")
Back to Journals » Eye and Brain » Volume 8
Promoting vascular repair in the retina: can stem/progenitor cells help?
Authors Trinh TLP, Li Calzi S, Shaw L, Yoder M, Grant M
Received 1 September 2015
Accepted for publication 4 December 2015
Published 26 May 2016 Volume 2016:8 Pages 113—122
DOI https://doi.org/10.2147/EB.S94451
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Margaret Wong-Riley
Thao Le Phuong Trinh,1 Sergio Li Calzi,1 Lynn C Shaw,1 Mervin C Yoder,2–4 Maria B Grant1
1Department of Ophthalmology, 2Department of Pediatrics, 3Herman B. Wells Center for Pediatric Research, 4Department of Biochemistry and Molecular Biology, Indiana University – Purdue University Indianapolis, Indianapolis, IN, USA
Abstract: Since its first epidemic in the 1940s, retinopathy of prematurity (ROP) has been a challenging illness in neonatology. Higher than physiological oxygen levels impede the development of the immature retinal neuropil and vasculature. Current treatment regimens include cryotherapy, laser photocoagulation, and anti-VEGF agents. Unfortunately, none of these approaches can rescue the normal retinal vasculature, and each has significant safety concerns. The limitations of these approaches have led to new efforts to understand the pathological characteristics in each phase of ROP and to find a safer and more effective therapeutic approach. In the era of stem cell biology and with the need for new treatments for ROP, this review discusses the possible future use of unique populations of proangiogenic cells for therapeutic revascularization of the preterm retina.
Keywords: retinopathy of prematurity, ROP therapy, endothelial progenitor cells, CD34+ cells, endothelial colony-forming cells, oxygen-induced retinopathy
Introduction
Retinopathy of prematurity (ROP) is a major cause of blindness in children worldwide.1,2 ROP causes approximately 14% of blindness in the US and more than 20% in developing countries.3 ROP only affects preterm newborns.4 Strikingly, the incidence of ROP has not changed significantly over the past 25 years, despite the fact that more resources have been devoted to the care of premature infants, such as pulse oximetry and surfactant antenatal steroids.1 ROP is associated with fluctuations in oxygen concentrations.4 The higher than physiological concentrations of oxygen that are needed to aid infant survival attenuate blood-vessel growth in the periphery, as well as the deep layers of the retina.5 Current treatments for ROP have targeted the more advanced stages, but none of the approaches is meant to resolve the underlying pathological defects, and inherently all current therapies carry adverse side effects. These therapies have been developed progressively with cryosurgery (1988) being developed first, then laser therapy (2003), and most recently intravitreal bevacizumab (2012).5
Mechanisms and current therapies for ROP
Mechanisms of vascular dysfunction
In order to understand the pathological events and how cell-based therapy works for ROP, the key features of normal retinal vasculature development need to be considered. Physiologic vasculogenesis of the retina begins in the posterior pole with the migration of precursor cells from the deep retina into inner layers, and is divided into different zones (Figure 1).1 Approximately between week 15 and week 22 of gestation, these precursor cells differentiate into angioblasts, forming the inner vascular plexus, which will almost reach zone I.4 This transforming process is relatively independent of VEGF.1 However, after week 22, blood vessels are believed to grow and proliferate through a VEGF-dependent “budding” process and to some extent by angioblast transformation to vascularize peripheral zones II and III.1,4
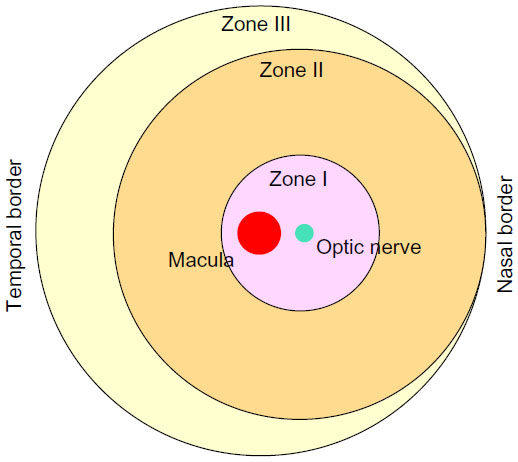 | Figure 1 Schematic of retinal zones. |
Phase I, or the vasocessation phase, begins with premature birth and supplemental oxygen given to premature infants at supraphysiological levels. The choroid is incapable of autoregulation, which results in the inner retina receiving excessive levels of oxygen. At this stage, the retinal outer segments, the most metabolically demanding cells in the body, have not appeared; therefore, the retina is under hyperoxia.1 The high oxygen concentration in turn downregulates hypoxia-induced VEGF, IGF-1, and CXCL-12 critical for normal vascularization.1,6,7 Diminished levels of vasogenic factors lead to delayed physiologic retinal vascular development, a key characteristic of the early phase of human ROP.4,8
Upon the return to ambient air, it is not clearly understood why one population of preterm infants do not develop intravitreous neovascularization (IVNV), also known as phase II or the neovascular phase of ROP, while the other group proceeds to abnormal neovascularization or serious ROP. However, it is appreciated that during this stage, the retina is likely to experience hypoxia, due to the tremendous rise in metabolism of the developing photoreceptor outer segments. Hypoxia itself can upregulate VEGF, which enhances angiogenesis and vasculogenesis.1
VEGF, formerly identified as “vascular permeability factor”, has increasingly been recognized to play a critical role in both physiologic as well as pathologic development of the vasculature, and is strongly associated with blood–retina barrier dysfuntion.4,9 In fact, one of the prominent features of ROP is the breakdown of the blood–retina barrier. Edema caused by ischemia and vascular damage may be the direct culprit of neural cell depletion and immune-cell activation.10 We also know that VEGF signaling regulates ischemia-induced hyperpermeability, endothelial cell division, and morphology of branching vessels.11,12 To reemphasize the significant role of hypoxia-induced factors (HIFs), such as VEGF, studies in animal models of oxygen-induced retinopathy (OIR) support the view that severe ROP is linked to different exogenous factors, including oxygen levels, inflammation, and nutritional status, but all through dysregulated signaling cascades involving HIFs.4
