeting CRIF1 or modulating SIRT1 activity could provide a novel approach for preventing AF, although further studies, including tissue-specific and long-term in vivo models, are required to confirm these protective effects and clarify their translational relevance.
Limitations
Our study has several limitations. First, although HL-1 cells are a well-established in vitro cardiomyocyte model, they may not fully recapitulate the phenotype of primary atrial cardiomyocytes or account for interactions with non-myocyte cell populations (eg, fibroblasts, immune cells, and endothelial cells) that contribute to atrial remodeling in vivo. Future studies using primary atrial cardiomyocytes or genetically modified mice with cardiomyocyte-specific CRIF1 overexpression would strengthen its translational relevance. Second, while our focus was on SIRT1/eNOS, CRIF1 may influence other signaling pathways that have not yet been explored. For instance, SIRT1 modulates mitochondrial dynamics (via PGC-1α and DRP1) in various contexts. In high-altitude cardiac dysfunction, SIRT1 inhibits mitochondrial fission via the PGC-1α-DRP1/MFF/FIS1 pathway.27 Investigating whether CRIF1 interacts with these pathways in atrial cardiomyocytes could yield additional mechanistic insight. Third, the in vivo model utilized systemic Ang II infusion, which not only increased atrial stress, but also affected blood pressure and vascular function. Distinguishing the direct cardiac effects of CRIF1 from systemic hemodynamic effects is important. Conditional, tissue-specific CRIF1-overexpression or loss-of-function models may help clarify these distinctions. Finally, only male rats were included in the present study. Sex-related differences in hormonal regulation may influence cardiovascular remodeling, oxidative stress responses, and susceptibility to atrial fibrillation. Therefore, future studies should incorporate female animals to determine whether CRIF1-mediated cardioprotective effects exhibit sex-specific differences.
Conclusion
In summary, our findings suggest that CRIF1 may play a protective role against Ang II–induced atrial remodeling and increased susceptibility to atrial fibrillation by activating SIRT1/eNOS signaling, preserving mitochondrial function, and potentially mitigating apoptosis, hypertrophy, inflammation, and oxidative stress (Figure 7). These results highlight the possibility that CRIF1 could serve as a molecular target for preventive strategies against AF, providing a foundation for future studies to further validate its therapeutic potential.
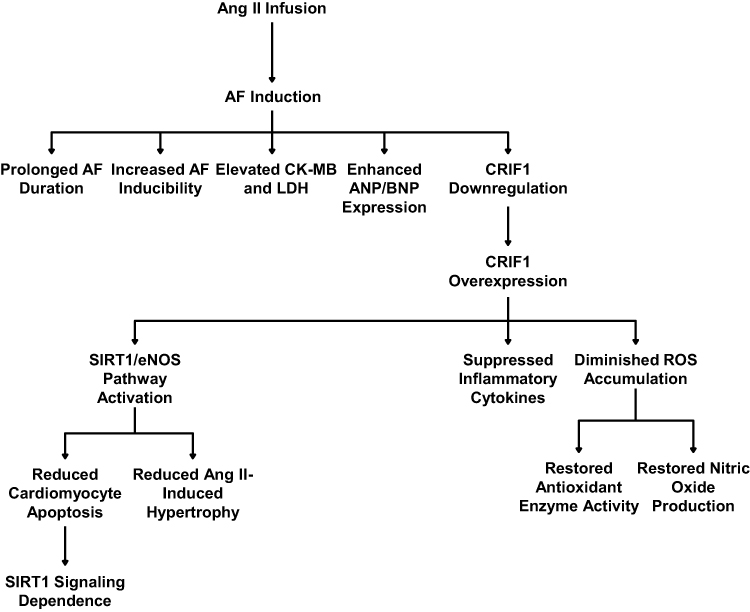 |
Figure 7 Schematic diagram illustrating the role of CRIF1 in angiotensin II–induced atrial remodeling and the protective effects of its overexpression. Chronic angiotensin II (Ang II) infusion, combined with atrial fibrillation (AF) induction, leads to increased AF duration and inducibility, elevated markers of cardiac injury (CK-MB, LDH) and hypertrophy (ANP/BNP). This pathological state is associated with enhanced CRIF1 expression. Overexpression of CRIF1 activates the SIRT1/eNOS signaling pathway, which mediates cardioprotective effects. These effects include suppression of inflammatory cytokines, diminished reactive oxygen species (ROS) accumulation, restoration of antioxidant enzyme activity, and restoration of nitric oxide production. Consequently, CRIF1 overexpression reduces cardiotoxicity, apoptosis, and Ang II–induced hypertrophy. These beneficial effects are dependent on SIRT1 signaling. |
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
This study was approved (2025-C-242-E01) by Shanghai Pudong New Area Zhoupu Hospital (Zhoupu Hospital affiliated to Shanghai Medical College of Health) Ethics Committee. The authors envisaged all standard protocols in accordance with the 1964 Declaration of Helsinki. This study was conducted and reported in accordance with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines to ensure rigorous and transparent reporting of animal research.
