")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
Prostanoid therapies in the management of pulmonary arterial hypertension
Authors LeVarge B
Received 29 September 2014
Accepted for publication 9 January 2015
Published 31 March 2015 Volume 2015:11 Pages 535—547
DOI https://doi.org/10.2147/TCRM.S75122
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Barbara L LeVarge
Department of Pulmonary and Critical Care Medicine, Beth Israel Deaconess Medical Center, Boston, MA, USA
Abstract: Prostacyclin is an endogenous eicosanoid produced by endothelial cells; through actions on vascular smooth-muscle cells, it promotes vasodilation. Pulmonary arterial hypertension (PAH) is characterized by elevated mean pulmonary artery pressure due to a high pulmonary vascular resistance state. A relative decrease in prostacyclin presence has been associated with PAH; this pathway has thus become a therapeutic target. Epoprostenol, the synthetic equivalent of prostacyclin, was first utilized as short-term or bridging therapy in the 1980s. Further refinement of its long-term use via continuous intravenous infusion followed. A randomized controlled trial by Barst et al in 1996 demonstrated functional, hemodynamic, and mortality benefits of epoprostenol use. This work was a groundbreaking achievement in the management of PAH and initiated a wave of research that markedly altered the dismal prognosis previously associated with PAH. Analogs of prostacyclin, including iloprost and treprostinil, exhibit increased stability and allow for an extended array of parenteral and non-parenteral (inhaled and oral) therapeutic options. This review further examines the pharmacology and clinical use of epoprostenol and its analogs in PAH.
Keywords: pulmonary arterial hypertension, prostacyclin analogs, epoprostenol, treprostinil, iloprost
Introduction
Pulmonary arterial hypertension (PAH) describes a particular subset of pulmonary hypertension fitting within Group 1 of the World Health Organization (WHO) classification system. Hemodynamically, patients exhibit elevation in mean pulmonary artery pressure (mPAP) of ≥25 mmHg – with normal pulmonary artery wedge pressure (PAWP) (≤15 mmHg), and elevation in pulmonary vascular resistance (PVR) of >3 Wood units.1 On a histologic level, PAH is characterized by abnormal remodeling of the pulmonary vasculature that may be idiopathic, heritable, or related to certain disease processes including connective tissue disease, congenital heart disease, portal hypertension, and drug exposure. Significant left heart, pulmonary parenchymal, and/or chronic thromboembolic disease is absent; patients with pulmonary hypertension of these types fall into separate WHO groups. Similarities in clinical manifestations and hemodynamics may exist between patients with PAH and other forms of pulmonary hypertension; however all prostaglandin-related therapies, as discussed in this review, are currently approved only for patients with WHO Group 1 PAH.
Prior to the introduction of advanced therapies, survival in PAH was poor. Early registry data following idiopathic PAH patients demonstrated overall survival of 2.8 years, with median 6-month survival in those with functional class IV disease (severe functional limitations and symptoms with any activity).2 Over the last 20 years three major classes of PAH therapies have emerged, with clear impact on the morbidity and mortality of the disease. Current registry data now supports 1- and 3-year survival of 91% and 74%, respectively, for patients with idiopathic or heritable PAH.3 While only one individual randomized controlled trial has shown a mortality benefit,4 meta-analyses have supported a statistically significant improvement in survival with the use of PAH-specific therapy.5,6
Choice of initial therapy for PAH depends on several factors, most importantly the WHO functional class of the patient. Similar to the New York Heart Association classification for congestive heart failure, WHO functional classes are graded I to IV, from most mild to most severe limitations. Recommended treatment algorithms from the Fifth World Symposium on Pulmonary Hypertension support initiating an oral agent for those with mild (WHO functional class II) symptoms. Options include phosphodiesterase type-5 inhibitors (PDE-5Is: sildenafil, tadalafil), the related soluble guanylate-cyclase stimulator riociguat, and the endothelin-receptor antagonists (ERAs: bosentan, ambrisentan, and macitentan).7
For patients with moderate to severe limitation (WHO functional class III or IV disease), prostacyclin analogs are often a key part of the treatment regimen. In the USA and Europe, options include intravenous epoprostenol, inhaled iloprost, and treprostinil, which can be administered via subcutaneous, intravenous, inhaled, and, most recently, oral routes. Combining data from all prostanoid trials, a recent meta-analysis supported improvements in mortality, clinical worsening, exercise capacity, and hemodynamics (Figure 1).8
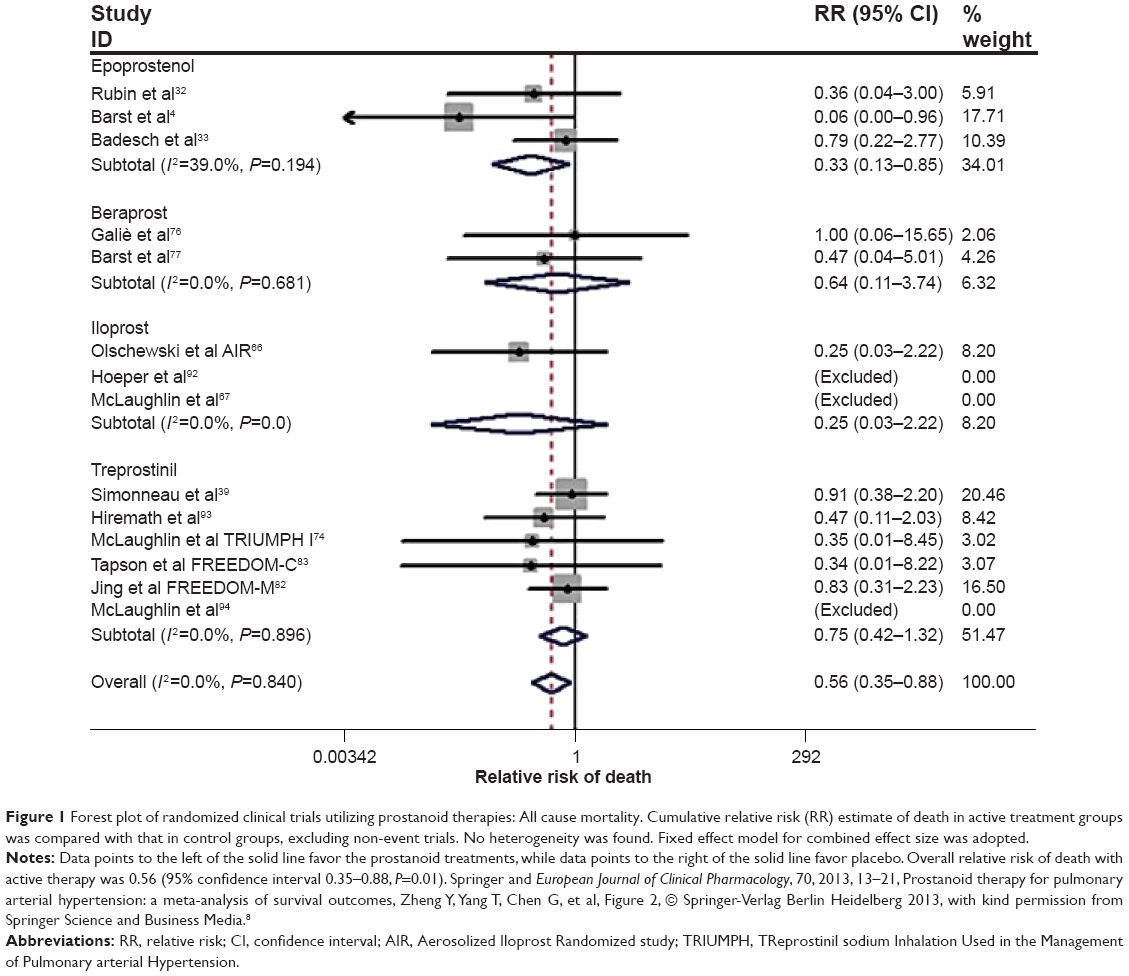 | Figure 1 Forest plot of randomized clinical trials utilizing prostanoid therapies: All cause mortality. Cumulative relative risk (RR) estimate of death in active treatment groups was compared with that in control groups, excluding non-event trials. No heterogeneity was found. Fixed effect model for combined effect size was adopted. |
Prostanoid therapy in pulmonary arterial hypertension
Prostacyclin, or prostaglandin I2 (IP), is an endogenous eicosanoid produced by endothelial cells. Epoprostenol is the synthetic equivalent of prostacyclin, and treprostinil and iloprost are both stable synthetic analogs.
