Back to Journals » Journal of Inflammation Research » Volume 18
Programmed Cell Death of Chondrocytes, Synovial Cells, Osteoclasts, and Subchondral Bone Cells in Osteoarthritis
Authors Huang J, Wu L, Zhao Y, Zhao H
Received 17 January 2025
Accepted for publication 31 May 2025
Published 8 September 2025 Volume 2025:18 Pages 12323—12360
DOI https://doi.org/10.2147/JIR.S514309
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tara Strutt
Jiwei Huang,1 Longfei Wu,1 Yuhao Zhao,1 Haiyan Zhao2
1The First Clinical College of Medicine, Lanzhou University, Lanzhou, 730000, People’s Republic of China; 2Department of Orthopedics, The First Hospital of Lanzhou University, Lanzhou, 730000, People’s Republic of China
Correspondence: Haiyan Zhao, Email [email protected]
Abstract: Osteoarthritis (OA) is a common and debilitating chronic disease characterized by severe inflammation and progressive damage to adjacent tissues and cartilage. Traditional risk factors such as obesity, gender, and aging have long been recognized as contributing factors to osteoarthritis. Emerging evidence highlights that the dysregulation of programmed cell death (PCD) plays a crucial role in the pathogenesis and progression of this disease. Numerous studies have shown that various forms of programmed cell death, including ferroptosis, pyroptosis, autophagy, cuproptosis, and apoptosis, are closely associated with osteoarthritis. Ferroptosis is an iron-dependent cell death driven by lipid peroxidation, which is related to iron overload and oxidative stress in osteoarthritis, leading to chondrocyte dysfunction and cartilage degradation. Pyroptosis, an inflammatory cell death, is triggered by the activation of inflammasomes, promoting the release of pro-inflammatory cytokines, exacerbating joint inflammation, and accelerating disease progression. Autophagy, a cellular self-degradation process, has a dual role in osteoarthritis: it acts as a protective mechanism against stress in the early stage, but when autophagy is dysregulated, it promotes cartilage degeneration. Cuproptosis is a newly discovered copper-dependent cell death pathway, and since copper metabolism dysregulation affects the function of bone and cartilage cells, it is associated with osteoarthritis. Apoptosis is an actively regulated cell death process controlled by genes and is mediated by two main pathways. The extrinsic pathway is activated when death ligands bind to receptors, triggering the activation of caspase-8 and caspase-3; the intrinsic pathway is initiated by cellular stress factors such as DNA damage, leading to mitochondrial damage and the activation of caspase-9 and caspase-3. In osteoarthritis, inflammatory factors and oxidative stress activate these two pathways, accelerating the apoptosis of chondrocytes and disease progression.This review systematically elaborates on these different types of programmed cell death and their specific roles in the development and progression of osteoarthritis. It also delves into the latest research on the molecular mechanisms of these programmed cell death pathways in the context of osteoarthritis, clarifying how they interact with other cellular processes to drive disease development. In addition, the review summarizes the clinical applications of therapeutic methods targeting programmed cell death in osteoarthritis. Ingredients from traditional Chinese medicine and other drugs show potential in regulating ferroptosis, pyroptosis, autophagy, cuproptosis, and apoptosis to alleviate the symptoms of osteoarthritis. For example, Icariin and Myristicin can prevent ferroptosis, while Matrine and metformin can reduce pyroptosis. Regarding cuproptosis, copper chelators and copper ion carriers are also under investigation. Therapeutic strategies targeting mitochondrial autophagy and copper balance also offer hope for the treatment of osteoarthritis. Currently, non-coding RNAs, phytochemicals, and some proteins have been explored for their ability to inhibit the apoptosis of chondrocytes. In conclusion, a deep understanding of the mechanisms of programmed cell death in osteoarthritis not only provides new perspectives on the pathogenesis of the disease but also points the way for the development of targeted treatment strategies and the improvement of the treatment outcomes for osteoarthritis.
Keywords: osteoarthritis, ferroptosis, pyroptosis, autophagy, cuproptosis, apoptosis
Introduction
OA is one of the most common chronic joint diseases and a major cause of disability in the elderly population worldwide. According to statistics, more than 500 million people worldwide are affected by OA.1 Its prevalence increases significantly with age, and it has become one of the major public health issues affecting the quality of life and imposing a health - related economic burden on people. OA was previously considered a simple degenerative disease of cartilage in its early stage.2 However, research in recent years has shown that OA is actually a whole - joint disorder. Its pathological process not only involves the destruction of articular cartilage and the remodeling of subchondral bone structure, but also includes meniscus degeneration, synovial inflammation, and changes in the structure and function of the infrapatellar fat pad.3–5 The clinical manifestations of OA mainly include joint pain, stiffness, swelling, and limited mobility, which severely affect the daily lives of patients. The pathogenesis of OA is complex and is affected by the combined action of multiple factors, including abnormal mechanical loading, genetic background, metabolic disorders, and chronic inflammatory responses.6 Common risk factors include aging, obesity, joint trauma, changes in female hormone levels, joint instability, and metabolic syndrome.7,8 During the progression of OA, chronic low - grade inflammation in the joint microenvironment plays an important role.9 Pro - inflammatory factors such as IL - 1β, TNF - α, and IL - 6 can induce enhanced catabolism in chondrocytes while inhibiting anabolism, thus disrupting joint homeostasis.10 In addition, the increased expression of matrix metalloproteinases (such as MMP13) and aggrecanases accelerates the degradation of the extracellular matrix (ECM), leading to irreversible damage to cartilage.11 Currently, there is no definite cure for OA. Clinical treatment mainly focuses on relieving symptoms, and commonly used medications include non - steroidal anti - inflammatory drugs (NSAIDs), analgesics, and intra - articular injection of glucocorticoids.12 However, these treatment methods cannot reverse the course of the disease, and long-term use may also lead to side effects such as hepatotoxicity, nephrotoxicity, and adverse cardiovascular reactions.13 Therefore, there is an urgent need to deeply explore the molecular mechanisms of OA in order to identify new therapeutic targets and intervention strategies.