Author Contributions
Luoning Zhu: Conceptualization, Methodology, Data curation, Investigation, and Writing - original draft. Yuying Gu and Zhongping Ning: Project administration, Supervision, Funding acquisition, Visualization, Validation, Resources, and Writing - review & editing. All authors took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was financially supported by 1. Key Discipline Group of Pudong New Area Health Commission (PWZxq2022-11); 2. Shanghai Key Discipline Construction Cardiovascular Disease Discipline (2024ZDXK0019); 3. Pudong New Area Health Commission New Quality Specialty (Specialized Disease) (2024-PWXZ-04).
Disclosure
During paper preparation, the AI tool ChatGPT (GPT-5) was used to assist with language editing and to enhance the clarity and readability of the text. The authors declare no conflicts of interest in this study.
References
1. Bordignon S, Chiara Corti M, Bilato C. Atrial fibrillation associated with heart failure, stroke and mortality. J Atr Fibrillation. 2012;5(1):467. doi:10.4022/jafib.467
2. Youn JY, Zhang J, Zhang Y, et al. Oxidative stress in atrial fibrillation: an emerging role of NADPH oxidase. J Mol Cell Cardiol. 2013;62:72–14. doi:10.1016/j.yjmcc.2013.04.019
3. Sygitowicz G, Maciejak-Jastrzębska A, Sitkiewicz D. A review of the molecular mechanisms underlying cardiac fibrosis and atrial fibrillation. J Clin Med. 2021;10(19):4430. doi:10.3390/jcm10194430
4. Karakasis P, Theofilis P, Vlachakis PK, et al. Atrial fibrosis in atrial fibrillation: mechanistic insights, diagnostic challenges, and emerging therapeutic targets. Int J Mol Sci. 2024;26(1):209. doi:10.3390/ijms26010209
5. Zheng L, Jia X, Zhang C, et al. Angiotensin II in atrial structural remodeling: the role of Ang II/JAK/STAT3 signaling pathway. Am J Transl Res. 2015;7(6):1021–1031.
6. Hou A, Shi D, Huang H, Liu Y, Zhang Y. Inflammation pathways as therapeutic targets in angiotensin II induced atrial fibrillation. Front Pharmacol. 2025;16:1515864. doi:10.3389/fphar.2025.1515864
7. Madamanchi NR, Runge MS. Redox signaling in cardiovascular health and disease. Free Radic Biol Med. 2013;61:473–501. doi:10.1016/j.freeradbiomed.2013.04.001
8. Wallace DC. Mitochondrial genetic medicine. Nat Genet. 2018;50(12):1642–1649. doi:10.1038/s41588-018-0264-z
9. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94(3):909–950. doi:10.1152/physrev.00026.2013
10. Kim SJ, Kwon MC, Ryu MJ, et al. CRIF1 is essential for the synthesis and insertion of oxidative phosphorylation polypeptides in the mammalian mitochondrial membrane. Cell Metab. 2012;16(2):274–283. doi:10.1016/j.cmet.2012.06.012
11. Jiang Y, Xiang Y, Lin C, et al. Multifunctions of CRIF1 in cancers and mitochondrial dysfunction. Front Oncol. 2022;12:1009948. doi:10.3389/fonc.2022.1009948
12. Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24(8):464–471. doi:10.1016/j.tcb.2014.04.002
13. Man AWC, Li H, Xia N. The role of sirtuin1 in regulating endothelial function, arterial remodeling and vascular aging. Front Physiol. 2019;10:1173. doi:10.3389/fphys.2019.01173
14. Nagar H, Jung SB, Ryu MJ, et al. CR6-interacting factor 1 deficiency impairs vascular function by inhibiting the Sirt1-endothelial nitric oxide synthase pathway. Antioxid Redox Signal. 2017;27(4):234–249. doi:10.1089/ars.2016.6719
