Back to Journals » Drug Design, Development and Therapy » Volume 9
Promoting endothelial function by S-nitrosoglutathione through the HIF-1α/VEGF pathway stimulates neurorepair and functional recovery following experimental stroke in rats
Authors Khan M, Dhammu T, Matsuda F, Baarine M, Dhindsa T, Singh I, Singh A
Received 13 November 2014
Accepted for publication 29 January 2015
Published 17 April 2015 Volume 2015:9 Pages 2233—2247
DOI https://doi.org/10.2147/DDDT.S77115
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Wei Duan
Mushfiquddin Khan,1 Tajinder S Dhammu,1 Fumiyo Matsuda,1,2 Mauhammad Baarine,3 Tejbir Singh Dhindsa,1 Inderjit Singh,1 Avtar K Singh3,4
1Department of Pediatrics, Medical University of South Carolina, Charleston, SC, USA; 2School of Health Sciences, Kagoshima University, Kagoshima, Japan; 3Department of Pathology and Laboratory Medicine, Medical University of South Carolina, Charleston, SC, USA; 4Ralph H Johnson VA Medical Center, Charleston, SC, USA
Background: For stroke patients, stimulating neurorepair mechanisms is necessary to reduce morbidity and disability. Our previous studies on brain and spinal cord trauma show that exogenous treatment with the S-nitrosylating agent S-nitrosoglutathione (GSNO) – a nitric oxide and glutathione metabolite of the human body – stimulates neurorepair and aids functional recovery. Using a rat model of cerebral ischemia and reperfusion (IR) in this study, we tested the hypothesis that GSNO invokes the neurorepair process and improves neurobehavioral functions through the angiogenic HIF-1α/VEGF pathway.
Methods: Stroke was induced by middle cerebral artery occlusion for 60 minutes followed by reperfusion in adult male rats. The injured animals were treated with saline (IR group, n=7), GSNO (0.25 mg/kg, GSNO group, n=7), and GSNO plus the HIF-1α inhibitor 2-methoxyestradiol (2-ME) (0.25 mg/kg GSNO + 5.0 mg/kg 2-ME, GSNO + 2-ME group, n=7). The groups were studied for either 7 or 14 days to determine neurorepair mediators and functional recovery. Brain capillary endothelial cells were used to show that GSNO promotes angiogenesis and that GSNO-mediated induction of VEGF and the stimulation of angiogenesis are dependent on HIF-1α activity.
Results: IR injury increased the expression of neurorepair mediators HIF-1α, VEGF, and PECAM-1 and vessel markers to a limited degree that correlate well with significantly compromised neurobehavioral functions compared with sham animals. GSNO treatment of IR not only remarkably enhanced further the expression of HIF-1α, VEGF, and PECAM-1 but also improved functioning compared with IR. The GSNO group also had a higher degree of vessel density than the IR group. Increased expression of VEGF and the degree of tube formation (angiogenesis) by GSNO were reduced after the inhibition of HIF-1α by 2-ME in an endothelial cell culture model. 2-ME treatment of the GSNO group also blocked not only GSNO’s effect of reduced infarct volume, decreased neuronal loss, and enhanced expression of PECAM-1 (P<0.001), but also its improvement of motor and neurological functions (P<0.001).
Conclusion: GSNO stimulates the process of neurorepair, promotes angiogenesis, and aids functional recovery through the HIF-1α-dependent pathway, showing therapeutic and translational promise for stroke.
Keywords: GSNO, IR, HIF-1α, VEGF, motor function, subtle behavior, neuroprotection, neurorepair, angiogenesis, S-nitrosylation, stroke
Introduction
Stroke is a leading cause of serious long-term disability and ranks fourth among all causes of death in the United States.1 Following stroke, direct trauma causes necrotic neuronal death; however, even greater apoptotic neuronal damage occurs hours and days later, caused by secondary injury due to inflammation and oxidative stress.2 These secondary injury mechanisms hamper the neurorepair process, leading to long-term chronic disabilities.3 Unfortunately, proven effective therapy for neuroprotection/neurorepair following stroke is presently not available, mainly because of the biphasic (acute and chronic) nature of the disease and limited understanding of the differential mechanisms involved in these phases.4 For functional recovery, clinical trials in human stroke show that neuroprotective drugs failed due to the lack of efficacy in the chronic phase.5,6 Therefore, an ideal therapy must ameliorate acute as well as chronic phases of the injury by well-understood mechanisms.
Recently, we and others identified an S-nitrosylating agent, S-nitrosoglutathione (GSNO),7,8 that provided robust neurovascular protection and blood–brain barrier (BBB) repair in a rat model of experimental stroke.7,9,10 This neurovascular protection was not associated with free nitric oxide (NO) donors.11 Later, we observed that GSNO was equally effective in aiding functional recovery in a rat model of ischemia and reperfusion (IR),12 spinal cord injury,13 and traumatic brain injury,14,15 indicating that GSNO could improve functional recovery following central nervous system injury. GSNO also improved learning and memory in a rat model of cerebral hypoperfusion, a vascular dementia model for experimental Alzheimer disease,16 suggesting that GSNO’s activities are associated with motor as well as cognitive function recovery. However, GSNO’s neurorepair mechanisms following central nervous system injury are not understood. The term “neurorepair” refers to the combination of neurochemical and cellular events, such as stimulation of angiogenic mediators (eg, PECAM-1 and vascular endothelial growth factor [VEGF]) and formation of vessels, leading to regenerative repair, neurorestoration, and functional recovery. In this study, we investigated the mechanisms of GSNO-mediated stimulation of the neurorepair process with a focus on the hypoxia-inducible factor-1 alpha (HIF-1α)/VEGF/angiogenesis pathway using a rat model of experimental stroke.
GSNO directly regulates several angiogenic and neurorepair mediators, including stabilization of HIF-1α17 and induction of neurorepair mechanisms.12 S-nitrosylation-mediated stabilization of HIF-1α has also been shown to protect against myocardial injury via the VEGF/angiogenesis pathway in GSNO reductase (GSNOR) knockout mice,18 indicating that HIF-1 is a key player in the repair processes. These studies provide a rationale for investigating S-nitrosylation-mediated modulation of HIF-1α for neurorepair in stroke.
HIF-1 is a nuclear transcription factor characterized as the master regulator of cellular oxygen homeostasis. HIF-1α is rapidly upregulated in response to hypoxia and is rapidly degraded upon reoxygenation/reperfusion.19 It activates the tissue survival pathways by inducing several key enzymes involved in cell metabolism (GLUT), angiogenesis (VEGF, VEGFR1, angiopoietin), and free radical scavenging (heme oxygenase-1 [HO-1]). HIF-1α knockout mice show impaired vascular development and embryonic lethality, indicating HIF-1’s protective role in vascular diseases.20 The HIF-1α pathway is deeply involved in both pathological (hypoxia) and neurorepair (normoxia) pathways following stroke.21 The HIF-1α stabilizers/inducers, such as desferrioxamine (an iron chelator approved for hemochromatosis treatment), promote a number of survival pathways, including neuroprotection, angiogenesis, and neurotrophins. These HIF-1α stabilizers also reduce brain infarctions when administered pre- or post-stroke.21 Prolyl hydroxylase (PHD) inhibitors, such as FG-4539, are presently in a Phase II anemia trial because of their activity to stabilize HIF-1α.22 However, inhibition of HIF-1α under hypoxic/ischemic conditions in the acute phase of IR injury has also been reported to be neuroprotective.22
Under normoxic conditions, studies are lacking on direct stabilization of HIF-1α by secondary modification and the induction of consequent protective genes. However, the S-nitrosylation reaction has been shown to stabilize HIF-1 protein expression and activity in normoxic endothelial cells.23 It was also confirmed that, while GSNO stabilizes HIF-1α by S-nitrosylation, reactive oxygen species (peroxynitrite, superoxide) destabilize it.24 The GSNO/S-nitrosylation-mediated stabilization of HIF-1α has been shown to involve upstream PI3K/Akt activity.25 GSNO also attenuates PHD activity during normoxia and inhibits proteasomal degradation of HIF-1α.26
GSNO is a natural component of the human body produced by the reaction of NO with glutathione (GSH) in the presence of oxygen.27 GSNO executes its action mainly via S-nitrosylation of target proteins.28 Under physiological conditions, GSNO and S-nitrosylated proteins are present in blood and brain.29–32 The concentration of GSNO in adult rat brain tissue is estimated to be 6–8 μM.30 A study on GSNO metabolism and its membrane crossing ability has been reported.33 Using an in vitro BBB model, we have also reported GSNO crossing the cellular membrane.7 Studies have also reported that GSNO inhibits platelet activation34 and protects both BBB integrity and epithelial permeability.15,35 Similarly, exogenous administration of GSNO36 also protects against cardiac ischemic injury,18,37 supporting the therapeutic potential of GSNO. Pharmacological inhibition of GSNOR has also been shown to improve endothelial functions,38 indicating a protective role of GSNO in neurovascular dysfunction-related diseases.
In this study, we demonstrate that GSNO-mediated HIF-1α/VEGF/PECAM-1-dependent mechanisms stimulate activation of angiogenesis-associated mediators, leading to increased vessel density and improved neurobehavioral functions during the delayed phases after IR. GSNO, via the stabilization of HIF-1α, thereby confers improved long-term outcomes with reduced IR injury and better functional recovery after stroke. The beneficial effects of GSNO are blocked by the HIF-1α inhibitor 2-methoxyestradiol (2-ME), thus supporting the conclusion that GSNO-stimulated beneficial effects are dependent on the activity of HIF-1α.