In recent years, programmed cell death (PCD), as an important mechanism for regulating cell fate, has gradually become a new research hotspot in the field of OA. PCD includes various forms, such as pyroptosis, ferroptosis, autophagy, the newly proposed cuproptosis, and apoptosis. These forms of cell death play a crucial role in maintaining the homeostasis and pathological changes of the joint tissues in OA.14 Studies have shown that the expression of NLRP3 inflammasome related to pyroptosis in the synovial fluid of patients with OA is significantly upregulated, which promotes the release of IL-1β and IL-18, thus exacerbating synovial inflammation and cartilage destruction.15 On the other hand, the P2X7 receptor can promote the assembly of the NLRP3 inflammasome, further inducing the process of pyroptosis.16 Ferroptosis is a process of cell death caused by lipid peroxidation damage driven by iron ions. The decrease in the expression of GPX4 in OA chondrocytes will increase their sensitivity to ferroptosis, leading to oxidative stress and matrix degradation.17 In addition, ferroptosis is closely related to the upregulation of MMP13 and the downregulation of type II collagen expression, leading to further degeneration of cartilage.18 In contrast, autophagy, as a protective mechanism for maintaining cellular homeostasis and responding to stress, a decrease in its activity is considered an important contributing factor to the progression of OA.19 Cuproptosis is a newly discovered type of cell death in recent years, and its mechanism is related to the protein stress caused by the excessive accumulation of copper ions in the mitochondria of cells. Although there is relatively little research on its role in OA, preliminary evidence shows that cell types related to immune regulation (such as dendritic cells, mast cells, and CCR2-positive monocytes) may participate in the immune response process of OA by regulating cuproptosis-related genes.20–23 Apoptosis also plays a crucial role in osteoarthritis. Exogenous inflammatory factors can activate the death receptor pathway, leading to apoptosis of chondrocytes.24 Endogenous factors such as DNA damage and oxidative stress trigger the mitochondrial apoptosis pathway, causing the release of cytochrome C and activating the caspase cascade, which accelerates the apoptosis of chondrocytes and promotes the progression of osteoarthritis.25 In conclusion, various forms of programmed cell death play an important role in the onset and progression of OA. They may not only mediate tissue damage but also serve as potential protective mechanisms. A thorough exploration of their molecular mechanisms and action pathways is expected to provide new strategies for the early intervention and targeted treatment of OA. This review aims to systematically sort out the latest research progress of pyroptosis, ferroptosis, autophagy, cuproptosis, and apoptosis in OA, and analyze their potential molecular targets and therapeutic values.
Research Method
This review systematically searched databases such as PubMed and Web of Science, using keywords including “Osteoarthritis”, “Programmed Cell Death”, “Pyroptosis”, “Ferroptosis”, “Autophagy”, and “Cuproptosis”. Relevant literatures published before April 2025 were screened. The research related to the pathogenesis of OA, the regulatory pathways of cell death, and potential therapeutic targets was mainly collected. Peer-reviewed research papers and the latest review articles were preferentially included.
The Mechanism of Osteoarthritis
The dysregulation of cytokines is a key factor in the progression of OA. Cytokines that promote inflammation can persistently damage chondrocytes and disrupt the metabolic balance of the extracellular matrix in cartilage.26 This mechanism triggers catabolic enzymes such as matrix metalloproteinases (MMPs) and aggrecanases (ADAMTS), ultimately leading to the degradation of cartilage and other joint structures.27 The primary inflammatory mediators that play a role in the onset of OA include IL-1β, TNF-α, and IL-6.28 They act as activators for different signaling pathways, which enables them to stimulate other cytokines and contribute to the advancement of the pathological processes linked to OA.29 During this process, chemokines play an important role as well. When stimulated by cytokines, they draw inflammatory cells to the joint, leading to an increased presence of these cells in the joint area.30 To sum up, chemokines can facilitate the release of inflammatory substances and contribute to the progression of OA.30 IL-1β is an important pro-inflammatory cytokine that significantly contributes to the progression of several diseases. It first binds to its receptor (IL-1RI), which activates signaling pathways like NF-κB and MAPK.31 This activation causes an increase in ADAMTS and MMPs, resulting in enhanced breakdown of proteoglycans and more extensive collagen damage in the joint, ultimately leading to increased joint injury.32 Moreover, IL-1β influences the same signaling pathway by lowering SOX-9 levels, which in turn decreases the production of type II collagen. It also enhances the production of COX-2, PGE-2, and NO, leading to a decline in proteoglycan synthesis. IL-1β is recognized for increasing the levels of several chemokines, such as IL-8, CCL2, and CCL5, in addition to cytokines like IL-6 and TNF-α.33 This mechanism establishes a positive feedback loop that intensifies the inflammatory response and leads to a higher production and release of IL-1β.27 In conjunction with IL-1β, TNF-α is also acknowledged as a key pro-inflammatory cytokine that plays a role in the progression of OA.34,35 The degeneration of cartilage and various joint components can be induced by these two cytokines, which in turn facilitates the progression of OA. TNF-α can bind to TNRF-1, resulting in the creation of two separate signaling complexes.36 Complex 1 supports cell survival and increases the levels of NF-κB, MAPK, and AP-1. This leads to the degradation of proteoglycans and subsequent harm to collagen, while also inhibiting the synthesis of both proteoglycans and collagen.37 The activation of complex 2 can initiate a series of reactions that produce FADD, which then activates procaspase 8/10 and caspase 3, ultimately resulting in cell apoptosis.38 Additionally, TNF-α engages with another receptor called TNRF-2, activating the inflammation-related signaling pathway JNK and resulting in the release of NF-κB. IL-6, a significant pro-inflammatory cytokine, operates via the mediator gp130 by attaching to either IL-6R or sIL-6R to produce its biological effects.39 As a result, Gp130 activates intracellular signaling pathways that regulate inflammatory responses, the production of enzymes, and the expression of collagen and proteoglycans. IL-6 activates the PI3K, JAK/STAT, and MAPK signaling pathways via traditional signal transduction and trans-signaling methods. This activation influences the production of enzymes like TIMP, MMPs, and ADAMTS, as well as type II collagen and proteoglycans.40