Deficiency of prostacyclin activity has long been identified as an important part of the pathobiology of PAH. Christman et al demonstrated decreased urinary excretion of prostacyclin metabolites in 34 patients with idiopathic or secondary pulmonary hypertension as compared with controls.9 Loss of expression of prostacyclin synthase has also been observed in lung tissue of patients with PAH.10
The primary target of prostacyclin and its analogs appears to be the IP receptor on vascular smooth-muscle cells. Activation of other vasodilatory prostaglandin receptors, such as EP2 and EP4 (prostaglandin E2 and E4 receptors, respectively), has also been observed, although this is of unclear importance.11,12 After prostacyclin binds target receptors on smooth-muscle cells, intracellular signaling leads to adenylate cyclase activation and an increase in cyclic adenosine monophosphate levels. Resulting smooth-muscle relaxation with vasodilation is one of the primary actions of prostacyclin. However, the important effects of the prostanoids expand well beyond pure vasodilation, targeting the pathologic vascular remodeling observed in PAH. Treprostinil and iloprost have been shown to suppress pulmonary artery smooth-muscle proliferation in vitro.13,14 Additional prostanoid effects include inhibition of platelet aggregation,15 inhibition of inflammation,16,17 and augmentation of ventricular inotropy.18
The pharmacokinetics, dosing, efficacy, and pertinent adverse effects for each agent are discussed following. Clinical efficacy data is further summarized in Table 1.
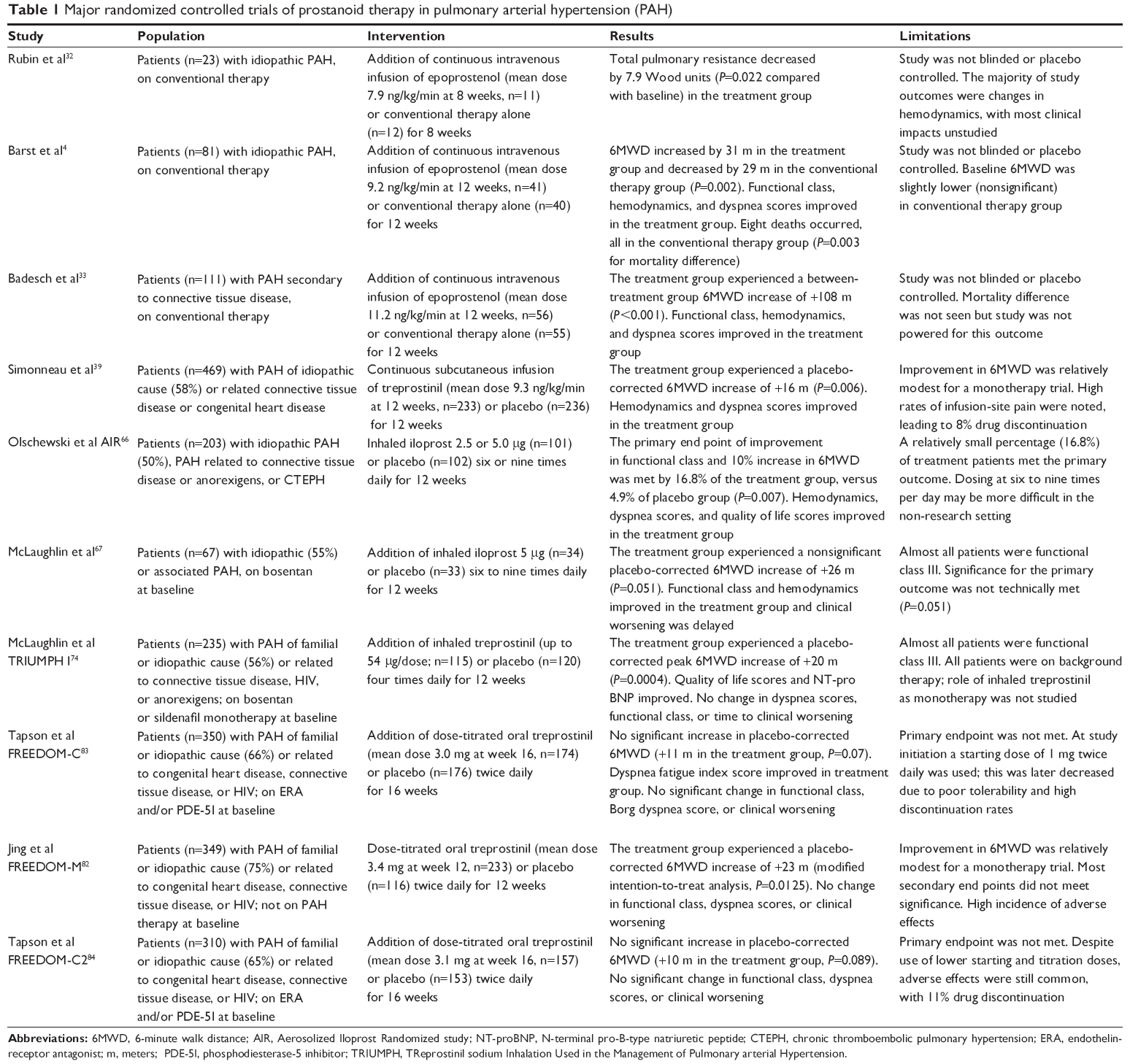 | Table 1 Major randomized controlled trials of prostanoid therapy in pulmonary arterial hypertension (PAH) |
Intravenous epoprostenol
Epoprostenol, the synthetically produced formulation of prostacyclin, was the first medication to be approved for the treatment of PAH by the US Food and Drug Administration (FDA) in 1995. Epoprostenol sodium for intravenous therapy is marketed as Flolan® (GlaxoSmithKline plc, London, UK) and is also available generic (Teva Pharmaceutical Industries Ltd, Petah Tikva, Israel). Since 2008 a room-temperature stable (RTS) formulation of epoprostenol (Veletri®, Actelion Pharmaceuticals Ltd, Allschwil, Switzerland) has also been available.