Current therapies and some reported pitfalls
Cryosurgery, used in the multicenter CRYO-ROP trial, works by ablating the avascular area of the peripheral retina to reduce the metabolic demand and the hypoxic level of retinal cells.1 Laser therapy was proved to be useful in the ET-ROP trial.1 The risk of poor prognosis in approximately 90% of eyes of severe ROP (or type 1 ROP), as in the ET-ROP study, may be lowered by laser treatment.4 However, generally ablation therapies tend to be costly, destructive, only reduce the risk of blindness by 25%, and may not prevent blindness particularly with zone I ROP.2,13 Laser photocoagulation was found to cause infectious ulcerative keratitis in a subgroup of ROP infants. The mechanism involved postoperative corneal epithelial defects that led to corneal haze.14 Another study of long-term ophthalmological outcomes in children treated for threshold ROP by indirect laser photocoagulation reported that these patients had higher risks of strabismus, astigmatism, nystagmus, myopia, and lowered visual acuity compared to the control subjects, infants with spontaneously regressed ROP.15
Most recently established (2012) is the use of anti-VEGF reagents like bevacizumab intravitreally. While injection of anti-VEGF into the vitreous body is not as an invasive procedure as the previous two therapies, the agent still raises significant concerns, especially in long-term outcome, because these agents can enter the systemic circulation.16 Intravitreally injected anti-VEGF agents have been found to diminish VEGF serum levels. Ironically, research has concluded that the use of anti-VEGF agents may cause intravitreal angiogenesis, retinal detachment, and continuing avascularized retina.3 In fact, late reactivation of ROP post-intravitreal anti-VEGF agents has been reported (Table 1).2,17–20
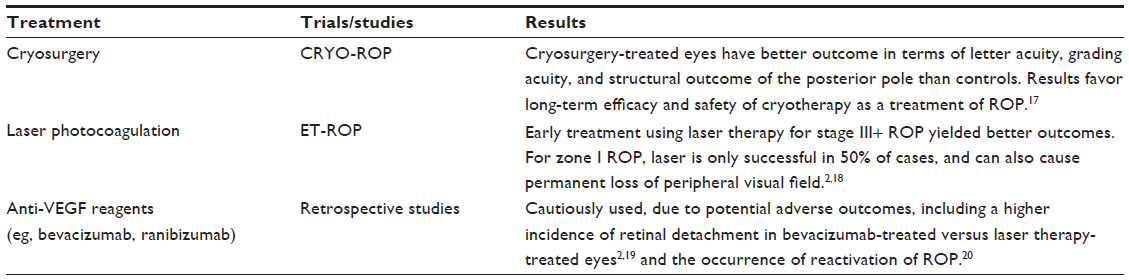 | Table 1 Conventional treatments for ROP and results from clinical studies |
Oxygen-induced retinopathy animal models
There are two commonly used OIR animal models: one using mice and the other rats in ROP research. It is noteworthy to remember that unlike humans, these species are born with an incomplete vascularized retina.4 In other words, for them the underdeveloped retinal vasculature is appropriate at birth. Comparison of the phases of OIR in the two models with human ROP stages is necessary. The early phase of human ROP is only reflected in the rat but not the mouse model, and is called phase I or “delayed physiologic retinal vascularization”.4 The vascular phase of stage III ROP with plus disease corresponds to phase II or the IVNV vasoproliferation phase in both models.4 A few animal models, such as the beagle OIR model, also have a third phase – the fibrovascular phase – that reflects the retinal fibroplasia and eventual detachment characteristics occurring during stages IV and V of human ROP.4 Furthermore, the mouse model demonstrates vaso-obliteration in phase I instead of “delayed physiologic retinal vascularization”.4
In vivo and in vitro studies: new therapeutic targets
Aligning with the approach of inhibiting pathological IVNV are studies that target VEGF and related factors like Ang-2 in angiogenesis. Ang-2, a destabilizing factor capable of controlling vessel regression, is expressed by the resting endothelium at low levels under physiologic conditions. However, both VEGF and Ang-2 are strongly upregulated by hypoxia.21 Zhao et al21 found an interesting interaction between VEGF and Ang-2 that was dependent on microRNAs (miRNAs). Among the miRNAs screened, miR-351 was found to downregulate both VEGF and Ang-2 in vitro and in vivo. The mechanism is believed to involve competitive endogenous RNAs, Ang-2 and VEGF competing for miR-351 through a competitive endogenous RNA- and miRNA-response elements. Therefore, miR-351 can potentially become a new target for ablation of retinal angiogenesis in ROP as well as diabetic retinopathy.21 Other miRNA studies have also reported miR-200b being able to reduce VEGF level in STZ-induced diabetic rats and miR-126 overexpression suppressing VEGF, HIF-1α, and IGF-2 for these reasons. Therefore, both miR-200b and miR-126 can reduce pathological neovascularization.22,23 One of the important characteristics of OIR models that has not been mentioned previously is microgliosis. Microglia, also known as the resident macrophages in the central nervous system and retina, are observed to be activated and accumulated at sites of tissue damage to release proinflammatory cytokines (eg, TNFα and IL-6), which may contribute to vascular dysfunction.23 Infiltration of microglia and monocytes/macrophages has been linked to neovascularization in OIR,25–27 and is also correlated with upregulation of proangiogenic and proinflammatory molecules, including metalloproteinases, FGF, and cytokines (ie, TNFα).28–30 In parallel with these observations, inflammation is actually a key difference between physiological and pathological angiogenesis.31 In fact, many types of proinflammatory cytokines, including TNFα and IL-6, exert angiogenic effects on the vascular endothelium via the JAK–STAT pathway.31 For example, elimination of SOCS3, an endogenous inhibitor of the JAK–STAT cascade, drives angiogenesis through both growth factor and cytokine secretion in tumor growth and OIR mice.31 Because of this phenomenon, Miyazaki et al used a compound called calpastatin in attempt to inhibit the degradation of SOCS3, and as a result they were able to suppress pathological angiogenesis. Nevertheless, the mechanism by which calpastatin acted on endothelial cells was not clearly delineated.31 On the other hand, using an inhibitor of VEGFR2 was found to decrease the length and filopodial number of endothelial tip cells. As a result, intravitreal but not intraretinal vascularization is lowered.32
