Back to Journals » OncoTargets and Therapy » Volume 12
Progress in the identification of gene mutations involved in multiple myeloma
Received 19 February 2019
Accepted for publication 30 April 2019
Published 24 May 2019 Volume 2019:12 Pages 4075—4080
DOI https://doi.org/10.2147/OTT.S205922
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Takuya Aoki
Ying Hu,1 Wenming Chen,2 Jingbo Wang1
1Department of Hematology, Aerospace Central Hospital of Peking University, Beijing, People’s Republic of China; 2Department of Hematology, Beijing Chao-Yang Hospital, Capital Medical University, Beijing, People’s Republic of China
Abstract: Sequencing studies have been used to determine a spectrum of multiple myeloma (MM) mutations. Mutation of certain genes, including KRAS, NRAS, TP53, FAM46C, DIS3 and BRAF, have a high recurrence rate and may play important roles in the pathogenesis, progression and prognosis of MM. Mutations in DIS3, which encodes a highly conserved RNA exonuclease, lead to loss of function. The expression of FAM46C is highly correlated with the expression of ribosomal protein, but the exact function of FAM46C mutation is unclear. There are mutants of IRF4, which is considered an MM survival factor. Mutations in the gene coding for the DNA damage-binding protein (DDB1) may affect interactions with CUL4A, which is part of the cereblon (CRBN) ubiquitin ligase complex. IRF4is part of the complex, which binds to DNA. These findings might explain the resistance to immunomodulatory. TP53 deletion or mutation is often present in B-cell malignancies and is associated with low response rates. Myeloma pathogenic mutations in ATM have been found in adult lymphatic tumors. XBP1 and PSMB5 mutations may be related to bortezomib resistance. Multiple gene mutations (KRAS, NRAS and BRAF) involved in the same pathway were found a single patient. Identification of driver gene mutations has brought great hope to the field of individualized, targeted medicine for MM.
Keywords: multiple myeloma, cytogenetic abnormalities, gene mutations
Cytogenetic risk stratification has been established in multiple myelo ma (MM). However, cytogenetic abnormalities are not sufficient to fully explain the occurrence, progression and prognosis of MM. The study of gene mutations involved in MM has taken a leap forward with the recent publication of two sequencing studies of 270 cases that represent a spectrum of MM mutation.1 Mutations with a high recurrence rate, such as those seen in KRAS, NRAS, TP53, FAM46C, DIS3 and BRAF, may play important roles in the pathogenesis, progression and prognosis of MM. The identification of the roles of CRBN, IKZF1 and IKZF3 in treatment with immune modulators, and XBP1s and IRE1 in treatment with proteasome inhibitors, as well as drug targets such as BRAF, have evolved our understanding of diseases such as MM and emphasize the necessity of individualized treatment. The presence of clone heterogeneity at baseline, linear and branching clonal evolution, and therapeutic selection of resistant mutations point to an urgent need for cloning strategies to quickly, accurately and comprehensively assess the patient’s genetic mutation profile to guide accurate treatment. Second-generation sequencing has been instrumental in understanding the genetic spectrum and complexity of subclones. Individual gene mutation profiling before treatment and clonal evolution during treatment will form the basis of individualized treatment and comprise the standard of future treatment. The integration of MM cytogenetics and gene mutations can be used to improve MM classification, track clonal changes, generate a more accurate prognosis and guide treatment more effectively.2
Cytogenetic abnormalities associated with MM
MM is a hematologic malignancy marked by strong heterogeneity throughout the clinical course. Survival ranges from less than 1 year of invasive disease to more than 10 years of inert disease. Therefore, prognostic factors and risk stratification assessment are very important for determining treatment strategies and predicting survival. Cytogenetics and fluorescence in situ hybridization (FISH) have been recognized as important players in risk stratification, especially in the identification of high-risk MM. Nearly half of all monoclonal gammopathy of undetermined significance (MGUS) and MM tumors are hyperdiploid (HRD). Strikingly, HRD tumors rarely have a primary immunoglobulin (Ig)H translocation, whereas non-HRD tumors usually do. Although it has been proposed that non-HRD and HRD tumors represent different pathways of pathogenesis, the timing, mechanism