Pharmacokinetics
The in vivo half-life of epoprostenol is less than 6 minutes, thus necessitating drug delivery by continuous intravenous infusion. Epoprostenol is rapidly broken down, spontaneously or enzymatically, into two very weakly active primary metabolites. Metabolites are primarily excreted in the urine.19,20
Once infused, traditional and RTS preparations of epoprostenol are regarded as equivalent from biologic and efficacy standpoints.21–23
Dosing and administration
Epoprostenol is administered by continuous intravenous infusion using an ambulatory, battery-operated infusion pump. The drug is supplied as a sterile powder (0.5 or 1.5 mg per vial) that must be reconstituted and diluted to the desired concentration with a sterile diluent specific to epoprostenol. At room temperature, reconstituted epoprostenol loses stability and cannot be used for longer than 8 hours. Thus, epoprostenol for injection is administered with cold pouches to maintain solution temperature at 2°C to 8°C, extending use time to 24 hours. An extended-stability RTS epoprostenol has been subsequently introduced; this formulation combines epoprostenol with arginine (as opposed to glycine as in traditional epoprostenol), resulting in a more alkaline pH after reconstitution and consequently greater RTS.19,20 RTS epoprostenol must be reconstituted in sterile water or sodium chloride; following reconstitution and dilution it is stable at room temperature for 48 hours.20
Intravenous epoprostenol is initiated in a monitored hospital setting, with a recommended starting dose of 2 ng/kg/min and titration rates variable according to disease severity and side effects. Long-term dosing goals are similarly individualized. In one observational study of 178 patients started on intravenous epoprostenol, as an example, mean doses at 3 months and 1 year were 14 and 21 ng/kg/min, respectively.24
Clinical efficacy
The first published experience with epoprostenol in PAH occurred in 1980; an 8-year-old female with severe idiopathic PAH was administered prostacyclin with pulmonary artery catheter in place. Doses up to 44 ng/kg/min reduced mPAP and PVR and improved cardiac index.25 Previous to this, successful use of prostacyclin in a neonate with severe hypoxemia and persistent fetal circulation had been described.26 Further series of acute use in adults with PAH followed.27–29
Early adoption of epoprostenol as a therapy was limited by administration challenges. Higenbottam et al first described a 27-year-old female with idiopathic PAH and severe symptoms who noted significant improvement in symptoms and hemodynamics with acute epoprostenol while hospitalized.30 A tunneled catheter was placed with delivery of epoprostenol by electric infusion pump, and she was able to be discharged home with great improvement in quality of life. The further courses of this patient and nine others were described separately.31 These patients received long-term (1–25 months) epoprostenol infusions, with demonstrated improvements in exercise capacity and symptoms.
In the first randomized trial of continuous intravenous epoprostenol, Rubin et al randomized 23 patients to receive epoprostenol or conventional therapy.32 After 8 weeks, total pulmonary resistance decreased in the epoprostenol group (21.7 Wood units at trial start, to 13.9 Wood units at trial end), with no significant change seen in the conventional therapy group. Functional class improvements were also more frequent in the epoprostenol group, and there were three deaths among those receiving conventional therapy versus one death in those receiving epoprostenol.32
The landmark trial of epoprostenol in PAH randomized 81 patients with functional class III or IV idiopathic PAH to epoprostenol plus conventional therapy or conventional therapy alone.4 At 12 weeks, 6-minute walk distance (6MWD) improved by 31 meters in the epoprostenol group, and decreased by 29 meters in the conventional therapy group. There was a between-treatment decrease in mPAP (−6.7 mmHg) and PVR (−4.9 Wood units) and improvement in cardiac index in favor of epoprostenol. Benefits on functional class, dyspnea scores, and quality of life scores were also noted. Eight patients died in the conventional therapy group, versus none in the epoprostenol group (P=0.003).4 A similar randomized trial in 111 patients with scleroderma-related PAH showed that epoprostenol has positive impact on 6MWD, hemodynamics, and functional class in this population as well.33
Long-term data have also supported the benefits of intravenous epoprostenol. McLaughlin et al followed 162 patients with idiopathic PAH on epoprostenol for a mean of 36 months. Survival at 1, 2, and 3 years was 88%, 76%, and 63%, respectively, which was above expected survival based on registry data (59%, 46%, and 35%, respectively, P<0.001 for each comparison).2,34 A separate long-term study of 178 treated idiopathic PAH patients demonstrated improved mortality compared with controls.24
Safety and tolerability
Adverse effects associated with intravenous epoprostenol include flushing, headache, jaw pain (typically described with the first bite of food), nausea/vomiting, diarrhea, and flu-like symptoms.19,20 During initiation and dose escalation, the acute development of pulmonary edema suggests an underlying diagnosis of pulmonary veno-occlusive disease and can be fatal.24
The presence of a chronic indwelling catheter predisposes patients to additional risk, including infection and thrombosis. From the 12-week randomized study treating 41 patients with epoprostenol, there were four episodes of catheter-related sepsis and one episode of thrombosis with paradoxical embolus.4 Long-term observational work has found catheter-related infection/sepsis rates of 0.14 to 0.19 per patient-year.24,34 Drug interruption due to pump or catheter malfunction is another serious concern – due to rebound severe pulmonary hypertension and short drug half-life, this complication can be fatal if not immediately recognized and managed.34,35
Subcutaneous and intravenous treprostinil
The poor stability and short plasma half-life of epoprostenol stimulated interest in developing more stable analogs. Treprostinil, a benzindine prostanoid, was initially studied for potential use in congestive heart failure,36 transplantation,37 and peripheral vascular disease.38 Support for treprostinil’s utility in PAH was sealed with a large subcutaneous infusion clinical trial.39
Parenteral treprostinil (Remodulin®, United Therapeutics Corporation, Silver Spring, MD, USA) is supplied for administration via subcutaneous or intravenous route. Subcutaneous treprostinil was approved by the FDA in 2002; with scientific support for bioequivalence, intravenous use of treprostinil was subsequently approved in 2004.
Pharmacokinetics
Subcutaneous treprostinil is rapidly absorbed with complete bioavailability.40,41 As a result of this documented bioequivalence, from an efficacy standpoint both routes of administration are considered similar, though important differences in risks and adverse effects remain. The relationship between infused dose and measured plasma concentration is linear in healthy volunteers42 and in PAH patients treated intravenously or subcutaneously over a dosing range of 10 to 125 ng/kg/min.43
With chronic intravenous or subcutaneous therapy, the plasma half-life of treprostinil has ranged from 2.9 to 4.6 hours, significantly longer than that of epoprostenol.41,42 Metabolism of treprostinil occurs primarily in the liver by CYP2C8, with the majority of metabolites inactive and excreted in the urine. Medication interactions with cytochrome P450 inhibitors or inducers should be noted. Rifampin, a CYPC28 inducer, decreases exposure to treprostinil, while the inhibitor gemfibrozil increases exposure.44
Dosing and administration
Treprostinil sodium is supplied in vials containing four possible drug concentrations, from 1 to 10 mg/mL. For patients using subcutaneous therapy, treprostinil is continuously administered undiluted via a self-inserted subcutaneous catheter with an appropriate infusion pump. Undiluted treprostinil from a single syringe is used for up to 72 hours. For patients using intravenous therapy, treprostinil is diluted in sterile water, saline, or epoprostenol diluent, with administration similar to that of epoprostenol solution. After dilution, intravenous treprostinil solution at room temperature can be administered for up to 48 hours.44
Parenteral treprostinil is initiated at a dose of 1.25 ng/kg/min, with dose escalation dependent on efficacy and side effects. For subcutaneous treprostinil, a rapid escalation plan (starting dose of 2.5 ng/kg/min, with increases of 2.5 ng/kg/min twice in the first week and then weekly or biweekly thereafter) led to greater improvements in exercise capacity at 12 weeks and actually decreased infusion-site pain.45 Of patients involved in randomized clinical trials of subcutaneous treprostinil, those remaining on therapy at 1 and 3 years were treated with a mean dose of 26 and 42 ng/kg/min, respectively.46 Escalation of intravenous treprostinil dosing can typically be achieved more rapidly. In 16 treatment-naïve patients initiating intravenous treprostinil, the mean dose was 41 ng/kg/min at 12 weeks, and 98 ng/kg/min at 48 weeks.47