Recall that ROP is associated with fluctuations in oxygen concentrations, the reason being that the fluctuations tend to produce reactive oxygen species.4 Increases in lipid hydroperoxide production have been found in OIR rat retinas.33 Taking advantage of this observation, Saito et al33 tested the effect of antioxidant compound N-acetylcysteine and the NADPH oxidase inhibitor apocynin on IVNV using the 50/10 OIR rat model. Results suggested that apocynin but not N-acetylcysteine could lessen the avascularized area and apoptosis in the retina, possibly involving pathways upstream from lipid hydroperoxide. However, neither had any effect on IVNV.33 In contrast, propranolol, originally used in the treatment of hemangiomas, was reported to be able to inhibit OIR neovascularization based on a mouse study that used nonstandard assessments (fluorescent angiography being unable to detect all neovascular tufts, especially those not fully perfused).13 To reevaluate this finding, Chen et al used a standard evaluation of staining vessels using specific endothelial cell markers to test the effect of propranolol delivered in three different ways – oral gavage, intraperitoneal injection, and subcutaneous injection – on an OIR mouse model. Unfortunately, the latter study proved that neither the doses nor route of delivery of propranolol prevented the development of retinopathy, and higher doses of intraperitoneal injection even intensified pathological vascularization. At the molecular level, propranolol did not alter VEGF expression.13
Cell-based therapy: the next generation of ROP treatment?
Endothelial progenitor cells: early- outgrowth EPCs vs late-outgrowth EPCs
Unlike stem cells (SCs), progenitor cells (PCs) represent a larger variety of cell types, and are thought to be more numerous and more readily accessible. In 1997, Asahara et al identified endothelial PCs (EPCs).34 EPCs were thought to participate in angiogenesis and vasculogenesis.35 Growing evidence also suggests that other PCs are present in distinct organs in the body and contribute to tissue homeostasis, as well as tissue repair and regeneration.36
However, more recently the term “EPCs” has fallen out of use, because it is too vague in nature and actually is inaccurate, as some of these subpopulations do not become endothelial cells. Two types of progenitors – circulating angiogenic cells (CACs; also known as early-outgrowth EPCs or CD34+ cells) and endothelial colony-forming cells (ECFCs; or late-outgrowth EPCs) – differ in their characteristics. CACs originated from the myelomonocytic lineage, and first become noticeable in culture approximately 7 days after isolation. These spindle-shaped cells act mainly as paracrine secretors, are involved in regulation of vascular homeostasis, and initiate vasculogenesis without directly becoming part of the endothelial intima.37,38 Their counterparts, ECFCs, are circulating SCs/PCs that first appear in culture after 10 days of isolation, and can be harvested from umbilical cord blood, bone marrow, or even peripheral blood.39 ECFCs contribute to both angiogenesis and vasculogenesis by integrating directly into the developing vessels.35 They can form tubelike structures in vitro and perfused vessels in vivo if the cells come into contact with perivascular cells like mesenchymal SCs (MSCs).40 Patients with pulmonary arterial hypertension (PAH) have increased numbers of circulating ECFCs in peripheral blood; these ECFCs have been demonstrated to contribute to the vascular remodeling process in PAH.40 Interestingly, using a mouse model of ischemic acute kidney injury, Burger et al showed that human ECFCs, when administered at the time of reperfusion, significantly reduced macrophage infiltration, oxidative stress, and tubular necrosis, even without the cells being retained in the kidneys. Furthermore, exosomes from ECFCs inhibited hypoxia/reoxygenation-induced apoptosis of cocultured human umbilical vein endothelial cells.41
On the other hand, dysfunctional circulating progenitors are implicated in a number of diseases, including but not limited to cardiovascular pathologies, pulmonary hypertension, cancer, and diabetes mellitus.35 Based on the physiologic functions of these cells, it is not surprising to learn that they are able to promote neovascularization in early tumor formation and metastasis. In particular, one study has found that deguelin, a chemopreventive drug, can reduce the number of colony-forming units of bone marrow-derived c-Kit+/Sca-1+ mononuclear cell migration, adhesion, and proliferation capability. When cocultured with endothelial cells, the treated EPC were less likely to form tubelike vessels, because deguelin arrested the cells at the G1 checkpoint.42 In Moyamoya disease (MMD), a form of childhood stroke, the expression of retinaldehyde dehydrogenase 2 is significantly reduced in ECFCs, rendering them less efficient in forming capillary networks in vitro as well as in vivo.43 Teofili et al studied patients with myelodysplastic syndromes, and found that their ECFCs adhered to normal mononuclear cells more strongly compared to those of healthy controls.44 In PAH, ECFCs, located between pulmonary arterial endothelial cells, were found to be more proliferative than healthy ECFCs, and were proposed to contribute to the proliferative pulmonary vascular remodeling process, the key pathological event of PAH.45
Because of such important roles under both physiologic and pathological conditions, these circulating progenitors have been an appealing topic in the field of vascular biology, and have been considered as the next generation of treatment regimen for ROP.