Methods
Reagents
GSNO was purchased from World Precision Instruments (Sarasota, FL, USA). 2-ME and all other chemicals and reagents were purchased from Sigma-Aldrich Co. (St Louis, MO, USA), unless stated otherwise.
Animals and experimental design
Animals were male Sprague Dawley rats (n=121) weighing 250–290 g at the time of surgery. All animals received humane care in compliance with the Medical University of South Carolina (MUSC)’s guidelines and the National Research Council’s criteria for humane care. Animal procedures were approved by the MUSC Institutional Animal Care and Use Committee. The animals were allowed to acclimatize for 3–5 days before the experiments. They were randomly divided into four groups: 1) IR for 7 and 14 days (IR group); 2) IR + GSNO treatment for 7 and 14 days (GSNO group); 3) IR + GSNO + 2-ME (GSNO + 2-ME group) for 7 days; and 4) sham-operated control without treatment (Sham group) for 14 days. The number of animals used in each experiment is indicated in Figures 1–7.
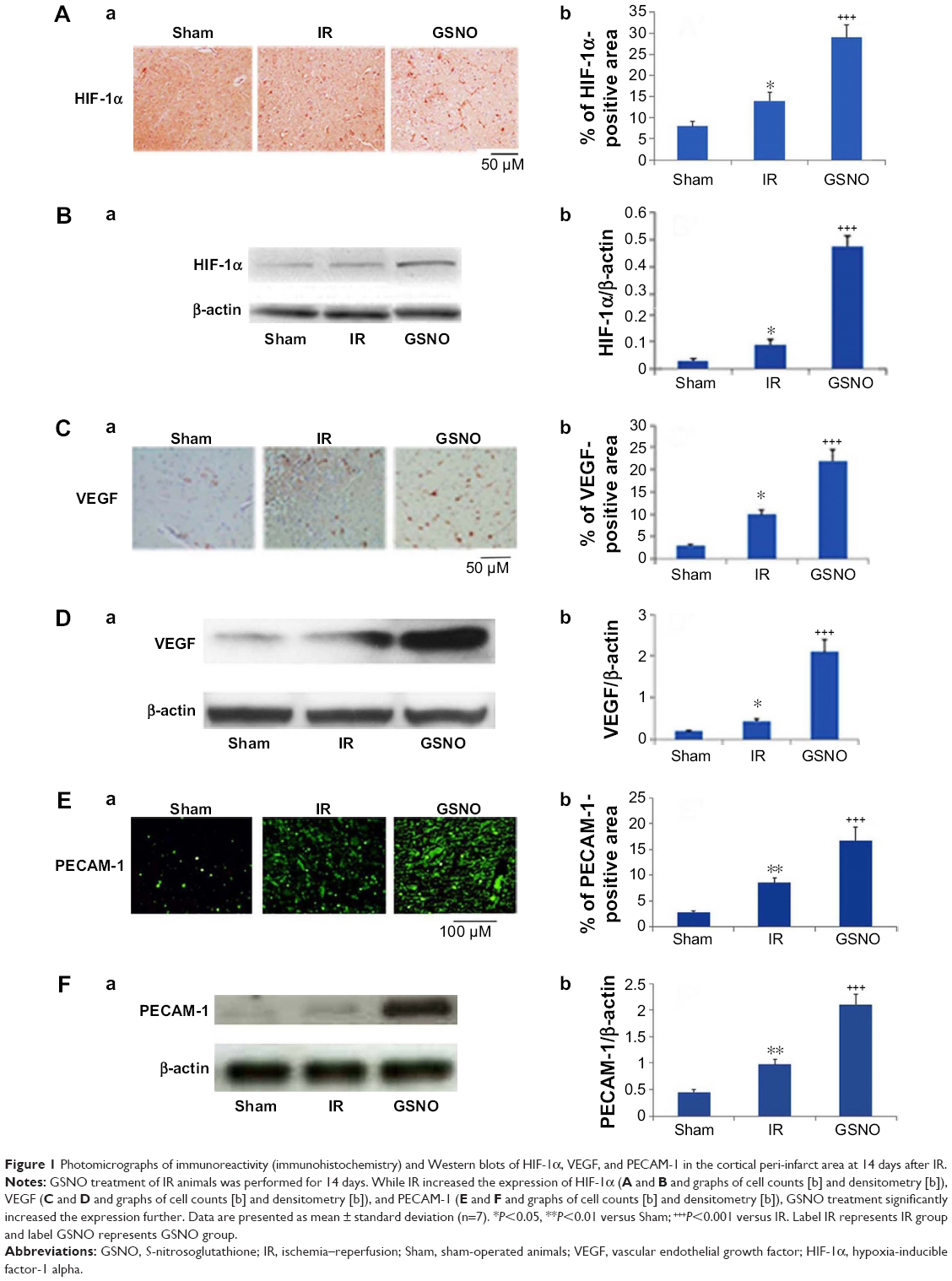 | Figure 1 Photomicrographs of immunoreactivity (immunohistochemistry) and Western blots of HIF-1α, VEGF, and PECAM-1 in the cortical peri-infarct area at 14 days after IR. |
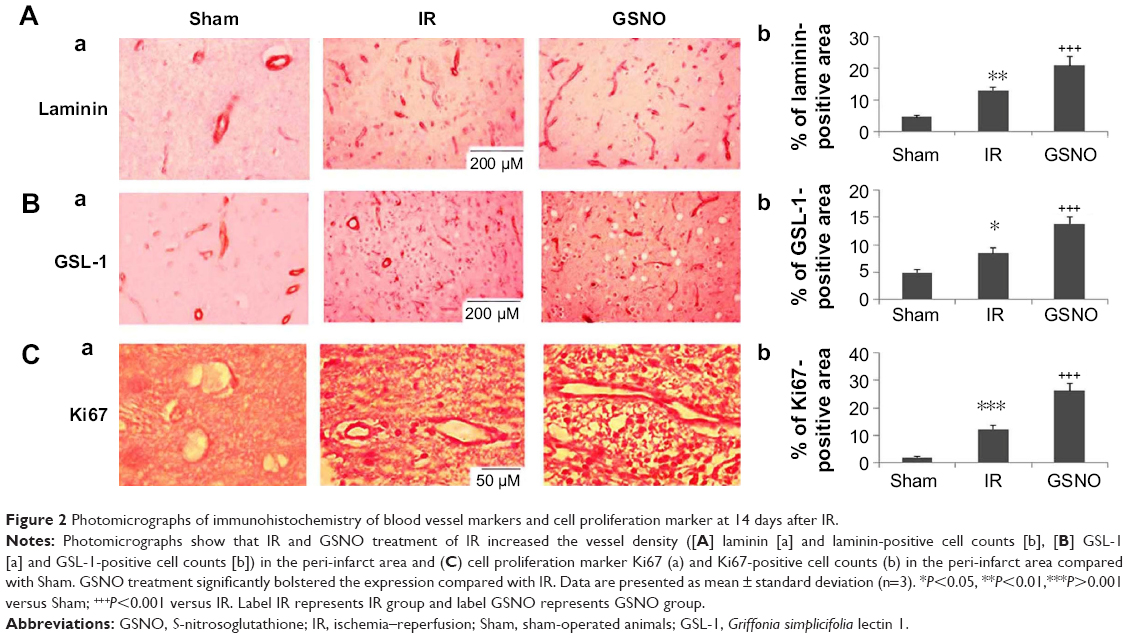 | Figure 2 Photomicrographs of immunohistochemistry of blood vessel markers and cell proliferation marker at 14 days after IR. |
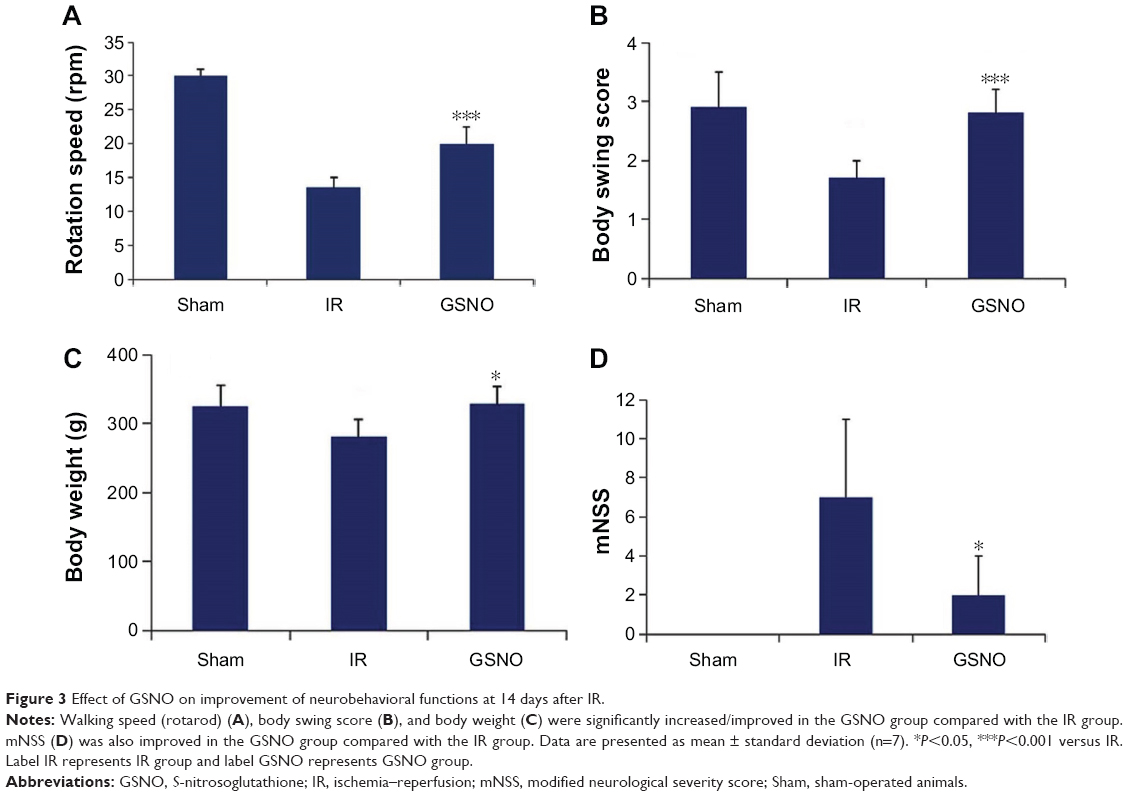 | Figure 3 Effect of GSNO on improvement of neurobehavioral functions at 14 days after IR. |