A novel, completely implantable delivery system for intravenous treprostinil has been under investigation, with prospects of improving patient convenience and safety.48,49
Clinical efficacy
The pivotal study of subcutaneous treprostinil in PAH was reported by Simonneau et al in 2002.39 A total of 470 patients from 40 international centers were randomized to receive subcutaneous treprostinil, starting at a dose of 1.25 ng/kg/min, or infused placebo. The primary outcome was 6MWD at 12 weeks, with secondary outcomes including signs and symptoms of pulmonary hypertension, hemodynamics, dyspnea scores, and quality of life measures. After 12 weeks, the median between-treatment difference in 6MWD was 16 meters, in favor of treprostinil (95% confidence interval 4.4–27.6 meters, P=0.006). The mean treprostinil dose was 9.3 ng/kg/min, and there was a significant relationship between achieved treprostinil dose and improvement in 6MWD (those in the highest quartile of treprostinil dosing had a median 6MWD improvement of 36 meters). Secondary outcomes were also in favor of treprostinil.39 Further analysis of the subset with connective tissue disease-associated PAH also confirmed benefits in this population.50
An open-label study described outcomes of 860 PAH patients (including subjects from the randomized clinical trial) treated long-term with subcutaneous treprostinil. Survival at 1, 2, 3, and 4 years was 87%, 78%, 71%, and 68%, respectively, which was well above anticipated survival rates from historical data. Fifteen percent of patients had other PAH-specific agents added during the study period.46 Another long-term study of 99 patients with PAH and 23 with chronic thromboembolic pulmonary hypertension (CTEPH) using subcutaneous treprostinil reported similar survival – 89% at 1 year and 71% at 3 years. Significant improvements in 6MWD (305–445 meters, P=0.0001), dyspnea scores, and functional class were noted.51
Efficacy of intravenous treprostinil has also been independently supported by clinical studies. Tapson et al treated 16 PAH patients with open-label intravenous treprostinil (mean dose 41 ng/kg/min at end of study) over 12 weeks, with noted improvements in 6MWD, dyspnea scores, and hemodynamics.52 Subjects were then followed on therapy for a 48-week period; further improvements in exercise capacity and hemodynamics were demonstrated. In the same study an additional 31 patients were transitioned from intravenous epoprostenol (mean baseline epoprostenol dose 41 ng/kg/min), of whom 23 remained on treprostinil at 1 year (mean treprostinil dose 111 ng/kg/min). Exercise parameters and hemodynamics were similar at 48 weeks compared with baseline on epoprostenol.47
Safety and tolerability
Diarrhea, jaw pain, flushing, and edema were all more common with treprostinil in comparison to placebo.39 Delivery system problems (pump malfunction or infusion-set complications) occurred in 30% during the long-term subcutaneous study; no serious complications developed as a result of these.46
Infusion-site pain was common (85%, versus 27% with placebo) in patients administered subcutaneous treprostinil in the large randomized trial;39 this led to drug discontinuation in 8%. In the long-term continuation study, site pain was responsible for almost all of the drug withdrawals due to adverse events (23% of the total study population).46 Infusion-site pain and reactions can be decreased with appropriate site selection; as pain peaks in the first few days after a site change, use of a single site for 4 weeks or more can be helpful and safe. Additional topical and/or systemic therapies, including topical anesthetics and steroids, analgesics, and histamine antagonists, are often employed to improve tolerance.53
While intravenous therapy eliminates the problem of infusion-site pain, as with intravenous epoprostenol, the risks of infection and thrombosis must be considered. Bloodstream infections, with Gram-negative infections in particular, occur more commonly in patients receiving intravenous treprostinil compared with intravenous epoprostenol.54–56 Data from 1,146 patients who received intravenous prostanoid therapy enrolled in the REVEAL Registry™ were reviewed by Kitterman et al.56 Treatment with treprostinil was associated with a significant increase in bloodstream infection rates in comparison with epoprostenol (0.36 vs 0.12 infections per 1,000 treatment days, P<0.001); for Gram-negative bloodstream infections a 6.86-fold increased incidence was found.56 Diluting treprostinil with the basic pH epoprostenol diluent instead of sterile water or saline was associated with a decreased risk of Gram-negative infections in one study.57
Inhaled iloprost
Due to the complexities and risks associated with parenteral prostanoids, development of alternative modes of administration, including oral and inhaled routes, has additionally been pursued. Inhalational therapy also has the benefit of reducing systemic side effects. Success with two nebulized prostanoids, iloprost and treprostinil, was achieved. Iloprost for inhalation (Ventavis®, Actelion Pharmaceuticals Ltd) was approved by the FDA for the treatment of PAH in 2004.
Pharmacokinetics
After inhalation, an early peak in iloprost levels in the serum is seen with undetectable levels 30 to 60 minutes after 5 μg dosing.58,59 Half-life is reported to be 20 to 30 minutes.58 In one clinical study, however, serum half-life was shorter (6.5 to 9.4 minutes); the half-life of physiologic effect (PVR decrease) was considerably longer, from 21 to 25 minutes.59 The discrepancy between molecular half-life and physiologic effect is also noted with inhaled treprostinil and may suggest storage of the drug in the perivascular lung tissue.60 Duration of molecular and cellular effects after receptor binding also likely outlasts the prostanoid’s presence within the microvasculature.
Iloprost is metabolized by β-oxidation and the major metabolite is excreted primarily through the urine.58
Dosing and administration
Inhaled iloprost is introduced at a dose of 2.5 μg and up-titrated to 5 μg if well tolerated. Inhalations are recommended six to nine times daily (at least 2 hours apart), inhaled via a specific nebulizer system, the I-neb® Adaptive Aerosol Delivery (AAD®) system (Philips Healthcare, Amsterdam, the Netherlands).
Clinical efficacy
Several uncontrolled case reports and series described substantial benefits of inhaled iloprost in PAH.61–65 In the largest, a 1-year study of inhaled iloprost in 24 idiopathic PAH patients, significant improvements in 6MWD (278 to 363 meters) and hemodynamics (mPAP 59 to 52 mmHg, with cardiac output improvement) were noted.65
Positive uncontrolled data led the way for the first placebo-controlled study of inhaled iloprost, Aerosolized Iloprost Randomized (AIR). AIR studied the efficacy of iloprost (2.5 or 5.0 μg per dose, six to nine times daily, median daily dose 30 μg) versus inhaled placebo in 203 patients with PAH or inoperable CTEPH. The primary endpoint, requiring a 10% increase in 6MWD, improvement in functional class, and absence of clinical deterioration, occurred in 16.8% versus 4.9% of the iloprost and placebo groups, respectively (P=0.007).66 A subsequent randomized controlled trial using iloprost as add-on therapy to bosentan demonstrated near-significant improvements in 6MWD (placebo-adjusted difference 26 meters, P=0.051) and improvement in functional class and hemodynamics.67 However, long-term stability on iloprost monotherapy has been questioned. In one study, only 42% of 76 patients on monotherapy were still stable on iloprost alone after 1 year.68
Safety and tolerability
Safety data from AIR suggested several relevant adverse effects, including flushing (27%), cough (39%), headache (30%), and jaw pain (12%). Serious syncopal episodes were also more common in the iloprost group (five patients [5%]).66 A 2-year, prospective, open-label study of iloprost in 63 patients with PAH showed similar findings; during the study only two patients discontinued therapy – one due to back pain, headache, and vertigo, and the other due to throat irritation.69
Inhaled treprostinil
Relative to iloprost, the half-life of treprostinil is prolonged, and inhalations of treprostinil can therefore be dosed on a less frequent basis (four times daily versus six to nine times daily). Treprostinil for oral inhalation (Tyvaso®, United Therapeutics Corporation) was approved by the FDA for the treatment of PAH in 2009.