Stem/progenitor cells: do they have therapeutic potential in the retina?
Over the past few decades, studies of therapeutic SCs have become increasingly more popular in several different diseases, such as cardiovascular illnesses and osteonecrosis. SCs can be of embryonic origin or derived from adult organs. Although embryonic SCs possess tremendous potential for differentiation, their use has been restricted due to ethical issues, limited sources, and higher risk of malignant transformation compared to other types of SCs.46 Fortunately, various kinds of adult SCs have been studied extensively and used in clinical trials throughout the world.47 As of today, the sources of adult SCs include: 1) MSCs derived from bone marrow, human umbilical cord blood, and other tissues, which have been demonstrated to have neuroprotective effects, and may differentiate into other cell types, including neural cells; 2) CACs, which are bone marrow-derived, and provide primarily paracrine support to the vasculature to foster vessel repair; 3) ECFCs, which are isolated from peripheral blood, cord blood, or the stromal vascular fraction, and can form endothelial cells; 4) neural precursor cells, which are multipotent cells found in the developing as well as the adult central nervous system and are heterogeneous, self-renewing, and mitotically active; and 5) induced pluripotent SCs, which can be generated from somatic cells after being treated with a defined cocktail of transcription factors, but tend to be time-consuming and costly.47,48
Research has aimed to use a variety of SC types to study drug delivery or gene therapy.47 In a 5-year follow-up study of femoral osteonecrosis in patients with sickle-cell disease, implantation of autologous bone marrow-derived mononuclear cells was reported to relieve pain significantly and slow the progressive early stages of femoral osteonecrosis. Additionally, MSCs from the same patients exhibited physiological characteristics that may have also played a role in the results observed.49 Interestingly, patients with osteonecrosis had better improvements on receiving a higher amount of PCs.50
Particularly in the field of vascular biology, evidence has also shown promising therapeutic applications of SCs/PCs and their associated factors. Indeed, in a genetic analysis of hypertensive rats and old mice, gene transfer of the longevity-associated variant BPIFB4 was able to enhance endothelial nitric oxide synthase and restore endothelial function.51 Most importantly, BPIFB4 is highly expressed in bloodstream CD34
Is therapeutic revascularization of the ROP retina by proangiogenic progenitor cells a feasible strategy for the future?
While current therapies mostly target the IVNV phase of ROP, scientists have become more interested in the early stage or phase I in OIR models. In fact, it has been seen in clinics that infants with zone I ROP are at higher risk of developing severe ROP and have poorer prognosis compared to those with zone II ROP.4 Therefore, restoring the vasculature of the immature retina is critical to reducing or preventing IVNV.
Using the OIR mouse model, we have investigated the concept of combination cell therapy for vascular repair. Specifically, we tested exogenous administration of CD34
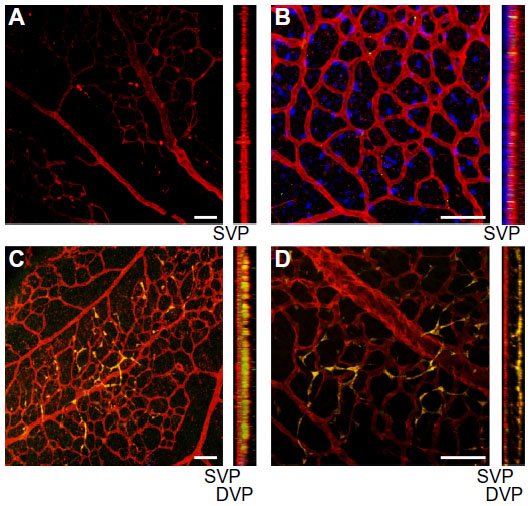 | Figure 2 Injection of ECFCs on P5 stimulates the development of the deep vascular plexus in the OIR pups prior to return to normoxic conditions on postnatal day 12. |
Hypoxic preconditioning (HPC) can further enhance SC/PC function. The rationale rests upon a number of observations. First, bone marrow SCs naturally reside under an oxygen tension of approximately 1%–7%,56,57 and long-term repopulating hematopoietic SCs in the mouse also exist under hypoxic conditions.58 It has been known that when the oxygen level falls below 5%, HIFs will be activated and continue to rise in concentration directly with the decreased level of oxygen.59 In a hypoxic environment, HIF-1α and HIF-2α are protected from ubiquitination and proteasomal degradation, and as a result HIF-1α and HIF-2α concentrations rise, along with other hypoxia-induced messenger RNAs, including those that are important for angiogenesis, apoptosis, and energy metabolism.60–63 In particular, HIF-1α can regulate MSC proliferation by increasing TWIST expression, and consequently downregulates the inhibitory effect of the E2A–p21 pathway on senescence to enhance proliferation.