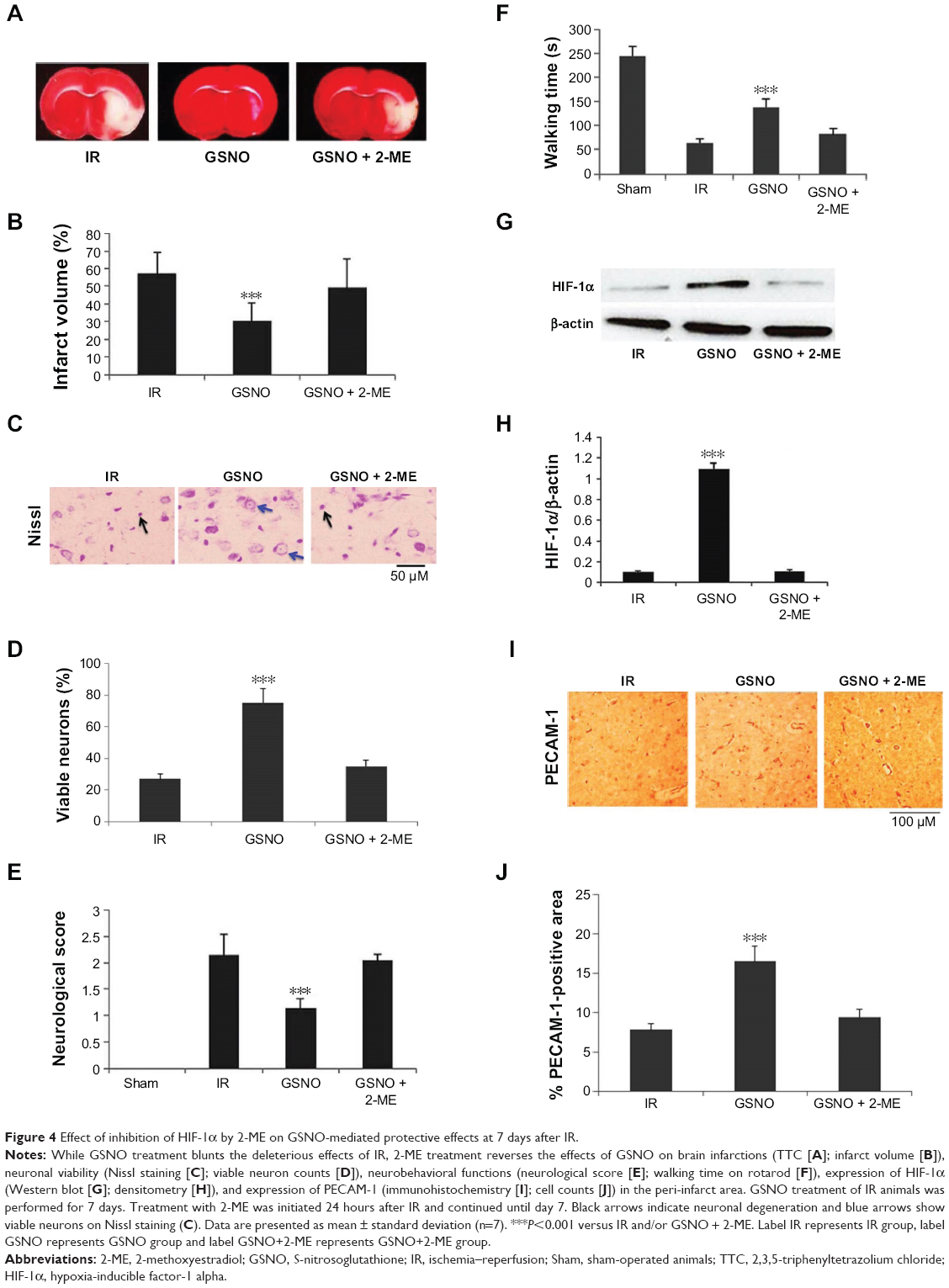 | Figure 4 Effect of inhibition of HIF-1α by 2-ME on GSNO-mediated protective effects at 7 days after IR. |
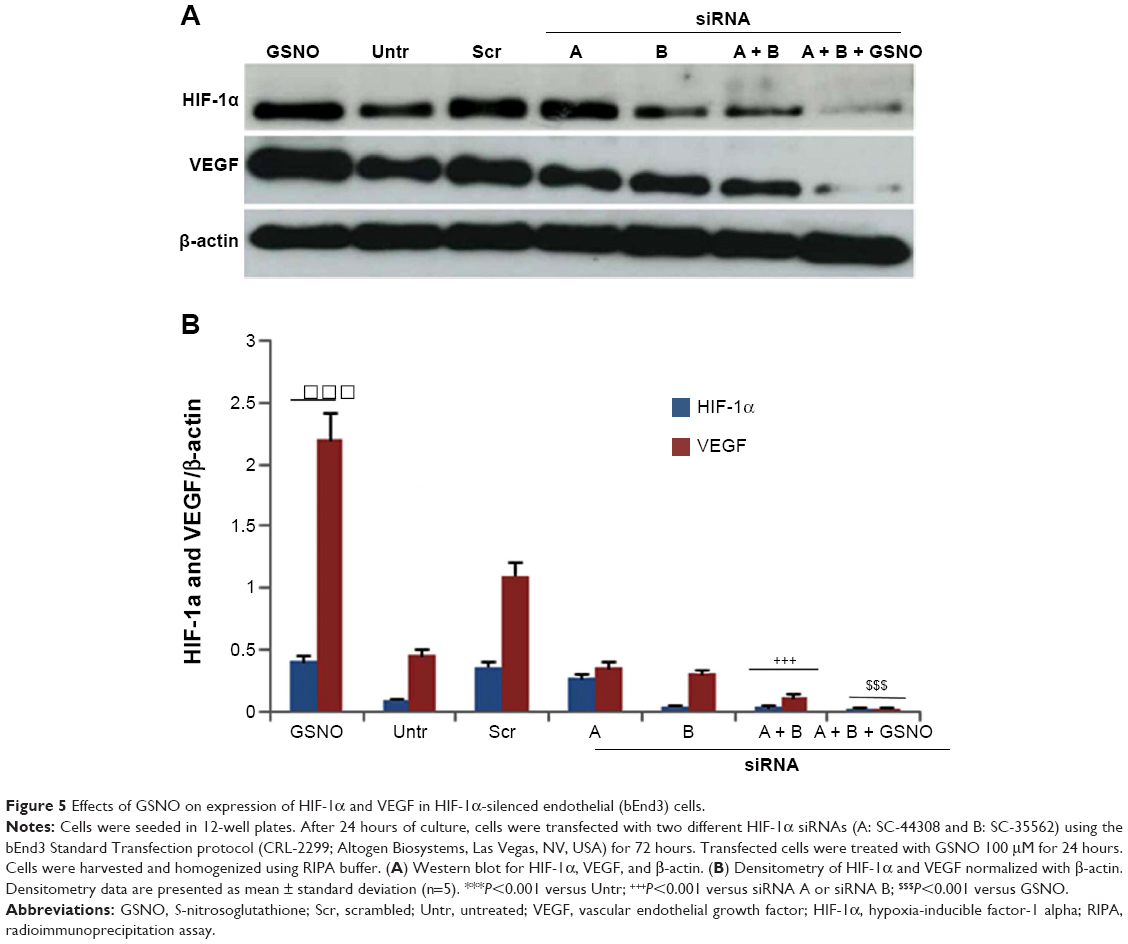 | Figure 5 Effects of GSNO on expression of HIF-1α and VEGF in HIF-1α-silenced endothelial (bEnd3) cells. |
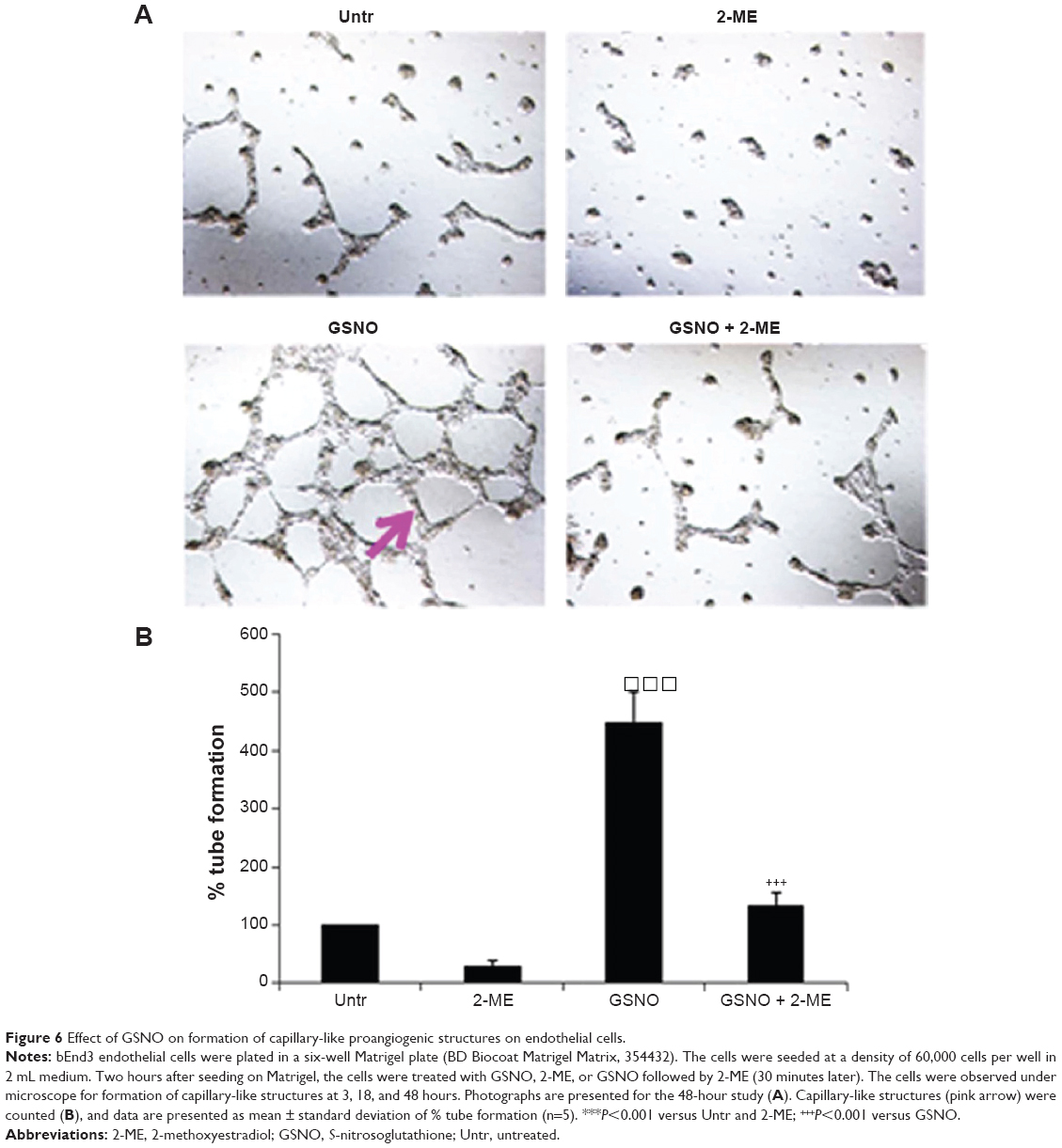 | Figure 6 Effect of GSNO on formation of capillary-like proangiogenic structures on endothelial cells. |
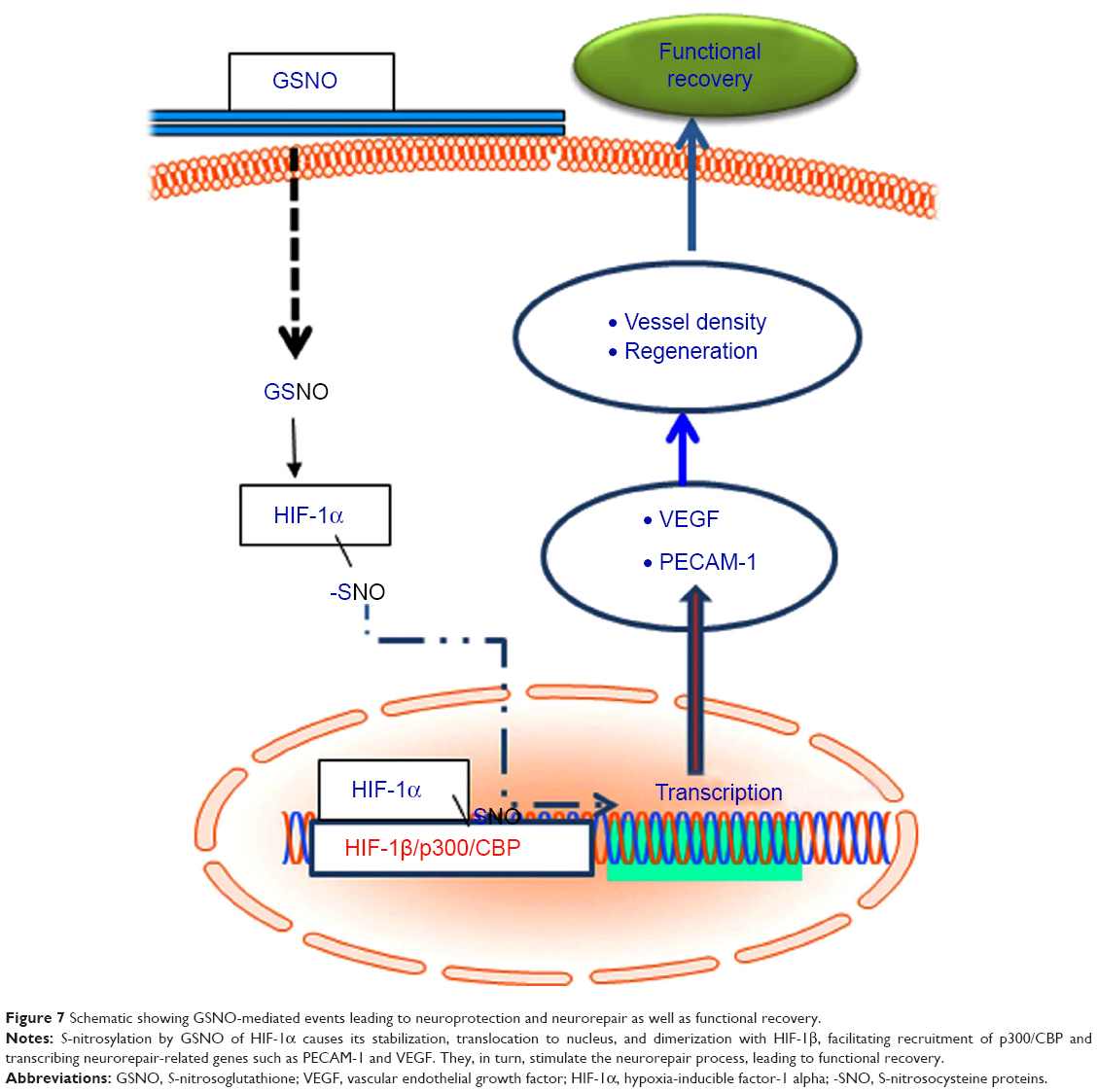 | Figure 7 Schematic showing GSNO-mediated events leading to neuroprotection and neurorepair as well as functional recovery. |
Middle cerebral artery occlusion (MCAO) rat model and GSNO treatment
Rats were anesthetized by ketamine hydrochloride (90 mg/kg body weight) and xylazine (10 mg/kg body weight) administered intraperitoneally. Stroke was induced by 60-minute left middle cerebral artery occlusion (MCAO) using an intraluminal filament as previously described.11 Regional cerebral blood flow was monitored during the occlusion and early reperfusion to ensure the obstruction of blood flow, as previously described.11,12 Reperfusion was established by withdrawal of the filament. After 7 or 14 days, animals were sacrificed with an overdose of Nembutal. The brain, including the peri-infarct region, was sliced for histological and immunohistochemical analyses, and 2,3,5-triphenyltetrazolium chloride (TTC) staining.
