Back to Journals » OncoTargets and Therapy » Volume 11
Progranulin modulates cholangiocarcinoma cell proliferation, apoptosis, and motility via the PI3K/pAkt pathway
Authors Daya M, Loilome W, Techasen A, Thanee M, Sa-Ngiamwibool P, Titapun A ![]() , Yongvanit P, Namwat N
, Yongvanit P, Namwat N ![]()
Received 29 October 2017
Accepted for publication 5 December 2017
Published 18 January 2018 Volume 2018:11 Pages 395—408
DOI https://doi.org/10.2147/OTT.S155511
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yao Dai
Minerva Daya,1–3 Watcharin Loilome,1,3 Anchalee Techasen,3,4 Malinee Thanee,3 Prakasit Sa-Ngiamwibool,4,5 Attapol Titapun,5,6 Puangrat Yongvanit,3 Nisana Namwat1,3
1Department of Biochemistry, Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand; 2Department of Biochemistry, Faculty of Pharmacy, University of Santo Tomas, Sampaloc, Manila, Philippines; 3Cholangiocarcinoma Research Institute, 4Faculty of Associated Medical Science, 5Department of Pathology, 6Department of Surgery, Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand
Abstract: Progranulin (PGRN) is a growth factor normally expressed in rapidly cycling epithelial cells for growth, differentiation, and motility. Several studies have shown the association of PGRN overexpression with the progression of numerous malignancies, including cholangiocarcinoma (CCA). However, the underlying mechanisms on how PGRN modulates CCA cell proliferation and motility is not clear. In this study, we investigated the prognostic significance of PGRN expression in human CCA tissue and the mechanisms of PGRN modulation of CCA cell proliferation and motility. We found that CCA tissues with high PGRN expression were correlated with poor prognosis and likelihood of metastasis. PGRN knockdown KKU-100 and KKU-213 cells demonstrated a reduced rate of proliferation and colony formation and decreased levels of phosphatidyl inositol-3-kinase (PI3K) and phosphorylated Akt (pAkt) proteins. Accumulation of cells at the G1 phase was observed and was accompanied by a reduction of cyclin D1 and CDK4 protein levels. Knockdown cells also induced apoptosis by increasing the Bax-to-Bcl-2 ratio. Increased cell apoptosis was confirmed by annexin V-FITC/PI staining. Moreover, suppression of PGRN reduced CCA cell migration and invasion in vitro. Investigating the biomarkers in epithelial–mesenchymal transition (EMT) revealed a decrease in the expression of vimentin, snail, and metalloproteinase-9. In conclusion, our findings imply that PGRN modulates cell proliferation by dysregulating the G1 phase, inhibiting apoptosis, and that it plays a role in the EMT affecting CCA cell motility, possibly via the PI3K/pAkt pathway.
Keywords: progranulin, cholangiocarcinoma, proliferation, migration, invasion, EMT
Introduction
Cholangiocarcinoma (CCA) is a cancer arising from the epithelial cells lining bile ducts, and its prevalence is increasing worldwide.1 In Thailand, CCA is the major public health problem, particularly in the Northeast Thailand where the etiology of the disease is strongly associated with liver fluke (Opisthorchis viverrini) infection. Infected individuals develop a persistent bile duct inflammation that can progress to CCA.2 The incidence rates of CCA in this region are ~93–318 per 100,000 people per year, affecting males more than females, with an estimated 20,000 deaths per year.3,4 The differences in the development of CCA between genders have been previously investigated in experimental animal infected with O. viverrini. These results showed no gender differences in individual’s responses to the infection and in the development of CCA, an implication that the higher prevalence of opisthorchiasis among males than that in females may depend on individual exposure to risk factors rather than gender difference.5 Tobacco smoking and alcohol consumption are among the risk factors associated with the production of free radical intermediates causing several types of DNA lesions leading to the development of cancer.6 Although habitual smoking and heavy alcohol consumption are more common among males in the region, there is no clear evidence for gender differences that associate smoking and drinking in the progression of CCA.7,8 Study on gender differences remains a challenge, elucidating the differences in hormonal expressions could possibly provide better understanding on gender differences in opisthorchiasis and the development of CCA.9–11 CCA progression is relatively slow, and patients present at the hospital mostly with late-stage disease when the cancer has metastasized to other organs. Chemotherapy in combination with surgery, rather than surgery alone, can reduce the tumor size and prolong the patients’ survival.12 Therefore, the underlying mechanisms promoting tumor cell function, particularly the changes in molecular pathways during CCA progression, need to be investigated. This will contribute to the improvement of CCA treatment guidelines.
Progranulin (PGRN) is a secreted cysteine-rich glycoprotein growth factor that is involved in inflammation and wound response. It is also an important mediator of tumor cell functions. It is expressed not only in rapidly cycling epithelial cells but also in leukocytes, neurons, and chondrocytes.13 Overexpression of PGRN has been observed in numerous tumors of epithelial origin, including breast, ovary, prostate, renal, liver, and bile duct cancers.14,15 These tumors show a strong correlation among high PGRN expression, a poor prognosis, and tumor severity. PGRN mediates tumor cell functions by regulating the rate of epithelial cell division and promotes the transformation to an invasive phenotype of these cells. PGRN activates oncogenic signaling pathways such as the extracellular-regulated kinase (ERK), mitogen-activated protein kinase (MAPK), phosphatidyl inositol-3-kinase (PI3K), and focal adhesion kinase (FAK).15
Activation of PI3K/Akt pathway is commonly observed in tumors overexpressing PGRN. The downstream effectors of the Akt pathway induce the cells to proliferate and transform into the metastatic phenotype.15–18 The epithelial–mesenchymal transition (EMT) is recognized as an important event in tumor metastasis in which the epithelial cells lose their apicobasal polarity, leading to reduced cell–cell adhesions and the acquisition of mesenchymal invasive characteristics.19 The Akt pathway promoting EMT in tumors confers the motility required for invasion and metastasis. Moreover, EMT is induced by various cytokines and growth factors.20 Among the growth factors associated with tumor metastasis, the possible involvement of PGRN in the EMT has not been previously addressed.
Persistent inflammation in the biliary tract strongly predisposes individuals to the development of CCA.3 The inflammatory cytokine, interleukin-6 (IL-6), is upregulated in CCA, which drives the overexpression of PGRN mediating cell proliferation by inactivating the forkhead box protein O1 (FoxO1), a downstream target of Akt signaling.18 Other molecular changes underlying PGRN-induced cell proliferation need further investigation to provide a better understanding of the role of PGRN in the progression of CCA.
In this study, we demonstrated the prognostic significance of PGRN expression and the changes in the molecular pathway underlying the involvement of PGRN in CCA cell proliferation, migration, and metastasis.
Materials and methods
Clinical samples
CCA tissues were collected from 50 patients admitted at Srinagarind Hospital, Khon Kaen University (Khon Kaen, Thailand) from January 2004 to December 2010. Of the 50 CCA patients investigated, 72% were male and 28% were female, resulting in a male-to-female ratio of 2.6:1. The age of patients ranged from 39 to 74 years (mean±SD, 59±7.7 years). The patients were at an advanced stage of the disease with 48% presenting with metastases. In this study, the histological types were classified as papillary type CCA (50%) and non-papillary type CCA (50%). The protocols for the collection of tissues and the study were approved by the Ethics Committee for Human Research, Khon Kaen University (no HE571283), and written informed consent was provided by all patients. The 50 CCA formalin-fixed, paraffin-embedded tissues stored at the specimen bank of the Liver Fluke and Cholangiocarcinoma Research Center (Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand) were cut into 4-μm-thick sections.
CCA cell lines and cell cultivation
The human CCA cell lines KKU-100 and KKU-213 were obtained from the Japanese Collection of Research Bioresources (JCRB) Cell Bank. The human cholangiocyte MMNK1 cell line (transduced with SV40T and hTERT)21 was a gift from Professor Naoya Kobayashi (Okayama University, Japan). Cells were cultured in HAM-F12 (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/mL penicillin, and 100 mg/mL streptomycin (Thermo Fisher Scientific) at 37°C with 5% CO2.
