Back to Journals » Drug Design, Development and Therapy » Volume 15
Prompt Agalsidase Alfa Therapy Initiation is Associated with Improved Renal and Cardiovascular Outcomes in a Fabry Outcome Survey Analysis
Authors Hughes D, Linhart A, Gurevich A, Kalampoki V, Jazukeviciene D, Feriozzi S ![]()
Received 13 April 2021
Accepted for publication 28 July 2021
Published 16 August 2021 Volume 2021:15 Pages 3561—3572
DOI https://doi.org/10.2147/DDDT.S313789
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Tin Wui Wong
Derralynn Hughes,1 Aleš Linhart,2 Andrey Gurevich,3 Vasiliki Kalampoki,3 Dalia Jazukeviciene,3 Sandro Feriozzi4 On behalf of the FOS Study Group
1University College London and Royal Free London NHS Foundation Trust, London, UK; 2Charles University, First Faculty of Medicine, Prague, Czech Republic; 3Takeda Pharmaceuticals International AG, Zurich, Switzerland; 4Nephrology and Dialysis Unit, Belcolle Hospital, Department of Nephrology and Dialysis, Viterbo, Italy
Correspondence: Derralynn Hughes
University College London and Royal Free London NHS Foundation Trust, Pond Street, London, NW3 2QG, UK
Email [email protected]
Background: The timing of enzyme replacement therapy initiation in patients with Fabry disease is hypothesized to be critical. In this study, we used Fabry Outcome Survey data to assess the impact of prompt versus delayed initiation of treatment with agalsidase alfa on cardiovascular and renal events in patients with Fabry disease.
Methods: Available genetic data at baseline were used to define patients with mutations associated with classical versus late-onset Fabry disease. Time to cardiovascular or renal events, from treatment initiation until 120 months, was compared for patients in prompt versus delayed groups. “Prompt” was defined as treatment initiation < 24 months from symptom onset (analysis A) or diagnosis (analysis B), and “delayed” was defined as ≥ 24 months from symptom onset (analysis A) or diagnosis (analysis B). Kaplan–Meier curves and Log rank tests compared event-free probabilities and time to first event. Multivariate Cox regression estimated hazard ratios (HRs).
Results: Analysis by time from symptom onset included 1374 patients (172 prompt, 1202 delayed). In a multivariate Cox regression analysis, prompt versus delayed treatment initiation significantly reduced the probability of cardiovascular (HR=0.62; P< 0.001) and renal (HR=0.57; P=0.001) events. History of cardiovascular or renal events was associated with increased risk of respective events. Analysis by time from diagnosis included 2051 patients (1006 prompt, 1045 delayed). In a multivariate Cox regression analysis, prompt treatment initiation significantly reduced the probability of cardiovascular events (HR=0.83; P=0.003) after adjusting for history of cardiovascular events, sex, and age at treatment initiation. Univariate analysis showed that the probability of renal events was significantly lower in the prompt group (P=0.018); this finding was attenuated in the multivariate Cox regression analysis.
Conclusion: This analysis suggests that prompt treatment initiation with agalsidase alfa provided better renal and cardiovascular outcomes than delayed treatment in patients with Fabry disease.
Keywords: cardiomyopathies, nephrology, mutation, therapeutics, early diagnosis
Introduction
Fabry disease is an X-linked lysosomal storage disorder caused by deficiency of the alpha-galactosidase A enzyme.1 The disease is characterized by progressive systemic involvement, with heterogeneous manifestations including acroparesthesia and abdominal pain, hypohidrosis, development of angiokeratomas, cardiomyopathy, cerebrovascular complications, and impaired renal function.2
Enzyme replacement therapy (ERT) with agalsidase alfa or agalsidase beta has been shown to stabilize and, in some cases, improve several signs and symptoms of Fabry disease.3–8 However, there is ongoing discussion in the field as to what the earliest timepoint should be for ERT treatment start in Fabry disease. Prior to 2001 and ERT availability, renal disease was the most common cause of death in patients with Fabry disease. However, after 2001 when ERT became available, the primary cause of death in both male and female patients became cardiovascular involvement, reflecting changes in the outcome of the underlying Fabry disease and especially improvements in the supportive management of renal disease.1 However, challenges in diagnosis owing to high variability in organ involvement, severity of symptoms, and age of onset can result in delayed initiation of therapy after the occurrence of substantial and irreversible organ damage or its more subtle precursors, which commit the organs to irreversible change.9,10
Recent evidence suggests that early initiation of ERT, prior to the onset of severe organ damage, may improve outcomes.4,10–14 ERT has been shown to stabilize renal function, especially when it is initiated before severe renal disease has developed.4,14–17 ERT is also associated with attenuated progression,3,4,13,14 or even some regression,5,17 of Fabry-associated hypertrophic cardiomyopathy; and in patients without LVH at baseline, ERT with agalsidase alfa stabilizes left ventricular mass indexed to height (LVMI).3,17
Although clinical trials are unsuited to the evaluation of long-term outcomes after delayed initiation of therapy, disease registries can provide a valuable source of longitudinal data from patients treated in real-world clinical practice. The Fabry Outcome Survey (FOS; ClinicalTrials.gov NCT03289065; sponsored by Shire, a Takeda company) is an ongoing worldwide disease registry that has over 20 years of data from treated and untreated patients with a confirmed diagnosis of Fabry disease. Until 2016, patients either untreated or treated with agalsidase alfa were eligible to participate in FOS. A protocol amendment in 2016, however, allowed any patients with Fabry disease, irrespective of treatment status (ie, no treatment or any approved Fabry treatment), to be eligible for enrolment in FOS.