In the GSNO group, GSNO (0.25 mg/kg body weight) in saline (~250 μL) was slowly infused by jugular vein cannulation at 1 hour after reperfusion. From 1 day after MCAO and thereafter, the same dose of GSNO was gavage fed until 7 or 14 days following IR. Physiological parameters did not alter after GSNO treatment. Details of the study on physiologic parameters in IR and GSNO-treated rats have been reported earlier.11 In GSNO + 2-ME group, GSNO was treated for 7 days as described above. 2-ME (5 mg/kg, intraperitoneally) administration to the GSNO group was initiated 24 hours after the first dose of GSNO treatment and continued until the 7th day after IR.
Evaluation of ischemic infarct
Coronal sections (2 mm) were immersed in 1% solution of TTC in phosphate-buffered saline (pH 7.4) at 37°C for 15 minutes, as described previously.12 After staining, infarctions were measured using Scion Image software (Scion Corp., Frederick, MD, USA). Total infarct area was multiplied by the thickness of the brain sections to obtain infarct volume as described previously.7 To minimize the error introduced by edema and liquefaction after infarction, an indirect method for calculating infarct volume was also used.39,40 The non-infarcted area in the ipsilateral hemisphere was subtracted from that in the contralateral hemisphere, and infarct volume was calculated using the following formula:
|
|

Evaluation of neurological score and motor behavior
Animals in each group were evaluated for neurological scores, subtle behavior (using body swing test), and motor coordination and balance (using rotarod task) before and at the 7th and 14th days after reperfusion. The body weight of each animal was determined before surgery and at days 7 and 14 after IR.