Immunohistochemical (IHC) staining of CCA tissues
The tissue sections (4 μm) of formalin-fixed, paraffin-embedded samples were deparaffinized in xylene and rehydrated in a series of graded alcohol solutions. Antigen retrieval was performed in 10 mM citrate buffer (pH 6.0) at high power for 10 min using a microwave oven. Then, the sections were incubated with 0.3% (v/v) hydrogen peroxide (H2O2) in phosphate-buffered saline (PBS) for 1 h, and non-specific bindings were blocked with 10% (w/v) skimmed milk in PBS for 1 h. The tissue sections were incubated with mouse monoclonal anti-human PGRN (1:50; Cat no PG359-7; Adipogen Inc., Yeonsu-gu, Incheon, South Korea) in a humidified chamber overnight at 4°C. The following day, the sections were washed with 0.1% (v/v) Tween 20 in PBS and were incubated with peroxidase-conjugated Envision™ (Agilent Technologies, Santa Clara, CA, USA) for 1 h. The immunostaining was developed with a 3,3′-diaminobenzidine tetrahydrochloride (DAB) substrate kit (Vector Laboratories, Burlingame, CA, USA) for 5 min and then counterstained with Mayer’s hematoxylin. The sections were dehydrated stepwise in increasing ethanol concentrations, cleared with xylene, and finally mounted with permount. The stained sections were viewed under a light microscope. The staining frequency of these proteins was semiquantitatively scored based on the percentage of positive cells as follows: 0%=negative, 1%–25%=+1, 26%–50%=+2, or >50%=+3. The staining intensity in the tumor cells was scored as negative=0, weak=1, moderate=2, or strong=3. Qualitative expression patterns were calculated by multiplying the frequency score with the intensity score and classified into three categories: negative; 1–3, low expression; or 4–9, high expression.
Transient knockdown of PGRN expression
The small interfering RNAs (siRNAs) of human PGRN and control siRNA were purchased from Dharmacon (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA). Briefly, 9.0×104 cells were cultured in a six-well plate, and the cells were transfected with a final concentration of 50 nM of siRNA duplexes using Lipofectamine transfection reagent (Thermo Fisher Scientific). Then, transfected cells were incubated at 37°C in 5% CO2 for 48 h. Silencing of PGRN expression was confirmed with Western blot analysis.
Western blot analysis
Transfected cells were lysed using radioimmunoprecipitation assay (RIPA) buffer containing protease K inhibitor cocktail, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, and deionized water (Thermo Fisher Scientific), and the total proteins were extracted by centrifugation. The extracted proteins were measured using the bicinchoninic acid assay (Thermo Fisher Scientific). Equal amounts of extracts were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene fluoride (PVDF) membrane (EMD Millipore, Billerica, MA, USA). The membrane was blocked with 5% skimmed milk in PBS with 0.5% Tween-20 (PBST) or tris-buffered saline (TBS) and incubated with the appropriate antibody. The following primary antibodies were used: mouse monoclonal anti-PGRN (1:3,000, Adipogen Inc.), anti-PI3K (1:2,000, Cat no 611342; BD Biosciences, San Diego, CA, USA), anti-phosphorylated Akt (pAkt) (ser473) (1:1,000, Cat no 560397; BD Biosciences), anti-CDK4 (DCS156) (1:500, Cat no 2906; Cell Signaling Technology Inc., Danvers, MA, USA), anti-vimentin (1;1,000, Cat no ab92457; Abcam, Cambridge, England, UK), anti-Bax (1:1,000, Cat no 810983, BD Biosciences), rabbit polyclonal anti-cyclin D1 (1:500, Cat no 29225; Cell Signaling Technology Inc.), anti-Bcl-2 (1:1,000, Cat no 2876; Cell Signaling Technology Inc.), anti-snail (1:100, Cat no 82846; Abcam), and goat polyclonal anti-human metalloproteinase (MMP)-9 (1:1,000, Cat no AF911; R&D Systems, Inc., Minneapolis, MN, USA). Mouse monoclonal anti-beta actin antibody (1:20,000, Cat no ab6276; Abcam) was used as the loading control. Horseradish peroxidase (HRP)-conjugated, secondary antibodies (Bio-Rad Laboratories Inc., Hercules, CA, USA) were used at an appropriate dilution. Finally, the membrane was exposed to SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific): luminal-based enhanced chemiluminescence HRP substrate (product code:10308449; Thermo Fisher Scientific) for chemiluminescent detection. Band density on the membranes was captured using the ImageQuant Imager (GE Healthcare Life Sciences, Chicago, IL, USA) and was calculated using ImageJ free software.
Cell growth assay
The sulforhodamine B (SRB) assay was used to determine growth inhibition, as described previously.22 In brief, transfected cells (1×103 cells per well) were seeded in five replicates into 96-well flat bottom microtiter plates and were incubated for 24, 48, 72, and 96 h at 37°C in a humidified 5% CO2 atmosphere. The culture medium was removed, and a 200 μL aliquot of 10% (w/v) ice-cold trichloroacetic acid (TCA) was added to each culture well. The plates were incubated at 40°C for 60 min, and the TCA-treated cells were stained for 30 min with 0.4% (w/v) SRB in 1% (v/v) acetic acid for 30 min and subsequently washed with 1% (v/v) acetic acid. The plates were air dried, and the protein-bound dye was solubilized with 200 μL of 10 mM Tris base (pH 10.5) for 60 min. Absorbance was measured at 540 nm using a microplate reader (Tecan, GmbH, Salzburg, Austria).
Clonogenic cell survival assay
The clonogenic assay determines the ability of a cell to proliferate indefinitely to form a large colony or a clone, as previously described with modifications.23 In brief, transfected cells were seeded (2×102 per well) in a six-well plate. After 10 days of observation, the colonies formed were washed once with PBS, then fixed with 1 mL 4% paraformaldehyde in PBS, and incubated at room temperature for 20 min. After incubating, the colonies were washed once with PBS, and the cells were stained with crystal violet diluted in ethanol. After 10 min of incubation, the dye was removed carefully by washing the plate under running tap water and allowed to air dry. The colonies were counted manually using ImageJ free software.
Scratch wound-healing assay
Transfected cells (50×104 cells) were seeded in a 24-well plate and grown until reaching ~70% confluence. The confluent cell monolayers were scratched using a sterile p200 pipette tip and rinsed several times with PBS to remove cell debris. Cell migration to close the wound was examined at 0, 12, 24, 48, and 96 h by microscopy and digitally photographed. The breadth of the wound area was measured on the images, and the migration area was calculated using the following formula:
Migration area = (Area of original wound − Area of wound during healing)/Area of original wound |
Cell migration assay
A cell migration assay was performed using a 6.5-mm Transwell® with an 8.0-μm Pore Polycarbonate Membrane Insert (Corning Incorporated, Corning, NY, USA). In brief, transfected cells (4×104 cells) were seeded into the upper chamber with serum-free medium, and aliquots of complete HAM’s F12 media were placed in the lower chamber. After 72 h of incubation for KKU-100 and 24 h for KKU-213, non-migrating cells residing at the upper filter were removed. Migrating cells that were attached to the underside of the filter were fixed with methanol and stained with hematoxylin overnight at room temperature. The number of migratory cells was quantified by counting them under a light microscope. The mean value of five fields at ×200 magnification was calculated.
Cell invasion assay
A cell invasion assay was performed using Corning® BioCoat™ Matrigel® Invasion Chambers with an 8.0-μm polyethylene terephthalate (PET) membrane (Discovery Labware, Inc., Bedford, MA, USA). In brief, a total of 4×104 transfected cells in serum-free media (HAM-F12) were seeded into the upper chamber of Transwell precoated Matrigel culture inserts (Becton Dickinson, Franklin Lakes, NJ, USA), and HAM-F12 medium supplemented with 10% (v/v) FBS was placed into the lower chamber. After incubation at 37°C for 72 h (KKU-100) and 24 h (KKU-213), cells at the upper surface of the filter were removed and those cells that had transversed through the Matrigel and invaded to the underside of the filter were fixed with absolute ethanol for 30 min and stained with hematoxylin. The invading cells (mean value of five fields at ×200 magnification) were counted under the microscope.
Cell cycle analysis
Transfected cells were collected and fixed with 70% precooled ethanol overnight. The cell cycle phases were examined by flow cytometry (FACSCanto II™, BD Biosciences) after staining with propidium iodide (Sigma-Aldrich Co., St Louis, MO, USA) in the dark for 30 min at 37°C. Quantitation of cell cycle distribution was performed using BDFACSDiva™ Software (BD Biosciences). The percentage of cells in G0, G1, S, and G2 phases was calculated.