The question remains whether there is benefit to prompt treatment in patients with Fabry disease. Thus, we sought to answer whether there is a greater effect of ERT in patients who start treatment promptly after diagnosis or at detection of the first symptoms in comparison with those patients with delayed treatment. The present retrospective study therefore uses data from the FOS registry to determine the potential benefits of prompt versus delayed initiation of ERT on cardiovascular and renal outcomes in Fabry disease.
Methods
Patients enrolled in FOS who had been treated with agalsidase alfa only and had available dates for agalsidase alfa initiation and Fabry disease symptom onset and/or diagnosis were included in this analysis. Patients were enrolled in FOS on a voluntary basis and were managed under the direction of their physician in accordance with routine clinical practice. FOS data used for these analyses were extracted for the period from database inception in 2001 to August 3, 2019. FOS was approved by the ethics institutional review boards of the participating centers. All participants gave written informed consent.
Two analyses were conducted: analysis A was based on the time of Fabry disease symptom onset, and analysis B was based on the time of Fabry disease diagnosis. For each analysis, patients were stratified by the magnitude of time delay between Fabry disease symptom onset or diagnosis and agalsidase alfa initiation. The prompt treatment cohorts included patients who had initiated agalsidase alfa within 24 months of the recorded date of symptom onset or diagnosis; the delayed treatment cohorts included patients who had initiated agalsidase alfa ≥24 months after the recorded date of symptom onset or diagnosis.
Assessments
Data on patient demographics, family history, clinical characteristics, and renal and cardiovascular endpoints were collected via the FOS registry’s web-based electronic case report form. To explore the impact of phenotype on time to treatment initiation, classical versus late-onset Fabry disease was determined for those patients with both available genetic data and a signed genetic informed consent form (ICF). Renal events included dialysis (peritoneal dialysis, hemodialysis, or unspecified dialysis), renal transplantation, renal failure, and proteinuria (recorded as a sign or symptom in FOS). Cardiovascular events included heart failure, arrhythmia, cardiac surgery, conduction abnormality, LVH, and myocardial infarction (recorded as a sign or symptom in FOS). LVM was calculated from linear measurements of septum, posterior wall, and cavity diameter as evaluated by investigators using the Devereux-modified American Society of Echocardiography cube formula.18 LVMI was calculated by correcting LVM to height2.7.19,20 eGFR was estimated by using the Chronic Kidney Disease Epidemiology Collaboration equation.21,22 History of cardiovascular and/or renal events refers to respective events that occurred prior to the date, or at the latest date, of agalsidase alfa initiation.
Statistical Analyses
Patient demographics, family history, and clinical characteristics at baseline were compared between prompt and delayed treatment cohorts using the Chi-square test and t-test, as appropriate. We also analyzed the patient population by phenotype group: classical versus late-onset Fabry disease. Baseline was defined as the date closest to agalsidase alfa initiation within a window of –6 to +3 months. Analysis of time to an event, from agalsidase alfa initiation until 120 months, was performed separately for each type of event (cardiovascular or renal). Kaplan-Meier curves and Log rank tests were used to compare event-free probabilities and time to first cardiovascular or renal event between the two treatment cohorts. Patients without an event reported during the study were censored at 120 months from agalsidase alfa initiation, and patients without a visit date recorded after agalsidase alfa initiation were censored either at the date of their last visit or at 120 months from agalsidase alfa initiation, whichever came first. In addition, multivariate Cox regression analyses were applied to examine the association between key study parameters—namely, age at agalsidase alfa initiation, sex, prompt versus delayed cohort, history of cardiovascular or renal events, and the risk of a cardiovascular or renal event. Hazard ratios (HRs) and their 95% confidence intervals (CIs) were estimated. The level of statistical significance was set at 0.05. All statistical analyses were performed using SAS v9.4 (SAS Institute Inc., Cary, NC, USA).
Results
Analysis A: Prompt and Delayed Treatment Since Symptom Onset
A total of 1374 patients had available data for the dates of both symptom onset and treatment initiation and were included in this analysis (Table 1). Of these, 172 (12.5%) patients started treatment within 24 months of the date of symptom onset (prompt treatment cohort), and 1202 (87.5%) patients started treatment ≥24 months after symptom onset (delayed treatment cohort). The prompt and delayed treatment cohorts had a similar distribution of male and female patients and a similar proportion of patients with a family history of Fabry disease, but patients in the prompt treatment cohort were older at symptom onset than the delayed treatment cohort (P<0.001), younger at treatment initiation (P=0.008), and had a shorter duration between symptom onset and treatment initiation (P<0.001; Table 1). A total of 31.4% of patients in the prompt treatment cohort had a history of a cardiovascular event versus 48.3% of patients in the delayed treatment cohort, whereas 19.8% of patients in the prompt treatment cohort had a history of a renal event versus 39.0% of patients in the delayed treatment cohort (both P<0.001).