Chemokines are tiny signaling molecules that can initiate chemotaxis, helping immune effector cells move and orient themselves towards sites of infection or inflammation. Consequently, chemokines play a vital role in maintaining joint inflammation linked to OA.30 Chemokines from the CC family, including CCL2, CCL3, CCL4, and CCL5, are important contributors to the development of OA.30 Monocyte chemoattractant protein-1 (MCP-1/CCL2) is a strong attractant for monocytes, aiding in the recruitment of memory T cells and natural killer (NK) cells. Its main function is linked to its engagement with the CCR2 receptor.41 CCL2 increases the levels of MMP-3, leading to a reduction in proteoglycans and the degradation of cartilage tissue.42 CCL3 (MIP-1α), CCL4 (MIP-1β), and CCL5 (RANTES) are other members of the CC chemokine family that exhibit heightened expression in OA. Studies show that people with radiographic knee OA before surgery have significantly different levels of CCL3 in their blood compared to a control group.43 As the disease progresses, the levels of CCL3 in plasma increase. Similar studies have shown that CCL5 levels in the synovial fluid of OA patients are considerably elevated compared to the control group.44 It is noteworthy that all three chemokines act as ligands for CCR5. Additionally, the CXC family of chemokines, particularly CXCL8 (also referred to as IL-8) and CXCL12, play important roles in the progression of OA.45 CXCL8 engages with the CXCR1 and CXCR2 receptors present on white blood cells and also affects chondrocytes, osteoclasts, fibroblasts, epithelial cells, and several other cell types.45–47 Studies using the human chondrocyte cell line (CHON-002) have shown that TNF-α can enhance the expression of CXCL8.48 Furthermore, advanced glycation end products (AGEs) are capable of activating the signaling route of NF-κB, leading to an increased production of CXCL8. This chemokine tends to build up in cartilage as people age and encourages the enhancement of catabolic activities in chondrocytes.49 Furthermore, studies show that in chondrocytes from individuals with OA, IL-8 can increase the levels of collagen I, MMP1, and MMP-13 proteins, and it also facilitates the phosphorylation of STAT3 and the p65 subunit of NF-kB.48 It affects the shape of chondrocytes by lowering internal GTP-Cdc42 levels and increasing the formation of stress fibers. IL-8 promotes the growth of chondrocytes and aids in the mineralization process of the extracellular matrix.46 CXCL12, also known as stromal cell-derived factor-1 (SDF-1), is a chemokine crucial for tissue repair. It binds to CXCR4 and draws mesenchymal stem cells (MSCs) to sites of injury.50 CCL21 is also an important inflammatory mediator, which is secreted by cartilage, meniscus, and synovial membrane. In the joint microenvironment, CCL21 contributes to the recruitment of immune cells and the regulation of local inflammatory responses. Its role in the diseased joint tissues may significantly affect the progression and severity of inflammation, and it has a crucial impact on the overall health of the joints.51 The inflammation of the infrapatellar fat pad and synovium, as important aspects in the pathogenesis of OA, has a complex relationship with the dysregulation of cytokines and chemokines. The inflammation of the infrapatellar fat pad can release a variety of cytokines and adipocytokines, further exacerbating the inflammatory microenvironment within the joint.3–5 This not only aggravates the damage to chondrocytes and the extracellular matrix but also promotes the infiltration of inflammatory cells. Similarly, synovial inflammation is a key feature of OA. Synovial fibroblasts and macrophages will release a series of cytokines, chemokines, and proteases, leading to cartilage degradation and joint destruction.52 Articular cartilage consists of chondrocytes along with the extracellular matrix (ECM) that surrounds them. Chondrocytes play an essential role in preserving the stability of collagen metabolism at a low turnover rate, provided there is no pathological damage.53 At the same time, a growing body of research indicates that the deterioration of cartilage is a major pathological alteration in OA, likely resulting from the breakdown of the ECM due to metabolic imbalances in chondrocytes.54
The above-mentioned inflammatory processes do not occur in isolation but are closely related to the elaborated mechanisms of programmed cell death. Programmed cell death, including ferroptosis, pyroptosis, autophagy, cuproptosis, and apoptosis can be triggered by inflammatory cytokines and chemokines.55 Conversely, it can also regulate the production and release of these factors. For example, in an inflamed joint, elevated levels of IL-1β and TNF-α can induce pyroptosis in chondrocytes, further disrupting the cartilage structure. On the other hand, the activation of autophagy triggered by inflammatory stress may attempt to maintain intracellular homeostasis and protect chondrocytes.14
The Research Progress of Programmed Cell Death in Osteoarthritis
Ferroptosis and Osteoarthritis
Overview of Ferroptosis
Ferroptosis is a new kind of programmed cell death which is caused by the accumulation of lipid peroxides dependent on iron. The mechanisms involved are mainly linked to disruptions in iron metabolism, the buildup of lipid peroxides, and an imbalance in the amino acid antioxidant system. Fe³⁺ attaches to transferrin (TF) in the blood and transports the complex into the cell’s nuclear endosome via transferrin receptor 1 (TfR1) located on the cell membrane.56 Within the endoplasmic reticulum, trivalent iron ions (Fe³⁺) are separated from their complex and converted to Fe²⁺ by the enzyme prostate stromal epithelial antigen 3 (STEAP3). These ferrous ions are subsequently transported into the cytoplasm via divalent metal transporter 1 (DMT1) or some members of the zinc-iron regulatory protein family, namely ZIP8/14.57 In the cytoplasm, a small amount of Fe²⁺ exists in the soluble iron pool (LIP), while the majority binds to the light chain of ferritin (FTL) and heavy chain 1 (FTH1) to form ferritin for storage. Ferritin (also designated as FPN or SLC40A1) has the ability to export excessive Fe²⁺ from the cell and convert it to Fe³⁺, thereby facilitating the regulation of iron levels within the cytoplasm.58 The accumulation of active Fe²⁺ has the potential to initiate ferroptosis through the generation of ROS by the Fenton reaction or by directly promoting lipid oxidation processes while serving as a cofactor for lipoxygenase (LOX).59 Two important lipid peroxidation metabolites are reactive oxygen species (ROS) and lipids. ROS are mainly generated by the Fenton reaction, while the other two methods involve oxidative processes occurring in mitochondria and enzymatic actions carried out by the NADPH-dependent oxidase (NOX) family.60 Arachidonic acid (AA), an important form of polyunsaturated fatty acids (PUFAs), acts as the main substrate for lipid peroxidation during ferroptosis. Polyunsaturated fatty acyls (PL-PUFAs) serve as key substrates for lipid peroxidation and play a role in the following processes of iron movement,17,61,62 the process can occur through either an enzymatic method or a non-enzymatic method. In the non-enzymatic process, hydroxyl radicals (HO−) produced by the Fenton reaction involving Fe²⁺ can remove hydrogen atoms from lipids, leading to the formation of lipid radicals. These lipid radicals then generate peroxyl radicals, which play a role in non-enzymatic lipid peroxidation.63 Researchers have discovered at least two separate pathways in the enzymatic process. One pathway features iron-dependent lipoxygenases (LOXs), which can transform polyunsaturated fatty acids (PUFAs) into peroxides, leading to the formation of several derivatives such as malondialdehyde (MDA) and 4-hydroxy nonadecenoic acid (4-HNE), among others.60 Studies have shown that cytochrome P450 oxidoreductase (POR) plays a role in lipid peroxidation through the alternative enzyme pathway, potentially leading to ferroptosis.64 Certainly, this process requires iron to act as a catalyst. Lipid peroxidation can cause enzymes at the membrane to malfunction, potentially altering the fluidity and permeability of the plasma membrane.65 When lipid peroxidation occurs, cells initiate an antioxidant response to counteract its effects and maintain balance. This response mainly involves cysteine/glutamate antiporters, referred to as the Xc-system, and GPX4.66 The Xc-system consists of solute carrier family 7 member 11 (SLC7A11) and solute carrier family 3 member 2 (SLC3A2). It enables the transport of glutamate (Glu) out of the cell and permits the entry of cysteine.67 Cysteine is then converted into cysteine, which serves as a building block for the synthesis of glutathione (GSH). Cells absorb glutamine (Gln) through the SLC1A5 transporter protein, and it is then converted into glutamate (Glu) by the enzyme glutaminase (GLS), which affects the consumption rate of Glutathione (GSH). GSH is a tripeptide that consists of glutamate, cysteine, and glycine.68 It is produced by the enzyme GCL (γ-glutamylcysteine synthetase) and is crucial as a cofactor for GPX4 in the process of detoxifying lipid peroxides. GPX4 is the sole enzyme capable of transforming GSH into its oxidized form, known as GSSG, while also reducing lipid peroxides to their respective alcohols,59 As a result, GPX4 and SLC7A11 can be regarded as crucial regulators of ferroptosis. Figure 1.