Cell apoptosis analysis
The Bax-to-Bcl-2 ratio of the transfected cells were determined by the Western blot analysis. Cell apoptosis was confirmed by annexin V staining. After treatment, cells were washed and resuspended in the staining buffer containing 1 μg/mL PI (Sigma-Aldrich Co.) and 0.025 μg/mL annexin V-FITC (BD Biosciences) at room temperature for 15 min in the dark. The stained cells were immediately determined using FACScanto II and analyzed using FACSDiva software (BD Biosciences).
Statistical analysis
The quantitative data are expressed as mean±SD. The SPSS statistical package for Windows version 19 (SPSS Inc., Chicago, IL, USA) and GraphPad Prism 5 software (GraphPad Software, Inc., La Jolla, CA, USA) were used for the statistical analysis. Results obtained from cell growth, clonogenic, cell migration, cell invasion assays, and cell cycle analysis were done in three independent trials with duplicates. The results from Western blotting were done in three independent trials. The significance was determined by the Student’s t-test. The survival curve for low and high PGRN expression was constructed by the Kaplan–Meier method, and the log-rank test was used for analyzing the differences. Fisher’s exact test was used to compare nominal data. The variables that correlated significantly with one of the parameters were included in the multivariate analysis. P<0.05 was defined as statistically significant.
Results
Upregulation of PGRN expression was correlated with a poor prognosis and overall metastasis
IHC results in this study revealed positive staining of PGRN protein in the cytoplasm of hyperplastic and tumor cells, whereas no or very weak staining was observed in the adjacent normal bile ducts (Figure 1A). Among the 50 CCA tissues investigated, high expression of PGRN was observed in 26 (52%) cases and low expression in 24 (48%) cases. Patients with higher expressions of PGRN had a significantly shorter survival when compared to those patients with lower expressions of PGRN (P=0.04; Figure 1B). There was a significant positive association between high PGRN expression levels and overall metastasis (Fisher’s exact test: P=0.002). Age, gender, and histological grade did not show any association with PGRN protein levels (Table 1). Metastasis and persistent PGRN expression significantly favored survival in multivariate analysis (Table 2).
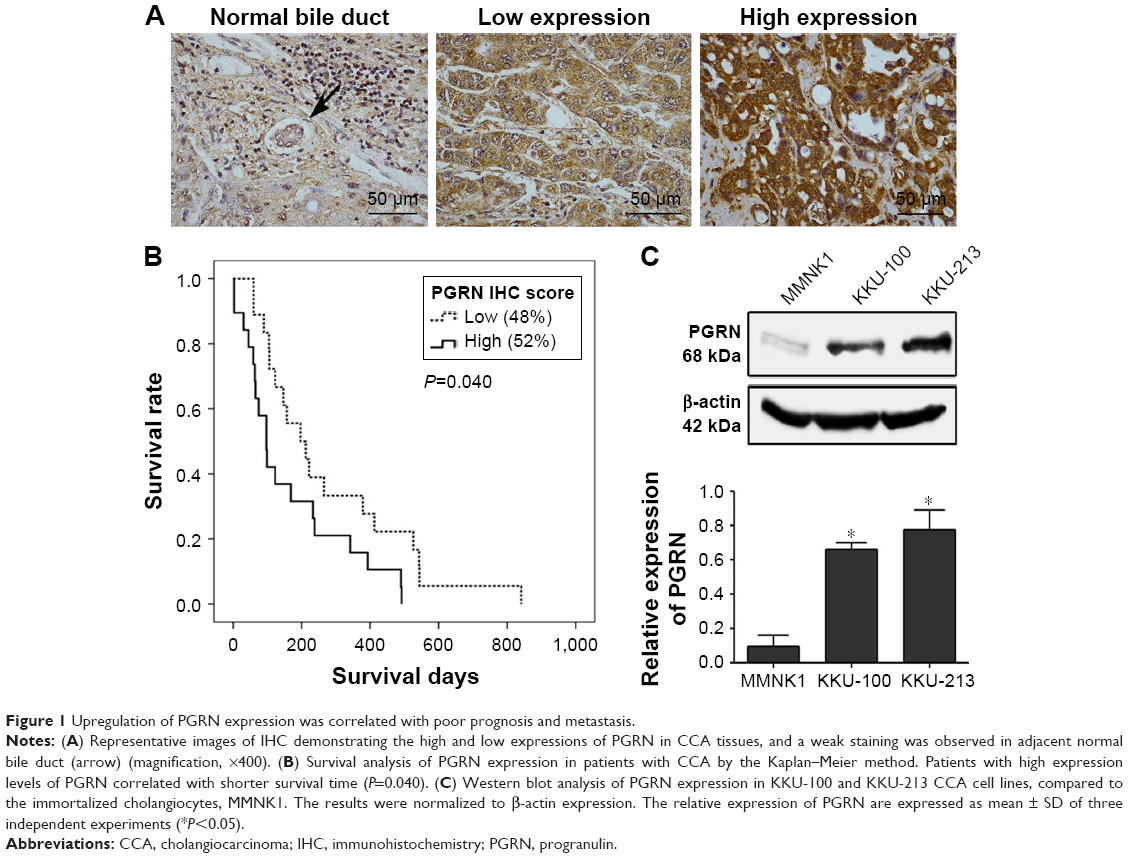 | Figure 1 Upregulation of PGRN expression was correlated with poor prognosis and metastasis. |
 | Table 1 Univariate analysis for PGRN expression in human CCA tissues and the patient’s clinicopathological data |
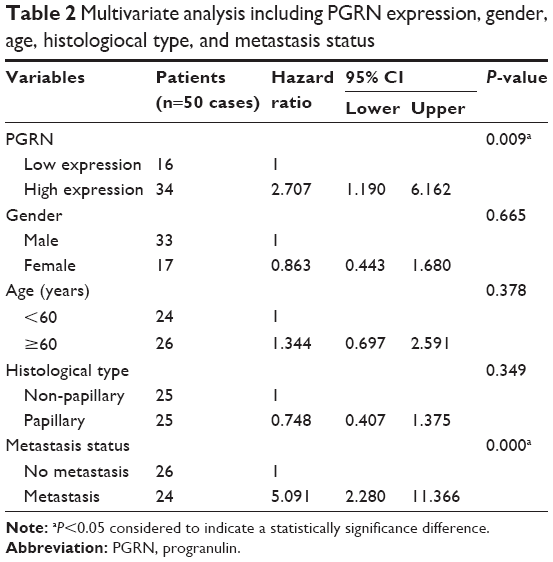 | Table 2 Multivariate analysis including PGRN expression, gender, age, histologiocal type, and metastasis status |
Knockdown of PGRN expression reduced CCA cell proliferation, colony formation, migration, and invasion
Western blotting results showed that both KKU-100 and KKU-213 cell lines expressed a high level of PGRN as opposed to immortalized cholangiocyte MMNK1 cells that showed a low-PGRN level (Figure 1C). The PGRN expression was significantly suppressed by transfecting the CCA cells with siRNA targeting PGRN (siPGRN) compared with the siRNA non-targeting control (siControl; Figure 2A). We then observed the viability and proliferative activity of these transfected cells using the SRB assay. Results showed that the cell viability of both transfected cells decelerated the rate of cell proliferation compared with the siControls. Reduced cell proliferation was significantly observed starting at 72 h post-transfection in KKU-100 and at 48 h posttransfection in KKU-213 (both P<0.05; Figure 2B). We also performed a clonogenic assay to observe the ability of the transfected cells to form colonies. Our results showed 25% reduction of colony formation on day 10 post-transfection for KKU-100 and a 50% reduction of colony formation on day 7 of post-transfection for KKU-213 compared with the siControls (both P<0.05; Figure 3).