Pharmacokinetics
Systemic concentrations of treprostinil peak within 15 minutes after inhalation, with the maximal concentration proportional to the inhaled dose.70,71 In a pilot study of PAH patients administered a single 60 μg dose of inhaled treprostinil, peak concentration (Cmax) was 1.59 ng/mL, similar to the plasma concentration in healthy subjects administered intravenous treprostinil at a dose of 15 ng/kg/min.40,71 As serum concentrations decrease steadily following inhalation (less than 0.5 ng/mL by 120 minutes),71 total drug exposure (area under the curve [AUC]) will be considerably lower with inhalational therapy than with most clinically relevant doses of subcutaneous or intravenous treprostinil. Plasma half-life, at 44 to 52 minutes, is longer than that of iloprost after inhalation.72
Similar to inhaled iloprost, plasma concentration and physiologic effect profiles are offset with inhaled treprostinil. Two studies described mPAP and PVR profiles following a single inhaled dose of treprostinil (15, 30, or 45 mcg) in PAH patients.60,72 The hemodynamic effects of treprostinil continued out to 180 minutes after inhalation, a time when serum concentrations are negligible based on the data by Voswinckel et al.71
Similar to parenteral therapy, inhaled treprostinil is metabolized predominantly in the liver by CYP2C8.70
Dosing and administration
Treprostinil for inhalation is administered using a specific ultrasonic pulsed delivery system, the Tyvaso® Inhalation System, with either the newer TD-100 or older Optineb models (United Therapeutics Corporation). Doses are administered four times daily, spaced throughout waking hours, at least 4 hours apart. At initiation, patients are delivered three breaths (18 μg) per session; this is serially increased to the full dose of nine breaths (54 μg) four times daily.
Clinical efficacy
Acute hemodynamic benefits of treprostinil inhalation were first described in three patients with PAH; two went on to be treated long-term, with benefits on functional class and 6MWD seen at 3 months.60 Positive effects of inhaled treprostinil on mPAP were also noted acutely in PAH patients on background bosentan72 or sildenafil73 therapy. Channick et al further continued the former study out to 12 weeks, with improvements in mPAP, 6MWD, and functional class also found at that time point.72
The TReprostinil sodium Inhalation Used in the Management of Pulmonary arterial Hypertension (TRIUMPH I) study randomized 235 PAH patients at 31 centers to inhaled treprostinil (escalated up to 54 μg/dose, four times daily) or placebo. The majority of subjects were functional class III and all were on background therapy with bosentan or sildenafil. After 12 weeks, the median between-treatment increase in post-dose 6MWD was 20 meters (P=0.0004). Quality of life measures and brain natriuretic peptide levels also improved compared with placebo.74 Extension data from TRIUMPH I, out to 24 months, were also presented. Improvements in 6MWD at 12 and 24 months were 31 and 18 meters, respectively. Survival in those on therapy was 97% at 12 months and 91% at 24 months.75
Safety and tolerability
Cough (54% vs 29% taking placebo in TRIUMPH I), headache (41%), nausea (19%), dizziness (19%), flushing (15%), and throat irritation (14%) were the more common side effects noted in the clinical trial. Adverse effects lead to study withdrawal in 6%.74 In the 24-month extension study, 19% discontinued treprostinil due to adverse events.75
Due to inhibition of platelet function, bleeding is a concern with all formulations of prostanoids. Most relevant to inhaled treprostinil, during an open-label study, hemoptysis occurred in three patients and was fatal in one case.70
Oral treprostinil
Potential for an oral prostacyclin derivative has long been of great interest. Investigations using oral beraprost, another prostacyclin analog, were initially encouraging, with a 25-meter improvement in 6MWD, the primary outcome, noted.76 However, a subsequent 12-month study failed to show such efficacy beyond the 6-month mark, and beraprost was never approved in the USA or Europe as a result.77
Oral treprostinil (Orenitram®, United Therapeutics Corporation) is the most recent of the prostanoid formulations to gain approval in the USA.
Pharmacokinetics
Treprostinil diolamine is provided in sustained-release tablets designed to supply the systemic circulation with a more constant concentration of the drug. In patients with PAH receiving chronic twice-daily dosing, Cmax increased with increasing doses of oral treprostinil, with time to maximum concentration within 8 hours; median 4 hours. Total exposure to treprostinil (AUC) increases linearly with increasing oral treprostinil dose.78 The oral bioavailability is approximately 17%.79 In comparison to the fasted state, Cmax and AUC are increased and time to maximum concentration delayed when the medication is ingested with a high-fat, high-calorie meal, though no significant difference in absorption kinetics was seen in healthy volunteers taking in four different meal types.80
Treprostinil is metabolized primarily by the liver, with similar metabolic pathways as in parenteral therapy. Administration to patients with mild, moderate, and severe hepatic impairment resulted in significantly increased drug exposure.81 Oral treprostinil is contraindicated in those with severe hepatic impairment.79
Dosing and administration
The recommended starting dose for oral treprostinil is 0.25 mg twice daily, 12 hours apart, with food, with dose escalations of 0.25 to 0.50 mg twice daily every 3 to 4 days. However, patients not tolerant of initial dosing or escalation can be provided with 0.125 mg tablets and can be dosed three times daily (every 8 hours).79 Similar to parenteral formulations, the maximum dose is limited by tolerability. The mean dose at 12 weeks in the monotherapy clinical trial was 3.4 mg twice daily.82
Clinical efficacy
The FREEDOM-C study investigated effect of oral treprostinil in 350 patients with PAH on background therapy with a PDE-5I, an ERA, or both, over the 16-week study period. At study initiation the starting dose of treprostinil was 1 mg twice daily with escalation of 1 mg per dose; protocol amendments subsequently allowed for lower starting doses and incremental increases due to difficulties with tolerance of side effects. Lower discontinuation rates were observed with availability of these lower doses. A nonsignificant between-treatment improvement in 6MWD of 11 meters (P=0.07) occurred in those on treprostinil; suggestion of improved efficacy in those achieving higher treprostinil doses and with access to smaller dosing increments was observed.83 Due to the described dosing and tolerance problems, FREEDOM-C2 enrolled 310 further subjects on background therapy; subjects were initiated at 0.25 mg twice daily, with incremental increases of 0.25 or 0.50 mg. Using the new protocol, the mean dose in the treatment group at 16 weeks was 3.1 mg twice daily, and difference in 6MWD between groups was again not statistically significant (10.0 meters, 95% confidence interval −2.2 to 22.0 meters, P=0.089).84
Efficacy in a population not on background therapy was studied in FREEDOM-M, a randomized, double-blind, placebo-controlled trial enrolling 349 subjects with PAH not on other advanced therapies. Subjects were WHO functional class II (33%) and III (66%) and the majority (75%) had idiopathic or heritable PAH. Similar protocol amendments were enacted to allow access to lower-dose tablets for 228 patients. In the intention-to-treat analysis, at 12 weeks a mean improvement in 6MWD of 26 meters (P=0.0001) was noted.82
Safety and tolerability
The most common noted adverse reactions in FREEDOM-M included headache (69% in the intention-to-treat population), diarrhea (37%), nausea (39%), jaw pain (25%), and flushing (21%). In subjects with access to the lower-dose tablets, 4% discontinued due to adverse effects.82
As with other prostanoid formulations, due to platelet aggregation inhibition, an increased risk of bleeding is anticipated. Specific to the oral formulation, caution should be taken in patients with diverticulosis, as the tablet shell does not dissolve and can lodge in a diverticulum.79
Transitioning between prostanoids
As the number of available PAH therapies has expanded in recent years, transitions from one line of therapy to another have become more familiar scenarios. Within prostanoid-based therapy, switching from one modality to another may be needed due to progression of symptoms (eg, change from inhaled therapy to subcutaneous or intravenous) or intolerance of mode of delivery (cough with inhaled therapy, site pain with subcutaneous, or bloodstream infections with intravenous). As newer agents have been introduced, transitions for patient safety or convenience are also recognized.