 |
Figure 1 The impacts of iron overload and lipid peroxidation on OA during ferroptosis. Ferroptosis is initiated due to excessive lipid peroxidation (LPO). The regulation of the efficacy of cysteine and reduced glutathione (GSH) is carried out by the cystine-glutamate reverse transport system Xc- or the transsulfuration pathway. The transsulfuration route is a metabolic way which converts cysteine into homocysteine, generating amino acids containing cysteine (Cys) and synthesizing GSH. Glutamine can be transported into the cell via the SLC1A5 transporter protein and subsequently transformed into glutamate by glutamine synthetase (GLS), thereby influencing the utilization rate of GSH. GSH serves as a crucial cofactor for GPX4 in eliminating lipid peroxides. On the one hand, it is believed that the oxidative effects of polyunsaturated fatty acids and excess iron constitute an important factor. The uptake of iron through transferrin receptor 1 (TFR1) or the degradation of ferritin iron reserves can enhance an unstable iron pool, therefore, cells are susceptible to ferroptosis due to the production of lipid hydroperoxides by the Fenton reaction. Ferroptosis can gradually intensify inflammatory responses, resulting in the augmented expression of enzymes like MMP-13 in chondrocytes and the reduced expression of collagen II, thereby accelerating the advancement of OA. Created in BioRender. Jiwei, H (2025) https://BioRender.com/9n3xifo. Abbreviations: POR, Cytochrome P450 reductase; LOX, Lipoxygenase; STEAP3, Prostate-specific Membrane Antigen 3; DMT1, Divalent metal transporter 1; Glu, Glutamate; Cys,Cysteine; GCL, Glutamylcysteine ligase; Gly, Glycine; GSH, Glutathione; GPX4, Glutathione peroxidase 4; Gln, Glutamine; SLC1A5, Transport protein; Gls, Glutaminase; PUFAs, Polyunsaturated fatty acids. |
The Role of Ferroptosis in Osteoarthritis
Iron Overload and Osteoarthritis
The basic pathological mechanisms of OA include damage to the cartilage at the joint margins and the subchondral bone, accompanied by synovitis, in which chondrocytes play an important role. In addition, the meniscus and the infrapatellar fat pad are also of great significance.69 The meniscus is a key structure for the distribution of joint loads. In OA, it undergoes degeneration, disrupting the normal joint mechanics and imposing abnormal pressure on the cartilage and subchondral bone. When the infrapatellar fat pad is inflamed, it secretes inflammatory mediators, affecting the function of chondrocytes and leading to synovitis, which further exacerbates the progression of OA.69,70 Research has shown that the iron levels in the synovial fluid of individuals with OA are elevated.71 However, when compared to the iron levels in the synovial fluid of patients with rheumatoid arthritis (RA), the increase in OA patients is less pronounced.71 Compared to the healthy control group, patients with OA showed higher serum ferritin levels, indicating more severe joint imaging results.72 Hereditary hemochromatosis is characterized by an excess buildup of iron in the body, which often results in patients suffering from OA.73 Jing and their team developed a mouse model exhibiting iron overload (IO) to explore the role of iron in the progression of OA.71 In the knee joint, compared to the control group, the IO model showed greater cartilage degradation, harm to the subchondral bone, and heightened sclerosis, leading to higher OARSI scores for OA. They observed an increase in the expression levels of ADAMTS5 and MMP13, as well as a rise in the number of osteoclasts. To explore the possible molecular mechanisms connecting inflammation and iron overload in OA, scientists exposed chondrocytes to IL-1β or TNF-α. They observed an increase in TfR1 and DMT1, which are transporters for metal ions, while FPN, the protein that facilitates iron export, was downregulated.71 This change could lead to an increase in iron accumulation within the cells.71 Despite the augmented apoptosis and the upregulated levels of MMP3 and MMP13 in chondrocytes treated with Ferric Ammonium Citrate (FAC), which establish a connection between iron overload and chondrocyte apoptosis, this might disclose the role of chondrocyte ferroptosis in OA.71 In individuals with OA, synovial cells produce 4-HNE (4-hydroxyheptenaldehyde), a byproduct from the oxidative degradation of polyunsaturated fatty acids, alongside interleukins and chemokines which play a significant role in the inflammatory process. Furthermore, there is an increase in the concentration of 4-HNE protein adducts present in both synovial fluid and osteoblasts.74–76 Studies have shown that high levels of 4-HNE are detrimental to human chondrocytes and osteoblasts.75–77 Research has shown that mitochondrial DNA damage in OA negatively affects chondrocytes.78 Studies show that ferroptosis in chondrocytes can play a major role in the advancement of OA.79 Yao and his team utilized IL-1b to mimic an inflammatory response and employed fluorescence-activated cell sorting (FAC) technology to replicate the conditions of iron overload in chondrocytes.79 In murine knee joint chondrocytes after simulating inflammation with interleukin-1 beta (IL-1β) and iron overload with FAC in vitro, the buildup of ROS and lipid peroxides, along with alterations in apoptosis-related proteins like GPX4, SLC7A11, ACSL4, P53, and NRF2ARE, can trigger ferroptosis.79 Conversely, Fer-1, which is a known inhibitor of iron-induced ferroptosis, can help diminish this process. In mouse chondrocytes, exposure to Erastin, known to trigger ferroptosis, caused an increase in MMP13 expression in cells, while also reducing the levels of type II collagen.79 Both of these markers are linked to OA. The results were additionally validated in OA mouse models that received treatment with FER-1.79 In the OA mouse model, there is a direct correlation between the levels of iron in synovial fluid and the advancement of OA.80 Excessive iron levels in the body can cause synovitis and hyperplasia, result in the death of chondrocyte cells, and impair the function of subchondral osteogenic cells.73 Research has indicated that excessive iron can disturb the iron balance in chondrocytes, resulting in oxidative stress and eventually triggering cell apoptosis.81 As a result, it is essential to maintain strict monitoring of the iron levels in the organization.82 Studies show that rats with iron overload had higher microCTOA and OARSI histological scores in their knee joints than those in a control group.71 This implies that excess iron may play a role in worsening the pathological damage linked to OA. Additional analysis indicated that rats exposed to the iron overload model exhibited markedly elevated levels of iron and mRNA expression for FTH-1, FPN, IL-1β, and TNF in both the cartilage of the knee and the fat pad of the patella.83 Conversely, there was a significant reduction in the mRNA levels of TFR, DMT1, Col2, and aggrecan. Research indicates that excess iron is linked to increased ferritin levels, heightened generation of inflammatory cytokines, and a reduction in the synthesis of cartilage tissue.84 The findings indicate that a high level of iron could play a major role in the onset of OA. In conclusion, an excess of iron can trigger the production of ROS and pro-inflammatory cytokines in chondrocytes. This process results in the breakdown of cartilage tissue and hinders its formation, leading to joint cartilage deformities and worsening the advancement of OA.85 Figure 1.