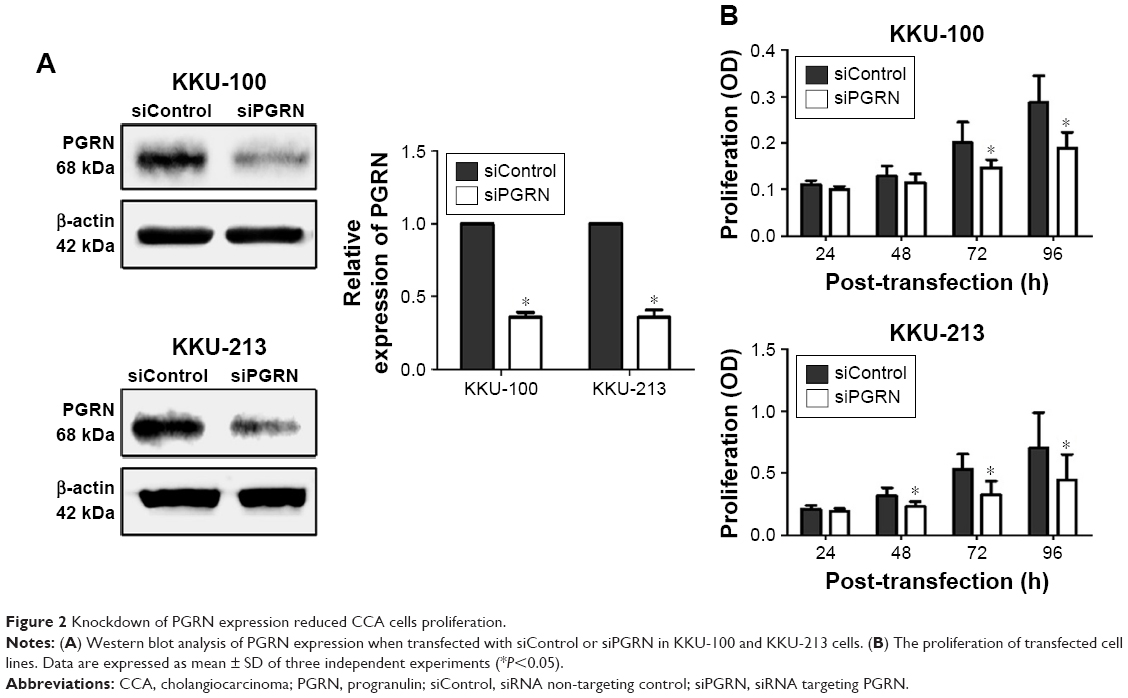 | Figure 2 Knockdown of PGRN expression reduced CCA cells proliferation. |
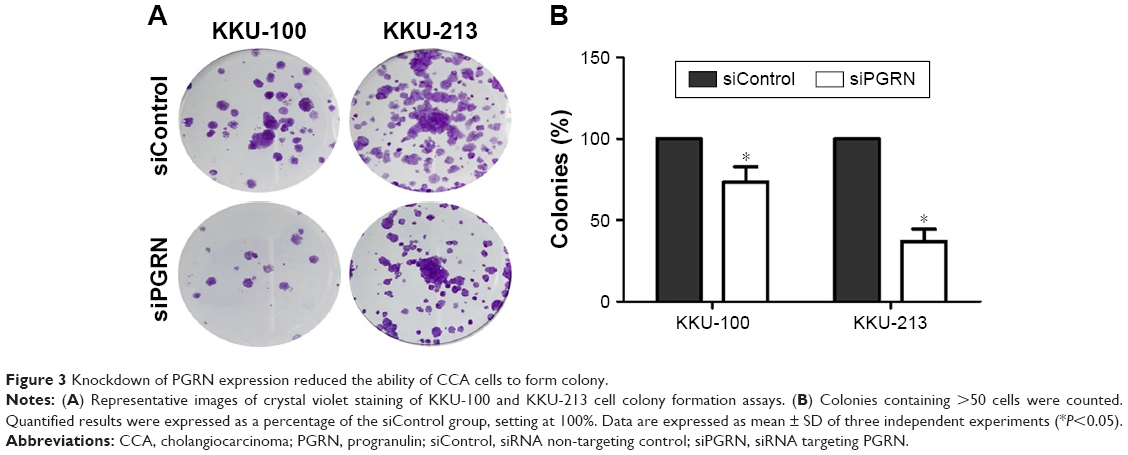 | Figure 3 Knockdown of PGRN expression reduced the ability of CCA cells to form colony. |
The lateral migration ability of siPGRN-transfected cells was also investigated using the in vitro wound-healing assay. Cell migration activity was observed after every 6 h until the siControl-transfected cells completely spread to close the wound. The siPGRN-transfected KKU-100 cells showed slower migration and an inability to completely close the wound at 96 h compared with the siControl-transfected cells (P<0.05). The siPGRN-transfected KKU-213 cells also showed slower migration and an inability to completely close the wound at 24 h compared with the siControl cells (P<0.05; Figure 4).
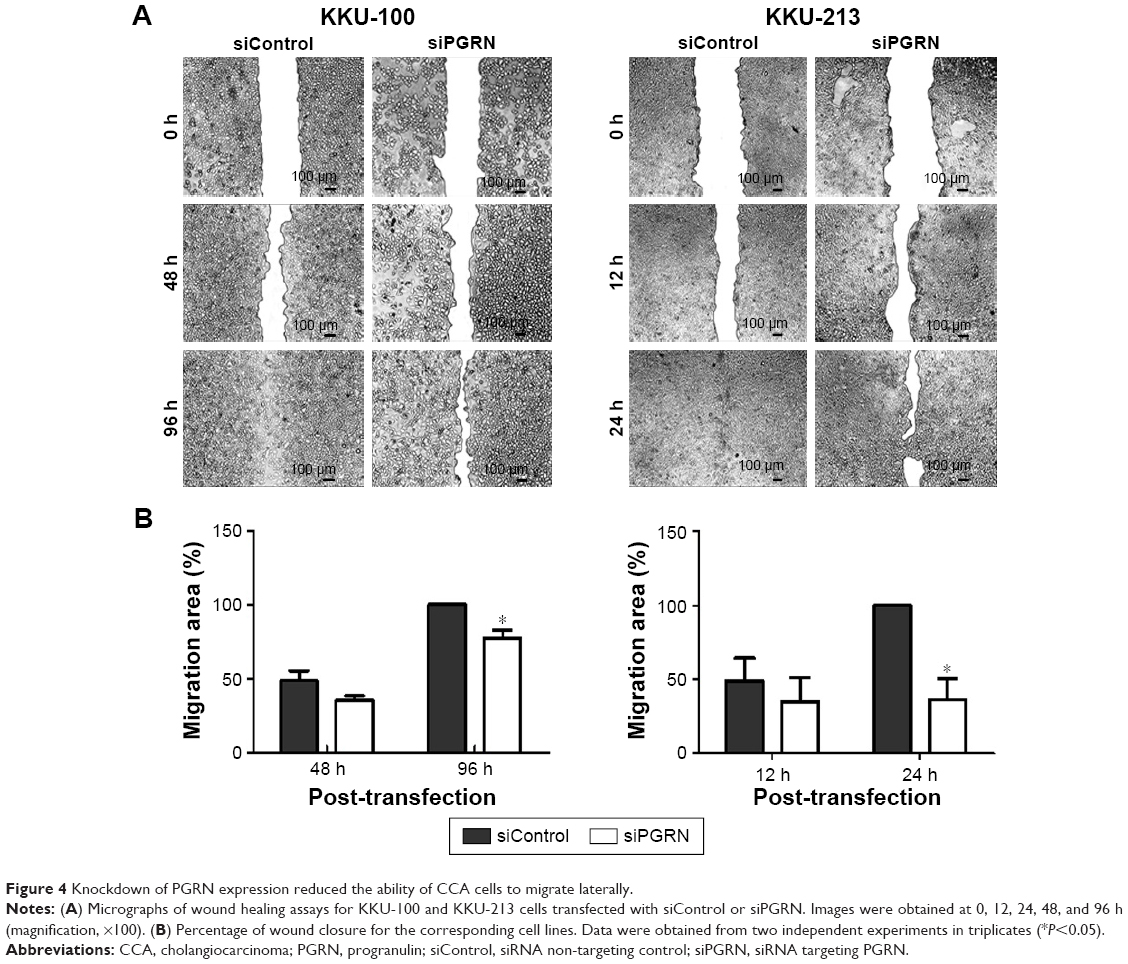 | Figure 4 Knockdown of PGRN expression reduced the ability of CCA cells to migrate laterally. |
The modified Boyden chamber transwell was performed to investigate the transmigration activity of the transfected cells. Results showed lesser number of migrating cells when KKU-100 cells were transfected with siPGRN, and a 65% reduction of cell migration was significantly observed (P<0.05). The siPGRN-transfected KKU-213 cells resulted in 70% reduction of cell migration compared with the siControl cells (P<0.05; Figure 5A).