Neurological examination was performed by an observer masked from the identity of the groups. A neurological grading system with a 4-point scale (0–3), as described previously,7 was used: 0, no observable neurological deficit (normal); 1, failure to extend right forepaw on lifting the whole body by tail (mild); 2, circling to the contralateral side (moderate); and 3, leaning to the contralateral side at rest or no spontaneous motor activity (severe). The animals not showing paralysis at 1 hour after MCAO were excluded from the study because the reduction of blood flow may not have produced an infarction of adequate size to cause quantifiable neurological deficits in those animals.
Modified neurological severity score (mNSS) measurement was performed as described previously.41 The test is sensitive to unilateral cortical injury because it reflects multiple asymmetries, including postural, sensory, and forelimb and hind limb use. A detailed description of this functional test has been previously reported.42 In our studies, mNSS was scaled from 0 to 12, with 0 as normal and 12 as the maximal deficit.15
The body swing test reflects symmetry of striatal function.43 A normal rat typically has an equal number of swings to both sides. Each rat was held along the vertical axis (defined as no more than 10° to either the left or the right side) approximately 2.5 centimeters from the base of its tail and elevated 2.5 centimeters above a table surface. A swing was recorded whenever the rat moved its head out of the vertical axis to either side. The rats have to return to the vertical position for the next swing to be counted. Thirty total swings (score 3) were counted.
In the motor coordination and balance test, rats were examined on an accelerating rotarod task by trained personnel blinded to animal groups as described previously.44 Walking time and speed on rotarod was recorded as previously described from our laboratory.12 Speed was increased from 0 rpm to 30 rpm, with an increment of 2 rpm every 5 seconds. Each rat was placed on the rotarod cylinder, and the speed (rpm) at which the animal fell off the drum was recorded. The trial ended if the animal fell off the drum. Each animal was given three trials, and the mean speed (rpm) of three trials was calculated for each animal.
Immunohistochemistry and histology
Paraffin-embedded sections from the formalin-fixed brain tissues, processed at 7 or 14 days after reperfusion, were stained using antibodies against angiogenesis markers/mediators HIF-1α, VEGF, and PECAM-1 (Santa Cruz Biotechnology Inc., Dallas, TX, USA) and vessel markers laminin (Sigma-Aldrich Co.) and GSL-1 (Griffonia simplicifolia lectin 1) (Vector Lab, Burlingame, CA, USA). Cell proliferation was assessed using Ki67 (Abcam, Cambridge, MA, USA). Secondary anti-rabbit and mouse IgG, conjugated with Alexa Fluor 488, were incubated on slides for 60 minutes. HIF-1α and VEGF sections were allowed to react with anti-mouse IgG conjugated to a peroxidase-labeled dextran polymer and then developed with diaminobenzidine substrate. All sections were examined for immunoreactivity in the peri-infarct area using an Olympus microscope as previously described.9,14 Three areas in the peri-infarct area of each immunostained section were digitized by a 40× microscope objective with microscope and camera without visual field overlap. Semiquantitative cell counting was performed using Scion Image software as described previously.12
The degree of neuronal damage (loss of viable neurons) was evaluated by cresyl violet (Nissl) staining. The staining highlights important structural features of neurons. The brain sections described above were used to stain with Nissl, and the staining was performed according to classical histology methods.12 Cells that contained Nissl substance were considered to be viable neurons. Condensed fragmented staining shows neuronal degeneration. The viable neurons were quantified by manually counting cells with visible nucleoli, as described previously,45 in the peri-infarct area using an Olympus microscope.
Western blot analysis
In the peri-infarct area from the ipsilateral injured brain tissue, Western blot was performed using antibodies against HIF-1α and PECAM-1 (Abcam), VEGF (Santa Cruz Biotechnology Inc.), and β-actin, as described earlier.9 Protein concentrations were determined using protein assay dye from Bio-Rad Laboratories Inc. (Hercules, CA, USA). Densitometry of protein expression was performed using a GS800 calibrated densitometer from Bio-Rad Laboratories Inc.
siRNA silencing of HIF-1α expression and treatment with GSNO in a mouse brain capillary endothelial cell line (bEnd3)
bEnd3, an immortalized mouse brain endothelial cell line originally generated and characterized,46 and lately used in our studies,47 is commercially available at American Type Culture Collection ([ATCC] CRL-2299; Manassas, VA, USA). It is a well-characterized endothelial cell line that shows the expression of ICAM-1 and VCAM-1.47 Cells were grown according to the supplier’s instructions and were allowed to grow to ~70%–80% confluence. HIF-1α expression was silenced for 72 hours by two different predesigned siRNA sequence primers from Santa Cruz Biotechnology Inc. (A: SC-44308 and B: SC-35562) and the corresponding scrambled siRNA (negative control) into the cells using the bEnd3 standard transfection reagent (CRL-2299; Altogen Biosystems, Las Vegas, ND, USA) according to the manufacturer’s instructions and as elsewhere described.48 Transfected cells were treated with freshly prepared GSNO (100 μM) for 24 hours. Western blots for HIF-1α (ab65979; Abcam), VEGF (SC507; Santa Cruz Biotechnology Inc.), and β-actin (4967; Cell Signaling, Danvers, MA, USA) were performed using 20 μg protein samples.
Matrigel analysis
Matrigel assay was performed as described previously.49 Briefly, bEnd3 endothelial cells were plated in a six-well Matrigel plate (BD Biocoat Matrigel Matrix, 354432). The cells were seeded at a density of 105 cells per well in 2 mL medium. At 2 hours after seeding in Matrigel, the cells were treated with GSNO (100 μM), 2-ME (5 μM), or GSNO followed by 2-ME (30 minutes later). The cells were observed under a microscope for formation of tube-like structures at 3, 18, and 48 hours. Tubule length ratio, branch number per field, and aggregation area were analyzed using ImageJ software.
Statistical evaluation
Statistical analysis was performed as described previously50 using GraphPad Prism 5.01 software. Unless otherwise stated, all values are expressed as mean ± standard deviation of n determinations or as mentioned. The results were examined by unpaired Student’s t-test. Multiple comparisons were performed using the Kruskal–Wallis test, or using analysis of variance followed by the Bonferroni test as appropriate. A P-value less than 0.05 was considered significant.
Results
GSNO treatment of IR for 2 weeks enhanced the expression of angiogenic mediators
Essential factors for the neurorepair process following stroke include the transcriptional activity of HIF-1α and enhanced expression of the angiogenic factor VEGF along with the endothelial cell proliferation mediator PECAM-1 (angiogenesis). S-Nitrosylation/GSNO has been shown to stabilize HIF-1α, leading to increased VEGF expression, angiogenesis, and protection against myocardial injury.18 In this study, we observed that immunohistochemical staining 14 days after IR shows an increased expression of HIF-1α (Figure 1A), VEGF (Figure 1C), and PECAM-1 (Figure 1E) in the peri-infarct area. GSNO treatment of IR significantly increased the expression further of HIF-1α, VEGF, and PECAM-1. Similar results of increased expression in the GSNO group from the peri-infract area at the 14th day after IR were confirmed for HIF-1α (Figure 1B), VEGF (Figure 1D), and PECAM-1 (Figure 1F) using Western blot. In summary, GSNO treatment of IR increased the expression of angiogenic mediators.
GSNO treatment of IR for 2 weeks increased the number of blood vessels and enhanced the expression of a cell proliferation marker
Quantitatively comparing vessel density using markers for vessels and a cell proliferation marker provides a measure of angiogenic vessel formation.51 GSNO treatment of IR for 2 weeks increased the expression of markers of vessel density, including laminin (Figure 2A) and GSL-1 (Figure 2B), indicating that GSNO may have promoted new vessel formation. Supporting the notion that the increased number of vessels present in the peri-infarct area resulted from angiogenic processes, we determined the expression of Ki67, a marker of cell proliferation. The expression of Ki67 (Figure 2C) was significantly increased in GSNO-treated animals compared with IR animals. We anticipate that proliferating cells include endothelial cells also because vessel density is increased; however, we did not identify Ki67-positive cell types in this study. Sham animals had significantly lower expressions of angiogenic markers (Figure 2).
GSNO treatment of IR for 2 weeks improved motor function and accelerated the recovery of subtle behavior
Compromised neurobehavioral functions, especially motor coordination and balance, and neurological functions are the leading causes of disability and morbidity following stroke. We determined the efficacy of GSNO in improving motor and subtle behaviors. GSNO treatment significantly improved tolerated rotation speed (rotarod task) (Figure 3A) compared with the IR group measured at the 14th day after IR. The treatment with GSNO also improved body swing behavior (Figure 3B) and mNSS measured at day 14 after IR (Figure 3D). The body weight of animals in the GSNO group was also significantly improved compared with animals in the IR group (Figure 3C).
Beneficial effects of GSNO on neuroprotection and functional recovery were blocked by HIF-1α inhibitor 2-ME
2-ME is a natural metabolite of estradiol and has been documented to inhibit HIF-1α stabilization and thus its transcriptional activity.52 Therefore, we investigated whether 2-ME reverses GSNO’s HIF-1α-dependent activities (Figure 4). Administration of 2-ME to GSNO-treated animals for 6 days (first dose of 2-ME was administered 24 hours after IR to investigate its effect after the acute phase) blocked GSNO’s protective effect on brain infarctions and functional recovery as evaluated by TTC staining (Figure 4A and B), Nissl staining (Figure 4C and D), and neurological score (Figure 4E). Furthermore, 2-ME treatment completely blocked the beneficial effect of GSNO on motor function recovery evaluated by rotarod performance (Figure 4F). As expected, 2-ME treatment robustly inhibited HIF-1α protein expression (Figure 4G and H) as well as significantly reversed GSNO-mediated enhanced expression of PECAM-1 (Figure 4I and J). Reversal of GSNO’s effect by 2-ME was remarkably significant at day 7 after IR; therefore, the experiment was stopped at the 7th day after the injury.