Lipid Peroxidation and Osteoarthritis
When ROS target the lipids in cell membranes, they generate peroxidized lipids like malondialdehyde (MDA). These compounds can interact with DNA or proteins within the organism, resulting in additional harm to the cell membrane’s structure and composition.86,87 Research indicates that in a rat model of OA induced by anterior cruciate ligament transection (ACLT) surgery, there is an increase in serum levels of MDA and lipid peroxides. Furthermore, reducing the concentrations of MDA and lipid peroxides may help slow the progression of OA.88 GPX4 is an enzyme that plays a role in repairing lipids and is involved in the oxidation of liposomes, with its activity being affected by GSH.89 Studies have shown that in regions of OA lesions with increased lipid peroxidation, levels of GPX4, GSH, and the GSH to GSSG ratio in chondrocytes are significantly lower. This implies that lipid peroxidation takes place as OA progresses.90 In a different in vitro experiment designed to cause oxidative damage to cells using a similar concentration of tert-butyl hydroperoxide (TBHP), researchers found that reducing GPX4 levels in mouse chondrocytes (ATDC5) and primary mouse chondrocytes (MCC) made the cells more susceptible to oxidative stress, resulting in a significant rise in MDA levels.91,92 The findings suggest that lower levels of GPX4 expression make chondrocytes more vulnerable to oxidative stress by elevating lipid peroxidation in these cells.90 As a result, GPX4 could be an important target for affecting lipid peroxidation in relation to OA. Figure 1.
The Xc−system/GSH/GPX4 Regulatory Axis and Osteoarthritis
The regulatory pathway involving Xc-system/GSH/GPX4 is crucial for preventing ferroptosis in articular chondrocytes. Wang93 and his team analyzed cartilage samples taken from the loading (Acronym L) area and unloading (Acronym UL) area of patients with OA who had total knee arthroplasty (Acronym TKA). The researchers utilized transmission electron microscopy to investigate the changes in the shape of chondrocytes from the L-zone of osteoarthritic cartilage linked to ferroptosis, which included thickening of the mitochondrial membrane and contraction of the mitochondria.94 Furthermore, the findings from the mRNA microarray analysis show that the expression of the GPX4 biomarker associated with ferroptosis is lower in chondrocytes located in the loading region than in those in the unloading region.93 At the same time, research conducted on cells revealed that primary mouse chondrocytes subjected to a mechanical stress of 1 MPa exhibited alterations in their mitochondrial structure associated with iron imbalances, in contrast to those kept without any mechanical stress.95 The findings indicate that mechanical stimulation results in reduced levels of GPX4 mRNA and protein expression. The in vivo study provided additional evidence showing that the levels of GPX4 protein in the cartilage of mice with an OA model were notably reduced.93 The research findings suggest that GPX4 plays a crucial role in regulating ferroptosis in chondrocytes and cartilage cells located in the weight-bearing regions of joint cartilage in OA patients experiencing high mechanical stress. The level of GPX4 expression might be linked to the advancement of OA.93 Exosomes from osteoarthritic fibroblast-like synoviocytes promote cartilage ferroptosis and damage via delivering microRNA-19b-3p to target SLC7A11 in OA.96 The Xc-system’s precise control of SLC7A11 is crucial for the accumulation of iron storage proteins in OA.97 In vitro studies provide additional evidence that excess iron enhances the production of ROS and lipid peroxides in chondrocytes, while reducing the levels of Xc-system (xCT), GSH, GPX4, and the mitochondrial membrane potential in these cells. Consequently, the regulatory pathway involving Xc-system/GSH/GPX4 is crucial for managing iron-related mutations associated with OA.98
Pyroptosis and Osteoarthritis
The Molecular Mechanism of Pyroptosis
Cell pyroptosis, often referred to as inflammatory necrosis, is a controlled form of PCD that relies on the Gasdermin protein family to create pore-like formations in the plasma membrane.99 This process entails the gradual swelling of the cell until the membrane breaks, leading to the release of internal substances and the activation of an inflammatory response.100 Inflammasomes are protein complexes found in the cytoplasm, usually made up of sensors (PRRs), adapters (ASC), and effectors (caspase-1).101 Sensors are receptors for pattern recognition (PRRs) that mainly consist of nucleotide-binding oligomerization domain (NOD) and leucine-rich repeat (LRR) receptors (NLRs), AIM2-like receptors (ALR), and pyrin (protein interaction modules).102 Caspases that play a role in pyroptosis are commonly known as pro-inflammatory caspases. This group typically consists of human caspases 1, 4, and 5, as well as mouse caspase 11.103 Gasdermins are a newly identified family of proteins known for their ability to form pores in cell membranes, with gasdermin D (GSDMD) playing a central role in the process of pyroptosis.104 Certain caspases or granzymes trigger the activation of the protein’s N-terminal region, which reinstates its cytotoxic abilities and encourages the secretion of inflammatory substances and cytokines.105 At present, the mechanisms of pyroptosis can be broadly categorized into three groups: the standard inflammasome pathways that rely on caspase-1, the non-standard inflammasome pathways that depend on caspases-4/5/11, and additional pathways that may or may not