and molecular consequences of hyperdiploidy are unknown. In any case, patients with HRD tumors seem to have a better prognosis than those with non-NHRD tumors. The detection of t(4,14), t(14,16), and del(17p) by FISH is considered high-risk;3 t(11;14) and t(6;14) confer a good prognosis, while t(14,20) is rare and associated with a poor prognosis. Chromosome 1 abnormalities are common in MM, usually in progressive MM, and are associated with resistance to chemotherapy. Lai et al found that 1q21 amplification and del(p53) were related to disease progression after initial treatment, lgH rearrangement and chromosome 1 abnormality were related to the shortening of progression-free survival (PFS), and the survival period for patients with MM with t(14,16) was shorter after conventional chemotherapy.4
Treatment based on cytogenetic risk stratification has yielded clinical benefits, and new targeted drugs have improved outcomes in patients with high-risk cytogenetic abnormalities. Bortezomib (BTZ) can partially overcome the adverse prognosis associated with t(4,14) and del(17p), but is not effective for t(4,14) combined with del(17p).5 High-dose chemotherapy followed by maintenance with a new drug (BTZ or lenalidomide) can improve the poor prognosis associated with high-risk cytogenetic abnormalities, and the 5-year survival rate is similar to that of the standard-risk patients.6 Maintenance therapy with lenalidomide following autologous hematopoietic stem cell transplantation is considered to be the recommended regimen for patients at risk, especially those who did not achieve a very good partial response or above. Maintenance therapy with BTZ is recommended for patients with moderate or high risk.7
High-frequency gene mutations in MM
A powerful approach to understanding the molecular basis of cancer is whole-genome sequencing or exon sequencing, which compare the sequencing results of an individual’s tumor cells with their normal cells to identify acquired somatic mutations. DIS3 encodes the highly conserved RNA exonuclease, which is the catalytic part of the exosome complex, involved in regulating the processing and abundance of all RNA. The multiple DIS3 mutations identified thus far lead to loss of function, which suggests that abnormal regulatory protein translation via DIS3 mutation could be a canceration mechanism of MM.8 Further evidence for translational control in the pathogenesis of MM comes from the mutation of FAM46C. The expression of FAM46C is highly correlated with the expression of ribosomal proteins; however, its exact function remains unclear.9 The BRAF mutation has important clinical significance because such patients can benefit from BRAF inhibitors, which have shown great clinical activity in some studies and may have an effect on MM.10 PRDM1 encodes a transcriptional inhibitor involved in plasma cell differentiation and acts as a tumor suppressor gene in diffuse large B-cell lymphoma (DLBCL). Mutations affecting its function have been described in DLBCL, but the role of PRDM1 in MM is unknown. PRDM1 was found to have recurrent missense or truncated shift mutations, or splicing site mutations.11 Knockout of EGR1 in MM cells can remove JUN-induced MM growth inhibition and apoptosis, and has been reported to be a mechanism of drug resistance in myeloma cells.12 IRF4 is considered an MM survival factor, and RNA interference screening showed that inhibition of IRF4 transcripts resulted in the unviability of MM cell lines. A missense mutation of IRF4 has been identified, for which K123R is the repeated hot spot.13 SP140 is a lymphoid-restricted homologue of SP100 that is expressed in plasma cells. Studies have confirmed that SP140 is a sensitive site for chronic lymphocytic leukemia, mediated by a decrease in the level of SP140 mRNA.14 Two truncation mutations and one nonsense mutation of SP140 have been observed in MM, but the clinical effects are unclear. XBP1 mutations that have been identified in association with proteasome resistance may alter sensitivity to proteasome inhibitors; however, these effects are speculative.15 CYLD mutations, have been observed in MM through deletion and mutation inactivation.16 Recently, PTPRD, which dephosphorylates STAT3 and increases lL-6 levels, has been investigated as a tumor suppressor gene in MM.17 Mutations in the gene coding for the DNA damage-binding protein (DDB1) may affect interactions with CUL4A, which is part of the cereblon (CRBN) ubiquitin ligase complex. IRF4 is part of the complex, which binds to DNA. These findings might explain the resistance to immunomodulatory and steroid