Back to Journals » Journal of Inflammation Research » Volume 7
Prostacyclin mediates endothelial COX-2-dependent neuroprotective effects during excitotoxic brain injury
Authors An Y, Belevych N, Wang Y, Zhang H, Nasse J, Herschman H, Chen Q, Tarr A, Liu X ![]() , Quan N
, Quan N
Received 27 February 2014
Accepted for publication 2 April 2014
Published 21 May 2014 Volume 2014:7 Pages 57—67
DOI https://doi.org/10.2147/JIR.S63205
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Ying An,1,2 Natalya Belevych,1,2 Yufen Wang,1,2 Hao Zhang,1 Jason S Nasse,3 Harvey Herschman,4 Qun Chen,1,2 Andrew Tarr,1,2 Xiaoyu Liu,1,2 Ning Quan1,2
1Institute for Behavior Medicine Research, 2Department of Oral Biology, College of Dentistry, 3Neuroscience Graduate Studies Program, The Ohio State University, Columbus, OH, USA; 4Department of Molecular and Medical Pharmacology, UCLA, Los Angeles, CA, USA
Abstract: In a previous study, we found that intracerebral administration of excitotoxin (RS)-(tetrazole-5yl) glycine caused increased neural damage in the brain in an endothelial COX-2 deleted mouse line (Tie2Cre COX-2flox/flox). In this study, we investigated whether prostacyclin might mediate this endothelial COX-2-dependent neuroprotection. Administration of excitotoxin into the striatum induced the production of prostacyclin (PGI2) in wild type, but not in endothelial COX-2 deleted mice. Inhibition of PGI2 synthase exacerbated brain lesions induced by the excitotoxin in wild type, but not in endothelial COX-2 deleted mice. Administration of a PGI2 agonist reduced neural damage in both wild type and endothelial COX-2 deleted mice. Increased PGI2 synthase expression was found in infiltrating neutrophils. In an ex vivo assay, PGI2 reduced the excitotoxin-induced calcium influx into neurons, suggesting a cellular mechanism for PGI2 mediated neuroprotection. These results reveal that PGI2 mediates endothelial COX-2 dependent neuroprotection.
Keywords: neural injury, prostaglandins, neutrophil, conditional COX-2 deletion, PGI2
Introduction
Increased COX-2 expression occurs in acute brain injury conditions including cerebral ischemia,1 traumatic brain injury,2 spreading depression,3 and excitotoxic brain injury.4,5 We found recently in a model of excitotoxic brain injury that induced COX-2 expression mediates both neuroprotective and neurotoxic effects depending on the cell type of COX-2 expressing cells. Specifically, increased endothelial COX-2 expression confers a neuroprotective effect, whereas increased neuronal COX-2 expression confers a neurotoxic effect that is mediated by the neuronal production of prostaglandin E2.6
The COX enzyme catalyzes the production of prostaglandin H2 (PGH2) from free arachidonic acid. Arachidonic acid is first cyclized and oxygenated by COX to form prostaglandin G2, which is then reduced to form PGH2. PGH2 is the intermediate that serves as the substrate for the terminal prostanoid synthase enzymes. Besides prostaglandin E2, prostaglandin G2 (PGD2), prostacyclin (PGI2), and thromboxane can be produced downstream of COX.7 Some of the COX dependent prostanoids have been reported to provide neuroprotective effects during acute brain injury. These include PGI2 and PGD2. For example, physiological concentrations of PGD2 have been shown to rescue neurons in an in vitro model of excitotoxicity,8 and cerebral ischemia-induced brain damage can be reduced by a PGI2 agonist.9 In the periphery, PGI2 is known to have vasodilation properties and it is used to relieve pulmonary hypertension.10 In the central nervous system (CNS), endothelial PGI2 production has been shown to exhibit neuroprotective activity in a model of cerebral ischemia.11 Because our observed neuroprotective effects were associated with endothelial COX-2 expression, we sought to investigate the role of PGI2 in mediating this neuroprotection in the present study.
Material and methods
Animals
Tie2Cre COX-2flox/flox mice were generated by cross breeding Tie2Cre; COX-2+/+ transgenic mice (stock # 004128; The Jackson Laboratory, Bar Harbor, ME, USA) with COX-2flox/flox mice. LysMCre COX-2flox/flox mice were generously provided by Dr Reddy (Department of Medicine, University of California, Los Angeles; Los Angeles, CA, USA). In the Tie2Cre; COX-2+/+ mouse, the Tie2 promoter restricts Cre recombinase expression in endothelial cells and hematopoietic cells during embryogenesis and adulthood.12 Therefore, the COX-2 gene is selectively deleted in endothelial cells and hematopoietic cells in Tie2Cre COX-2flox/flox mice. In LysMCre; COX-2+/+ mice, transgenic expression of Cre recombinase is restricted to myeloid lineage cells; consequently, COX-2 is deleted specifically in myeloid cells in LysMCre COX-2flox/flox mice.13 Results in Tie2Cre COX-2flox/flox mice and LysMCre COX-2flox/flox mice were compared with their respective Cre-negative COX-2flox/flox littermates. Mice of 10–16 weeks of age, with body weights of 25–30 g, were used in experimental procedures. All procedures were approved by the Ohio State University Animal Care and Use Committee.
Genotyping
Genomic DNA was purified from mouse tail tissue. Briefly, tail samples were frozen for at least 15 minutes at −80°C. Each sample was incubated with 500 μL of lysis buffer (10 mM Tris HCl pH 8.0; 100 mM ethylenediaminetetraacetic acid; 0.5% sodium dodecyl sulfate; 0.2 mg/mL ribonuclease A; 1 mg/mL proteinase K) for 2 hours at 56°C with repeated agitation. Samples were then centrifuged at 13,000 rpm for 10 minutes to remove tissue residue from the lysate. Genomic DNA was precipitated by adding 500 μL isopropanol and was washed with 1 mL ice-cold 70% ethanol. DNA pellets were dissolved in 50 μL of 5 mM Tris HCl buffer (pH 8.5) by incubation at 65°C for 10 minutes.