involve caspases.106 The activation of intracellular receptors such as NLRP1, NLRP3, NLRP6, NLRP9, and NLRC4 by certain pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) is critical, as these processes are known to contribute to inflammatory responses.107 In the context of OA, this activation is implicated in exacerbating inflammatory pathways that contribute to joint degradation and disease progression.108 An example of this is the harmful substance produced by Bacillus anthracis, which can activate NLRP1b in mice.109 The activation of NLRP6/9 can take place as a reaction to viral or bacterial infections during the innate immune response.110,111 In contrast, NLRP3 is the intracellular sensor that has been studied the most extensively, NLRP3 is capable of reacting to infections caused by bacteria, viruses, and fungi, in addition to endogenous DAMPs that trigger sterile inflammation and various environmental factors, this is what sets it apart from the majority of PRRs that focus on just one or a limited number of PAMPs or DAMPs.112 It is important to highlight that the full activation of NLRP3 occurs in two steps: the initiation phase and then the activation phase. The first step in activation is the transcription of NLRP3, which requires the involvement of Toll-like receptors (TLRs), interleukin-1 receptors, and tumor necrosis factor receptors.113 As a result, the signals that are sent regulate NLRP3, procaspase-1β, and procaspase-18 via the NF-κB pathway. Furthermore, when the NAIP protein interacts with triggers like Salmonella enterica serovar Typhimurium, it activates NLRC4, leading to an inflammatory response.114 Besides NLRs, there are other intracellular sensors such as pyrin and AIM2 that primarily serve a function in detecting viral infections and dangerous pathogens. As a result, the activated pattern recognition receptors (PRRs) engage with each other via CARD-CARD domains, which leads to the recruitment of ASC (apoptosis-associated speck-like protein) and the binding of pro-caspase-1, ultimately forming the inflammasome.115 In response to transmitted danger signals, procaspase-1 is cleaved and activated, resulting in the formation of caspase-1 polymers known as p10/p20 tetramers. The active version of Caspase-1 is capable of identifying pro-IL-1β and pro-IL-18, even though these precursors lack biological activity.103 On the other hand, when caspase-1 is activated, it can cut GSDMD, which is crucial for initiating cell pyroptosis. This cleavage leads to the creation of openings in GSDMD, promoting cell pyroptosis and facilitating the release of biologically active substances, this encompasses IL-1β, IL-18, and high mobility group box 1 protein (HMGB1).104 Figure 2.
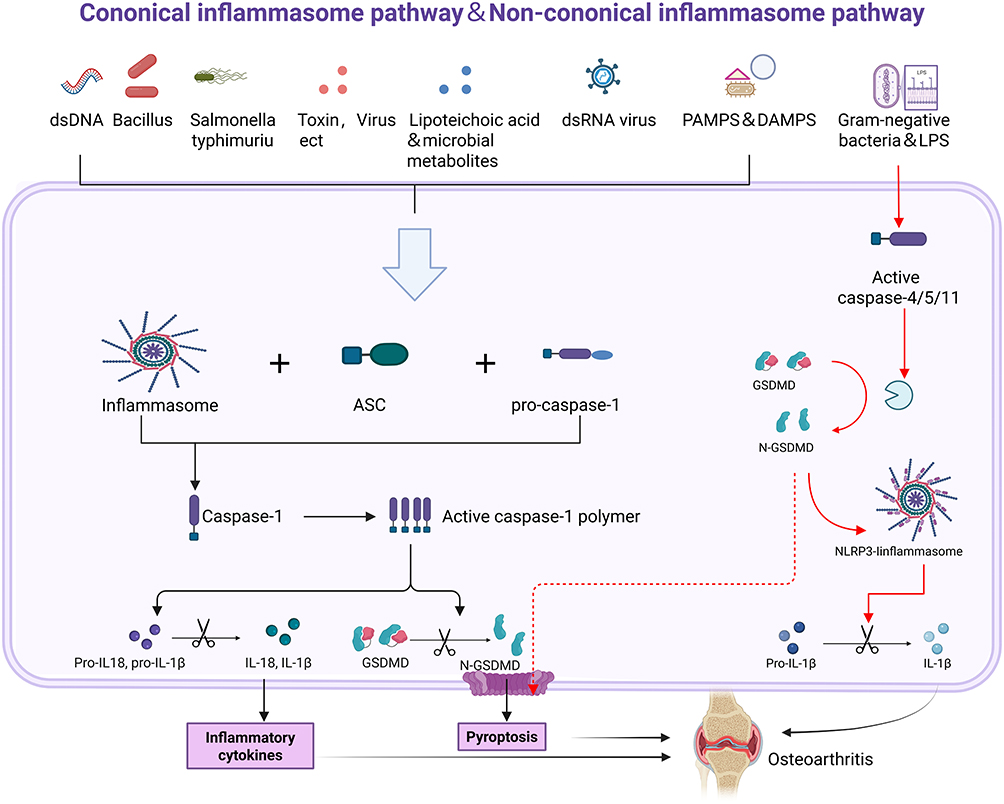 |
Figure 2 The molecular mechanism of pyroptosis and its impact on OA. Typical inflammatory pathways (black arrows). Atypical inflammatory body pathway (red arrow). Pyroptosis is mainly induced by the NLRP3 inflammasome. The NLRP3 inflammasome is capable of activating GSDMD or triggering the generation of IL-1β and IL-18, thereby intensifying the progression of OA. Created in BioRender. Jiwei, H (2025) https://BioRender.com/v25o168. |
The Role of Pyroptosis in Osteoarthritis
Chondrocyte Pyroptosis
It remains uncertain whether cartilage deterioration in OA is a contributing factor or a consequence of the disease, numerous research studies have indicated that various risk factors associated with OA, such as oxidized low-density lipoprotein (Ox-LDL), cholesterol related to obesity, hydroxyapatite crystals, and issues with intestinal circulation that result in the movement of lipopolysaccharides (LPS), can initiate pyroptosis via molecules associated with damage (DAMP) or molecules associated with pathogens (PAMP).116–118 These substances trigger pyroptosis in chondrocytes via the NLRP3 inflammasome pathway, interfere with the metabolism of different compounds, and lead to the release of important molecules that promote the advancement of OA. Key molecules involved include matrix metalloproteinases (MMPs), cysteine proteases with propeptide sequences (like cathepsins), ADAMTS metalloproteinases, and cytokines including IL-1β, IL-18, and TNF-α.119,120 Studies have shown that MMP-1, MMP-3, MMP-13, and ADAMTS-4/5 are the primary agents engaged in the disintegration of cartilage ECM components in the mentioned molecules,121,122 The rise in their levels can be somewhat attributed to the secretion of IL-1β and TNF-α from cartilage cells undergoing pyroptosis.123,124 As a result, the incidence of pyroptosis in chondrocytes disturbs the equilibrium between the synthesis and breakdown of the ECM in chondrocytes, causing a shift towards increased degradation metabolism as a consequence of chondrocyte pyroptosis. Furthermore, the released IL-1β, IL-18, and TNF-α disseminate to the cartilage, synovial fluid, and even the subchondral bone, leading to additional harm to the joints affected by OA.125 IL-1β could be associated with the breakdown of cartilage and the inflammatory process. The activities related to IL-1β in chondrocytes encompass gene expression, cell growth, and cell apoptosis. IL-1β influences the expression of MMP-1 and MMP-11,123 and it also triggers the generation of ROS,126 In the end, it leads to the death of chondrocytes and the breakdown of the ECM, creating a harmful cycle of positive feedback. When the feedback loop speeds up the breakdown of cartilage, pieces of cartilage may be released into the synovial fluid, leading to additional harm to the synovial membrane.127 Moreover, the presence