GSNO-mediated enhanced protein expression of VEGF was reversed by silencing HIF-1α in endothelial cells
Stabilization of HIF-1α results in increased HIF-1 transcriptional activity, leading to induction of HIF-1-dependent genes, including angiogenic VEGF. GSNO treatment resulted in greater protein expression of both HIF-1α and VEGF compared with untreated cells (Figure 5). Single siRNA (A or B) silencing for HIF-1α reduced the expression of HIF-1α as well as VEGF; however, silencing with both A and B siRNA remarkably reduced the expression of HIF-1α and VEGF. Moreover, cell transfection with two HIF-1α-targeted siRNAs markedly reduced GSNO-mediated HIF-1α stabilization and prevented the resultant expression of VEGF (Figure 5), consistent with a role of GSNO in HIF-1α stabilization and the stimulation of angiogenesis. However, a reduced level of both HIF-1α and VEGF in A + B + GSNO compared with A + B set was unanticipated and may be attributed to the stability of GSNO under bEnd3 cell transfection conditions.
GSNO-stimulated formation of capillary-like structures on endothelial cells was blunted by the inhibition of HIF-1α
We next examined the ability of GSNO to stimulate formation and stabilization of tube/capillary-like proangiogenic structures on endothelial cultured cells using Matrigel (in vitro angiogenesis). Tube/capillary-like proangiogenic structures were significantly increased after 48-hour GSNO treatment (Figure 6). Further, we assessed the effect of HIF-1α inhibition using 2-ME on GSNO-stimulated capillary-like structure formation. 2-ME is a specific HIF-1α inhibitor and has been widely used for the inhibition of angiogenesis.52 We observed that GSNO’s capillary-like structure formation ability was prevented by 2-ME, indicating that the GSNO-stimulated proangiogenic process was mediated by HIF-1α activity (Figure 6).
Discussion
To facilitate functional recovery, an ideal stroke therapy must ameliorate acute as well as chronic phases of the injury by well-understood mechanisms. Our previous studies showed that GSNO treatment of IR provided neuroprotection in the acute phase7,9 and improved neurobehavioral function in the chronic phase.12 However, the mechanisms of GSNO-mediated stimulation of the neurorepair process require elucidation in order to determine and then to widen its therapeutic potential.
Functional recovery in stroke is associated with vascular integrity and the stimulation of both angiogenesis and neurogenesis.53 Stroke patients who have higher cerebral blood vessel density recover better and survive longer than patients who have lower vascular density.54 A low degree of restorative angiogenesis after stroke results in limited and insufficient functional recovery. However, angiogenesis-promoting therapeutics confer not only a greater degree but also a faster rate of functional improvement.55,56 Because GSNO also produces effects that increase vessel density (Figure 2) and enhance functional recovery (Figure 3), GSNO likely promotes angiogenesis.54 Findings from other studies that observed GSNO-induced angiogenesis in ischemic heart18 and in a cell culture model57 support our observation of GSNO-mediated increased expression of angiogenic markers (Figure 1) and a cell proliferation marker (Figure 2C), as well as an increased number of vessels (Figure 2A and B).
The angiogenic marker VEGF is closely related to both angiogenesis and neurogenesis.53 Increased neurogenesis is accompanied by increased angiogenesis, whereas angiogenesis upregulates neurogenesis and NO-based drugs upregulate both.58 Angiogenesis itself is regulated by VEGF, mainly via HIF-1-based transcription. VEGF has been shown to modulate coupling of angiogenesis and neurogenesis; hence, it is an essential factor for regeneration.53,59 Therefore, we investigated the effect of GSNO on VEGF as well as PECAM-1 (markers of angiogenesis), finding that GSNO increased the expression of both following IR (Figure 1). This observation indicates that GSNO might be acting via the upregulation of VEGF. VEGF is transcriptionally regulated by HIF-1, and HIF-1α is stabilized by GSNO.25,26 Figure 1 shows that GSNO increased the expression of and thereby stabilized HIF-1α. Consistent with our findings, endogenous GSNO-induced stabilization of HIF-1α via S-nitrosylation was shown to induce the expression of VEGF, resulting in angiogenesis and myocardial protection.18
A potential concern of GSNO’s impact on VEGF is that HIF-1α-dependent upregulation of VEGF has been implicated in edema and BBB leakage in the acute phase of IR and cerebral hemorrhage.60,61 For VEGF-mediated BBB disruption to occur, MMP-9 and endothelial dysfunction are required.9,62 However, in addition to upregulating VEGF, GSNO transcriptionally downregulated MMP-9 as well as ICAM-1 and E-selectin, leading to reduced edema and increased BBB protection in the acute phase of IR and traumatic brain injury.9,15 Similarly, MMP-9-associated BBB disruption in experimental diabetes has been prevented by an exogenous treatment of GSNO.63 Exogenous inhibition of GSNOR and thus increased levels of GSNO have also been reported to improve endothelial function.38 Decreased levels of HIF-1α in cerebrospinal fluid from hypoxic/ischemic humans64 support the notion that HIF-1α-mediated mechanisms may ameliorate stroke injury. These observations indicate that HIF-1α is likely an important beneficial component of neuroprotection and the neurovascular repair process.
To show that GSNO invokes its neurorestorative/neurorepair effects via the HIF-1α/VEGF pathway, as depicted in Figure 7, we inhibited HIF-1α, anticipating that it would block the effect of GSNO on angiogenic mediators and functional recovery. The inhibition of HIF-1α in GSNO-treated animals reduced the expression of HIF-1α (Figure 4G and H) as well as PECAM-1 (Figure 4I and J), indicating that the angiogenic effect of GSNO was dependent on the activity of HIF-1α. 2-ME treatment of GSNO animals also blunted GSNO-mediated neuroprotection (Figure 4A–D), decreased neurological score (Figure 4E), and reduced motor function recovery (Figure 4F). These findings support the protective role of HIF-1α. Furthermore, endothelial cell culture studies showing reduced tube formation (Matrigel assay for angiogenesis) by 2-ME in GSNO-treated cells (Figure 6) provide evidence that GSNO’s regenerative repair effects are mediated by HIF-1α/VEGF pathway-induced angiogenesis. Reduced VEGF expression in GSNO-treated HIF-1α-silenced cells (Figure 5) confirms that the GSNO-stimulated VEGF/angiogenesis pathway is dependent on the transcriptional activity of HIF-1α. Recently, stabilization of HIF-1α by N-acetylcysteine has been documented to provide neuroprotection in stroke,65 and its inhibition by 2-ME reduces neuroprotection and downregulates neurorepair processes in cerebral IR.65,66 2-ME has also been reported to worsen outcomes after global ischemia.67 These observations further suggest that GSNO-induced angiogenic and neurorestorative processes are mediated through the stabilization of HIF-1α. The documented HIF-1α stabilization by GSNO-mediated S-nitrosylation (Cys-800) leading to stimulation of its transcriptional activity68 supports our finding of GSNO-mediated enhanced transcriptional activity of HIF-1α and the expression of VEGF (Figure 1). An enhanced level of GSNO has been shown to stimulate the HIF-1α-dependent angiogenic process, protecting against cardiac ischemic injury.18,37 In view of the above observations, we hypothesized that GSNO-mediated S-nitrosylation and stabilization of HIF-1α will enhance VEGF and PECAM-1, leading to increased vessel density and functional recovery (Figure 7).
Both deficient S-nitrosylation and a reduction in NO signaling pathways are observed in several neurodegenerative disease processes, including stroke.9,69–72 These pathologies occur due to decreased bioavailability of NO and, hence, reduced biosynthesis of GSNO. We have previously documented reduced levels of NO and decreased expression of S-nitrosocysteine proteins (-SNO) in the peri-infarct area. This decreased -SNO was normalized by an exogenous GSNO treatment.9 In short, reduced NO and -SNO are associated with IR pathophysiology. These observations are further supported by a report showing decreased levels of plasma NO and associated poor outcomes in stroke patients.73 Plasma S-nitrosothiols are also decreased in patients with endothelial dysfunction.74 These results indicate that S-nitrosylation is reduced in IR; therefore, exogenous GSNO supplementation in stroke therapy is a promising strategy. Although HIF-1α stabilization-based therapy using chemical inhibitors of PHDs is under investigation in chronic ischemic diseases, they also invoke HIF-1α-independent effects.22 Therefore, a direct stabilization by S-nitrosylation of HIF-1α through the endogenous signaling molecule GSNO is mechanistically a more specific therapeutic approach especially for IR stroke therapy. The role of the GSNO/HIF-1α/VEGF pathway in permanent ischemic stroke is less clear. Nonetheless, evidence from clinical studies suggests that administration of NO-based therapeutics in humans is safe and may be an effective treatment for stroke injury in human subjects.75,76
Conclusion
GSNO-mediated HIF-1α stabilization for neuroprotection and the stimulation of the neurorepair process provides a novel target mechanism for a therapy that could confer benefits in both the acute and chronic phases of stroke. The translational potential of GSNO is high because it occurs naturally in the human body and its exogenous administration to humans is not associated with any known side effects or toxicity. This study shows the feasibility of a new treatment paradigm for stroke, introducing the concept that stabilization of HIF-1α using GSNO may reduce disabilities in stroke survivors.