To detect the presence of Cre recombinase by polymerase chain reaction, the following primer set was used for the generation of a 300 bp amplicon: Cre300F: 5′-CGATGCAACGAGTGATGAGG-3′ and Cre300R: 5′-CGCATAACCAGTGAAACAGC-3′. To detect the knockout COX-2 alleles, the following primer set was used: COX-2E3F1: 5′-AATTACTGCTGAAGCCCACC-3′ and COX-2I5R1: 5′-GAATCTCCTAGAACTGACTGG-3′. The COX-2 floxed allele amplicon is 2,670 bp while the same primer set detects the COX-2 deleted allele as a 1,054 bp amplicon. Detailed description on how these primers can be used to differentiate different genotypes has been published previously.14
Reagents
(RS)-(Tetrazol-5-yl)glycine (5 μM, TZG; Tocris Bioscience, Ellisville, MO, USA), a potent N-Methyl-D-aspartate (NMDA) agonist, was dissolved in phosphate-buffered saline (PBS) (pH 7.4; Thermo Fisher Scientific, Waltham, MA, USA) to induce pharmacological excitotoxic lesions in the brain. NS-398 (2 mg/kg; Cayman Chemical Company, Ann Arbor, MI, USA) and MRE-269 (1 mg/kg; Cayman Chemical Company) were dissolved in dimethyl sulfoxide before being further diluted in PBS. Tranylcypromine (10 mg/kg; Enzo Life Sciences Inc., Farmingdale, NY, USA) was dissolved in PBS (pH 7.4). Antibodies used for immunohistochemistry were rabbit anti-mouse PGI2 synthase (PGIS) (Abcam, Cambridge, MA, USA), rat anti-mouse CD45 (BD Biosciences, San Jose, CA, USA), biotinylated anti-mouse Ly6G (BD Biosciences), and rat anti-mouse F4/80 (ABD Serotec, Raleigh, NC, USA). Antibodies used for flow cytometry were fluorescein isothiocyanate-conjugated anti-Ly6G and peridinin chlorophyll protein-conjugated anti-Gr1 (BD Biosciences).
Intrastriatal injection of NMDA agonist
Mice were anaesthetized by intraperitoneal injection (IP) of 100 mg/kg of Nembutal (Abbott Laboratories, Abbott Park, IL, USA) and then securely fastened onto a small animal stereotaxic frame with a mouse head holder (David Kopf Instruments, Tujunga, CA, USA). A burr hole was drilled and an injection cannula (28 gauge) was lowered into the center of the right striatum (anterior, 0.5 mm; lateral, 2.0 mm; ventral, 4.0 mm to bregma) according to a mouse brain atlas.15 Ten microliters of TZG (5 μM) or sterile PBS was injected at a rate of 0.8 μL/min using a microinjection system (KDS Scientific, Holliston, MA, USA). Needles were left unmoved for an additional 5 minutes after fluid injection, to prevent backflow. Wounds were then sutured and animals were allowed to recover from anesthesia. Animals were sacrificed and brain samples were collected at 4, 8, 12, or 24 hours after the intrastriatal injection.
Hematoxylin and eosin staining
Fresh-frozen brain sections were allowed to air-dry for at least 15 minutes before fixing in 95% alcohol for 5 minutes. Sections were then briefly rinsed in water and stained in Mayer’s hematoxylin solution (Sigma-Aldrich Co, St Louis, MO) for 15 minutes. After rinsing in water five times, slides were stained in 1% eosin Y solution (Thermo Fisher Scientific) for 3 minutes. Slides were then dipped five to ten times in 95% and 100% alcohol prior to cover-slipping using Permount (Thermo Fisher Scientific).
Determination of lesion volumes
Brains were sectioned on a cryostat to generate 20 μm sections throughout the entire striatum. Every third coronal section was collected and stained. Lesion areas on each section were quantified using an image analyzer (ImageJ; National Institutes of Health, Bethesda, MD, USA). The entire lesion area was calculated to determine the lesion volume. All lesion sizes were quantified by a person blinded to the treatment groups.
Immunohistochemistry (IHC)
Fresh-frozen coronal brain sections were fixed in an acetone/alcohol mixture (3:1) for 5 minutes. The sections were treated with glucose oxidase and sodium azide to reduce background interference. They were then incubated with rabbit anti-PGIS antibody (1:800) followed by an anti-rabbit secondary antibody (1:200). The labeling was amplified with the ABC solution (Vector Laboratories, Inc., Burlingame, CA, USA) and visualized with a DAB peroxidase substrate kit (Vector Laboratories, Inc.). To identify the types of cells expressing PGIS, double-label IHC was performed. PGIS-stained cells were double-labeled with antibodies directed against the cellular protein markers anti-CD45 (pan-leukocytes; 1:200), anti-Ly6G (neutrophils; 1:1000), or anti-F4/80 (monocytes; 1:600). The labeling of these cell-type markers was visualized with Cy-2 (1:400) or Cy3-streptavidin (1:400; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA). Negative controls (ie, omission of primary antibody) were used to determine specificity of antibody binding.
Semiquantitative analysis of PGIS and CD45 immunoreactivity
Relative values of PGIS and CD45 immunoreactivity are given as estimates of the density of immunohistochemical staining. Two coronal sections (0.5 mm anterior to bregma) per animal for three animals per group were analyzed (Table 1). Mice injected with saline were used as controls.