of IL-1β in synovial fluid encourages the production of inflammatory substances as well as various cytokines and chemokines, this includes inducible nitric oxide synthase (iNOS),127 Cyclooxygenase-2 (COX-2),128 IL-6,129 IL-8130 and CCL5,131 consequently, it plays a role in controlling inflammation in the synovial tissue. The biological impacts of TNF-α resemble those of IL-1β, and they have the ability to stimulate one another.132 TNF-α has the ability to elevate levels of inflammatory substances like PGE2 and VEGF, which subsequently enhances inflammatory reactions and promotes vascular infiltration.133,134 While IL-18 can trigger the synthesis of MMPs and lead to chondrocyte pyroptosis, these events are more likely to happen via pathways that depend on IL-1 and/or TNF-α.135,136 In general, the immediate consequence of chondrocyte apoptosis in articular cartilage is the removal of chondrocytes, which disturbs the previously stable architecture of the cartilage ECM and leads to the release of pro-inflammatory substances. This process indirectly speeds up cartilage deterioration and initiates joint inflammation. Consequently, treatments targeting IL-1β and TNF-α might remain some of the limited effective approaches for reducing chondrocyte pyroptosis, lessening the degradation of the ECM, and easing inflammatory reactions.
Synovial Cell Pyroptosis
Research indicates that alterations in the synovial membrane of joints affected by OA can be classified into three main categories: accumulation of debris, inflammatory responses, and fibrosis.137 Nonetheless, synovial macrophages and fibroblast-like synoviocytes (FLS) are significantly associated with the alterations occurring in the synovium of joints affected by OA.138 As previously stated, the components found in the synovial fluid consist of: fragments of degenerated cartilage, cytokines that are released, and proteases produced by dying chondrocytes. They function as DAMPs by engaging with the membrane TLRs found in synovial macrophages and FLS, which initiates pyroptosis and leads to harmful alterations in the synovial tissue.138,139 Certain studies indicate that chondrocytes in OA communicate with synovial cells via vesicles resembling exosomes.140 Regardless of the previously mentioned factors, MMPs, ADAMTS, IL-1β, IL-18, and TNF-α worsen cartilage damage and synovitis. During pyroptosis, synovial macrophages and FLS continue to generate high mobility group box 1 (HMGB1) and transforming growth TGF-β.141,142 HMGB1 engages with FLS and facilitates the clustering of pro-inflammatory substances, which contributes to the onset of synovitis,143 HMGB1 has the ability to attach to LPS and be carried to synovial macrophages via RNA, leading to pyroptosis.144 TGF-β elevated the levels of expression of fibrosis markers in OA FLS, for instance, the expression levels of collagen lysine, prolyl 4-hydroxylase 2 (PLOD2), type I collagen alpha 1 chain (COL1A1), and tissue inhibitor of metalloproteinases 1 (TIMP1) are being examined, this could ultimately cause an excess production of ECM, resulting in fibrosis.141 Moreover, the injured synovium triggers the production of HIF-1α and ATP, which prompts FLS to experience pyroptosis, resulting in synovitis and fibrosis.145,146 In summary, recent research indicates that the pyroptotic mechanism in synovial cells and the particular cytokines they generate are linked to the synovitis and fibrosis seen in OA.141 Simultaneously, the loss of cartilage and the development of fibrosis affect chondrocytes via cytokines, resulting in the depletion of cartilage and alterations in the synovial tissue. This establishes a harmful positive feedback loop akin to the process of cartilage degradation.147 Most synovial macrophages are classified as M2 type, and they play a role in managing inflammation when they are activated. M2 macrophages have the ability to convert into M1 macrophages, leading to an inflammatory reaction. In people with OA, macrophages accumulate crystals in the synovial tissue.148 When a cell dies, ATP is released, triggering the triggering of the NLRP3 inflammasome, which leads to the production of IL-1β and IL-18.130 The existence of sodium urate crystals in the joints is linked to the severity of OA and the decline of bone health. Lowering the levels of these crystals could help maintain joint function. Caspase-1 is essential for creating pores in GSDMD, which intensifies inflammation in the synovial tissue. Zhang and his team noted a rise in the components of the NLRP3 inflammasome found in the synovial tissue of rats with OA of the knee joint.149 Employing a caspase-1 inhibitor may enhance tissue structure, lower inflammation, reduce fibrosis, and decrease the amounts of pro-inflammatory substances. Nonetheless, inhibiting caspase-1 can decrease pyroptosis triggered by LPS, although the macrophages do not originate directly from the affected synovium.112 In line with the earlier statement, FLS cells displayed characteristics of pyroptosis and demonstrated elevated levels of fibrotic factors when co-cultured with macrophages undergoing pyroptosis.122 It is evident that macrophage pyroptosis contributes to the progression of OA synovium. Nevertheless, further experiments are required to investigate the various connections between the two, since in the advanced stages of OA synovium, pyroptosis is frequently linked to catabolic processes, whereas fibrosis is mainly associated with anabolic processes. The change in metabolic activity indicates that a broader variety of cells or cytokines may be participating in the process.