In addition, HPC causes reduced apoptosis and thereby increases capacity of implanted MSCs in fixing myocardial infarction or diabetic cardiomyopathy. HPC also promotes angiogenesis and vascularization through paracrine factors.64–66 When cultured in a hypoxic environment, human cord blood-derived CD34+ cells can reverse their senescence and become more proliferative again by higher HIF-1α-induced TWIST expression.67 It has been shown that the population-doubling time of marrow-isolated adult multilineage-inducible cells is decreased as oxygen tension is lowered, with the optimal oxygen tension being 3%.68 Therefore, there is ample evidence that preconditioning SCs may improve their viability and function when injected into an adverse and diseased environment, such as ROP or diabetic retina.
Combination cell therapy
A combination of ECFCs and CD34+ cells in nude mice was seen to promote revascularization in a synergistic manner in response to acute vascular injury.69 As shown in the schematic in Figure 3, there is considerable evidence to suggest that several SC–PC combinations can serve to enhance vascular repair. There are different populations that can become pericytes or smooth-muscle cells, and these include adipose-derived MSCs (ASC) and bone marrow-derived MSCs.70 ECFCs can serve as a source of endothelial cells, and can easily be combined with either MSC population. As shown in Figure 4, the combined use of CD34+ cells with ECFCs shows improved homing compared to either cell type alone in the ROP model, suggesting that CD34+ cells are likely secreting factors that enhance ECFC function.
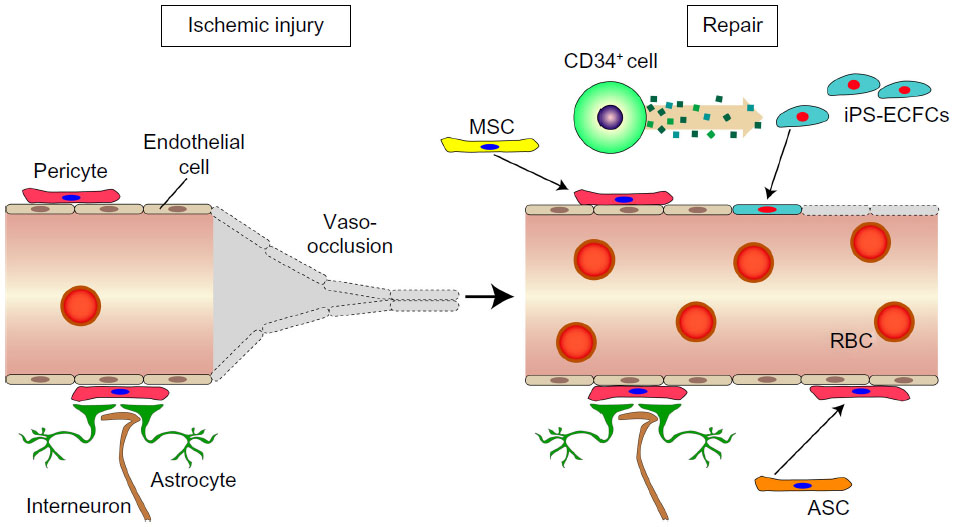 | Figure 3 Ischemic injury on retinal micro-blood vessel results in loss of pericytes and endothelial cells and vaso-occlusion. |
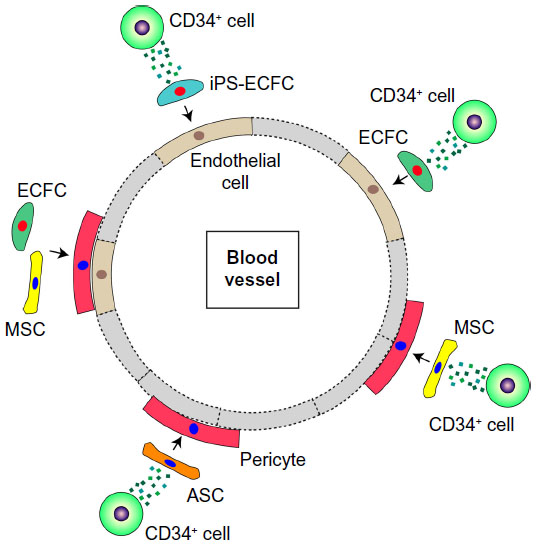 | Figure 4 Combination of various cell types that repair damage to ischemia-derived injuries. |
Moreover, understanding the molecular factors influencing these cells will likely reveal potential modulators of vascular formation and remodeling. Interestingly, despite the aforementioned important angiogenic property of VEGF, in the absence of IGF-1, VEGF is not sufficient to drive normal development of retinal vasculature. In fact, low levels of IGF-1 are linked to the pathogenesis of ROP.71 However, aberrant expression of IGF-1 can also contribute to pathological neovascularization.72
It is believed that IGF-1 exerts its effects via interactions with IGFBPs. Among these, IGFBP-3 is found to promote migration of CD34+ cells and differentiation of CD34+ cells to ECs; both are essential for endothelial repair after ischemic injury in the OIR model. The remodeling process occurs through downregulation of CD133 and upregulation of endothelial nitric oxide synthase expression.71 Intriguingly, proliferating ECs that express IGFBP-3 have been shown to be protected from hyperoxia-induced vascular ablation while reducing preretinal neovascularization.73,74 As already stated, restoration of a healthy endothelium not only involves CD34+ cells and ECFCs but also other bone marrow-derived cells, such as pericytes and astrocytes, for their supportive functions. Further investigation reveals that IGFBP-3 indeed enhances differentiation of bone marrow-derived cells into pericytes and astrocytes. Moreover, IGFBP-3 reduces pericyte apoptosis while attenuating activated microglia cells during the hypoxic phase of the OIR model.74
To summarize, angiogenesis is a highly dynamic and finely tuned process that engages both cellular and paracrine factors. By manipulating the right balance of key molecular factors, we will enable the angiogenic cells (ie, CD34+ cells, ECFCs, and supporting cells like pericytes) to fulfill their functions.