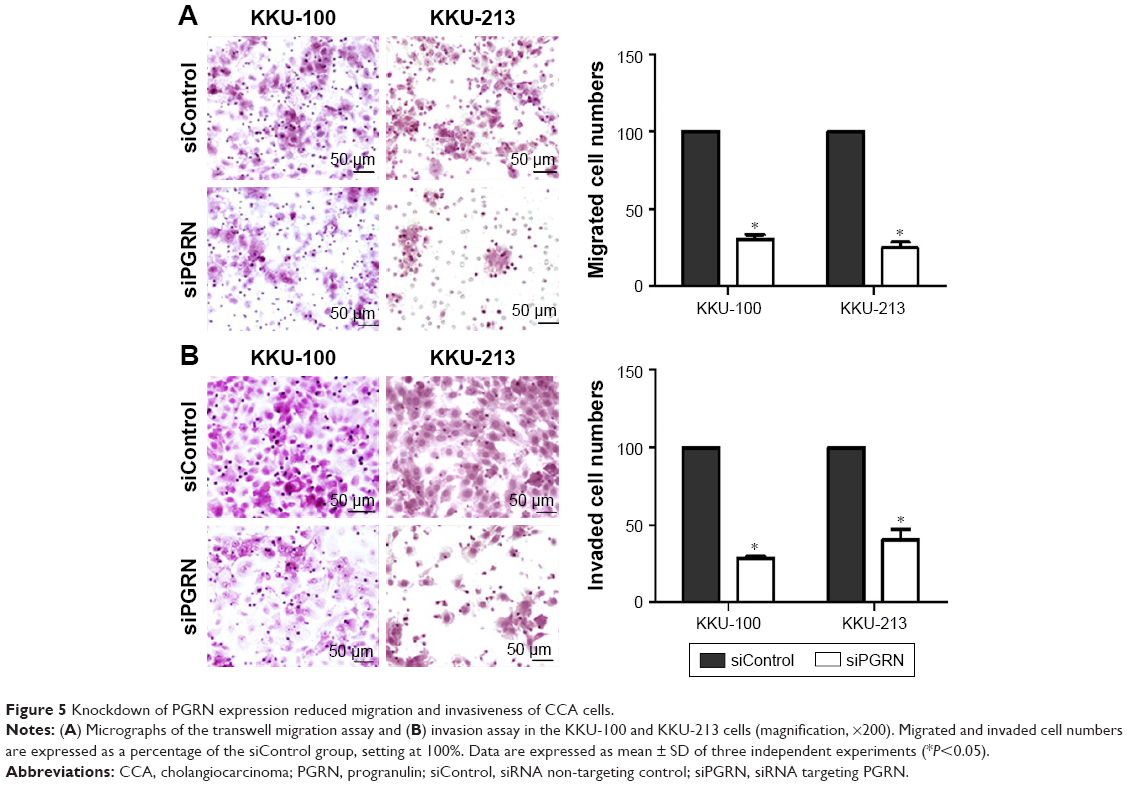 | Figure 5 Knockdown of PGRN expression reduced migration and invasiveness of CCA cells. |
The ability of the siPGRN-transfected CCA cells to digest the matrigel and invade the underside of the membrane was also performed, and the results showed reduction of the ability of KKU-100 transfected cells to invade by 70% (P<0.05) and KKU-213 by 60% compared with that of the siControl cells (P<0.05; Figure 5B).
Knockdown of PGRN expression downregulates the expression of PI3K and phosphorylation of Akt
The signaling pathway possibly involved in modulating cell proliferation, apoptosis, and motility was investigated in siPGRN-transfected cells using Western blotting. Results showed the downregulation of PI3K and pAkt in both siPGRN-transfected KKU-100 and KKU-213 cell lines compared with that in the siControl cells (both P<0.05; Figure 6).
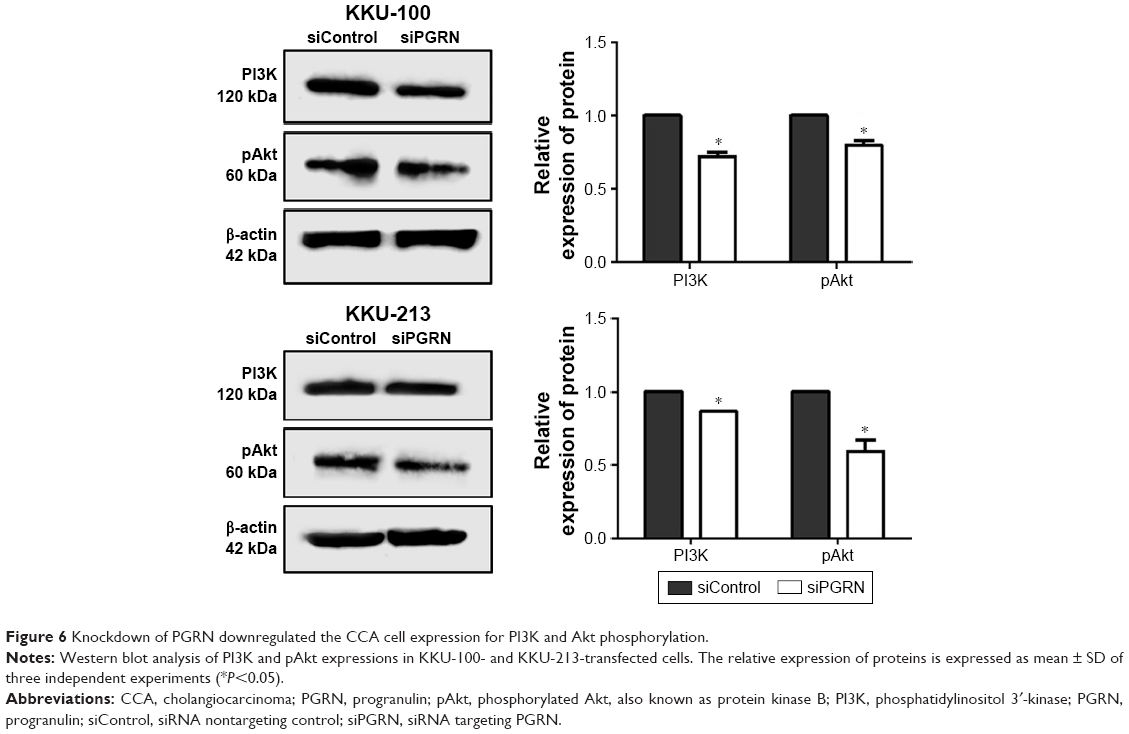 | Figure 6 Knockdown of PGRN downregulated the CCA cell expression for PI3K and Akt phosphorylation. |
Knockdown of PGRN expression dysregulates the CCA cell cycle at the G1 phase and promotes apoptosis
Cell cycle analysis of siPGRN-transfected cells showed an increase of 3% parental cells at the G1 phase in both KKU-100 and KKU-213 cell lines (both P<0.05; Figure 7A). Investigating the molecular changes at the G1 phase revealed a significant decrease in the expressions of cyclin D1 and CDK4 in KKU-100- and KKU-213-transfected cells compared with the siControl cells (both P<0.05; Figure 7B).