Transition from intravenous epoprostenol to parenteral treprostinil has been well described and is successful in most patients. Hospitalization is required during this change, which is typically done with stepwise increases in treprostinil combined with decreases in epoprostenol, over 1 to 2 days for change to intravenous treprostinil.44,85 Several days may be needed for conversion to subcutaneous therapy.86,87 At the end of the transition period, the treprostinil dose is typically equal to or greater than the initial epoprostenol dose and often requires further up-titration to a dosing range approximately double the starting epoprostenol dose. In one series of 31 patients making an epoprostenol to intravenous treprostinil transition, mean epoprostenol dose started at 40 ng/kg/min; at the time of hospital discharge treprostinil was administered at an average of 47 ng/kg/min, and by week 12 the mean dose was up to 83 ng/kg/min.85
Select patients on low to moderate doses of parenteral prostanoids who also display reasonable functional status and right ventricular function may be appropriate to consider for transition to inhaled prostanoid therapy. In a few small series this has been successful in many patients, though some do not tolerate such a change.88–90 In the largest, a multicenter observational study of 37 PAH patients on parenteral epoprostenol or treprostinil (mean dose 16.8 ng/kg/min), 29 (78.4%) successfully transitioned to and remained on inhaled iloprost at 90 days. Six of these patients also had changes made to their oral therapies; background use of an ERA or a PDE-5I was additionally associated with greater likelihood of persisting on inhaled therapy.89 Transitioning between inhaled therapies (iloprost and treprostinil) appears to be well tolerated and safe.91
Conclusion
Associated with deficiencies in production or action, prostacyclin has been identified as having an important role in the pathobiology of PAH. The treatment of PAH patients with epoprostenol and related molecules takes advantage of their vasodilatory properties; antiproliferative and other secondary actions of these prostanoids are also noteworthy and gaining increasing attention. In clinical trials, the prostanoid agents – namely epoprostenol, treprostinil, and iloprost delivered via intravenous, subcutaneous, inhaled, and now oral routes – confer benefits in function, morbidity, and mortality to PAH patients. Much about the use of these agents, from analog selection to route of delivery to dose titration, is highly individualized and requires experience and understanding on the part of both the patient and the provider. Particularly for patients with the most severe disease, prostanoid-based therapy remains a critical component of the optimal management of PAH.
Disclosure
The author declares no conflicts of interest in this work.
References
Hoeper MM, Bogaard HJ, Condliffe R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D42–D50. | ||
D’Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115(5):343–349. | ||
Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, McGoon MD. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest. 2012;142(2):448–456. | ||
Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996;334(5):296–301. | ||
Galiè N, Manes A, Negro L, Palazzini M, Bacchi-Reggiani ML, Branzi A. A meta-analysis of randomized controlled trials in pulmonary arterial hypertension. Eur Heart J. 2009;30(4):394–403. | ||
Ryerson CJ, Nayar S, Swiston JR, Sin DD. Pharmacotherapy in pulmonary arterial hypertension: a systematic review and meta-analysis. Respir Res. 2010;11:12. | ||
Galiè N, Corris PA, Frost A, et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D60–D72. | ||
Zheng Y, Yang T, Chen G, Hu E, Gu Q, Xiong C. Prostanoid therapy for pulmonary arterial hypertension: a meta-analysis of survival outcomes. Eur J Clin Pharmacol. 2014;70(1):13–21. | ||
Christman BW, McPherson CD, Newman JH, et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327(2):70–75. | ||
Tuder RM, Cool CD, Geraci MW, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159(6):1925–1932. | ||
Whittle BJ, Silverstein AM, Mottola DM, Clapp LH. Binding and activity of the prostacyclin receptor (IP) agonists, treprostinil and iloprost, at human prostanoid receptors: treprostinil is a potent DP1 and EP2 agonist. Biochem Pharmacol. 2012;84(1):68–75. | ||
Lai YJ, Pullamsetti SS, Dony E, et al. Role of the prostanoid EP4 receptor in iloprost-mediated vasodilatation in pulmonary hypertension. Am J Respir Crit Care Med. 2008;178(2):188–196. | ||
Clapp LH, Finney P, Turcato S, Tran S, Rubin LJ, Tinker A. Differential effects of stable prostacyclin analogs on smooth muscle proliferation and cyclic AMP generation in human pulmonary artery. Am J Respir Cell Mol Biol. 2002;26(2):194–201. | ||
Falcetti E, Hall SM, Phillips PG, et al. Smooth muscle proliferation and role of the prostacyclin (IP) receptor in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2010;182(9):1161–1170. | ||
Moncada S, Higgs EA, Vane JR. Human arterial and venous tissues generate prostacyclin (prostaglandin x), a potent inhibitor of platelet aggregation. Lancet. 1977;1(8001):18–20. | ||
Zhou W, Hashimoto K, Goleniewska K, et al. Prostaglandin I2 analogs inhibit proinflammatory cytokine production and T-cell stimulatory function of dendritic cells. J Immunol. 2007;178(2):702–710. | ||
Raychaudhuri B, Malur A, Bonfield TL, et al. The prostacyclin analogue treprostinil blocks NFkappaB nuclear translocation in human alveolar macrophages. J Biol Chem. 2002;277(36):33344–33348. | ||
Fontana M, Olschewski H, Olschewski A, Schluter KD. Treprostinil potentiates the positive inotropic effect of catecholamines in adult rat ventricular cardiomyocytes. Br J Pharmacol. 2007;151(6):779–786. | ||
Flolan® [package insert]. London: GlaxoSmithKline; 2011. Available from: https://http://www.gsksource.com/gskprm/htdocs/documents/FLOLAN.PDF. Accessed September 1, 2014. | ||
Veletri® [package insert]. Allschwil: Actelion Pharmaceuticals Ltd; 2012. Available from: http://www.veletri.com/pdf/veletri_full_prescribing_information_2nd_gen.pdf. Accessed September 1, 2014. | ||
Nicolas LB, Gutierrez MM, Dingemanse J. Comparative pharmacokinetic, pharmacodynamic, safety, and tolerability profiles of 3 different formulations of epoprostenol sodium for injection in healthy men. Clin Ther. 2013;35(4):440–449. | ||
Sitbon O, Delcroix M, Bergot E, et al. EPITOME-2: An open-label study assessing the transition to a new formulation of intravenous epoprostenol in patients with pulmonary arterial hypertension. Am Heart J. 2014;167(2):210–217. | ||
Chin KM, Badesch DB, Robbins IM, et al. Two formulations of epoprostenol sodium in the treatment of pulmonary arterial hypertension: EPITOME-1 (epoprostenol for injection in pulmonary arterial hypertension), a phase IV, open-label, randomized study. Am Heart J. 2014;167(2):218–225.e1. | ||