Conclusion
ROP continues to be a great concern, particularly in developing countries, where regulating oxygenation of preterm infants is not yet possible. Considering the currently available treatment options for ROP, new innovative approaches for better ROP therapies are definitely needed. More insights are being gained into the pathophysiology of the developing neuronal and vascular retina, and this knowledge will facilitate the translation of basic studies into clinical practice. PCs/SCs have been used therapeutically in other vascular diseases, providing a framework for their study in animal models of retinal disease and eventually for their translational to ocular clinical trials. To accelerate the translational process, more preclinical work defining the safety and function of each SC/PC population alone and in combination is needed.
Acknowledgments
This work was supported by the National Institutes of Health (NIH) grants. EY012011, EY007739, HL110170 to MBG, and by a Research to Prevent Blindness unrestricted grant awarded to the Department of Ophthalmology at Indiana University – Purdue University Indianapolis. This paper was presented in part at the Association for Research in Vision and Ophthalmology Annual Meeting in Orlando, FL, May 4–8, 2014, as an oral presentation with interim findings.
Disclosure
The authors report no conflicts of interest in this work.
References
Reynolds JD. Insights in ROP. Am Orthopt J. 2014;64(1):43–53. | |
Mintz-Hittner HA, Kennedy KA, Chuang AZ. Efficacy of intravitreal bevacizumab for stage 3+ retinopathy of prematurity. N Engl J Med. 2011;364(7):603–615. | |
Hartnett ME. Vascular endothelial growth factor antagonist therapy for retinopathy of prematurity. Clin Perinatol. 2014;41(4):925–943. | |
Hartnett ME. Pathophysiology and mechanisms of severe retinopathy of prematurity. Ophthalmology. 2015;122(1):200–210. | |
Hellström A, Smith LE, Dammann O. Retinopathy of prematurity. Lancet. 2013;382(9902):1445–1457. | |
Chan-Ling T, Gock B, Stone J. The effect of oxygen on vasoformative cell division: evidence that ‘physiological hypoxia’ is the stimulus for normal retinal vasculogenesis. Invest Ophthalmol Vis Sci. 1995;36(7):1201–1214. | |
Stone J, Chan-Ling T, Pe’er J, Itin A, Gnessin H, Keshet E. Roles of vascular endothelial growth factor and astrocyte degeneration in the genesis of retinopathy of prematurity. Invest Ophthalmol Vis Sci. 1996;37(2):290–299. | |
Chan-Ling T, Hughes S. NG2 can be used to identify arteries versus veins enabling the characterization of the different functional roles of arterioles and venules during microvascular network growth and remodeling. Microcirculation. 2005;12(7):539–540; author reply 540–541. | |
Nagy JA, Benjamin L, Zeng H, Dvorak AM, Dvorak HF. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis. 2008;11(2):109–119. | |
Xin X, Rodrigues M, Umapathi M, et al. Hypoxic retinal Müller cells promote vascular permeability by HIF-1-dependent up-regulation of angiopoietin-like 4. Proc Natl Acad Sci U S A. 2013;110(36):E3425–E3434. | |
Zeng G, Taylor SM, McColm JR, et al. Orientation of endothelial cell division is regulated by VEGF signaling during blood vessel formation. Blood. 2007;109(4):1345–1352. | |
Lang GE. Diabetic macular edema. Ophthalmologica. 2012;227 Suppl 1:21–29. | |
Chen J, Joyal JS, Hatton CJ, et al. Propranolol inhibition of β-adrenergic receptor does not suppress pathologic neovascularization in oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 2012;53(6):2968–2977. | |
Modi KK, Chu DS, Wagner R, Guo S, Zarbin MA, Bhagat N. Infectious ulcerative keratitis following retinopathy of prematurity treatment. J Pediatr Ophthalmol Strabismus. 2015;52(4):221–225. | |
Ziylan Ş, Öztürk V, Yabaş-Kiziloğlu Ö, Çiftçi F. Myopia, visual acuity and strabismus in the long term following treatment of retinopathy of prematurity. Turk J Pediatr. 2014;56(5):518–523. | |
Sato T, Wada K, Arahori H, et al. Serum concentrations of bevacizumab (Avastin) and vascular endothelial growth factor in infants with retinopathy of prematurity. Am J Ophthalmol. 2012;153(2):327–333. e1. | |
Multicenter trial of cryotherapy for retinopathy of prematurity. 3 1/2-year outcome--structure and function. Cryotherapy for Retinopathy of Prematurity Cooperative Group. Arch Ophthalmol. 1993;111(3):339–344. | |
Early Treatment For Retinopathy Of Prematurity Cooperative G. Revised indications for the treatment of retinopathy of prematurity: results of the early treatment for retinopathy of prematurity randomized trial. Arch Ophthalmol. 2003;121(12):1684–1694. | |
Ittiara S, Blair MP, Shapiro MJ, Lichtenstein SJ. Exudative retinopathy and detachment: a late reactivation of retinopathy of prematurity after intravitreal bevacizumab. J Aapos. 2013;17(3):323–325. | |
Wong RK, Hubschman S, Tsui I. Reactivation of retinopathy of prematurity after ranibizumab treatment. Retina. 2015;35(4):675–680. | |