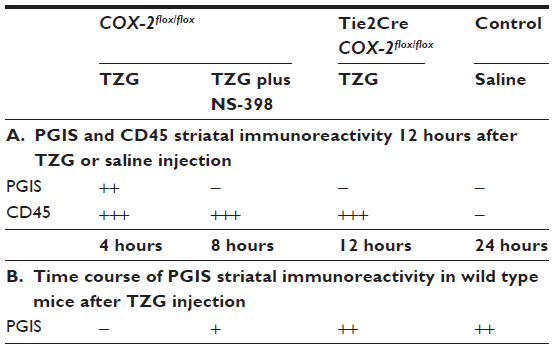 | Table 1 Semiquantitative analysis of TZG-induced PGIS and CD45 immunoreactivity |
A five-point scale was used to rate the data: 5) +++, high density of cellular signals; 4) ++, moderate density; 3) +, low density; 2) ±, single cellular signals in some cases; and 1) −, no signals. The values for two sections per animal were averaged, and the means of these averages for the three animals per group are presented. Quantification of the immunoreactivity data was performed by an investigator blinded to the treatment groups. Negative controls (ie, omission of primary antibody) were used to determine specificity of antibody binding.
Depletion of neutrophils
Mice were injected IP with 100 μg of an anti-neutrophil antibody (clone 1A8; BD Biosciences) 24 hours before intrastriatal injection of TZG. Control animals were injected IP with PBS. Twenty-four hours after TZG injection, blood and brain samples were collected. Neutrophil depletion was confirmed by analysis of the blood samples for Ly6G and Gr1 double-labeled cells using flow cytometry and by IHC labeling of Ly6G in brain sections.
Flow cytometry
Blood samples were collected by cardiac puncture. Red blood cells were lysed by using red blood cell lysis buffer (4.4 g NH4Cl, 0.5 g KHCO3, and 0.019 g ethylenediaminetetraacetic acid in 500 mL distilled water, pH 7.2–7.4) for 2 minutes at room temperature. Cells were washed and incubated with Fc receptor block (BD Biosciences) for 10 minutes at 4°C. Cells were then washed and incubated with fluorescein isothiocyanate-conjugated anti-Ly6G and peridinin chlorophyll protein-conjugated anti-Gr1 for 45 minutes at 4°C in the dark. Cells were washed and then resuspended in fluorescence activated cell sorting (FACS) buffer (2% fetal bovine serum [FBS] in PBS with 1 mg/mL sodium azide). Nonspecific binding was determined by using isotype-matched control antibodies. Analysis of stained cells was performed with a Becton-Dickinson FACS Caliber four color flow cytometer (BD, Franklin Lakes, NJ, USA). Ten thousand events were recorded for each sample and isotype match-conjugate. Flow cytometric data were analyzed with FlowJo software (TreeStar Inc., Ashland, OR, USA).
Measurement of PGI2 level
Mice received intrastriatal injection of TZG or saline. Animals were euthanized by decapitation 12 hours after injection. A 4 mm-thick coronal striatal brain section including the needle track (from 2.5 mm to −1.5 mm relative to bregma) was collected from each animal, placed in 1 mL ice-cold PBS, and homogenized thoroughly. All tested samples weighed 11.6 mg. Samples were centrifuged at 13,000 rpm at 4°C for 10 minutes to remove tissue residue, and supernatants were saved until assays were performed. The level of PGI2 was measured using an EIA kit (Cayman Chemical Company) to detect 6-keto prostaglandin f1α, the stable metabolite of PGI2. All samples were run in triplicates to determine variability. Relative abundant changes were expressed as the ratio of (6-keto prostaglandin f1α present in striatal samples from mice injected with TZG) divided by (6-keto prostaglandin f1α present in striatal samples from mice injected with saline).
Calcium imaging
Striatal slices were cut from P7 to P9 wild type rat pups to 350 μm-thick sections on a vibratome and were allowed to recover for 1 hour at 32°C. Sections were then incubated in an artificial cerebral-spinal fluid (ACSF) bath containing 25 μM Quest Fluo-8 AM (AAT-Bioquest, Inc., Sunnyvale, CA, USA) with 0.01% dimethyl sulfoxide and 0.002% Pluronic-F127 for 20 minutes, rinsed twice for 30 minutes each, and held at room temperature until the beginning of each experiment. Individual slices were imaged on a fixed-stage upright microscope equipped with a custom made perfusion chamber. Imaging experiments were conducted using a Visitech Infinity 3 confocal live-cell imaging system (Visitech International, Sunderland, UK) within the OSU campus microscopy and imaging facility (CMIF). During the imaging experiments, all slices were continuously perfused with warmed (32°C) ACSF at a rate of 2 mL/min. All drugs were delivered through a manifold system directly in line with the perfusion chamber. Neurons in a given slice preparation were first tested for baseline NMDA receptor activation by perfusing 5 μM TZG through the chamber for 30 seconds while the section was imaged at 6.7 frames/second. TZG induced fluorescence in individual neurons was measured off-line using Metamorph imaging software (Molecular Devices LLC, Sunnyvale, CA, USA) and set to be 100%. After a 2.5-minutes ACSF washout, the slice was tested again to determine the effect of PGI2 on TZG-induced calcium influx: the PGI2 agonist MRE-269 (0.1 μM) was perfused through the chamber for 20 minutes prior to a second 30 sec perfusion of TZG. Fluorescence intensity in individual neurons after the second TZG perfusion was normalized to the fluorescence intensity of the same neurons after the first TZG perfusion (similar to Huang et al).16 To test for the possibility of dye rundown or NMDA receptor desensitization, control experiments were performed where only ACSF was perfused for 20 minutes between the first and second application of TZG.
Statistical analysis
Data were analyzed, as appropriate, by one-way or two-way analysis of variance followed by post hoc Student’s t-tests where appropriate. An alpha level of P≤0.05 was used for the rejection of the null hypothesis.
Results
PGI2 mediates the endothelial COX-2- dependent neuroprotective effect following excitotoxin injection
We first measured the level of 6-keto prostaglandin f1α, the major stable metabolite of PGI2, in brain sections 12 hours after TZG injection. 6-Keto prostaglandin f1α levels in striatal sections from TZG-treated COX-2flox/flox mice were significantly increased (nearly twofold) relative to the 6-keto prostaglandin f1α levels in saline injected controls (Figure 1A). In contrast, a much smaller increase in 6-keto prostaglandin f1α levels was observed following TZG injection in Tie2Cre COX-2flox/flox mice relative to their saline-injected controls (Figure 1A); TZG induced 6-keto prostaglandin f1α was significantly higher in the wild type COX-2flox/flox mice than in the Tie2Cre COX-2flox/flox mice (P<0.01; Figure 1A).