drugs, respectively.7 CCND1 point mutation was found, and FGFR3 was initially considered to be a key driver of t(4,14) myeloma.18 Cereblon is a key therapeutic target for immunomodulators. Single nucleotide polymorphisms (SNPs) of CRBN were found in newly diagnosed refractory/relapse patients, but no CRBN mutations were found in those who were resistant to lenalidomide. SNPS were also found in DDB1, but only one patient had a heterozygous mutation. These findings suggest that CRBN and DDB1 mutations are rare and may have limited effects on CRBN-associated drug resistance.19 Sequencing of a multidrug-resistant, extramedullary recurrent tissue revealed frame-shift and point mutations in CRBN, as well as a point mutation in PSMG2 and a point mutation of NR3C1. These mutations may be associated with drug resistance.20 TP53 deletion or mutation is often present in B-cell malignancies and is associated with low response rates. Analyses of the p53 pathway and upstream signaling molecules have included MYC, RAS, ARF, MDM2, ATM and TP53. Deletion and mutation of ATM or TP53 are commonly seen in DLBCLs and mantle cell lymphomas, whereas RAS mutations only occur in MM and plasma cell leukemia (PCL).21 ATM mutations have been found in some adult lymphatic tumors. To study the incidence of ATM mutations in MM, 45 ATM mutations were screened, 2 of which were myeloma pathogenic.22 Downregulation of XBP1, which is highly expressed in malignant plasma cells, is associated with proteasome inhibitor resistance. Certain point mutations of XBP1 may be associated with the transcriptional activity of XBP1, and some studies have shown that low XBP1 protein levels can predict poor efficacy of bortezomib (BTZ). Studies have shown that BTZ-induced PSMB5 mutations lead to resistance to different proteasome inhibitors. However, PSMB5 mutations have not been confirmed in clinical specimens that are resistant to BTZ.23
Characteristics and clinical significance of MM gene mutation
Targeted sequencing analysis revealed that KRAS was the most common mutated gene (36%), followed by NRAS (20%), TP53 (16%), DIS3 (16%) and FAM46C (12%). Initial treatment for MM is usually the induction of high-quality remission, including complete response. However, there is recurrence in almost all patients, which is best explained by the presence of tumor clonal heterogeneity at the time of diagnosis, with differential sensitivity to different drugs leading to clonal selection and evolution. Successful treatment requires the targeting of a wide range of targets including tiny subclones. Therefore, it is necessary to monitor the gene changes of the tumor cell population under the pressure of treatment selection to evaluate the efficacy.15
Mutation diversity affects different nodes of the signal network and is an inherent feature of myeloma.24 Multiple gene mutations (KRAS, NRAS and BRAF) have been found in the same patient, with mutation of different genes located in the same pathway. Studies have found that FAM46C and DIS3 are likely to be the driver genes of MM.18 Other studies have found that BRAF, TRAF3, CYLD and RB1 are involved in the pathogenesis of MM.25 The identification of such driver gene mutations in MM has brought great hope to the field of individualized medicine. Patients with a unique set of mutations can now receive appropriate targeted therapy.
Some mutations are early molecular events, while others occur as the tumor progresses. Another complication is the coexistence of one or more mutations in KRAS, NRAS or BRAF in one master clone (ie, in all tumor cells).26 In a study of a group of refractory MM patients with multidrug resistance who were previously treated with a proteasome inhibitor/immunomodulator or both, the mutation rate of the RAS pathway (KRAS, NRAS, BRAF) was increased by 72% compared with newly diagnosed MM; the mutation rates of TP53 and CRBN were 26% and 12%, respectively.27 Genetic mutations were also detected in patients with recurrent MM who participated in a clinical trial of BTZ. Mutations in NRAS were associated with a low response rate and rapid progression after a single-drug regimen of BTZ. KRAS mutation did not decrease the sensitivity to BTZ.28