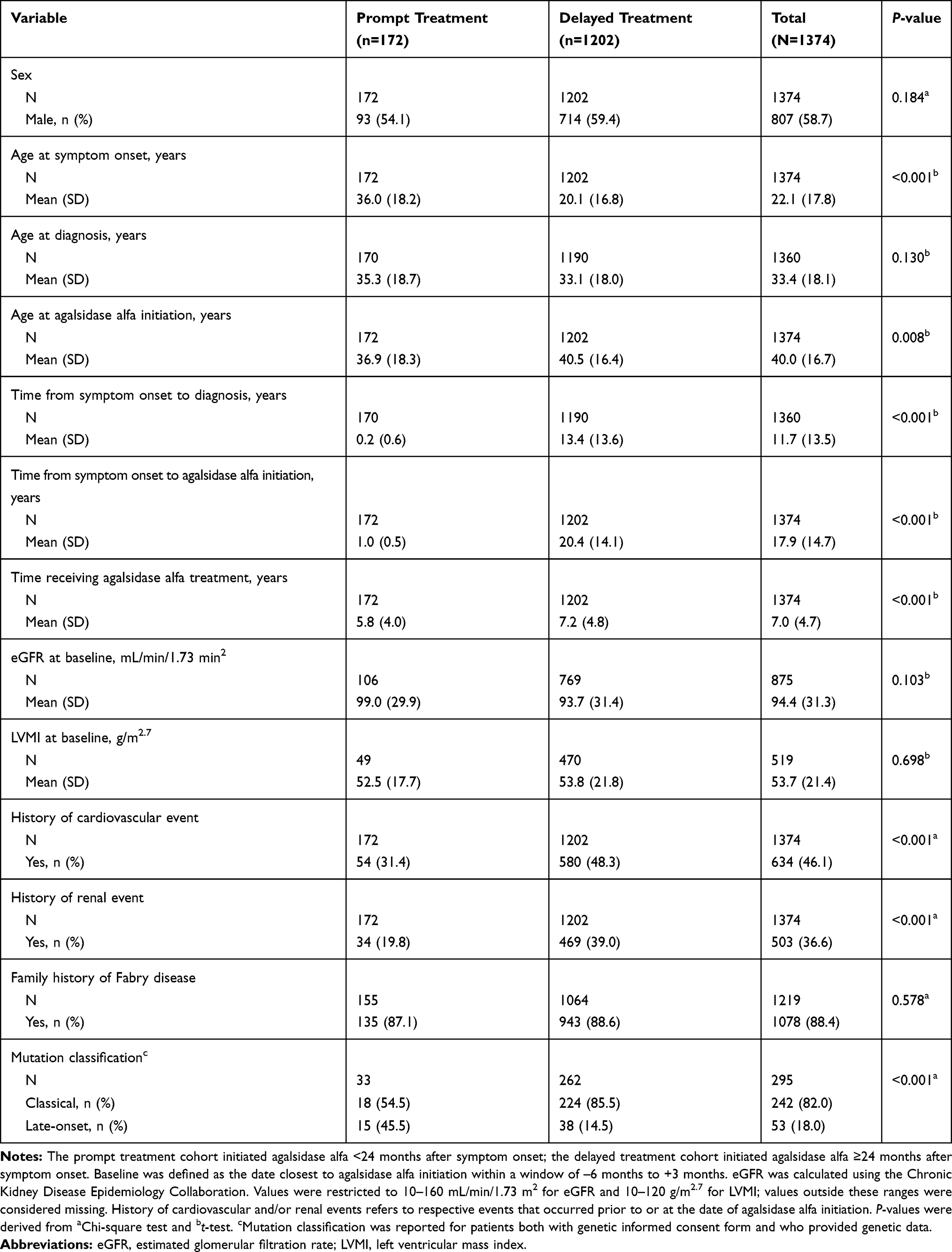 |
Table 1 Baseline Demographic and Clinical Characteristics for Prompt versus Delayed Agalsidase Alfa Initiation Cohorts, Based on Time from Symptom Onset (Analysis A) |
According to time-to-event analysis, median (95% CI) time to first cardiovascular event for patients in the prompt treatment cohort was 111.7 (72.9–not calculated) months compared with 31.6 (24.1–36.8) months for patients in the delayed treatment cohort. A Log rank test showed that prompt treatment initiation after symptom onset was associated with a significantly lower risk of cardiovascular events compared with delayed treatment initiation (log-rank P<0.001; Figure 1A). There were also significant differences for prompt versus delayed treatment in time to first cardiovascular event when male and female patients were analyzed separately (male P=0.0024; Supplemental Figure 1; female P<0.001; Supplemental Figure 2).
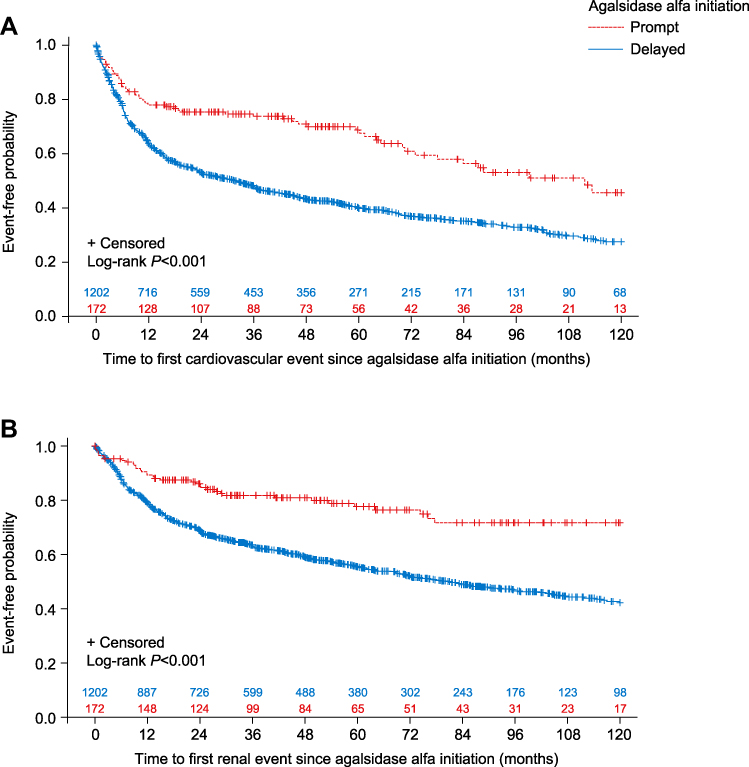 |
Figure 1 Kaplan-Meier curves with Log rank test showing (A) time to first cardiovascular event and (B) time to first renal event for prompt versus delayed agalsidase alfa initiation cohorts, based on time from symptom onset (analysis A). |
According to time-to-event analysis, median (95% CI) time to first renal event was not reached for patients in the prompt treatment cohort, whereas for the delayed treatment cohort it was estimated at 81.3 (69.1–95.7) months. A Log rank test indicated that prompt agalsidase alfa initiation after symptom onset was associated with a significantly lower risk of renal events compared with delayed treatment initiation (log-rank P<0.001; Figure 1B). Significant differences in prompt versus delayed treatment in time to first renal event were also observed when male (log-rank P<0.001; Supplemental Figure 3) and female patients (log-rank P=0.0033; Supplemental Figure 4) were analyzed separately.