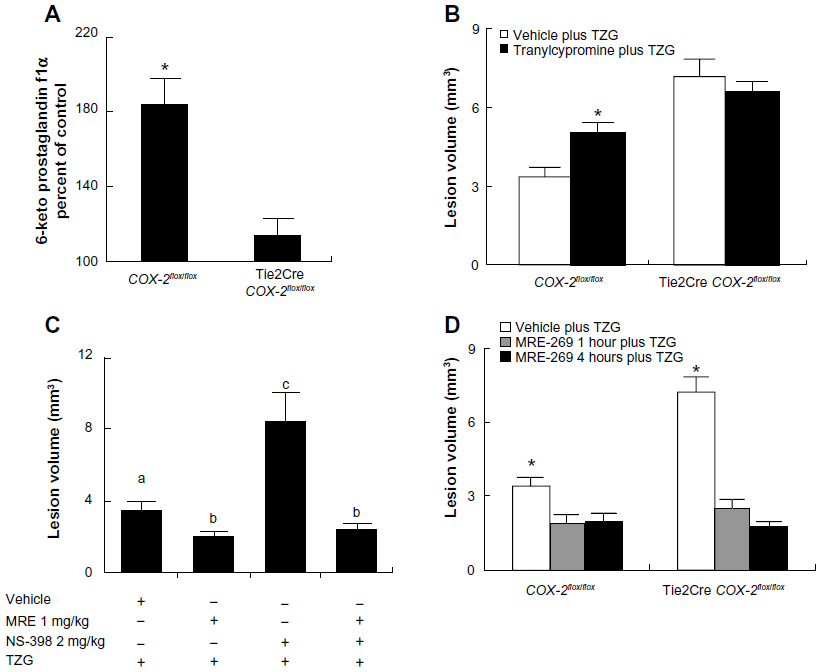 | Figure 1 PGI2 mediates the endothelial COX-2-dependent neuroprotective effect following excitotoxin injection. |
If PGI2 does indeed mediate the endothelial COX-2-dependent neuroprotection to excitotoxin treatment, then inhibition of PGIS should prevent the neuroprotective effect and lead to increased lesion volume. IP administration of tranylcypromine (10 mg/kg), an inhibitor of PGIS, 1 hour before the TZG injection resulted in an increased lesion volume in COX-2flox/flox mice (P<0.01; Figure 1B). In contrast, there was no change in excitotoxin-induced lesion volume in Tie2Cre COX-2flox/flox mice (Figure 1B) following tranylcypromine treatment.
If PGI2 mediates the endothelial COX-2-dependent neuroprotection to excitotoxin treatment, then administration of a stable PGI2 analogue should reduce TZG-induced lesions. IP administration of the stable PGI2 analogue MRE-269 (1 mg/kg) significantly reduced the injury volume following TZG injection relative to vehicle treated mice receiving TZG injections (P<0.05; Figure 1C). We showed previously that administration of a low dose (2 mg/kg) of the selective COX-2 inhibitor (NS-398) resulted in exacerbated neurotoxicity.6 We then tested whether administration of a PGI2 stable analogue would supplement the endogenously produced PGI2 and reduce the exaggerated neurotoxicity induced by TZG administration to wild type mice in the presence of the low level of NS-398.6 As predicted, treatment with MRE-269 strongly diminished the lesion-enlarging effect of the low-dose NS-398 for TZG induced neurotoxicity (P<0.05; Figure 1C).
Lastly, to explore the therapeutic potential of MRE-269, MRE-269 was injected 4 hours after the TZG injection, in both control COX-2flox/flox and in Tie2Cre COX-2flox/flox mice, in which the neuroprotective effect of the endothelial COX-2-expressing cells had been eliminated by targeted gene deletion in our previous study.6 Results show post-injury treatment with MRE-269 was able to significantly reduce neural damage in both wild type mice and Tie2Cre COX-2flox/flox mice (P<0.05 for both; Figure 1D). These results indicate that the stable PGI2 analogues had a relatively broad time-window in treating acute excitotoxic brain injury.
Presence of PGIS+ and CD45+ cells following TZG injection
The preceding results suggest that the terminal enzyme for PGI2 synthesis, PGIS, should be present in the PGI2-producing cells responsible for the neuroprotective effects. Results show PGIS+ cells were not detectable in the brain 12 hours after saline injection in COX-2flox/flox mice (Figure 2A) or Tie2Cre COX-2flox/flox mice (data not shown). PGIS+ cells were, however, scattered in the lesion core of COX-2flox/flox mice following TZG injection (Figure 2B). In contrast, PGIS+ cells were not present in Tie2Cre COX-2flox/flox mice after TZG injection (Figure 2C). Further, the low-dose NS-398 pretreatment in COX-2flox/flox mice, which exacerbates TZG induced neurotoxicity, also abolished the appearance of PGIS+ cells (Figure 2D).