Non-myeloma plasma cell disease mutations are significantly lower in patients with myeloma. The spectrum of mutations from MGUS and amyloidosis to MM represents a complex pattern of changes.29 The gene mutation rates among refractory/recurrent patients and newly diagnosed patients are 27.2% and 6.6%, respectively,30 while the rates in MM, primary PCL and secondary PCL are 59.8%, 41.7% and 63.6%, respectively.31 Among the newly diagnosed high-risk del(17p) MM patients, TP53 is the most common (27.8%) mutated gene.7 NRAS and KRAS mutations are less common in high-risk patients, including those with a del(17p) mutation, and more common in relapsed patients.32 In fact, TP53 mutations are more common in unselected MM patients than del(17p) patients. In the non-del(17p) group in one report, TP53 mutations were associated with shortened event-free survival and overall survival (OS).26 The most common mutations in patients with 1q21 included TP53 (38%) and KRAS (25%).33 One study showed that patients with mutations in the RAS pathway (NF-KB) had a neutral prognosis, while CCND1 and DNA repair pathway mutations (TP53, ATM, ATR and ZNFHX4) were associated with a poor prognosis. Mutations in IRF4 and EGR1 were related to good OS. The recurrence of adverse prognostic mutation factors and International Staging System scoring were used to generate an international staging mutation score to identify high-risk patients with recurrence and early death34 (Figure 1). A study was undertaken to compare the prognosis of patients receiving autologous hematopoietic stem cell transplantation induced by immunomodulators and/or proteasome inhibitors with or without TP53 mutations. Before transplantation, 62% of both groups (p=0.97) achieved part response above efficacy; the recurrence rates of the two groups after transplantation were 68% and 42%, respectively (p=0.01). The median PFS rates were 8 months and 28 months (P<0.001), and the median OS rates were 21 months and 56 months (P<0.001), in the patients with and without TP53 mutations, respectively. Hence, TP53 mutation is an independent prognostic factor for the progression of autologous transplantation.35
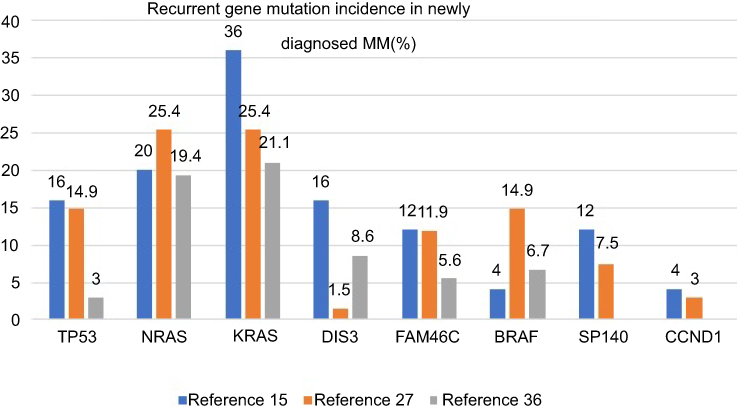 | Figure 1 Recurrent gene mutation incidence in the newly diagnosed MM.15,26,34 |
Persistent response to BRAF inhibitors in MM patients with a single BRAF mutation has been recently reported. It has been shown that BRAF inhibitors can be used to successfully treat recurrent/refractory myeloma with BRAF mutation. MDM2 inhibitors target TP53 deletion or mutation, block the interaction between MDM22-p53 proteins and play an anti-myeloma role. Inhibition of MAPK kinase (MEK) can be used for the treatment of KRAS/NRAS mutant clones. FGFR3 antibodies and MMSET inhibitors are being evaluated. KRAS and ATM mutations affect the downstream signals of MEK and can be used as therapeutic targets. The PI3K pathway plays a role in regulating downstream pathways such as AKT and MTOR, and a large number of clinical trials are studying PIK3 inhibitors20 (Table 1).
 | Table 1 Clinical significance of mutant genes |
The emergence of drug-resistant subclones is one of the root causes of MM recurrence. Whole-genome sequencing at the time of onset and recurrence in 56 MM patients showed that those with complete remission relapsed mainly through the form of branching evolution, which was characterized by the addition of new mutations and changes in the mutant spectrum. Patients with partial remission had similar mutant profiles at onset and recurrence. There was no significant difference in the gene mutation profile at the time of recurrence between the patients observed and those who received lenalidomide for maintenance.36 Analysis of the 60 driver mutations that were identified was used to determine the corresponding pathways for use as therapeutic targets. Drugs that target survival pathways, such as venetoclax, a BCL2 inhibitor, have been shown to be effective in treating myeloma.37
Conclusion
The spectrum of mutants identified in recent studies is insufficient to define their role and place in the individualized treatment of MM. It is not yet clear whether drugs that target these mutational changes will produce a meaningful or lasting response in patients.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lohr JG, Stojanov P, Carter SL. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25(1):91–101. doi:10.1016/j.ccr.2013.12.015
2. Kortüm KM, Langer C, Monge J. Targeted sequencing using a 47 gene multiple myeloma mutation panel (M 3 P) in −17p high risk disease. Br J Haematol. 2015;168(4):507–510. doi:10.1111/bjh.13171
3. Munshi NC, Anderson KC, Bergsagel PL. Consensus recommendations for risk stratification in multiple myeloma: report of the International myeloma workshop consensus panel 2. Blood. 2011;117(18):4696–4700. doi:10.1182/blood-2010-10-300970
4. Lai YY, Huang XJ, Cai Z. Prognostic power of abnormal cytogenetics for multiple myeloma: a multicenter study in China. Chin Med J. 2012;125(15):2663–2670.