In accordance with the univariate findings of the Log rank testing, multivariate Cox regression analyses demonstrated that prompt treatment initiation was associated with a significant benefit in the reduction of both cardiovascular (HR=0.62; 95% CI 0.48–0.81; P<0.001) and renal (HR=0.57; 95% CI 0.41–0.80; P=0.001) events compared with delayed treatment initiation (Table 2). The risk of a cardiovascular or renal event was found to increase significantly in patients with a history of respective cardiovascular or renal events (P<0.001 for each). Male patients and patients with older age at agalsidase alfa initiation had a higher risk of experiencing a cardiovascular event, but not a renal event.
 |
Table 2 Multivariate Cox Regression Analyses to Examine the Risk of Cardiovascular and/or Renal Events for Prompt versus Delayed Agalsidase Alfa Initiation Cohorts, Based on Time from Symptom Onset (Analysis A) |
At baseline, 295 patients had both available genetic data and a signed genetic ICF to assess classical versus late-onset Fabry disease; 242 (82.0%) patients had classical Fabry disease and 53 (18.0%) had late-onset Fabry disease (Table 1). Patients receiving prompt treatment had a higher mean age at symptom onset than patients receiving delayed treatment for both classical (30.3 vs 16.7 years, P<0.001) and late-onset phenotypes (54.4 vs 43.7 years, P=0.025), and a mean time from symptom onset to treatment initiation versus those with delayed treatment of 0.9 vs 22.4 years (P<0.001) for the classical phenotype and 0.9 vs 13.4 years (P=0.002) for the late-onset phenotype (Supplementary Table 1).
At baseline, patients with classical disease had similar eGFR and LVMI compared with patients with late-onset disease, irrespective of treatment timing. A greater proportion of late-onset patients than classical patients had a history of cardiovascular events, but not renal events, at baseline. There were no differences in the number of patients with family history of Fabry disease by genotype or treatment group.
Analysis B: Prompt and Delayed Treatment Since Diagnosis
A total of 2051 patients had available data for the dates of both diagnosis and treatment initiation and were included in this analysis (Table 3). Of these patients, 1006 (49.0%) started treatment within 24 months of diagnosis (prompt treatment cohort) and 1045 (51.0%) started treatment ≥24 months after diagnosis (delayed treatment cohort). Male and female patients were similarly distributed in the prompt and delayed treatment cohorts, and although mean (SD) age at treatment initiation was similar for both cohorts, mean age at symptom onset and mean age at diagnosis were both significantly higher in the prompt versus the delayed treatment cohort (both P<0.001). A total of 38.3% of patients in the prompt treatment cohort had a history of a cardiovascular event versus 44.9% of patients in the delayed treatment cohort, whereas 27.1% of patients in the prompt treatment cohort had a history of a renal event versus 33.8% of patients in the delayed treatment cohort.
 |
Table 3 Baseline Demographic and Clinical Characteristics for Prompt versus Delayed Agalsidase Alfa Initiation Cohorts, Based on Time from Diagnosis (Analysis B) |
Median (95% CI) time to first cardiovascular event, as assessed by time-to-event analysis, was 60.3 (50.3–72.9) months for patients in the prompt treatment cohort versus 43.0 (34.4–54.8) months for those in the delayed treatment cohort. A Log rank test indicated a significant cardiovascular risk difference between patients given prompt versus delayed treatment (log-rank P=0.002; Figure 2A). There were also significant differences for prompt versus delayed treatment in time to first cardiovascular event for female patients (P=0.0015; Supplemental Figure 6) but not for male patients (P=0.2047; Supplemental Figure 5). Multivariate Cox regression analysis also found that prompt treatment initiation was associated with a significant effect in reducing the risk of cardiovascular events (HR=0.83; 95% CI 0.74–0.94; P=0.003). Being male, having a history of cardiovascular events, and older age at agalsidase alfa initiation were all found to significantly increase the risk of experiencing a cardiovascular event (Table 4).