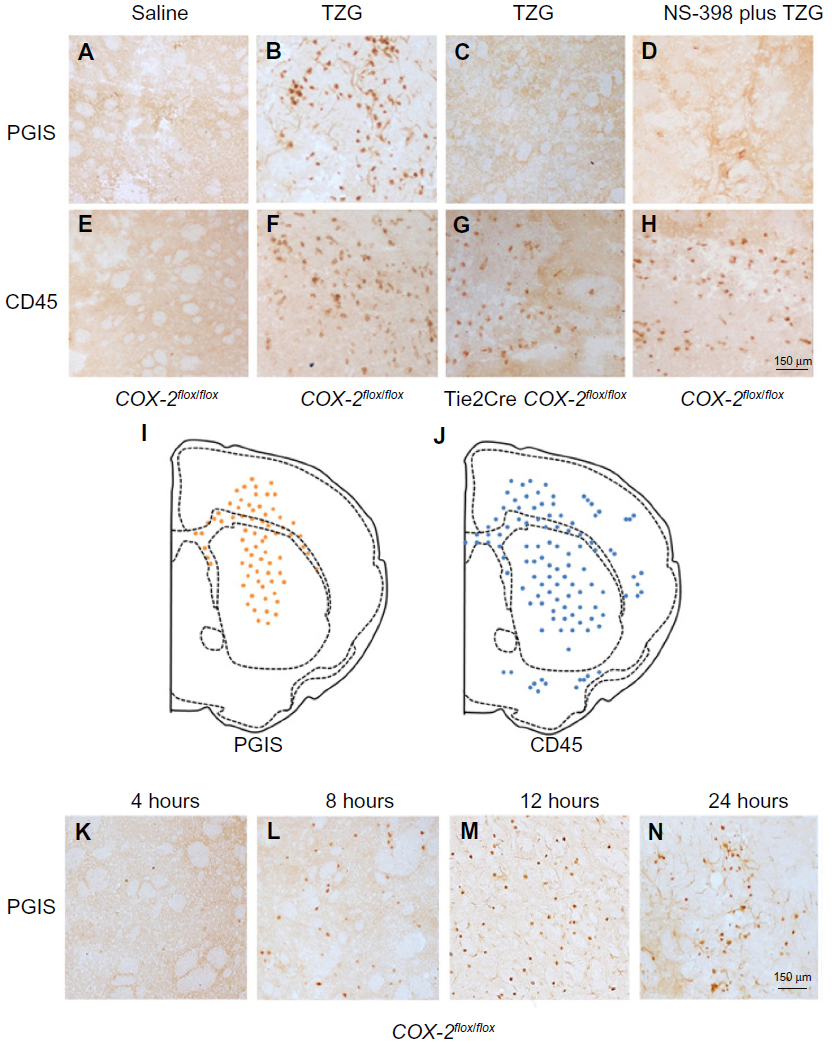 | Figure 2 Presence of PGIS and CD45 IHC-positive cells following TZG injection. |
Because the pattern of PGIS+ cells observed in the parenchyma of TZG-treated mice resembles that of the infiltrating leukocytes in the brain, we used IHC with the pan-leukocyte marker CD45 to compare the distribution of PGIS+ cells (Figure 2A–D) with the distribution of CD45+ leukocytes (Figure 2E–H). Unlike PGIS+ cells, CD45+ cells were still present in Tie2Cre COX-2flox/flox mice (Figure 2G) and in low-dose NS-398 pretreated COX-2flox/flox mice (Figure 2H). The distributions of PGIS+ and CD45+ cells in TZG treated COX-2flox/flox mice are illustrated in Figure 2I and J. PGIS+ cells were distributed in a more restricted pattern in the injured striatum and corpus callosum (Figure 2I); the distribution of CD45+ cells was spread further outside of the lesion core (Figure 2J). A semiquantitative analysis of the density for this IHC labeling is presented in Table 1. To further characterize PGIS expression, the time course of PGIS+ labeling in COX-2flox/flox mice was examined. Results revealed that these cells were observed in the brain 8 hours after the TZG injection, and plateaued at 12 hours and 24 hours post-TZG (Figure 2K–N and Table 1).
Identification of PGIS-expressing cell types in the lesion core
The data suggests that the PGI2 observed in response to excitotoxin administration requires the induction of endothelial COX-2 (and presumably downstream prostanoid/prostanoids), but that the PGI2 is not produced in the COX-2-expressing endothelial cells. We used double-label IHC to identify the PGIS+ cells that were present in the brain parenchyma. PGIS+ cells were found in affected brain parenchyma as early as 8 hours after injury (Figure 2K–N). Using fluorescent IHC, PGIS+ cells were further analyzed for the coexpression of CD45 (a leukocyte marker), F4/80 (a macrophage marker), or Ly6G (a neutrophil marker) to determine cell type. Results show essentially all the PGIS labeling colocalized with CD45 labeling (Figure 3A–C); 59% of CD45+ cells are PGIS+. PGIS+ cells and F4/80 cells did not colocalize (Figure 3D–F). In contrast, PGIS+ cells were also positive for Ly6G expression (Figure 3G–I); 57% Ly6G+ cells are PGIS+. To our surprise, COX-2 expression did not colocalize with Ly6G expression (data not shown). These data suggest that the PGIS+ parenchymal-infiltrating cells are a neutrophil population that does not express COX-2.