Acknowledgments
This work was supported by grants from NIH (NS-72511) and VA merit award (BX001062). This work was also supported by the NIH, Grants C06 RR018823 and No C06 RR015455 from the Extramural Research Facilities Program of the National Center for Research Resources. We thank Ms Joyce Bryan and Ms Terry Hope for their technical help and secretarial assistance. We are grateful to Dr Harutoshi Sakakima and Dr Yoshihiro Yoshida for their valuable input. We also acknowledge Dr Tom Smith and Dr Lisa Kerr from the MUSC Writing Center for their valuable editing of the manuscript.
Author background
M Khan (PhD) is a research associate professor at the MUSC with a strong background in neuroprotection and functional recovery in animal models of stroke, traumatic brain injury, and spinal cord injury. He has published more than 80 articles in the field of neuroprotection and neuropharmacology. TS Dhammu (PhD) and M Baarine (PhD) are postdoctoral fellows with expertise in cell culture and biochemical techniques. TS Dhammu has expertise in creating MCAO animal model of stroke and evaluating neurobehavioral functions of rodents. F Matsuda (PhD) is an assistant professor at the Kagoshima University, Kagoshima, Japan, and she has a strong background in evaluation of animals’ neurobehavioral functions. TS Dhindsa is a medical student at the MUSC and works in our group on projects related to neurodegeneration. He is an enthusiastic scientist with a special interest in neuropharmacology of stroke and traumatic brain injury. I Singh is a PhD and a distinguished university professor and Scientific Director of the Children’s Research Institute at the MUSC. He has published more than 280 articles in the field of neuroprotection and functional recovery. AK Singh is an MD and a practicing pathologist. She is professor at the MUSC and Director of the Pathology Laboratory at the VA Medical Center, Charleston, SC, USA. Her major research interest is traumatic brain injury and neurodegenerative diseases including stroke and Alzheimer’s. She has published more than 130 articles related to neurodegenerative diseases in peer-reviewed journals.
Author contributions
This study is based on an original idea of M Khan and AK Singh. All authors have contributed to conception and design of the study, analysis and interpretation of data and drafting the manuscript. Drs. TS Dhammu, F Matsuda, M Baarine, M Khan and Mr. TS Dhindsa are responsible for acquisition of data. All authors hereby approve the content of the article and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics – 2014 update: a report from the American Heart Association. Circulation. 2014;129(3):e28–e292. | ||
Rosamond W, Flegal K, Furie K, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics – 2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008;117(4):e25–e146. | ||
Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67(2):181–198. | ||
Maki T, Hayakawa K, Pham LD, Xing C, Lo EH, Arai K. Biphasic mechanisms of neurovascular unit injury and protection in CNS diseases. CNS Neurol Disord Drug Targets. 2013;12(3):302–315. | ||
Cramer SC. Repairing the human brain after stroke. II. Restorative therapies. Ann Neurol. 2008;63(5):549–560. | ||
Cramer SC. Repairing the human brain after stroke: I. Mechanisms of spontaneous recovery. Ann Neurol. 2008;63(3):272–287. | ||
Khan M, Sekhon B, Giri S, et al. S-Nitrosoglutathione reduces inflammation and protects brain against focal cerebral ischemia in a rat model of experimental stroke. J Cereb Blood Flow Metab. 2005;25(2):177–192. | ||
Liu DH, Yuan FG, Hu SQ, et al. Endogenous nitric oxide induces activation of apoptosis signal-regulating kinase 1 via S-nitrosylation in rat hippocampus during cerebral ischemia-reperfusion. Neuroscience. 2013;229:36–48. | ||
Khan M, Dhammu TS, Sakakima H, et al. The inhibitory effect of S-nitrosoglutathione on blood-brain barrier disruption and peroxynitrite formation in a rat model of experimental stroke. J Neurochem. 2012;123 Suppl 2:86–97. | ||
Yin XH, Yan JZ, Hou XY, Wu SL, Zhang GY. Neuroprotection of S-nitrosoglutathione against ischemic injury by down-regulating Fas S-nitrosylation and downstream signaling. Neuroscience. 2013;248:290–298. | ||
Khan M, Jatana M, Elango C, Paintlia AS, Singh AK, Singh I. Cerebrovascular protection by various nitric oxide donors in rats after experimental stroke. Nitric Oxide. 2006;15(2):114–124. | ||
Sakakima H, Khan M, Dhammu TS, et al. Stimulation of functional recovery via the mechanisms of neurorepair by S-nitrosoglutathione and motor exercise in a rat model of transient cerebral ischemia and reperfusion. Restor Neurol Neurosci. 2012;30(5):383–396. | ||
Chou PC, Shunmugavel A, El Sayed H, et al. Preclinical use of longitudinal MRI for screening the efficacy of S-nitrosoglutathione in treating spinal cord injury. J Magn Reson Imaging. 2011;33(6):1301–1311. | ||
Khan M, Sakakima H, Dhammu TS, et al. S-nitrosoglutathione reduces oxidative injury and promotes mechanisms of neurorepair following traumatic brain injury in rats. J Neuroinflammation. 2011;8:78. | ||
Khan M, Im YB, Shunmugavel A, et al. Administration of S-nitrosoglutathione after traumatic brain injury protects the neurovascular unit and reduces secondary injury in a rat model of controlled cortical impact. J Neuroinflammation. 2009;6:32. | ||
Won JS, Kim J, Annamalai B, Shunmugavel A, Singh I, Singh AK. Protective role of S-nitrosoglutathione (GSNO) against cognitive impairment in rat model of chronic cerebral hypoperfusion. J Alzheimers Dis. 2013;34(3):621–635. | ||
Kimura H, Ogura T, Kurashima Y, Weisz A, Esumi H. Effects of nitric oxide donors on vascular endothelial growth factor gene induction. Biochem Biophys Res Commun. 2002;296(4):976–982. | ||
Lima B, Lam GK, Xie L, et al. Endogenous S-nitrosothiols protect against myocardial injury. Proc Natl Acad Sci U S A. 2009;106(15):6297–6302. | ||
Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1). Mol Pharmacol. 2006;70(5):1469–1480. | ||
Iyer NV, Kotch LE, Agani F, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12(2):149–162. | ||
Kasivisvanathan V, Shalhoub J, Lim CS, Shepherd AC, Thapar A, Davies AH. Hypoxia-inducible factor-1 in arterial disease: a putative therapeutic target. Curr Vasc Pharmacol. 2011;9(3):333–349. | ||
Harten SK, Ashcroft M, Maxwell PH. Prolyl hydroxylase domain inhibitors: a route to HIF activation and neuroprotection. Antioxid Redox Signal. 2010;12(4):459–480. | ||
Palmer LA, Gaston B, Johns RA. Normoxic stabilization of hypoxia-inducible factor-1 expression and activity: redox-dependent effect of nitrogen oxides. Mol Pharmacol. 2000;58(6):1197–1203. | ||
Wellman TL, Jenkins J, Penar PL, Tranmer B, Zahr R, Lounsbury KM. Nitric oxide and reactive oxygen species exert opposing effects on the stability of hypoxia-inducible factor-1alpha (HIF-1alpha) in explants of human pial arteries. FASEB J. 2004;18(2):379–381. | ||
Carver DJ, Gaston B, Deronde K, Palmer LA. Akt-mediated activation of HIF-1 in pulmonary vascular endothelial cells by S-nitrosoglutathione. Am J Respir Cell Mol Biol. 2007;37(3):255–263. | ||
Metzen E, Zhou J, Jelkmann W, Fandrey J, Brune B. Nitric oxide impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Mol Biol Cell. 2003;14(8):3470–3481. | ||
Singh SP, Wishnok JS, Keshive M, Deen WM, Tannenbaum SR. The chemistry of the S-nitrosoglutathione/glutathione system. Proc Natl Acad Sci U S A. 1996;93(25):14428–14433. | ||
Hess DT, Stamler JS. Regulation by S-nitrosylation of protein post-translational modification. J Biol Chem. 2012;287(7):4411–4418. | ||
Stamler JS, Jaraki O, Osborne J, et al. Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proc Natl Acad Sci U S A. 1992;89(16):7674–7677. | ||
Kluge I, Gutteck-Amsler U, Zollinger M, Do KQ. S-nitrosoglutathione in rat cerebellum: identification and quantification by liquid chromatography-mass spectrometry. J Neurochem. 1997;69(6):2599–2607. | ||
Bryan NS, Rassaf T, Maloney RE, et al. Cellular targets and mechanisms of nitros(yl)ation: an insight into their nature and kinetics in vivo. Proc Natl Acad Sci U S A. 2004;101(12):4308–4313. | ||
Tsikas D, Sandmann J, Holzberg D, Pantazis P, Raida M, Frölich JC. Determination of S-nitrosoglutathione in human and rat plasma by high-performance liquid chromatography with fluorescence and ultraviolet absorbance detection after precolumn derivatization with o-phthalaldehyde. Anal Biochem. 1999;273(1):32–40. | ||