Zhao R, Qian L, Jiang L. miRNA-dependent cross-talk between VEGF and Ang-2 in hypoxia-induced microvascular dysfunction. Biochem Biophys Res Commun. 2014;452(3):428–435. | |
McArthur K, Feng B, Wu Y, Chen S, Chakrabarti S. MicroRNA-200b regulates vascular endothelial growth factor-mediated alterations in diabetic retinopathy. Diabetes. 2011;60(4):1314–1323. | |
BaiY, Bai X, Wang Z, Zhang X, Ruan C, Miao J. MicroRNA-126 inhibits ischemia-induced retinal neovascularization via regulating angiogenic growth factors. Exp Mol Pathol. 2011;91(1):471–477. | |
Rivera JC, Sitaras N, Noueihed B, et al. Microglia and interleukin-1β in ischemic retinopathy elicit microvascular degeneration through neuronal semaphorin-3A. Arterioscler Thromb Vasc Biol. 2013; 33(8):1881–1891. | |
Deliyanti D, Miller AG, Tan G, Binger KJ, Samson AL, Wilkinson-Berka JL. Neovascularization is attenuated with aldosterone synthase inhibition in rats with retinopathy. Hypertension. 2012;59(3):607–613. | |
Vessey KA, Wilkinson-Berka JL, Fletcher EL. Characterization of retinal function and glial cell response in a mouse model of oxygen- induced retinopathy. J Comp Neurol. 2011;519(3):506–527. | |
Zhao L, Ma W, Fariss RN, Wong WT. Retinal vascular repair and neovascularization are not dependent on CX3CR1 signaling in a model of ischemic retinopathy. Exp Eye Res. 2009;88(6):1004–1013. | |
Ishida S, Usui T, Yamashiro K, et al. VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization. J Exp Med. 2003;198(3):483–489. | |
Davies MH, Eubanks JP, Powers MR. Microglia and macrophages are increased in response to ischemia-induced retinopathy in the mouse retina. Mol Vis. 2006;12:467–477. | |
Davies MH, Stempel AJ, Powers MR. MCP-1 deficiency delays regression of pathologic retinal neovascularization in a model of ischemic retinopathy. Invest Ophthalmol Vis Sci. 2008;49(9):4195–4202. | |
Miyazaki T, Taketomi Y, Saito Y, et al. Calpastatin counteracts pathological angiogenesis by inhibiting suppressor of cytokine signaling 3 degrada- tion in vascular endothelial cells. Circ Res. 2015;116(7):1170–1181. | |
Budd S, Byfield G, Martiniuk D, Geisen P, Hartnett ME. Reduction in endothelial tip cell filopodia corresponds to reduced intravitreous but not intraretinal vascularization in a model of ROP. Exp Eye Res. 2009; 89(5):718–727. | |
Saito Y, Geisen P, Uppal A, Hartnett ME. Inhibition of NAD(P)H oxidase reduces apoptosis and avascular retina in an animal model of retinopathy of prematurity. Mol Vis. 2007;13:840–853. | |
Asahara T, Murohara T, Sullivan A, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275(5302):964–967. | |
Vaughan EE, O’Brien T. Isolation of circulating angiogenic cells. Methods Mol Biol. 2012;916:351–356. | |
Mace KA, Braun KM. Progenitor Cells: Methods and Protocols. Heidelberg: Springer; 2012. | |
Sieveking DP, BuckleA, Celermajer DS, Ng MK. Strikingly different angio- genic properties of endothelial progenitor cell subpopulations: insights from a novel human angiogenesis assay. J Am Coll Cardiol. 2008;51(6):660–668. | |
Hirschi KK, Ingram DA, Yoder MC. Assessing identity, phenotype, and fate of endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2008;28(9):1584–1595. | |
Yoder MC, Mead LE, Prater D, et al. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. 2007;109(5):1801–1809. | |
Smadja DM, Levy M, Huang L, et al. Treprostinil indirectly regulates endothelial colony forming cell angiogenic properties by increasing VEGF-A produced by mesenchymal stem cells. Thromb Haemost. 2015;114(4):735–747. | |
Burger D, Viñas JL, Akbari S, et al. Human endothelial colony-forming cells protect against acute kidney injury: role of exosomes. Am J Pathol. 2015;185(8):2309–2323. | |
Nguyen MP, Lee D, Lee SH, Lee HE, Lee HY, LeeYM. Deguelin inhibits vasculogenic function of endothelial progenitor cells in tumor progression and metastasis via suppression of focal adhesion. Oncotarget. 2015;6(18):16588–16600. | |
Lee JY, MoonYJ, Lee HO, et al. Deregulation of retinaldehyde dehydrogenase 2 leads to defective angiogenic function of endothelial colony- forming cells in pediatric moyamoya disease. Arterioscler Thromb Vasc Biol. 2015;35(7):1670–1677. | |
Teofili L, Martini M, Nuzzolo ER, et al. Endothelial progenitor cell dysfunction in myelodysplastic syndromes: possible contribution of a defective vascular niche to myelodysplasia. Neoplasia. 2015; 17(5):401–409. | |
Duong HT, Comhair SA, Aldred MA, et al. Pulmonary artery endothelium resident endothelial colony-forming cells in pulmonary arterial hypertension. Pulm Circ. 2011;1(4):475–486. | |
Kim KS, Park JM, Kong T, et al. Retinal angiogenesis effects of TGF-β1, and paracrine factors secreted from human placental stem cells in response to a pathological environment. Cell Transplant. Epub 2015 Jun 10. | |
Bhere D, Shah K. Stem cell-based therapies for cancer. Adv Cancer Res. 2015;127:159–189. | |