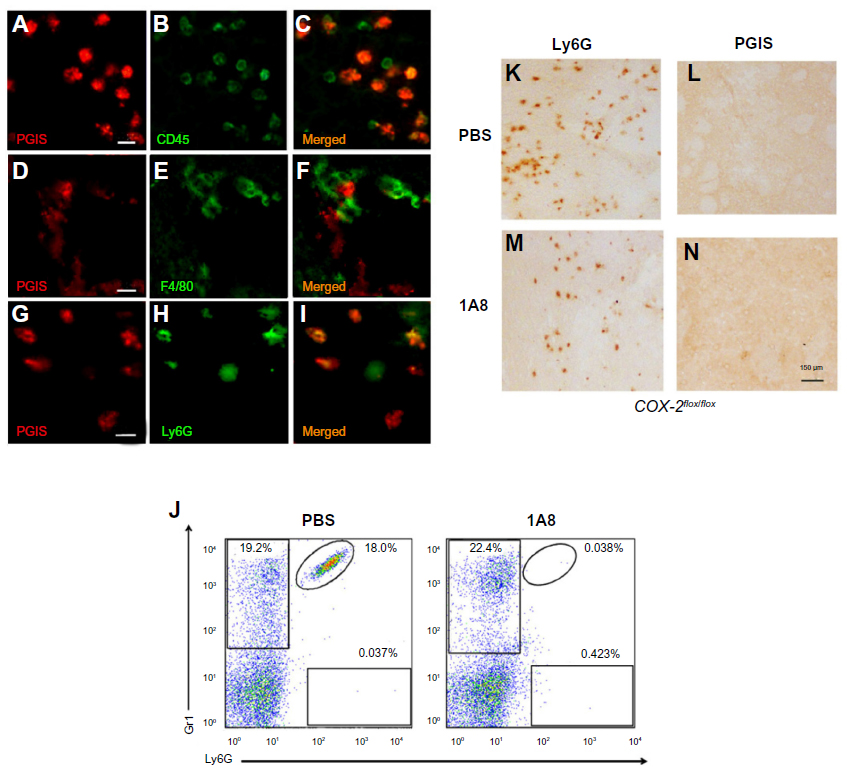 | Figure 3 Identification of PGIS expressing cell types in the lesion core. |
In an independent examination of PGIS+ neutrophil migration into the parenchyma following excitotoxin administration, we used the monoclonal antibody 1A8 to selectively deplete neutrophils and determined the consequences on PGIS+ cell migration into the lesion area.17 Figure 3J is a representative bivariate dot plot of Ly6G- and Gr1-stained blood cells from TZG-injected mice that had received prior systemic injections of either 1A8 antibody or PBS. Flow cytometric results showed that in the PBS treated control group, 24 hours after intrastriatal TZG injection, neutrophils (Ly6G+/Gr1+ cells) were found in the blood and 1A8 antibody treatment depleted these neutrophils (Figure 3J). IHC results further showed that 1A8 antibody treatment also successfully depleted both Ly6G-labeled cells (Figure 3K and L) and PGIS+ cells (Figure 3M and N) in brains from TZG-treated COX-2flox/flox mice.
PGI2 attenuates TZG-induced calcium influx into neurons
We next explored the molecular mechanism responsible for the protective effect of PGI2 on excitotoxin-induced neurotoxicity. The PGI2-producing cells in our study were mostly in the lesion core rather than in the vicinity of blood vessels. Whether PGI2 can affect neuronal cells independent of blood flow to exert neuroprotective effects is not known. To this end, we tested whether PGI2 confers neuroprotection by attenuating TZG induced Ca2+ influx, using calcium imaging to monitor neuronal Ca2+ transients in rat striatum slices. Brain slices loaded with Fluo-8, a Ca2+ labeling dye, were exposed to TZG for 30 seconds. Data from representative neurons demonstrated that TZG induced a marked elevation in intracellular Ca2+ (Figure 4A and B, blue lines). Pretreatment with MRE-269 (the stable PGI2 analogue) for 20 minutes before TZG stimulation significantly reduced Ca2+ influx in the same neuron (Figure 4A, red line). In contrast, pretreatment with ACSF (Figure 4B, red line) had no significant effect on TZG-induced Ca2+ influx. In three repeated experiments, significant differences were demonstrated in both amplitude (Figure 4C) and the area under the curve (Figure 4D) for cells that received TZG in the presence of MRE-269 (P<0.01 for both). Cells perfused with MRE-269 alone did not display changes in Ca2+ homeostasis (data not shown). Thus, PGI2 protected neurons from TZG-induced toxicity by attenuating the increased Ca2+ influx elicited by the excitotoxin.
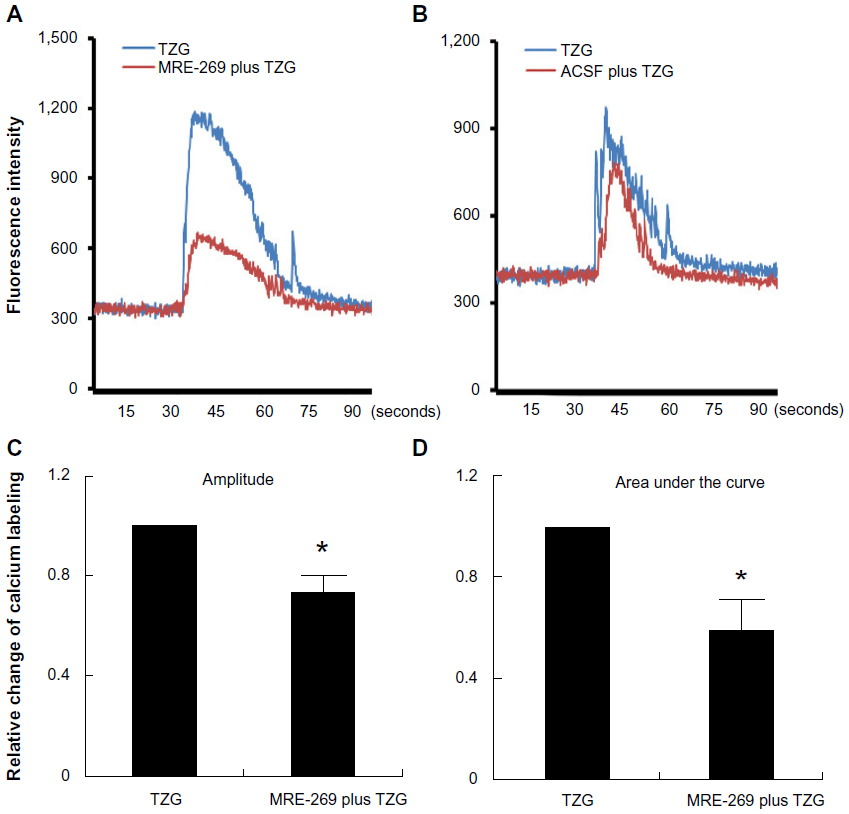 | Figure 4 PGI2 attenuates TZG-induced calcium influx into neurons. |
Deletion of the COX-2 gene in myeloid cells does not prevent endogenous PGI2 dependent protection from TZG induced neurotoxicity
LysMCre expression deletes “floxed” DNA sequences in both macrophages and neutrophils.18 To further evaluate the cell type/types responsible for the production of the neuroprotective PGI2 following excitotoxin administration, we examined the consequences of TZG injection into LysMCre COX-2flox/flox mice. The level of 6-keto prostaglandin f1α in LysMCre COX-2flox/flox mice induced by the TZG injection was markedly higher than that in Tie2Cre COX-2flox/flox mice (P<0.01; Figure 5A). IHC results also confirmed that PGIS cells were present in the TZG-induced injured striatum in LysMCre COX-2flox/flox mice (Figure 5B) and that low-dose NS-398 pretreatment abrogated the appearance of PGIS cells (Figure 5C) without preventing the arrival of infiltrating leukocytes (Figure 5D and E). Furthermore, NS-398 treatment increased the size of TZG-induced lesions in LysMCre COX-2flox/flox mice (P<0.01; Figure 5F), just as it did in wild type mice. Tranylcypromine treatment also increased the size of TZG-induced lesions in LysMCre COX-2flox/flox mice (P<0.01; Figure 5F). These results are consistent with the suggestion that endothelial COX-2 is essential for the neuroprotective effect.