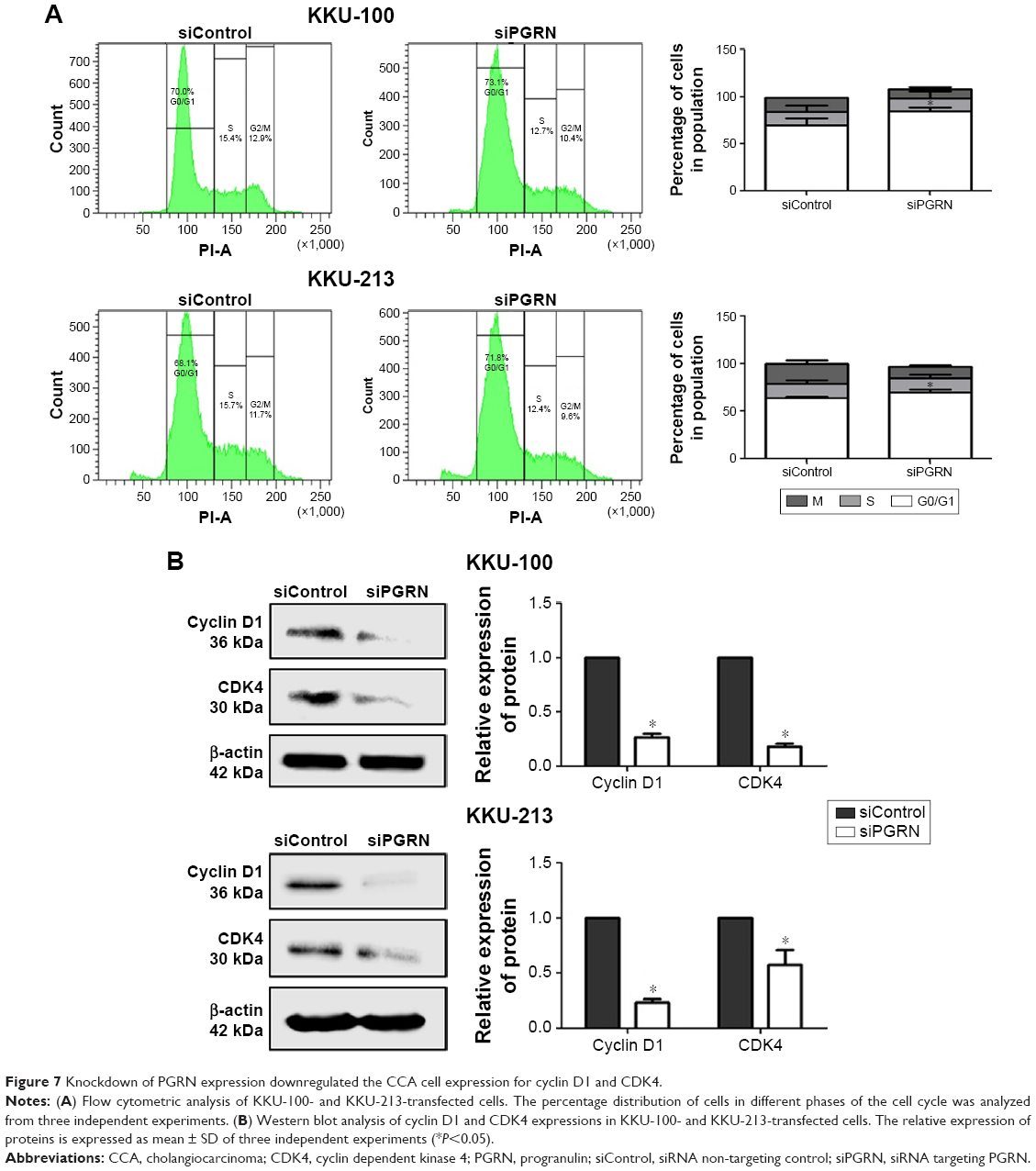 | Figure 7 Knockdown of PGRN expression downregulated the CCA cell expression for cyclin D1 and CDK4. |
The involvement of PGRN in the apoptotic activity of CCA cells was investigated by examining the molecular changes in the expression of Bax and Bcl-2. Results showed an increase in the ratio of Bax-to-Bcl-2 protein observed in siRNA-PGRN-transfected KKU-100 and KKU-213 cells compared with the siControl cells (both P<0.05; Figure 8A). Increased apoptotic cells were also observed in siRNA-PGRN-transfected cells using the annexin V/PI staining (P<0.05; Figure 8B).
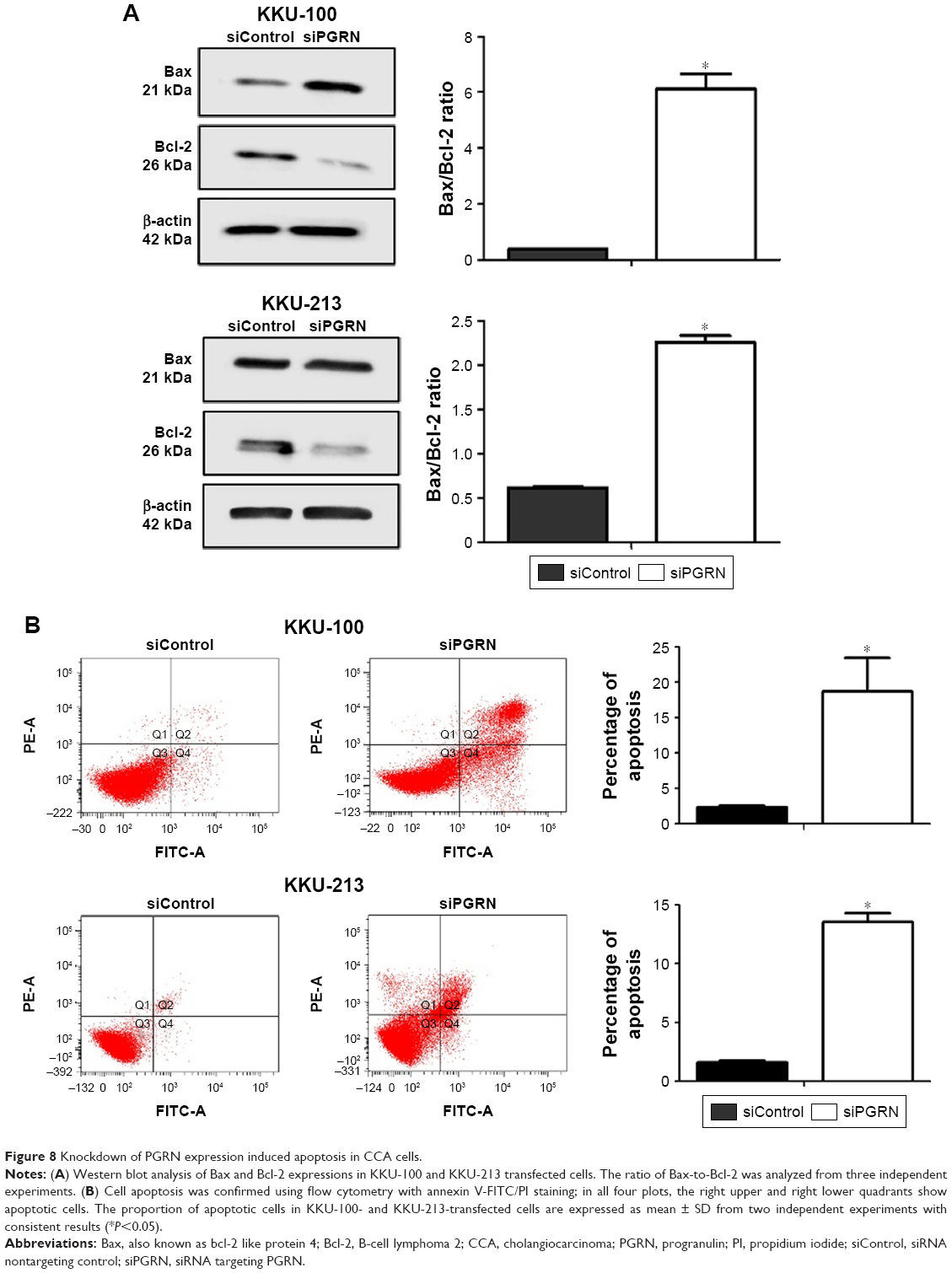 | Figure 8 Knockdown of PGRN expression induced apoptosis in CCA cells. |
Knockdown of PGRN expression decreased the expression of mesenchymal biomarkers in CCA cells
Most tumor cells express the mesenchymal-like characteristics that enable the cells to migrate and invade the surrounding tissues. The expression of mesenchymal biomarkers was investigated in this study. Our results showed expressions of vimentin, snail, and MMP-9 in CCA cell lines, and a reduction in the expression of these biomarkers was observed in siRNA-PGRN-transfected KKU-100 and KKU-213 cells compared with that in the siControl cells (both P<0.05; Figure 9).