5. Sonneveld P, Avet-Loiseau H, Lonial S. Treatment of multiple myeloma with high-risk cytogenetics: a consensus of the International myeloma working group. Blood. 2016;127(24):2955–2962. doi:10.1182/blood-2016-01-631200
6. Kaufman GP, Gertz MA, Dispenzieri A. Impact of cytogenetic classification on outcomes following early high-dose therapy in multiple myeloma. Leukemia. 2016;30(3):633–639. doi:10.1038/leu.2015.287
7. Rajkumar SV. Multiple myeloma: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91(7):719–734. doi:10.1002/ajh.24402
8. Dziembowski A, Lorentzen E, Conti E, Séraphin B. A single subunit, Dis3, is essentially responsible for yeast exosome coreactivity. Nat Struct Mol Biol. 2007;14(1):15–22. doi:10.1038/nsmb1184
9. Tanay A, Regev A, Shamir R. Conservation and evolvability in regulatory networks: the evolution of ribosomal regulation in yeast. Proc Natl Acad Sci U S A. 2005;102(20):7203–7208. doi:10.1073/pnas.0502521102
10. Kim K, Kong S-Y, Fulciniti M, et al. Blockade of the MEK/ERK signalling cascade by AS703026, a novel selective MEK1/2 inhibitor, induces pleiotropic anti-myeloma activity in vitro and in vivo. Br J Haematol. 2010;149(4):537–549. doi:10.1111/bjh.2010.149.issue-4
11. Lin FR, Kuo HK, Ying HY. Induction of apoptosis in plasma cells by B lymphocyte-induced maturation protein-1 knockdown. Cancer Res. 2007;67(24):11914–11923. doi:10.1158/0008-5472.CAN-07-1868
12. Chen L, Wang S, Zhou Y. Identification of early growth response protein 1 (EGR-1) as a novel target for JUN-induced apoptosis in multiple myeloma. Blood. 2010;115(1):61–70. doi:10.1182/blood-2009-12-255992
13. Shaffer AL, Emre NC, Lamy L. IRF4 addiction in multiple myeloma. Nature. 2008;454(7201):226–231. doi:10.1038/nature07064
14. Di Bernardo MC, Crowther-Swanepoel D, Broderick P. A genome-wide association study identifies six susceptibility loci for chronic lymphocytic leukemia. Nat Genet. 2008;40(10):1204–1210. doi:10.1038/ng.126
15. Kortüm KM, Langer C, Monge J. Longitudinal analysis of 25 sequential sample-pairs using a custom multiple myeloma mutation sequencing panel (M 3 P). Ann Hematol. 2015;94(7):1205–1211. doi:10.1007/s00277-015-2344-9
16. Demchenko YN, Glebov OK, Zingone A. Classical and/or alternative NF-kappaB pathway activation in multiple myeloma. Blood. 2010;115(17):3541–3552. doi:10.1182/blood-2009-12-255992
17. Kamada Y, Sakata-Yanagimoto M, Sanada M. Identification of unbalanced genome copy number abnormalities in patients with multiple myeloma by single-nucleotide polymorphism genotyping microarray analysis. Int J Hematol. 2012;96(4):492–500. doi:10.1007/s12185-012-1171-1
18. Chapman MA, Lawrence MS. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471(7339):467–472. doi:10.1038/nature09833
19. Thakurta A, Gandhi AK, Waldman MF. Absence of mutations in cereblon (CRBN) and DNA damage-binding protein 1 (DDB1) genes and significance for IMiD therapy. Leukemia. 2014;28(5):1129–1131. doi:10.1038/leu.2013.315
20. Egan JB, Kortuem KM, Kurdoglu A. Extramedullary myeloma whole genome sequencing reveals novel mutations in cereblon, proteasome subunit G2 and the glucocorticoid receptor in multi drug resistant disease. Br J Haematol. 2013;161(5):748–751. doi:10.1111/bjh.12291
21. Tessoulin B, Eveillard M, Lok A. p53 dysregulation in B-cell malignancies: more than a single gene in the pathway to hell. Blood Rev. 2017;31(4):251–259. doi:10.1016/j.blre.2017.03.001
22. Austen B, Barone G, Reiman A. Pathogenic ATM mutations occur rarely in a subset of multiple myeloma patients. Br J Haematol. 2008;142(6):925–933. doi:10.1111/j.1365-2141.2008.07281.x
23. Verbrugge SE, Assaraf YG, Dijkmans BA. Inactivating PSMB5 mutations and P-glycoprotein (multidrug resistance-associated protein/ATP-binding cassette B1) mediate resistance to proteasome inhibitors: ex vivo efficacy of (immuno)proteasome inhibitors in mononuclear blood cells from patients with rheumatoidarthritis. J Pharmacol Exp Ther. 2012;341(1):174–182.