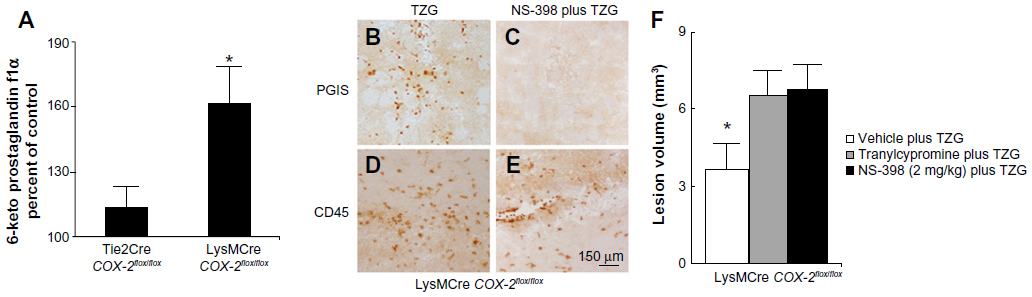 | Figure 5 Deletion of the COX-2 gene in myeloid cells does not prevent endogenous PGI2 dependent protection from TZG-induced neurotoxicity. |
Discussion
Our search for a COX-2 dependent neuroprotective mechanism led us to several important observations regarding PGI2. Consistent with previous studies,11,19 we found that PGI2 was induced by excitotoxic neural injury and that exogenous PGI2 administration affords neuroprotection. The new findings in the present study are: 1) endogenous PGI2 production mediates nonneuronal COX-2-dependent neuroprotection; 2) PGI2 is produced by a subset of infiltrating neutrophils that populate the lesion core in response to excitotoxin administration; and 3) PGI2 can exert neuroprotective effects by directly reducing NMDA-mediated excess calcium influx. There is a significant clinical implication resulting from the observation that endogenous PGI2 has a neuroprotective effect: the PGIS inhibitor tranylcypromine is also an inhibitor of monoamine oxidase, and this drug is used clinically as an antidepressant;20 consequently, patients on tranylcypromine therapy may have an increased risk of exacerbated neural damage in response to acute brain injury.
The identification of PGIS-producing cells as a subset of neutrophils following a neurologic insult is, to our knowledge, a new observation. Previous studies have suggested that vascular cells are a major source of the PGI2 produced in response to neural injury.11 PGI2 is an effective vasodilator; one therapeutic mechanism suggested for the efficacy of PGI2-like agonists in treating acute brain injury has been their ability to improve cerebral blood flow to the injured tissue.21 Our findings add a previously undescribed, and unexpected, mechanism for the neuroprotective role of PGI2: the PGI2 produced near the injured neurons by infiltrating neutrophils may directly participate in neuronal rescue. This suggestion is supported by the observation that PGI2 can reduce TZG-induced neuronal Ca2+ influx in neurons in the slice preparation, where access to nutrients as a result of blood flow modulation by PGI2 agonists is not limiting. This suggestion is also supported by a previous report that a ligand for the neural specific PGI2 receptor can provide neuroprotection without affecting cerebral vascular PGI2 receptors.19 It should be noted that PGI2 could affect multiple calcium channels, to effect NMDA-induced calcium increases. Elucidating the mechanisms by which the direct effects of PGI2 on neurons are mediated will require extensive future studies. In addition, PGI2 may exert neuroprotective effect indirectly via its anti-inflammatory effects.22
The relationship, following excitotoxin administration, between the nonneuronal COX-2-expressing cells and the PGIS-expressing cells found in this study has not previously been observed and was, to us, surprising. Because we first found that PGI2 production in response to neurotoxin administration depended upon endothelial COX-2 expression, we presumed – as most current models suggest – that the elevated PGI2 must also be produced in these cells, as a consequent downstream product of COX-2 induction. However, our subsequent observations demonstrated that, instead, PGIS/PGI2 is produced by infiltrating neutrophils elicited by TZG administration. Thus, the endothelial COX-2-expression is indirectly connected to the PGI2 production; COX-2 in the endothelial cells plays a required inductive role to elicit PGI2 production by a second cell type, the infiltrating neutrophil. Moreover, COX-2 expression was not detected in these PGIS+ infiltrating neutrophils. These data suggest that, in the infiltrating neutrophils, COX-1, not COX-2, may synthesize the PGH2 precursor of PGI2. Neutrophils are a member of the myeloid series; their COX-2 expression should be eliminated by targeted deletion of the COX-2 gene in LysMCre COX-2flox/flox mice.23 Recall that TZG-induced neuroprotection is abrogated in Tie2Cre COX-2flox/flox mice, presumably as a consequence of COX-2 gene deletion in endothelial cells. In contrast, deletion of the COX-2 gene in myeloid cells in LysMCre COX-2flox/flox mice had no effect on the neuroprotection to excitotoxin administration that is eliminated either in Tie2Cre COX-2flox/flox mice or by low dose selective COX-2-inhibitor administration. These data support the suggestions that: 1) COX-1, rather than COX-2, is the cyclooxygenase necessary for neutrophil PGI2 production in response to excitotoxin; and 2) COX-2 expression in cells of the myeloid series is not necessary for neuroprotection in TZG-treated mice.