Matsumoto A, Gow AJ. Membrane transfer of S-nitrosothiols. Nitric Oxide. 2011;25(2):102–107. | ||
Radomski MW, Rees DD, Dutra A, Moncada S. S-nitroso-glutathione inhibits platelet activation in vitro and in vivo. Br J Pharmacol. 1992;107(3):745–749. | ||
Savidge TC, Newman P, Pothoulakis C, et al. Enteric glia regulate intestinal barrier function and inflammation via release of S-nitrosoglutathione. Gastroenterology. 2007;132(4):1344–1358. | ||
Rassaf T, Poll LW, Brouzos P, et al. Positive effects of nitric oxide on left ventricular function in humans. Eur Heart J. 2006;27(14):1699–1705. | ||
Konorev EA, Tarpey MM, Joseph J, Baker JE, Kalyanaraman B. S-nitrosoglutathione improves functional recovery in the isolated rat heart after cardioplegic ischemic arrest-evidence for a cardioprotective effect of nitric oxide. J Pharmacol Exp Ther. 1995;274(1):200–206. | ||
Chen Q, Sievers RE, Varga M, et al. Pharmacological inhibition of S-nitrosoglutathione reductase improves endothelial vasodilatory function in rats in vivo. J Appl Physiol (1985). 2013;114(6):752–760. | ||
Matsuda F, Sakakima H, Yoshida Y. The effects of early exercise on brain damage and recovery after focal cerebral infarction in rats. Acta Physiol (Oxf). 2011;201(2):275–287. | ||
Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10(2):290–293. | ||
Mahmood A, Goussev A, Lu D, et al. Long-lasting benefits after treatment of traumatic brain injury (TBI) in rats with combination therapy of marrow stromal cells (MSCs) and simvastatin. J Neurotrauma. 2008;25(12):1441–1447. | ||
Li Y, Chen J, Chen XG, et al. Human marrow stromal cell therapy for stroke in rat: neurotrophins and functional recovery. Neurology. 2002;59(4):514–523. | ||
Borlongan CV, Sanberg PR. Elevated body swing test: a new behavioral parameter for rats with 6-hydroxydopamine-induced hemiparkinsonism. J Neurosci. 1995;15(7 Pt 2):5372–5378. | ||
Monville C, Torres EM, Dunnett SB. Comparison of incremental and accelerating protocols of the rotarod test for the assessment of motor deficits in the 6-OHDA model. J Neurosci Methods. 2006;158(2):219–223. | ||
Zhu Y, Bu Q, Liu X, Hu W, Wang Y. Neuroprotective effect of TAT-14-3-3ε fusion protein against cerebral ischemia/reperfusion injury in rats. PLoS One. 2014;9(3):e93334. | ||
Montesano R, Pepper MS, Möhle-Steinlein U, Risau W, Wagner EF, Orci L. Increased proteolytic activity is responsible for the aberrant morphogenetic behavior of endothelial cells expressing the middle T oncogene. Cell. 1990;62(3):435–445. | ||
Prasad R, Giri S, Nath N, Singh I, Singh AK. GSNO attenuates EAE disease by S-nitrosylation-mediated modulation of endothelial-monocyte interactions. Glia. 2007;55(1):65–77. | ||
Bove PF, Hristova M, Wesley UV, Olson N, Lounsbury KM, van der Vliet A. Inflammatory levels of nitric oxide inhibit airway epithelial cell migration by inhibition of the kinase ERK1/2 and activation of hypoxia-inducible factor-1 alpha. J Biol Chem. 2008;283(26):17919–17928. | ||
He P, Philbrick MJ, An X, Wu J, Messmer-Blust AF, Li J. Endothelial differentiation gene-1, a new downstream gene is involved in RTEF-1 induced angiogenesis in endothelial cells. PLoS One. 2014;9(2):e88143. | ||
Jatana M, Giri S, Ansari MA, et al. Inhibition of NF-kappaB activation by 5-lipoxygenase inhibitors protects brain against injury in a rat model of focal cerebral ischemia. J Neuroinflammation. 2006;3:12. | ||
Valable S, Montaner J, Bellail A, et al. VEGF-induced BBB permeability is associated with an MMP-9 activity increase in cerebral ischemia: both effects decreased by Ang-1. J Cereb Blood Flow Metab. 2005;25(11):1491–1504. | ||
Mooberry SL. Mechanism of action of 2-methoxyestradiol: new developments. Drug Resist Updat. 2003;6(6):355–361. | ||
Lok J, Gupta P, Guo S, et al. Cell-cell signaling in the neurovascular unit. Neurochem Res. 2007;32(12):2032–2045. | ||
Wei L, Erinjeri JP, Rovainen CM, Woolsey TA. Collateral growth and angiogenesis around cortical stroke. Stroke. 2001;32(9):2179–2184. | ||
Chen J, Zhang ZG, Li Y, et al. Statins induce angiogenesis, neurogenesis, and synaptogenesis after stroke. Ann Neurol. 2003;53(6):743–751. | ||
Chen J, Cui X, Zacharek A, et al. Niaspan increases angiogenesis and improves functional recovery after stroke. Ann Neurol. 2007;62(1):49–58. | ||
Kawasaki K, Smith RS Jr, Hsieh CM, Sun J, Chao J, Liao JK. Activation of the phosphatidylinositol 3-kinase/protein kinase Akt pathway mediates nitric oxide-induced endothelial cell migration and angiogenesis. Mol Cell Biol. 2003;23(16):5726–5737. | ||
Zhang RL, Zhang ZG, Chopp M. Targeting nitric oxide in the subacute restorative treatment of ischemic stroke. Expert Opin Investig Drugs. 2013;22(7):843–851. | ||
Teng H, Zhang ZG, Wang L, et al. Coupling of angiogenesis and neurogenesis in cultured endothelial cells and neural progenitor cells after stroke. J Cereb Blood Flow Metab. 2008;28(4):764–771. | ||
Lee CZ, Xue Z, Zhu Y, Yang GY, Young WL. Matrix metalloproteinase-9 inhibition attenuates vascular endothelial growth factor-induced intracerebral hemorrhage. Stroke. 2007;38(9):2563–2568. | ||
Bauer AT, Bürgers HF, Rabie T, Marti HH. Matrix metalloproteinase-9 mediates hypoxia-induced vascular leakage in the brain via tight junction rearrangement. J Cereb Blood Flow Metab. 2010;30(4):837–848. | ||
Lee CZ, Xue Z, Hao Q, Yang GY, Young WL. Nitric oxide in vascular endothelial growth factor-induced focal angiogenesis and matrix metalloproteinase-9 activity in the mouse brain. Stroke. 2009;40(8):2879–2881. | ||
Aggarwal A, Khera A, Singh I, Sandhir R. S-nitrosoglutathione prevents blood–brain barrier disruption associated with increased matrix metalloproteinase-9 activity in experimental diabetes. J Neurochem. Epub 2014 Sep 4. | ||
Ke XJ, Zhang JJ. Changes in HIF-1α, VEGF, NGF and BDNF levels in cerebrospinal fluid and their relationship with cognitive impairment in patients with cerebral infarction. J Huazhong Univ Sci Technolog Med Sci. 2013;33(3):433–437. | ||
Zhang Z, Yan J, Taheri S, Liu KJ, Shi H. Hypoxia-inducible factor 1 contributes to N-acetylcysteine’s protection in stroke. Free Radic Biol Med. 2014;68:8–21. | ||
Wang Z, Tsai LK, Munasinghe J, et al. Chronic valproate treatment enhances postischemic angiogenesis and promotes functional recovery in a rat model of ischemic stroke. Stroke. 2012;43(9):2430–2436. | ||
Zhou D, Matchett GA, Jadhav V, Dach N, Zhang JH. The effect of 2-methoxyestradiol, a HIF-1 alpha inhibitor, in global cerebral ischemia in rats. Neurol Res. 2008;30(3):268–271. | ||
Yasinska IM, Sumbayev VV. S-nitrosation of Cys-800 of HIF-1alpha protein activates its interaction with p300 and stimulates its transcriptional activity. FEBS Lett. 2003;549(1–3):105–109. | ||
Schonhoff CM, Matsuoka M, Tummala H, et al. S-nitrosothiol depletion in amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2006;103(7):2404–2409. | ||
Ju TC, Chen SD, Liu CC, Yang DI. Protective effects of S-nitrosoglutathione against amyloid beta-peptide neurotoxicity. Free Radic Biol Med. 2005;38(7):938–949. | ||
Rauhala P, Mohanakumar KP, Sziraki I, Lin AM, Chiueh CC. S-nitrosothiols and nitric oxide, but not sodium nitroprusside, protect nigrostriatal dopamine neurons against iron-induced oxidative stress in vivo. Synapse. 1996;23(1):58–60. | ||
Terpolilli NA, Moskowitz MA, Plesnila N. Nitric oxide: considerations for the treatment of ischemic stroke. J Cereb Blood Flow Metab. 2012;32(7):1332–1346. | ||
Rashid PA, Whitehurst A, Lawson N, Bath PM. Plasma nitric oxide (nitrate/nitrite) levels in acute stroke and their relationship with severity and outcome. J Stroke Cerebrovasc Dis. 2003;12(2):82–87. | ||
Heiss C, Lauer T, Dejam A, et al. Plasma nitroso compounds are decreased in patients with endothelial dysfunction. J Am Coll Cardiol. 2006;47(3):573–579. | ||
Roberts BW, Mitchell J, Kilgannon JH, Chansky ME, Trzeciak S. Nitric oxide donor agents for the treatment of ischemia/reperfusion injury in human subjects: a systematic review. Shock. 2013;39(3):229–239. | ||
Pluta RM, Oldfield EH, Bakhtian KD, et al. Safety and feasibility of long-term intravenous sodium nitrite infusion in healthy volunteers. PLoS One. 2011;6(1):e14504. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.