Sitbon O, Humbert M, Nunes H, et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol. 2002;40(4):780–788. | ||
Watkins WD, Peterson MB, Crone RK, Shannon DC, Levine L. Prostacyclin and prostaglandin E1 for severe idiopathic pulmonary artery hypertension. Lancet. 1980;1(8177):1083. | ||
Lock JE, Olley PM, Coceani F, Swyer PR, Rowe RD. Use of prostacyclin in persistent fetal circulation. Lancet. 1979;1(8130):1343. | ||
Szczeklik J, Szczeklik A, Nizankowski R. Prostacyclin for pulmonary hypertension. Lancet. 1980;2(8203):1076. | ||
Guadagni DN, Ikram H, Maslowski AH. Haemodynamic effects of prostacyclin (PGI2) in pulmonary hypertension. Br Heart J. 1981;45(4):385–388. | ||
Rubin LJ, Groves BM, Reeves JT, Frosolono M, Handel F, Cato AE. Prostacyclin-induced acute pulmonary vasodilation in primary pulmonary hypertension. Circulation. 1982;66(2):334–338. | ||
Higenbottam T, Wheeldon D, Wells F, Wallwork J. Long-term treatment of primary pulmonary hypertension with continuous intravenous epoprostenol (prostacyclin). Lancet. 1984;1(8385):1046–1047. | ||
Jones DK, Higenbottam TW, Wallwork J. Treatment of primary pulmonary hypertension intravenous epoprostenol (prostacyclin). Br Heart J. 1987;57(3):270–278. | ||
Rubin LJ, Mendoza J, Hood M, et al. Treatment of primary pulmonary hypertension with continuous intravenous prostacyclin (epoprostenol). Results of a randomized trial. Ann Intern Med. 1990;112(7):485–491. | ||
Badesch DB, Tapson VF, McGoon MD, et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann Intern Med. 2000;132(6):425–434. | ||
McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation. 2002;106(12):1477–1482. | ||
Barst RJ, Rubin LJ, McGoon MD, Caldwell EJ, Long WA, Levy PS. Survival in primary pulmonary hypertension with long-term continuous intravenous prostacyclin. Ann Intern Med. 1994;121(6):409–415. | ||
Patterson JH, Adams KF Jr, Gheorghiade M, et al. Acute hemodynamic effects of the prostacyclin analog 15AU81 in severe congestive heart failure. Am J Cardiol. 1995;75(3):26A–33A. | ||
Dumble LJ, Gibbons S, Tejpal N, et al. 15 AU81, a prostacyclin analog, potentiates immunosuppression and mitigates renal injury due to cyclosporine. Transplantation. 1993;55(5):1124–1128. | ||
Mohler ER 3rd, Klugherz B, Goldman R, Kimmel SE, Wade M, Sehgal CM. Trial of a novel prostacyclin analog, UT-15, in patients with severe intermittent claudication. Vasc Med. 2000;5(4):231–237. | ||
Simonneau G, Barst RJ, Galiè N, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165(6):800–804. | ||
Wade M, Baker FJ, Roscigno R, DellaMaestra W, Hunt TL, Lai AA. Absolute bioavailability and pharmacokinetics of treprostinil sodium administered by acute subcutaneous infusion. J Clin Pharmacol. 2004;44(1):83–88. | ||
Laliberte K, Arneson C, Jeffs R, Hunt T, Wade M. Pharmacokinetics and steady-state bioequivalence of treprostinil sodium (Remodulin) administered by the intravenous and subcutaneous route to normal volunteers. J Cardiovasc Pharmacol. 2004;44(2):209–214. | ||
Wade M, Baker FJ, Roscigno R, et al. Pharmacokinetics of treprostinil sodium administered by 28-day chronic continuous subcutaneous infusion. J Clin Pharmacol. 2004;44(5):503–509. | ||
McSwain CS, Benza R, Shapiro S, et al. Dose proportionality of treprostinil sodium administered by continuous subcutaneous and intravenous infusion. J Clin Pharmacol. 2008;48(1):19–25. | ||
Remodulin® [package insert]. Silver Spring, MD: United Therapeutics Corporation; 2013. Available from: https://http://www.remodulin.com/downloads/remodulin-prescribinginformation.pdf. Accessed September 1, 2014. | ||
Skoro-Sajer N, Lang IM, Harja E, Kneussl MP, Sing WG, Gibbs SJ. A clinical comparison of slow- and rapid-escalation treprostinil dosing regimens in patients with pulmonary hypertension. Clin Pharmacokinet. 2008;47(9):611–618. | ||
Barst RJ, Galie N, Naeije R, et al. Long-term outcome in pulmonary arterial hypertension patients treated with subcutaneous treprostinil. Eur Respir J. 2006;28(6):1195–1203. | ||
Benza RL, Tapson VF, Gomberg-Maitland M, Poms A, Barst RJ, McLaughlin VV. One-year experience with intravenous treprostinil for pulmonary arterial hypertension. J Heart Lung Transplant. 2013;32(9):889–896. | ||
Desole S, Velik-Salchner C, Fraedrich G, Ewert R, Kahler CM. Subcutaneous implantation of a new intravenous pump system for prostacyclin treatment in patients with pulmonary arterial hypertension. Heart Lung. 2012;41(6):599–605. | ||
Ewert R, Halank M, Bruch L, Ghofrani HA. A case series of patients with severe pulmonary hypertension receiving an implantable pump for intravenous prostanoid therapy. Am J Respir Crit Care Med. 2012;186(11):1196–1198. | ||
Oudiz RJ, Schilz RJ, Barst RJ, et al. Treprostinil, a prostacyclin analogue, in pulmonary arterial hypertension associated with connective tissue disease. Chest. 2004;126(2):420–427. | ||
Lang I, Gomez-Sanchez M, Kneussl M, et al. Efficacy of long-term subcutaneous treprostinil sodium therapy in pulmonary hypertension. Chest. 2006;129(6):1636–1643. | ||
Tapson VF, Gomberg-Maitland M, McLaughlin VV, et al. Safety and efficacy of IV treprostinil for pulmonary arterial hypertension: a prospective, multicenter, open-label, 12-week trial. Chest. 2006;129(3):683–688. | ||
Mathier MA, McDevitt S, Saggar R. Subcutaneous treprostinil in pulmonary arterial hypertension: Practical considerations. J Heart Lung Transplant. 2010;29(11):1210–1217. | ||
Bloodstream infections among patients treated with intravenous epoprostenol or intravenous treprostinil for pulmonary arterial hypertension – seven sites, United States, 2003–2006. MMWR Morb Mortal Wkly Rep. 2007;56(8):170–172. | ||
Kallen AJ, Lederman E, Balaji A, et al. Bloodstream infections in patients given treatment with intravenous prostanoids. Infect Control Hosp Epidemiol. 2008;29(4):342–349. | ||
Kitterman N, Poms A, Miller DP, Lombardi S, Farber HW, Barst RJ. Bloodstream infections in patients with pulmonary arterial hypertension treated with intravenous prostanoids: insights from the REVEAL REGISTRY®. Mayo Clinic Proc. 2012;87(9):825–834. | ||
Rich JD, Glassner C, Wade M, et al. The effect of diluent pH on bloodstream infection rates in patients receiving IV treprostinil for pulmonary arterial hypertension. Chest. 2012;141(1):36–42. | ||
Ventavis® [package insert]. Allschwil: Actelion Pharmaceuticals Ltd; November 2013. Available from: http://www.4ventavis.com/pdf/Ventavis_PI.pdf. Accessed September 1, 2014. | ||
Olschewski H, Rohde B, Behr J, et al. Pharmacodynamics and pharmacokinetics of inhaled iloprost, aerosolized by three different devices, in severe pulmonary hypertension. Chest. 2003;124(4):1294–1304. | ||
Voswinckel R, Ghofrani HA, Grimminger F, Seeger W, Olschewski H. Inhaled treprostinil [corrected] for treatment of chronic pulmonary arterial hypertension. Ann Intern Med. 2006;144(2):149–150. | ||