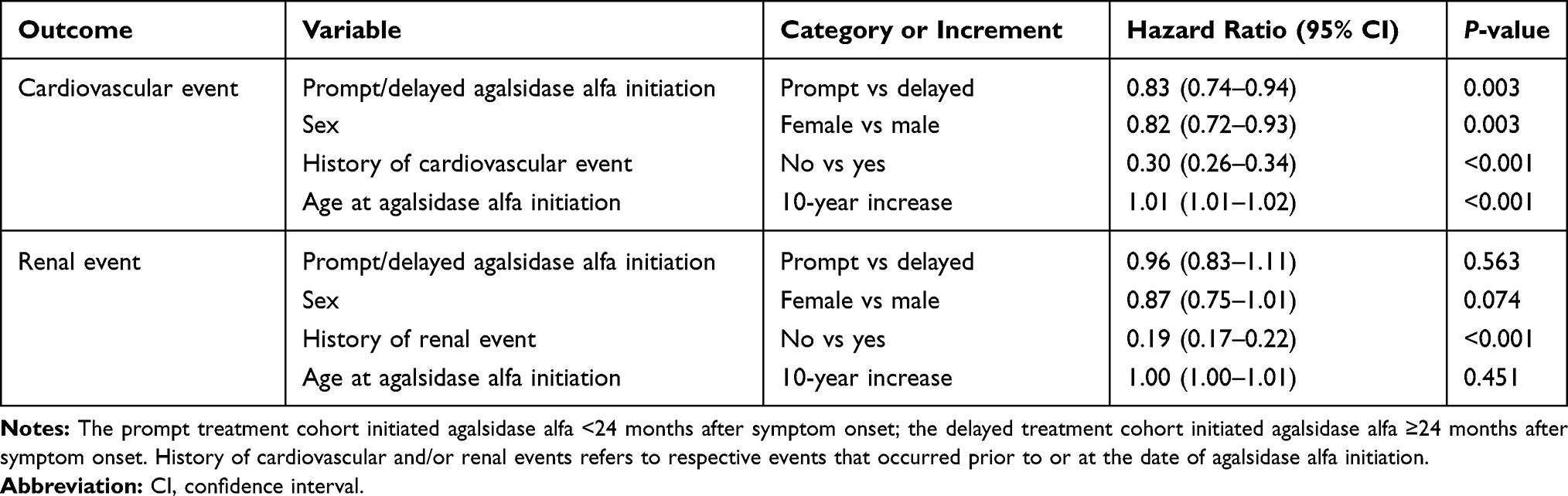 |
Table 4 Multivariate Cox Regression Analyses to Examine the Risk of Cardiovascular and/or Renal Events for Prompt versus Delayed Agalsidase Alfa Initiation Cohorts, Based on Time from Diagnosis (Analysis B) |
 |
Figure 2 Kaplan-Meier curves with Log rank test showing (A) time to first cardiovascular event and (B) time to first renal event for prompt versus delayed agalsidase alfa initiation cohorts, based on time from diagnosis (analysis B). |
Median time to first renal event was not reached for the prompt treatment cohort compared with a median (95% CI) of 104.0 (88.4–not calculated) months for the delayed treatment cohort. However, a Log rank test indicated a significant difference between prompt versus delayed treatment cohorts (log-rank P=0.018; Figure 2B). There were also significant differences for prompt versus delayed treatment in time to first renal event in both male (P=0.0182; Supplemental Figure 7) and female (P=0.0184; Supplemental Figure 8) patients. Univariate Cox regression analysis found a significantly lower risk of a renal event in the prompt treatment cohort versus the delayed treatment cohort (HR=0.84; 95% CI 0.73–0.97; P=0.018; data not shown). However, this finding was attenuated in the multivariate Cox regression analysis (HR=0.96; 95% CI 0.83–1.11; P=0.563; Table 4), possibly indicating that the overall approximate 6-year median delay between symptom onset and diagnosis of Fabry disease can negatively influence long-term patient outcomes. History of renal events was highly associated with an increasing risk of further renal events, whereas sex and age at agalsidase alfa initiation were not found to have any significant effect.
At baseline, 445 patients had both available genetic data and a signed genetic ICF to assess classical versus late-onset Fabry disease—224 in the prompt treatment group and 221 in the delayed treatment group (Table 3). Among patients with classical Fabry disease, mean time from diagnosis to treatment initiation was 13.0 years for the delayed treatment group versus 0.8 years for the prompt treatment group (P<0.001) compared with 5.1 and 1.0 years, respectively, for patients with late-onset Fabry disease (P<0.001; Supplementary Table 2).
LVMI was similar for patients with classical and late-onset disease at baseline, irrespective of treatment timing (P=0.386 and 0.168, respectively; Supplementary Table 2). For patients with classical disease, there was no difference in baseline eGFR between prompt and delayed treatment groups, but a greater proportion of patients in the delayed treatment group had a history of cardiovascular or renal events versus the prompt treatment group (P<0.001 and P=0.027, respectively). For patients with late-onset disease, those receiving prompt treatment had lower eGFR at baseline than patients receiving delayed treatment, but there were no differences in the proportion of patients having a history of cardiovascular or renal events. As with analysis A, there were no differences in the number of patients with a family history of Fabry disease by genotype or treatment group (Supplementary Table 2).
Discussion
The present analysis from the FOS has shown that prompt initiation of agalsidase alfa ERT (<24 months after Fabry disease symptom onset or diagnosis) was associated with a significant reduction in the risk of cardiovascular and renal events compared with delayed initiation (≥24 months after symptom onset or diagnosis). Significant differences for prompt versus delayed treatment were also observed when analyzing male and female patients separately.