15. Lee I, Kim S, Nagar H, et al. CR6-interacting factor 1 deficiency reduces endothelial nitric oxide synthase activity by inhibiting biosynthesis of tetrahydrobiopterin. Sci Rep. 2020;10(1):842. doi:10.1038/s41598-020-57673-9
16. Piao S, Lee JW, Nagar H, et al. CR6 interacting factor 1 deficiency promotes endothelial inflammation by SIRT1 downregulation. PLoS One. 2018;13(2):e0192693. doi:10.1371/journal.pone.0192693
17. Piao S, Lee I, Jin SA, et al. SIRT1 activation attenuates the cardiac dysfunction induced by endothelial cell-specific deletion of CRIF1. Biomedicines. 2021;9(1):52. doi:10.3390/biomedicines9010052
18. Zhang A, Li H, Song Q, et al. High-fat stimulation induces atrial neural remodeling by reducing NO production via the CRIF1/eNOS/P21 axis. Lipids Health Dis. 2023;22(1):189. doi:10.1186/s12944-023-01952-7
19. Lu H, Howatt DA, Balakrishnan A, et al. Subcutaneous angiotensin II infusion using osmotic pumps induces aortic aneurysms in mice. J Vis Exp. 2015;103:53191.
20. da Silva Pinheiro P, Vilar-Pereira G, Castaño-Barrios L, et al. Benznidazole therapy improves pressure overload and cardiac electrical profile in an experimental model of Angiotensin II infusion-induced hypertension: mechanistic insights. PLoS One. 2026;21(1):e0340280. doi:10.1371/journal.pone.0340280
21. Zhan Y, Abe I, Nakagawa M, et al. A traditional herbal medicine rikkunshito prevents angiotensin II-Induced atrial fibrosis and fibrillation. J Cardiol. 2020;76(6):626–635. doi:10.1016/j.jjcc.2020.07.001
22. Jin X, Zhang Y, Zhou Y, et al. Sirt1 deficiency promotes age-related AF through enhancing atrial necroptosis by activation of RIPK1 acetylation. Circ Arrhythm Electrophysiol. 2024;17(7):e012452. doi:10.1161/CIRCEP.123.012452
23. Di Bartolo BA, Straub AC. Highlights from vascular discovery: from genes to medicine scientific sessions 2018. J Am Heart Assoc. 2018;7(15):e009470. doi:10.1161/JAHA.118.009470
24. Zablocki D, Sadoshima J. Angiotensin II and oxidative stress in the failing heart. Antioxid Redox Signal. 2013;19(10):1095–1109. doi:10.1089/ars.2012.4588
25. Ma S, Feng J, Zhang R, et al. SIRT1 activation by resveratrol alleviates cardiac dysfunction via mitochondrial regulation in diabetic cardiomyopathy mice. Oxid Med Cell Longev. 2017;2017(1):4602715. doi:10.1155/2017/4602715
26. Wang Y, Zhao R, Wu C, et al. Activation of the sirtuin silent information regulator 1 pathway inhibits pathological myocardial remodeling. Front Pharmacol. 2023;14:1111320. doi:10.3389/fphar.2023.1111320
27. Xu H, Song X, Zhang X, et al. SIRT1 regulates mitochondrial fission to alleviate high altitude hypoxia inducedcardiac dysfunction in rats via the PGC-1α-DRP1/FIS1/MFF pathway. Apoptosis. 2024;29(9–10):1663–1678. doi:10.1007/s10495-024-01954-5
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Bilirubin Elevation During Hospitalization Post Radiofrequency Catheter Ablation of Persistent Atrial Fibrillation: Variation Trend, Related Factors, and Relevance to 1-Year Recurrence
Shao JM, Shen B, Zhou ZX, D'Angelo L, James SM, Lin JF, Zheng C
Clinical Interventions in Aging 2024, 19:817-825
Published Date: 13 May 2024