Two possibilities exist to explain why endothelial COX-2 expression is necessary for recruitment of PGIS-expressing neutrophils: 1) COX-2 expression in the injured vasculature gates the entry of PGIS-expressing cells to the brain parenchyma; or 2) COX-2 expression in the vasculature provides the necessary signal to induce PGIS expression in the neutrophils after they have entered the brain in response to excitotoxin. For either option, without COX-2 expression in the endothelial cells, PGIS-expressing neutrophils are not present and neuroprotection is abrogated. It remains to be determined which of these mechanisms is correct.
In summary, the endothelial COX-2 expression in the lesion core provides neuroprotection by facilitating the arrival of PGIS-expressing neutrophils; the PGI2 produced by these cells reduces excitotoxin-mediated excess calcium influx into affected neurons.
Acknowledgment
This study was supported by NIH funding RO1 AI076926 to NQ.
Disclosure
The authors report no conflicts of interest in this work.
References
Hara K, Kong DL, Sharp FR, Weinstein PR. Effect of selective inhibition of cyclooxygenase 2 on temporary focal cerebral ischemia in rats. Neurosci Lett. 1998;256:53–56. | |
Strauss KI, Barbe MF, Marshall RM, Raghupathi R, Mehta S, Narayan RK. Prolonged cyclooxygenase-2 induction in neurons and glia following traumatic brain injury in the rat. J Neurotrauma. 2000;17:695–711. | |
Koistinaho J, Chan PH. Spreading depression-induced cyclooxygenase-2 expression in the cortex. Neurochem Res. 2000;25:645–651. | |
Adams J, Collaço-Moraes Y, de Belleroche J. Cyclooxygenase-2 induction in cerebral cortex: an intracellular response to synaptic excitation. J Neurochem. 1996;66:6–13. | |
Kaufmann WE, Worley PF, Pegg J, Bremer M, Isakson P. COX-2, a synaptically induced enzyme, is expressed by excitatory neurons at postsynaptic sites in rat cerebral cortex. Proc Natl Acad Sci U S A. 1996;93:2317–2321. | |
An Y, Belevych N, Wang Y, et al. Neuronal and nonneuronal COX-2 expression confers neurotoxic and neuroprotective phenotypes in response to excitotoxin challenge. J Neurosci Res. 2014;92:486–495. | |
Sales KJ, Jabbour HN. Cyclooxygenase enzymes and prostaglandins in pathology of the endometrium. Reproduction. 2003;126:559–567. | |
Liang X, Wu L, Hand T, Andreasson K. Prostaglandin D2 mediates neuronal protection via the DP1 receptor. J Neurochem. 2005;92:477–486. | |
Cui Y, Takamatsu H, Kakiuchi T, et al. Neuroprotection by a central nervous system-type prostacyclin receptor ligand demonstrated in monkeys subjected to middle cerebral artery occlusion and reperfusion: a positron emission tomography study. Stroke. 2006;37:2830–2836. | |
Gryglewski RJ. Prostacyclin among prostanoids. Pharmacol Rep. 2008;60:3–11. | |
Fang YC, Wu JS, Chen JJ, et al. Induction of prostacyclin/PGI2 synthase expression after cerebral ischemia-reperfusion. J Cereb Blood Flow Metab. 2006;26:491–501. | |
Constien R, Forde A, Liliensiek B, et al. Characterization of a novel EGFP reporter mouse to monitor Cre recombination as demonstrated by a Tie2 Cre mouse line. Genesis. 2001;30:36–44. | |
Narasimha AJ, Watanabe J, Ishikawa TO, et al. Absence of myeloid COX-2 attenuates acute inflammation but does not influence development of atherosclerosis in apolipoprotein E null mice. Arterioscler Thromb Vasc Biol. 2010;30:260–268. | |
Ishikawa TO, Herschman HR. Conditional knockout mouse for tissue-specific disruption of the cyclooxygenase-2 (Cox-2) gene. Genesis. 2006;44:143–149. | |
Paxinos G, Franklin KB. The Mouse Brain in Stereotaxic Coordinates. Compact 2nd ed. Amsterdam; Boston: Elsevier Academic Press; 2004. | |
Huang Y, Smith DE, Ibáñez-Sandoval O, Sims JE, Friedman WJ. Neuron-specific effects of interleukin-1beta are mediated by a novel isoform of the IL-1 receptor accessory protein. J Neurosci. 2011;31:18048–18059. | |
Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol. 2008;83:64–70. | |
Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8(4):265–277. | |
Takamatsu H, Tsukada H, Watanabe Y, et al. Specific ligand for a central type prostacyclin receptor attenuates neuronal damage in a rat model of focal cerebral ischemia. Brain Res. 2002;925:176–182. | |
Frieling H, Bleich S. Tranylcypromine: new perspectives on an “old” drug. Eur Arch Psychiatry Clin Neurosci. 2006;256:268–273. | |
Lundblad C, Grände PO, Bentzer P. Increased cortical cell loss and prolonged hemodynamic depression after traumatic brain injury in mice lacking the IP receptor for prostacyclin. J Cereb Blood Flow Metab. 2008;28:367–376. | |
Wilson SM, Shen P, Rider CF, et al. Selective prostacyclin receptor agonism augments glucocorticoid-induced gene expression in human bronchial epithelial cells. J Immunol. 2009;183:6788–6799. | |
Luyendyk JP, Flanagan KC, Williams CD, et al. Tissue factor contributes to neutrophil CD11b expression in alpha-naphthylisothiocyanate-treated mice. Toxicol Appl Pharmacol. 2011;250:256–262. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.