24. Leich E, Weißbach S, Klein HU. Multiple myeloma is affected by multiple and heterogeneous somatic mutations in adhesion- and receptor tyrosine kinase signaling molecules. Blood Cancer J. 2013;3:e102. doi:10.1038/bcj.2012.47
25. Keats JJ, Fonseca R, Chesi M. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12(2):131–144. doi:10.1016/j.ccr.2007.07.003
26. Bolli N, Avet-Loiseau H, Wedge DC. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun. 2014;5:2997. doi:10.1038/ncomms5972
27. Kortüm KM, Mai EK, Hanafiah NH. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood. 2016;128(9):1226–1233. doi:10.1182/blood-2016-02-698092
28. Mulligan G, Lichter DI, Di Bacco A. Mutation of NRAS but not KRAS significantly reduces myeloma sensitivity to single-agent bortezomib therapy. Blood. 2014;123(5):632–639. doi:10.1182/blood-2013-05-504340
29. Rossi A, Voigtlaender M, Janjetovic S. Mutational landscape reflects the biological continuum of plasma cell dyscrasias. Blood Cancer J. 2017;7(2):e537. doi:10.1038/bcj.2017.19
30. Mithraprabhu S, Khong T, Ramachandran M. Circulating tumour DNA analysis demonstrates spatial mutational heterogeneity that coincides with disease relapse in myeloma. Leukemia. 2017;31(8):1695–1705. doi:10.1038/leu.2016.366
31. Lionetti M, Barbieri M, Todoerti K. Molecular spectrum of BRAF, NRAS and KRAS gene mutations in plasma cell dyscrasias: implication for MEK-ERK pathway activation. Oncotarget. 2015;6(27):24205–24217. doi:10.18632/oncotarget.4434
32. Chng WJ, Gonzalez-Paz N, Price-Troska T. Clinical and biological significance of RAS mutations in multiple myeloma. Leukemia. 2008;22(12):2280–2284. doi:10.1038/sj.leu.2404889
33. Shah GL, Landau H, Londono D. Gain of chromosome 1q portends worse prognosis in multiple myeloma despite novel agent-based induction regimens and autologous transplantation. Leuk Lymphoma. 2017;58(8):1823–1831. doi:10.1080/10428194.2016.1260126
34. Walker BA, Boyle EM, Wardell CP. Mutational spectrum, copy number changes, and outcome: results of a sequencing study of patients with newly diagnosed myeloma. J Clin Oncol. 2015;33(33):3911–3920. doi:10.1200/JCO.2014.59.1503
35. Gaballa S, Saliba RM, Srour S. Outcomes in patients with multiple myeloma with TP53 deletion after autologous hematopoietic stem cell transplant. Am J Hematol. 2016;91(10):E442–E447. doi:10.1002/ajh.24487
36. Jones JR, Weinhold N, Ashby C. Clonal evolution in myeloma: the impact of maintenance lenalidomide and depth of response on the genetics and sub-clonal structure of relapsed disease in uniformly treated newly diagnosed patients. Haematologica. 2019. doi:10.3324/haematol.2018.202200
37. Morgan GJ, Rasche L. Maintaining therapeutic progress in multiple myeloma by integrating genetic and biological advances into the clinic. Expert Rev Hematol. 2018;11(7):513–523. doi:10.1080/17474086.2018.1489718
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.