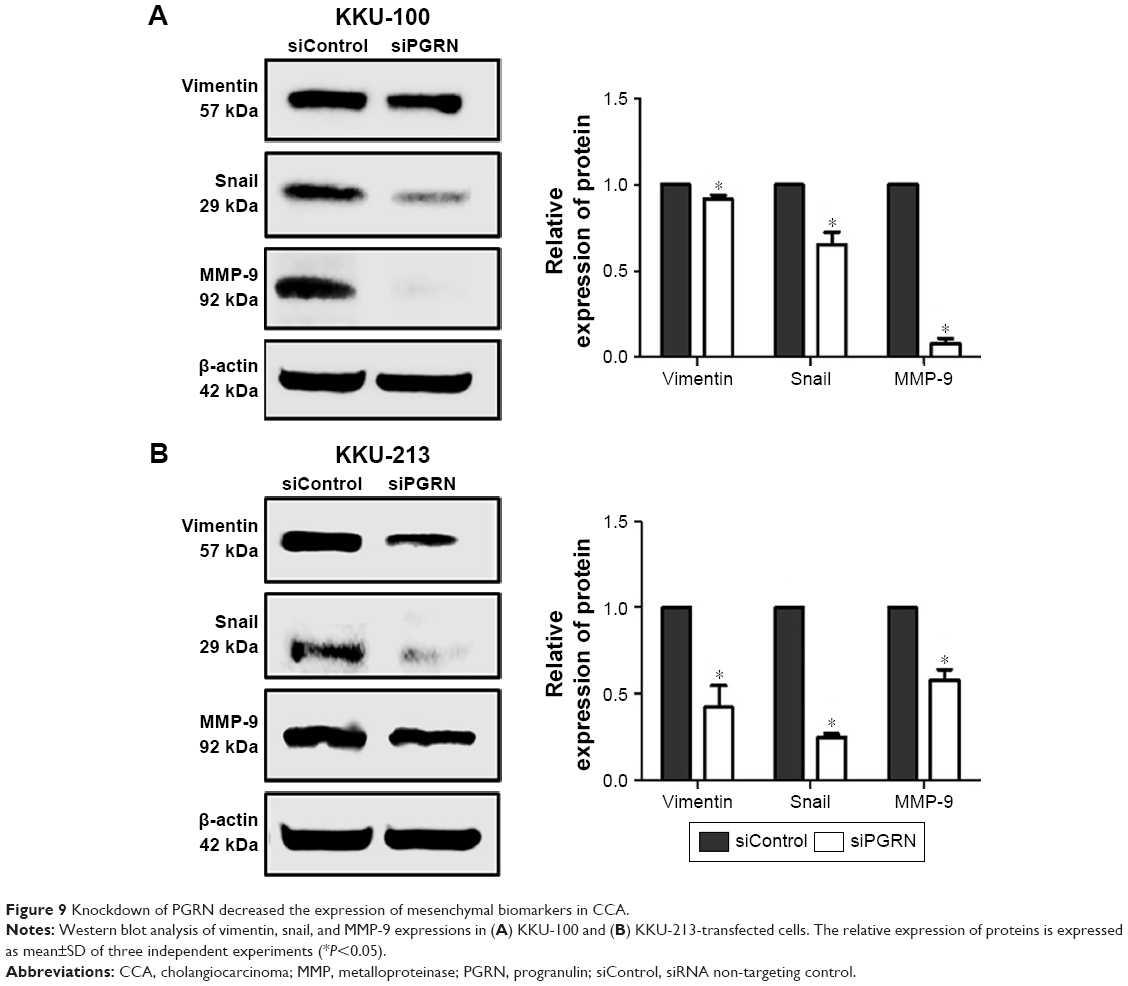 | Figure 9 Knockdown of PGRN decreased the expression of mesenchymal biomarkers in CCA. |
Discussion
PGRN, the growth factor, is a secreted glycoprotein known to play an important role in the regulation of normal tissue development, cell cycle progression, and cell motility.24 PGRN is expressed at higher levels in numerous cancers than in normal tissues, and this overproduction is correlated with a poor prognosis and high tumor severity.25 The reduction of tumor formation after PGRN knockdown provides considerable evidence for its crucial role in tumor progression.26,27 Previous studies on PGRN overproduction emphasize tumor cell growth and the signaling pathways involved in cell proliferation.16–18,28,29 Other studies demonstrate the involvement of PGRN in cell migration and invasion;16,28–30 however, the mechanisms of PGRN involvement in CCA cell proliferation and motility needed further investigation. Understanding the biological role of PGRN in the progression of tumors could provide a promising target for the development of new cancer therapies.
Our study revealed high expression of PGRN in human CCA tissues compared to adjacent non-tumor areas. This IHC finding is consistent with a previous study showing increased PGRN immunoreactivity in CCA tissues.18 Our study also demonstrated that high PGRN expression is significantly associated with patient’s shorter survival and the likelihood of finding metastasis. Metastasis and PGRN expression were the only independent prognostic variables in the multivariate analysis. This study supports the previous finding that PGRN expression could possibly serve as a potential prognostic biomarker for predicting the prognosis of patients.31
To understand the involvement of PGRN in the progression of CCA, we interfered with the expressions of PGRN in two CCA cell lines, KKU-213 and KKU-100, using siRNA transfection. Our results showed that cells transfected with siPGRN showed a significantly reduced rate of proliferation and colony formation, which is also consistent with previous findings for the crucial role of PGRN as a novel growth factor regulating tumor growth.17,18,32
PGRN knockdown in CCA cells also showed suppression of PI3K and pAkt in protein levels, indicating the involvement of PGRN in stimulating the activity of this signaling pathway in CCA cells. Our result is consistent with the study by Frampton et al, demonstrating the growth-promoting effect of PGRN in CCA cells via the activation of Akt signaling.18 The increased activation of PI3K/Akt signaling was also observed in CCA when compared with normal adjacent tissues.33,34 In many cancers, the PI3K/Akt pathway is overactive, and, as a consequence of this overactivity, there is enhanced proliferative activity of the tumor cells.16,35
We further investigated the changes in cell cycle activity in PGRN knockdown cells. Our results showed an accumulation of transfected cells at the G1 phase, indicating the involvement of PGRN in promoting cell proliferation at this phase. Considering the key molecular players at the G1 phase, cyclin D1 and CDK4 showed decreased levels in both knockdown cell lines. We investigated the apoptotic activity of CCA revealing that knockdown of PGRN-modulated apoptosis by reversing the Bax-to-Bcl-2 ratio. Increased cell apoptosis was confirmed from flow cytometry analysis stained with annexin V-FITC/PI. Our data suggest that PGRN might control CCA cell survival via the PI3K/Akt pathway and play an important role in the prevention of apoptosis.
The significant reduction in the abilities of PGRN knockdown cells for migration and invasion obtained from this study prompted us to investigate the role of PGRN in the EMT. Emerging evidence suggests that EMT plays an important role in tumorigenesis. This is a major mechanism responsible for mediating invasiveness and metastasis.36 Occurrence of this phenomenon has been previously observed in CCA tissues and cell lines where the biomarkers for mesenchymal phenotype, such as N-cadherin, S100A4, and the transcription factors Snail, Twist and ZEB2, were reported to express at higher levels compared to controls.37–39 Moreover, the expression levels of these biomarkers for the mesenchymal phenotype are inversely correlated with the expression of the epithelial biomarkers E-cadherin and CK-19.37
The results obtained from this study show that CCA cells expressing PGRN possess mesenchymal-like characteristics, expressing vimentin and the EMT-inducing transcription factor, snail. These CCA cells also expressed the MMP-9, a class of matrix MMP whose main function is to degrade and remodel the extracellular matrix (ECM). The present finding on MMP is consistent with previous results in which MMP-2 and MMP-9 were upregulated and inversely correlated to the reversion-inducing cysteine-rich protein with kazal motifs (RECK), a metastasis suppressor protein that inhibits the secretion and proteolytic activities of MMPs.40 Our findings demonstrate that siRNA-suppressed PGRN expression in CCA cells leads to decreased levels of mesenchymal biomarkers vimentin and snail, and hence a reduction in the abilities of cells to migrate and invade under the role of MMP-9. This strongly suggests that PGRN plays a critical role in tumor metastasis mediated by the EMT process. Understanding the mechanisms by which PGRN regulates tumor growth and metastasis needs to be further investigated.
Conclusion
The growth factor PGRN is upregulated in the tissues of patients with CCA. This upregulation is correlated with a poor prognosis, suggesting that PGRN could be a prognostic marker for CCA. The mechanism of action by which PGRN regulates CCA cell proliferation, apoptosis, and EMT might be mediated by the PI3K/Akt pathway.
Acknowledgments
This work was supported by a grant from Mid-Career Grant (RSA5980012), Thailand Research Fund (to NN), a grant from Khon Kaen University (to NN), a grant from CASCAP Program (to NN), and a grant from the Faculty of Medicine, Khon Kaen University for supporting the PhD program (to MD) (IN59339). We would like to acknowledge Prof Trevor N Petney for editing the manuscript via Publication Clinic KKU, Thailand.