Alonso-Alonso ML, Srivastava GK. Current focus of stem cell application in retinal repair. World J Stem Cells. 2015;7(3):641–648. | |
Daltro GC, Fortuna V, de Souza ES, et al. Efficacy of autologous stem cell-based therapy for osteonecrosis of the femoral head in sickle cell disease: a five-year follow-up study. Stem Cell Res Ther. 2015;6:110. | |
Hernigou P, Beaujean F. Treatment of osteonecrosis with autologous bone marrow grafting. Clin Orthop Relat Res. 2002;(405):14–23. | |
Villa F, Carrizzo A, Spinelli CC, et al. Genetic analysis reveals a longevity-associated protein modulating endothelial function and angiogenesis. Circ Res. 2015;117(4):333–345. | |
Medina RJ, O’Neill CL, O’Doherty TM, et al. Myeloid angiogenic cells act as alternative M2 macrophages and modulate angiogenesis through interleukin-8. Mol Med. 2011;17(9–10):1045–1055. | |
Caballero S, Sengupta N, Afzal A, et al. Ischemic vascular damage can be repaired by healthy, but not diabetic, endothelial progenitor cells. Diabetes. 2007;56(4):960–967. | |
Milbauer LC, Enenstein JA, Roney M, et al. Blood outgrowth endothelial cell migration and trapping in vivo: a window into gene therapy. Transl Res. 2009;153(4):179–189. | |
Prasain N, Lee MR, Vemula S, et al. Differentiation of human pluripotent stem cells to cells similar to cord-blood endothelial colony-forming cells. Nat Biotechnol. 2014;32(11):1151–1157. | |
Cipolleschi MG, Dello Sbarba P, Olivotto M. The role of hypoxia in the maintenance of hematopoietic stem cells. Blood. 1993;82(7):2031–2037. | |
Lennon DP, Edmison JM, Caplan AI. Cultivation of rat marrow-derived mesenchymal stem cells in reduced oxygen tension: effects on in vitro and in vivo osteochondrogenesis. J Cell Physiol. 2001;187(3):345–355. | |
Parmar K, Mauch P, Vergilio JA, Sackstein R, Down JD. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci U S A. 2007;104(13):5431–5436. | |
Pouysségur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441(7092):437–443. | |
Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399(6733):271–275. | |
Cockman ME, Masson N, Mole DR, et al. Hypoxia inducible factor-α binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem. 2000;275(33):25733–25741. | |
Tanimoto K, Makino Y, Pereira T, Poellinger L. Mechanism of regulation of the hypoxia-inducible factor-1α by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000;19(16):4298–4309. | |
Ohh M, Park CW, Ivan M, et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the β-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2(7):423–427. | |
Wang JA, He A, Hu X, et al. Anoxic preconditioning: a way to enhance the cardioprotection of mesenchymal stem cells. Int J Cardiol. 2009; 133(3):410–412. | |
Li JH, Zhang N, Wang JA. Improved anti-apoptotic and anti-remodeling potency of bone marrow mesenchymal stem cells by anoxic pre- conditioning in diabetic cardiomyopathy. J Endocrinol Invest. 2008; 31(2):103–110. | |
Hu X,Yu SP, Fraser JL, et al. Transplantation of hypoxia-preconditioned mesenchymal stem cells improves infarcted heart function via enhanced survival of implanted cells and angiogenesis. J Thorac Cardiovasc Surg. 2008;135(4):799–808. | |
Lee SH, Lee JH, Yoo SY, Hur J, Kim HS, Kwon SM. Hypoxia inhibits cellular senescence to restore the therapeutic potential of old human endothelial progenitor cells via the hypoxia-inducible factor-1α- TWIST- p21 axis. Arterioscler Thromb Vasc Biol. 2013;33(10):2407–2414. | |
D’Ippolito G, Diabira S, Howard GA, Roos BA, Schiller PC. Low oxygen tension inhibits osteogenic differentiation and enhances stemness of human MIAMI cells. Bone. 2006;39(3):513–522. | |
Yoon CH, Hur J, Park KW, et al. Synergistic neovascularization by mixed transplantation of early endothelial progenitor cells and late outgrowth endothelial cells: the role of angiogenic cytokines and matrix metalloproteinases. Circulation. 2005;112(11):1618–1627. | |
Shaw LC, Neu MB, Grant MB. Cell-based therapies for diabetic retinopathy. Curr Diab Rep. 2011;11(4):265–274. | |
Chang KH, Chan-Ling T, McFarland EL, et al. IGF binding protein-3 regulates hematopoietic stem cell and endothelial precursor cell function during vascular development. Proc Natl Acad Sci U S A. 2007; 104(25):10595–10600. | |
Grant M, Russell B, Fitzgerald C, Merimee TJ. Insulin-like growth factors in vitreous: studies in control and diabetic subjects with neovascularization. Diabetes. 1986;35(4):416–420. | |
Kielczewski JL, Jarajapu YP, McFarland EL, et al. Insulin-like growth factor binding protein-3 mediates vascular repair by enhancing nitric oxide generation. Circ Res. 2009;105(9):897–905. | |
Kielczewski JL, Hu P, Shaw LC, et al. Novel protective properties of IGFBP-3 result in enhanced pericyte ensheathment, reduced microglial activation, increased microglial apoptosis, and neuronal protection after ischemic retinal injury. Am J Pathol. 2011;178(4):1517–1528. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.