Several previous studies have reported treatment outcomes with early versus delayed initiation of therapy.11,13,14,23,24 Notably, these previous studies used an absolute age range as the reference point for treatment start. In contrast, the present analysis is the first study to use a relative time interval in relation to symptom start, and this method is potentially more precise to assess when to begin treatment because disease progression occurs at different ages in patients with Fabry disease. In the present analysis, the patient populations analyzed had overall mean ages of 40.0 (analysis A) and 41.0 (analysis B) years at ERT initiation. The prompt treatment group in analysis A, however, was significantly younger than the delayed treatment group (36.9 vs 40.5 years at ERT initiation; P=0.008). In comparison, a 2020 analysis of FOS data including 560 male patients found that cardiovascular and renal disease progression was attenuated when patients started ERT in childhood (≤18 years of age) or early adulthood (>18 to ≤30 years of age) versus patients who were >30 years of age when starting ERT.14 Likewise, a smaller study of 85 males with classical Fabry disease showed that ERT initiation before 25 years of age was associated with significantly greater reductions in plasma globotriaosylsphingosine levels than later initiation of ERT, indicating a notable biochemical response to early treatment.24 In a study of 52 patients with Fabry disease who had received ERT for approximately 10 years, patients with low renal involvement were found to have initiated ERT at an earlier age than those with high renal involvement (25 vs 38 years of age, respectively) and experienced lower rates of disease progression.11 Further, ERT appeared to stabilize the progression of myocardial involvement in patients who initiated treatment before 40 years of age. ERT was also shown to stabilize or improve LVH in male patients with Fabry disease when treatment was initiated at the age of 18–30 years, in contrast with progression in males who started ERT at ≥40 years of age, and the authors suggested that initiating ERT before proteinuria develops may be an important factor to prevent the progression of renal disease.23 Altogether, the published data along with the results of the present analysis suggest that treatment of Fabry disease should be initiated early (ie, at a younger age) and promptly (within 2 years of symptom onset or diagnosis) to mitigate disease burden.
To explore the impact of Fabry disease phenotype on prompt versus delayed treatment, we classified patients at baseline by the presence of mutations associated with classical or late-onset Fabry disease. We observed that significant delays in time from symptom onset to diagnosis and treatment initiation were apparent for both classical and late-onset patients in both analysis A and analysis B (Supplementary Tables 1 and 2). In analysis A, there was no difference in age at diagnosis between the prompt and delayed treatment groups, although patients with late-onset Fabry disease were >30 years older at diagnosis than those with classical Fabry disease (Supplementary Table 1). In analysis B (Supplementary Table 2), patients in the prompt treatment group were older at diagnosis than patients in the delayed treatment group for both classical and late-onset Fabry disease, possibly suggestive of a greater urgency to treat older patients promptly, although this group may not be as well defined. The differences in outcomes between analyses A and B suggest that “prompt” determined by time of diagnosis (ie, analysis B) is subject to many different influences, whereas “prompt” determined by time of first symptoms (ie, analysis A) better reflects the disease progression of the individual patient and thus reveals a more realistic view of what can be expected in the treatment of patients with Fabry disease.
The heterogeneity, severity, and variations in age at disease manifestation complicate the prompt diagnosis and treatment of Fabry disease. Initiation of treatment is typically in response to signs and symptoms, including pain and gastrointestinal symptoms, and test results such as increased LVM or reduced GFR. Age and gender may also influence the patient’s willingness to start treatment, and country and regional guidelines may dictate the decision to treat.25 A previous study has shown the significant disease burden in younger patients and the potential early emergence of signs and symptoms of Fabry disease; however, not all children are symptomatic.25 Rather, factors such as family history and mutation analysis of individuals with possible disease may inform the decision of when to treat. Family history of Fabry disease was present in more than 88% of the patient population in this analysis. Furthermore, studies have shown that progression of Fabry disease occurs despite the use of ERT when organ damage, indicative of advanced Fabry disease, is present at ERT start.26–28 In this analysis, 40% and 30% of patients had a history of cardiovascular or renal events, respectively, and a history of an event significantly increased the risk of a subsequent event. Early investigation and prompt treatment initiation in patients with Fabry disease may decrease or delay the occurrence of an initial cardiovascular or renal event thereby potentially improving renal and cardiovascular outcomes in these patients.
There are several limitations of the current analysis that should be considered. This was a retrospective analysis of data from the FOS disease registry database. As such, the patients in the FOS registry were not randomly selected, which may have led to selection bias. As a result, the findings may not be generalizable to all patients with Fabry disease. In FOS, because there is no centralized reading of echocardiograms, their interpretation by the investigator may introduce bias. Additionally, this analysis did not investigate other factors involved in renal or cardiovascular disease progression, such as the use of angiotensin-converting enzyme inhibitors, use of angiotensin II receptor blockers, or blood pressure control. Determining the timings of symptom onset and diagnosis can be susceptible to inaccuracy; for example, identification of the date of symptom onset may be dependent on patient memory, whereas diagnosis date can be influenced by family screening programs that may lead to diagnosis before symptom onset in some patients. Another consideration is the heterogeneity of phenotypes in Fabry disease—patients with the late-onset phenotype may not report signs and symptoms until a relatively older age, and so may still be at an early stage of disease progression at an older age than a patient with the classical form of the disease. Lastly, there may be many reasons for a delay in treatment (eg, patients may not have been referred to a specialist center or there may have been a need to see a clinical worsening before treatment was initiated), which are not captured in this retrospective database. Despite these limitations, this analysis of real-world data in a large population of patients with Fabry disease has shown that prompt treatment with agalsidase alfa significantly reduces the probability of cardiovascular and renal events.