Olschewski H, Walmrath D, Schermuly R, Ghofrani A, Grimminger F, Seeger W. Aerosolized prostacyclin and iloprost in severe pulmonary hypertension. Ann Intern Med. 1996;124(9):820–824. | ||
Olschewski H, Ghofrani HA, Walmrath D, Temmesfeld-Wollbruck B, Grimminger F, Seeger W. Recovery from circulatory shock in severe primary pulmonary hypertension (PPH) with aerosolization of iloprost. Intensive Care Med. 1998;24(6):631–634. | ||
Stricker H, Domenighetti G, Fiori G, Mombelli G. Sustained improvement of performance and haemodynamics with long-term aerosolised prostacyclin therapy in severe pulmonary hypertension. Schweiz Med Wochenschr. 1999;129(24):923–927. | ||
Olschewski H, Ghofrani HA, Schmehl T, et al. Inhaled iloprost to treat severe pulmonary hypertension. An uncontrolled trial. German PPH Study Group. Ann Intern Med. 2000;132(6):435–443. | ||
Hoeper MM, Schwarze M, Ehlerding S, et al. Long-term treatment of primary pulmonary hypertension with aerosolized iloprost, a prostacyclin analogue. N Engl J Med. 2000;342(25):1866–1870. | ||
Olschewski H, Simonneau G, Galiè N, et al; Aerosolized Iloprost Randomized Study Group. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347(5):322–329. | ||
McLaughlin VV, Oudiz RJ, Frost A, et al. Randomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;174(11):1257–1263. | ||
Opitz CF, Wensel R, Winkler J, et al. Clinical efficacy and survival with first-line inhaled iloprost therapy in patients with idiopathic pulmonary arterial hypertension. Eur Heart J. 2005;26(18):1895–1902. | ||
Olschewski H, Hoeper MM, Behr J, et al. Long-term therapy with inhaled iloprost in patients with pulmonary hypertension. Respir Med. 2010;104(5):731–740. | ||
Tyvaso® [package insert]. Silver Spring, MD: United Therapeutics Corporation; 2014. Available from: https://http://www.tyvaso.com/pdf/Tyvaso-PI.pdf. Accessed September 1, 2014. | ||
Voswinckel R, Enke B, Reichenberger F, et al. Favorable effects of inhaled treprostinil in severe pulmonary hypertension: results from randomized controlled pilot studies. J Am Coll Cardiol. 2006;48(8):1672–1681. | ||
Channick RN, Olschewski H, Seeger W, Staub T, Voswinckel R, Rubin LJ. Safety and efficacy of inhaled treprostinil as add-on therapy to bosentan in pulmonary arterial hypertension. J Am Coll Cardiol. 2006;48(7):1433–1437. | ||
Voswinckel R, Reichenberger F, Enke B, et al. Acute effects of the combination of sildenafil and inhaled treprostinil on haemodynamics and gas exchange in pulmonary hypertension. Pulm Pharmacol Ther. 2008;21(5):824–832. | ||
McLaughlin VV, Benza RL, Rubin LJ, et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: a randomized controlled clinical trial. J Am Coll Cardiol. 2010;55(18):1915–1922. | ||
Benza RL, Seeger W, McLaughlin VV, et al. Long-term effects of inhaled treprostinil in patients with pulmonary arterial hypertension: the Treprostinil Sodium Inhalation Used in the Management of Pulmonary Arterial Hypertension (TRIUMPH) study open-label extension. J Heart Lung Transplant. 2011;30(12):1327–1333. | ||
Galiè N, Humbert M, Vachiéry JL, et al. Effects of beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol. 2002;39(9):1496–1502. | ||
Barst RJ, McGoon M, McLaughlin V, et al. Beraprost therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2003;41(12):2119–2125. | ||
White RJ, Torres F, Allen R, et al. Pharmacokinetics of oral treprostinil sustained release tablets during chronic administration to patients with pulmonary arterial hypertension. J Cardiovasc Pharmacol. 2013;61(6):474–481. | ||
Orenitram® [package insert]. Silver Spring, MD: United Therapeutics Corporation; 2013. Available from: http://www.orenitram.com/dtc/pdf/Orenitram_Full_Prescribing_Information.pdf. Accessed September 1, 2014. | ||
Lim A, Wang-Smith L, Kates J, Laurent A, Kumar P, Laliberte K. The effect of different meal compositions on the oral bioavailability of treprostinil diolamine in healthy volunteers. J Clin Pharm Ther. 2013;38(6):450–455. | ||
Peterson L, Marbury T, Marier J, Laliberte K. An evaluation of the pharmacokinetics of treprostinil diolamine in subjects with hepatic impairment. J Clin Pharm Ther. 2013;38(6):518–523. | ||
Jing ZC, Parikh K, Pulido T, et al. Efficacy and safety of oral treprostinil monotherapy for the treatment of pulmonary arterial hypertension: a randomized, controlled trial. Circulation. 2013;127(5):624–633. | ||
Tapson VF, Torres F, Kermeen F, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C study): a randomized controlled trial. Chest. 2012;142(6):1383–1390. | ||
Tapson VF, Jing ZC, Xu KF, et al; FREEDOM-C2 Study Team. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients receiving background endothelin receptor antagonist and phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C2 study): a randomized controlled trial. Chest. 2013;144(3):952–958. | ||
Gomberg-Maitland M, Tapson VF, Benza RL, et al. Transition from intravenous epoprostenol to intravenous treprostinil in pulmonary hypertension. Am J Respir Crit Care Med. 2005;172(12):1586–1589. | ||
Vachiery JL, Hill N, Zwicke D, Barst R, Blackburn S, Naeije R. Transitioning from i.v. epoprostenol to subcutaneous treprostinil in pulmonary arterial hypertension. Chest. 2002;121(5):1561–1565. | ||
Rubenfire M, McLaughlin VV, Allen RP, et al. Transition from IV epoprostenol to subcutaneous treprostinil in pulmonary arterial hypertension: a controlled trial. Chest. 2007;132(3):757–763. | ||
de Jesus Perez VA, Rosenzweig E, Rubin LJ, et al. Safety and efficacy of transition from systemic prostanoids to inhaled treprostinil in pulmonary arterial hypertension. Am J Cardiol. 2012;110(10):1546–1550. | ||
Channick RN, Frantz RP, Kawut SM, et al. A multicenter, retrospective study of patients with pulmonary arterial hypertension transitioned from parenteral prostacyclin therapy to inhaled iloprost. Pulm Circ. 2013;3(2):381–388. | ||
Enderby CY, Soukup M, Al Omari M, Zeiger T, Burger C. Transition from intravenous or subcutaneous prostacyclin therapy to inhaled treprostinil in patients with pulmonary arterial hypertension: a retrospective case series. J Clin Pharm Ther. 2014;39(5):496–500. | ||
Bourge RC, Tapson VF, Safdar Z, et al. Rapid transition from inhaled iloprost to inhaled treprostinil in patients with pulmonary arterial hypertension. Cardiovasc Ther. 2013;31(1):38–44. | ||
Hoeper MM, Leuchte H, Halank M, et al. Combining inhaled iloprost with bosentan in patients with idiopathic pulmonary arterial hypertension. Eur Respir J. 2006;28(4):691–694. | ||
Hiremath J, Thanikachalam S, Parikh K, et al. Exercise improvement and plasma biomarker changes with intravenous treprostinil therapy for pulmonary arterial hypertension: a placebo-controlled trial. J Heart Lung Transplant. 2010;29(2):137–149. | ||
McLaughlin VV, Gaine SP, Barst RJ, et al. Efficacy and safety of treprostinil: an epoprostenol analog for primary pulmonary hypertension. J Cardiovasc Pharmacol. 2003;41(2):293–299. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.