Author contributions
MD, WL, AT, and NN planned experiments; MD performed experiments; MD, WL, AT, MT, PS-N, AT, PY, and NN analyzed the data; WL, AT, PY, and NN contributed reagents and/or other essential materials; MD and NN wrote the manuscript. All authors read and revised the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Doherty B, Nambudiri VE, Palmer WC. Update on the diagnosis and treatment of cholangiocarcinoma. Curr Gastroenterol Rep. 2017;19(1):2. | ||
Yongvanit P, Pinlaor S, Loilome W. Risk biomarkers for assessment and chemoprevention of liver fluke-associated cholangiocarcinoma. J Hepatobiliary Pancreat Sci. 2014;21(5):309–315. | ||
Khuntikeo N, Loilome W, Thinkhamrop B, Chamadol N, Yongvanit P. A comprehensive public health conceptual framework and strategy to effectively combat cholangiocarcinoma in Thailand. PLoS Negl Trop Dis. 2016;10(1):e0004293. | ||
Kirstein MM, Vogel A. Epidemiology and risk factors of cholangiocarcinoma. Visc Med. 2016;32(6):395–400. | ||
Sudsarn P, Wongchalee N, Boonmars T, et al. Sex differences in opisthorchiosis and the development of cholangiocarcinoma in Syrian hamster model. Parasitol Res. 2014;113(3):829–835. | ||
Songserm N, Promthet S, Pientong C, Ekalaksananan T, Chopjitt P, Wiangnon S. Gene-environment interaction involved in cholangiocarcinoma in the Thai population: polymorphisms of DNA repair genes, smoking and use of alcohol. BMJ Open. 2014;4(10):e005447. | ||
Wakabayashi M, McKetin R, Banwell C, et al. Alcohol consumption patterns in Thailand and their relationship with non-communicable disease. BMC Public Health. 2015;15:1297. | ||
Miwa M, Honjo S, You G, et al. Genetic and environmental determinants of risk for cholangiocarcinoma in Thailand. World J Gastrointest Pathophysiol. 2014;5(4):570–578. | ||
Mancino A, Mancino MG, Glaser SS, et al. Estrogens stimulate the proliferation of human cholangiocarcinoma by inducing the expression and secretion of vascular endothelial growth factor. Dig Liver Dis. 2009;41(2):156–163. | ||
DeMorrow S. Cholangiocarcinoma: estrogen-induced autocrine effects of VEGF on cell proliferation. Dig Liver Dis. 2009;41(2):164–165. | ||
Marzioni M, Torrice A, Saccomanno S, et al. An oestrogen receptor beta-selective agonist exerts anti-neoplastic effects in experimental intrahepatic cholangiocarcinoma. Dig Liver Dis. 2012;44(2):134–142. | ||
Banales JM, Cardinale V, Carpino G, et al. Expert consensus document: Cholangiocarcinoma: current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat Rev Gastroenterol Hepatol. 2016;13(5):261–280. | ||
Ong CH, Bateman A. Progranulin (granulin-epithelin precursor, PC-cell derived growth factor, acrogranin) in proliferation and tumorigenesis. Histol Histopathol. 2003;18(4):1275–1288. | ||
Demorrow S. Progranulin: a novel regulator of gastrointestinal cancer progression. Transl Gastrointest Cancer. 2013;2(3):145–151. | ||
Tanimoto R, Lu KG, Xu SQ, et al. Mechanisms of progranulin action and regulation in genitourinary cancers. Front Endocrinol (Lausanne). 2016;7:100. | ||
Liu F, Zhang W, Yang F, et al. Interleukin-6-stimulated progranulin expression contributes to the malignancy of hepatocellular carcinoma cells by activating mTOR signaling. Sci Rep. 2016;6:21260. | ||
Lu Y, Zheng L, Zhang W, et al. Growth factor progranulin contributes to cervical cancer cell proliferation and transformation in vivo and in vitro. Gynecol Oncol. 2014;134(2):364–371. | ||
Frampton G, Invernizzi P, Bernuzzi F, et al. Interleukin-6-driven progranulin expression increases cholangiocarcinoma growth by an Akt-dependent mechanism. Gut. 2012;61(2):268–277. | ||
Ye X, Weinberg RA. Epithelial-mesenchymal plasticity: a central regulator of cancer progression. Trends Cell Biol. 2015;25(11):675–686. | ||
Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–196. | ||
Maruyama M, Kobayashi N, Westerman KA, et al. Establishment of a highly differentiated immortalized human cholangiocyte cell line with SV40T and hTERT. Transplantation. 2004;77(3):446–451. | ||
Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1(3):1112–1116. | ||
Munshi A, Hobbs M, Meyn RE. Clonogenic cell survival assay. Methods Mol Med. 2005;110:21–28. | ||
He Z, Bateman A. Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J Mol Med (Berl). 2003;81(10):600–612. | ||
Wang M, Li G, Yin J, Lin T, Zhang J. Progranulin overexpression predicts overall survival in patients with glioblastoma. Med Oncol. 2012;29(4):2423–2431. | ||
Zhang H, Serrero G. Inhibition of tumorigenicity of the teratoma PC cell line by transfection with antisense cDNA for PC cell-derived growth factor (PCDGF, epithelin/granulin precursor). Proc Natl Acad Sci U S A. 1998;95(24):14202–14207. | ||
Lu R, Serrero G. Inhibition of PC cell-derived growth factor (PCDGF, epithelin/granulin precursor) expression by antisense PCDGF cDNA transfection inhibits tumorigenicity of the human breast carcinoma cell line MDA-MB-468. Proc Natl Acad Sci U S A. 2000;97(8):3993–3998. | ||
He Z, Ismail A, Kriazhev L, Sadvakassova G, Bateman A. Progranulin (PC-cell-derived growth factor/acrogranin) regulates invasion and cell survival. Cancer Res. 2002;62(19):5590–5596. | ||
Lovat F, Bitto A, Xu SQ, et al. Proepithelin is an autocrine growth factor for bladder cancer. Carcinogenesis. 2009;30(5):861–868. | ||
Tangkeangsirisin W, Serrero G. PC cell-derived growth factor (PCDGF/GP88, progranulin) stimulates migration, invasiveness and VEGF expression in breast cancer cells. Carcinogenesis. 2004;25(9):1587–1592. | ||
Han JJ, Yu M, Houston N, Steinberg SM, Kohn EC. Progranulin is a potential prognostic biomarker in advanced epithelial ovarian cancers. Gynecol Oncol. 2011;120(1):5–10. | ||
Frampton G, Ueno Y, Quinn M, et al. The novel growth factor, progranulin, stimulates mouse cholangiocyte proliferation via sirtuin-1-mediated inactivation of FOXO1. Am J Physiol Gastrointest Liver Physiol. 2012;303(11):G1202–G1211. | ||
Yothaisong S, Dokduang H, Techasen A, et al. Increased activation of PI3K/AKT signaling pathway is associated with cholangiocarcinoma metastasis and PI3K/mTOR inhibition presents a possible therapeutic strategy. Tumour Biol. 2013;34(6):3637–3648. | ||
Dokduang H, Juntana S, Techasen A, et al. Survey of activated kinase proteins reveals potential targets for cholangiocarcinoma treatment. Tumour Biol. 2013;34(6):3519–3528. | ||
Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627–644. | ||
Son H, Moon A. Epithelial-mesenchymal transition and cell invasion. Toxicol Res. 2010;26(4):245–252. | ||
Techasen A, Namwat N, Loilome W, et al. Tumor necrosis factor-alpha (TNF-alpha) stimulates the epithelial-mesenchymal transition regulator Snail in cholangiocarcinoma. Med Oncol. 2012;29(5):3083–3091. | ||
Techasen A, Namwat N, Loilome W, et al. Tumor necrosis factor-alpha modulates epithelial mesenchymal transition mediators ZEB2 and S100A4 to promote cholangiocarcinoma progression. J Hepatobiliary Pancreat Sci. 2014;21(9):703–711. | ||
Duangkumpha K, Techasen A, Loilome W, et al. BMP-7 blocks the effects of TGF-beta-induced EMT in cholangiocarcinoma. Tumour Biol. 2014;35(10):9667–9676. | ||
Namwat N, Puetkasichonpasutha J, Loilome W, et al. Downregulation of reversion-inducing-cysteine-rich protein with Kazal motifs (RECK) is associated with enhanced expression of matrix metalloproteinases and cholangiocarcinoma metastases. J Gastroenterol. 2011;46(5):664–675. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.