Conclusions
This is the first analysis in Fabry disease that assesses the influence of prompt treatment as defined by time between symptom onset/diagnosis and treatment start. Our analysis of real-world data of patients from the FOS registry shows that prompt initiation of agalsidase alfa treatment can result in improved cardiovascular and renal outcomes in patients with Fabry disease, in alignment with previous, smaller studies, which focused on age at treatment start. Moreover, time between symptom onset and diagnosis, sex, history of cardiovascular or renal events prior to treatment initiation, and age at symptom onset may be important factors contributing to long-term outcomes. However, further analyses are needed to better understand these relationships and confirm these findings. The results of the current analysis suggest that there may be significant benefits with prompt initiation of agalsidase alfa after symptom onset and/or diagnosis of Fabry disease.
Abbreviations
CI, confidence interval; eGFR, estimated glomerular filtration rate; ERT, enzyme replacement therapy; FOS, Fabry Outcome Survey; HR, hazard ratio; ICF, informed consent form; LVH, left ventricular hypertrophy; LVM, left ventricular mass; LVMI, left ventricular mass index.
Data Sharing Statement
The datasets, including the redacted study protocol, redacted statistical analysis plan, and individual participant data supporting the results reported in this article, will be made available within three months from initial request, to researchers who provide a methodologically sound proposal. The data will be provided after its de-identification, in compliance with applicable privacy laws, data protection and requirements for consent and anonymization.
Ethics Approval and Informed Consent
FOS was approved by the ethics institutional review boards of the participating centers. Further, this registry was compliant with relevant global and local regulations and best practices: Good Pharmacoepidemiological Practice and Good Research for Comparative Effectiveness principles. The relevant principles of the International Council for Harmonisation Good Clinical Practice (ICH GCP) guidelines (ICH E6) were followed as appropriate for an observational registry, consistent with the Declaration of Helsinki. All participants gave written informed consent.
Patient Consent for Publication
Patient consent for publication was not required.
Acknowledgments
Jaco Botha, employee of Takeda Pharmaceuticals International AG, provided additional statistical support. Under the direction of the authors, Latoya M. Mitchell, PhD, CMPP, employee of Excel Medical Affairs, provided writing assistance for this article. Editorial assistance in formatting, proofreading, copy editing, fact-checking, and preparation of the manuscript for submission was provided by Excel Medical Affairs. Takeda Development Center Americas, Inc., provided funding to Excel Medical Affairs for support in writing and editing this article and participated in data collection and analysis. Portions of these data were previously presented at the 16th Annual WORLDSymposium 2020, February 10–13, 2020, Orlando, FL, USA; the 13th Annual WORLDSymposium 2017, February 13–17, 2017, San Diego, CA, USA; the European Society of Cardiology (ESC) Annual Congress, August 26–30, 2017, Barcelona, Spain; and the Society for the Study of Inborn Errors of Metabolism (SSIEM) Annual Symposium, September 6–9, 2016, Rome, Italy.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
The Fabry Outcome Survey (FOS) is funded by Shire Human Genetic Therapies, a Takeda Company.
Disclosure
DH reports personal fees from Takeda, Sanofi Genzyme, Amicus Therapeutics, Idorsia, Protalix Biotherapeutics, and Freeline Therapeutics, outside the submitted work. AL reports personal fees from Amicus Therapeutics, Avrobio, Sanofi Genzyme, and Takeda, outside the submitted work. AG and VK were employees of Takeda at the time of the study. DJ is an employee of Takeda and is a stockholder of Takeda Pharmaceuticals Company Limited. SF reports personal fees from Takeda, Sanofi Genzyme, and Amicus Therapeutics, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Mehta A, Clarke JTR, Giugliani R, et al. Natural course of Fabry disease: changing pattern of causes of death in FOS – Fabry Outcome Survey. J Med Genet. 2009;46(8):548–552. doi:10.1136/jmg.2008.065904
2. Mehta A, Ricci R, Widmer U, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest. 2004;34(3):236–242. doi:10.1111/j.1365-2362.2004.01309.x
3. Mehta A, Beck M, Elliott P, et al. Enzyme replacement therapy with agalsidase alfa in patients with Fabry’s disease: an analysis of registry data. Lancet. 2009;374(9706):1986–1996. doi:10.1016/S0140-6736(09)61493-8
4. Beck M, Hughes D, Kampmann C, et al. Long-term effectiveness of agalsidase alfa enzyme replacement in Fabry disease: a Fabry Outcome Survey analysis. Mol Genet Metab Rep. 2015;3:21–27. doi:10.1016/j.ymgmr.2015.02.002
5. Hughes DA, Elliott PM, Shah J, et al. Effects of enzyme replacement therapy on the cardiomyopathy of Anderson–Fabry disease: a randomised, double-blind, placebo-controlled clinical trial of agalsidase alfa. Heart. 2008;94(2):153–158. doi:10.1136/hrt.2006.104026
6. Fellgiebel A, Gartenschläger M, Wildberger K, Scheurich A, Desnick RJ, Sims K. Enzyme replacement therapy stabilized white matter lesion progression in Fabry disease. Cerebrovasc Dis. 2014;38(6):448–456. doi:10.1159/000369293
7. Schiffmann R, Kopp JB, Austin HA, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001;285(21):2743–2749. doi:10.1001/jama.285.21.2743
8. West M, Nicholls K, Mehta A, et al. Agalsidase alfa and kidney dysfunction in Fabry disease. J Am Soc Nephrol. 2009;20(5):1132–1139. doi:10.1681/ASN.2008080870
9. Reisin R, Perrin A, García‐Pavía P. Time delays in the diagnosis and treatment of Fabry disease. Int J Clin Pract. 2017;71:e12914. doi:10.1111/ijcp.12914
10. Ortiz A, Abiose A, Bichet DG, et al. Time to treatment benefit for adult patients with Fabry disease receiving agalsidase β: data from the Fabry Registry. J Med Genet. 2016;53(7):495–502. doi:10.1136/jmedgenet-2015-103486
11. Germain DP, Charrow J, Desnick RJ, et al. Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J Med Genet. 2015;52(5):353–358. doi:10.1136/jmedgenet-2014-102797
12. Hughes DA. Fabry disease: will markers of early disease enable early treatment and better outcomes? Curr Opin Cardiol. 2016;31(4):434–439. doi:10.1097/HCO.0000000000000308
13. Feriozzi S, Linhart A, Ramaswami U, et al. Effects of baseline left ventricular hypertrophy and decreased renal function on cardiovascular and renal outcomes in patients with Fabry disease treated with agalsidase alfa: a Fabry Outcome Survey Study. Clin Ther. 2020;42(12):2321–2330. doi:10.1016/j.clinthera.2020.10.007
14. Parini R, Pintos-Morell G, Hennermann JB, et al. Analysis of renal and cardiac outcomes in male participants in the Fabry Outcome Survey starting agalsidase alfa enzyme replacement therapy before and after 18 years of age. Drug Des Devel Ther. 2020;14:2149–2158. doi:10.2147/DDDT.S249433
15. Beck M, Ricci R, Widmer U, et al. Fabry disease: overall effects of agalsidase alfa treatment. Eur J Clin Invest. 2004;34(12):838–844. doi:10.1111/j.1365-2362.2004.01424.x
16. Feriozzi S, Torras J, Cybulla M, Nicholls K, Sunder-Plassmann G, West M. The effectiveness of long-term agalsidase alfa therapy in the treatment of Fabry nephropathy. Clin J Am Soc Nephrol. 2012;7(1):60–69. doi:10.2215/CJN.03130411
17. Hughes DA, Barba Romero M-Á, Hollak CEM, Giugliani R, Deegan PB. Response of women with Fabry disease to enzyme replacement therapy: comparison with men, using data from FOS—the Fabry Outcome Survey. Mol Genet Metab. 2011;103(3):207–214. doi:10.1016/j.ymgme.2011.03.022
18. Devereux RB, Alonso DR, Lutas EM, et al. Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings. Am J Cardiol. 1986;57(6):450–458. doi:10.1016/0002-9149(86)90771-X
19. Kampmann C, Linhart A, Baehner F, et al. Onset and progression of the Anderson–Fabry disease related cardiomyopathy. Int J Cardiol. 2008;130(3):367–373. doi:10.1016/j.ijcard.2008.03.007
20. de Simone G, Devereux RB, Daniels SR, Koren MJ, Meyer RA, Laragh JH. Effect of growth on variability of left ventricular mass: assessment of allometric signals in adults and children and their capacity to predict cardiovascular risk. J Am Coll Cardiol. 1995;25(5):1056–1062. doi:10.1016/0735-1097(94)00540-7
21. Stevens LA, Schmid CH, Greene T, et al. Comparative performance of the CKD Epidemiology Collaboration (CKD-EPI) and the Modification of Diet in Renal Disease (MDRD) Study equations for estimating GFR levels above 60 mL/min/1.73 m2. Am J Kidney Dis. 2010;56(3):486–495. doi:10.1053/j.ajkd.2010.03.026
22. Rombach SM, Smid BE, Bouwman MG, Linthorst GE, Dijkgraaf MGW, Hollak CEM. Long term enzyme replacement therapy for Fabry disease: effectiveness on kidney, heart and brain. Orphanet J Rare Dis. 2013;8:47. doi:10.1186/1750-1172-8-47
23. Germain DP, Weidemann F, Abiose A, et al. Analysis of left ventricular mass in untreated men and in men treated with agalsidase-β: data from the Fabry Registry. Genet Med. 2013;15(12):958–965. doi:10.1038/gim.2013.53
24. Arends M, Wijburg FA, Wanner C, et al. Favourable effect of early versus late start of enzyme replacement therapy on plasma globotriaosylsphingosine levels in men with classical Fabry disease. Mol Genet Metab. 2017;121(2):157–161. doi:10.1016/j.ymgme.2017.05.001
25. Ramaswami U, Parini R, Pintos-Morell G, Kalkum G, Kampmann C, Beck M. Fabry disease in children and response to enzyme replacement therapy: results from the Fabry Outcome Survey. Clin Genet. 2012;81(5):485–490. doi:10.1111/j.1399-0004.2011.01671.x
26. Warnock DG, Ortiz A, Mauer M, et al. Renal outcomes of agalsidase beta treatment for Fabry disease: role of proteinuria and timing of treatment initiation. Nephrol Dial Transplant. 2012;27(3):1042–1049. doi:10.1093/ndt/gfr420
27. Banikazemi M, Bultas J, Waldek S, et al. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med. 2007;146(2):77–86. doi:10.7326/0003-4819-146-2-200701160-00148
28. Weidemann F, Niemann M, Breunig F, et al. Long-term effects of enzyme replacement therapy on Fabry cardiomyopathy: evidence for a better outcome with early treatment. Circulation. 2009;119(4):524–529. doi:10.1161/CIRCULATIONAHA.108.794529
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.