Pyroptosis in Subchondral Bone
Subchondral bone, an essential part of the knee joint’s composition and function, is a thin layer of dense bone situated beneath the calcified cartilage. In patients with OA, the thickness of the subchondral cortical bone is impacted, resulting in subchondral sclerosis, excess bone formation, bone spurs, and the development of bone cysts in later stages.150,151 When the inner meniscus (DMM) model is unstable, a rise in pyroptosis-related molecules results in a notable increase in the OARSI (Osteoarthritis Research Society International) score, Yan152 et al identified the effects of pyroptosis in the OA model, observing an increase in the levels of pyroptosis-related factors (including NLRP3, Caspase-1, GSDMD, and IL-1β) in chondrocytes, which corresponded with a rise in the OARIS score. During the early phase of the research, they noted a reduction in the mass of subchondral bone, an increase in trabecular separation (Tb.Sp), a rise in osteoclast numbers, and a decline in osteoprotegerin (OPG) levels.152 In the advanced stages of OA, they noted a rise in subchondral bone volume along with a reduction in trabecular bone (Tb).152 The results suggest a strong relationship between chondrocyte pyroptosis and alterations in subchondral bone during the progression of OA.
Autophagy and Osteoarthritis
The Molecular Mechanisms of Autophagy
Autophagy is a process that recycles cellular waste by encapsulating aging cells, damaged organelles, misfolded proteins, and other materials into autophagosomes, which are subsequently sent to lysosomes for breakdown.153 This mechanism plays a crucial role in sustaining energy balance, allowing the body to cope with stressors like nutrient shortages, low oxygen levels, and other challenges.154,155 Autophagy involves several important stages, such as the start of the process, the formation of vesicles (creating a double-membrane isolation membrane), the growth and development of these vesicles, the merging of the autophagosome with the lysosome, and the degradation of the autophagosome156–159 Figure 3. Autophagy starts with the ULK1 complex, which is made up of ULK1, ATG13 (a protein associated with autophagy), the 200kDa FIP200 protein that interacts with proteins from the adhesion-related kinase family, and ATG101. Once autophagy begins, the ULK1 complex detaches from the mTOR complex and moves to the location where autophagy starts. At this site, it phosphorylates ATG13 and FIP200, thereby triggering the autophagy process.160 Subsequently, the ULK1 complex brings in the Beclin1-Vps34 complex, which consists of Beclin1, Vps34, Vps15, and ATG14L, to the precursor structure of the autophagosome by phosphorylating Ambra1, a protein that plays a role in vesicle formation.161 During this process, Ambra1 interacts with the tumor necrosis factor receptor-associated factor (TRAF6) and functions as the E3 ubiquitin ligase for ULK1, adding K63 ubiquitin chains and enhancing the stability of ULK1. The activation of ULK1 can result in the phosphorylation of Beclin1, thereby increasing the activity of the Vps34 complex.162 Once the ULK1 and Beclin1-Vps34 complex is activated, the phosphorylation of downstream targets commences. This results in an increase in phosphatidylinositol-3-phosphate at the site of autophagosome formation, which is vital for recruiting proteins necessary for the development of autophagosomes. For instance, the WD-repeat domain phosphatidylinositol (PI) interaction protein and DFCP1 are involved in this process. This results in the formation and growth of autophagosomes, ultimately leading to the creation of fully developed autophagosomes.163,164
 |
Figure 3 The general process of autophagy and its role in cartilage and chondrocytes. In regular joint cartilage, HIF-1/AMPK/mTOR constitutes the primary pathway for the activation of autophagy in cartilage cells under hypoxic circumstances. Moreover, AMPK restrains the activation of mTOR, while mTOR hinders autophagy. At the initial phases of OA, autophagy temporarily increases in reaction to pathogenic elements like mechanical injury and aging, which cause oxidative stress. In the late stage of OA, the levels of oxidative stress and inflammation rise further, exceeding the repair capacity of autophagy, this brings about a reduction in autophagy, leading to an imbalance in the synthesis and degradation of cartilage. Furthermore, in cases where chondrocytes are overly exposed to stress stimuli, excessive autophagy might take place, which further intensifies chondrocyte apoptosis. Created in BioRender. Jiwei, H (2025) https://BioRender.com/1is3jwl. |
The process of elongating and maturing autophagosomes requires two systems that involve ubiquitin-like conjugation: the ATG12-ATG5-ATG16L complex and LC3-phosphatidylethanolamine.165 ATG12, in collaboration with ATG7 and ATG10, attaches LC3 to phosphatidylethanolamine (PE). ATG10 interacts with ATG5, which then associates with ATG16L to create the ATG12-ATG5-ATG16L complex.166 LC3 is processed by ATG4 to create LC3-i, which is subsequently linked to PE via the ATG3-ATG7 and ATG5-ATG12 complexes, resulting in the formation of LC3-ii. This version of LC3 attaches to the autophagosome and plays a role in its maturation.167 Once autophagosomes are formed, they travel along microtubules to the area near the lysosomes surrounding the nucleus and establish contact with them.168 Using the Rab-SNARE system along with various autophagy proteins, the outer membrane of the autophagosome merges with the lysosomal membrane. This fusion allows the release of degradation enzymes into the fully formed autolysosome, which then break down the contents.169,170
The Role of Autophagy in Osteoarthritis
Autophagy plays a crucial role in regulating the survival and lifecycle of chondrocytes by maintaining cellular homeostasis.171 Nonetheless, certain research indicates that too much autophagy could result in damage and death of cartilage cells.172 Next, we will concentrate on the dual function of autophagy in cartilage or chondrocytes. Figure 3.
Protective Autophagy in Chondrocytes and Cartilage
Autophagy and Hypoxia
OA is a condition that is not related to blood vessel inflammation, in which the joint cartilage exists in a low-oxygen setting for its entire life. It depends on HIF-1 and HIF-2 to adjust to this lack of oxygen. HIF-1 is found in both healthy cartilage and cartilage affected by OA.173 It supports the growth of healthy cartilage by enhancing the characteristics of cartilage cells, their survival, and the production of the cartilage matrix. HIF-2 is responsible for facilitating the metabolic process that results in the breakdown of the OA cartilage matrix, which ultimately causes damage to the joint cartilage.174 In contrast to cells that thrive in oxygen-abundant conditions, cartilage cells mainly depend on autophagy instead of cell turnover to preserve a healthy cartilage endothelium.175 In an environment lacking oxygen, chondrocytes show increased basal autophagy and generate a higher quantity of col-2 cells, leading to stronger collagen structures in the joint.176 On the contrary, inhibiting HIF-1 would negatively impact cell autophagy, resulting in the buildup of MMP13 and impairing mitochondrial function.177 The primary pathway for triggering autophagy in chondrocytes during low oxygen conditions is the HIF-1-adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK)-mTOR pathway.178 Under hypoxic conditions, cellular energy levels drop, prompting the activation of HIF-1, which enhances glycolysis. This process results in increased phosphorylation of AMPK, which subsequently suppresses mTOR activity, ultimately leading to the onset of autophagy.179 This guarantees that chondrocytes stay alive and finish their life cycle in their designated low-oxygen environment following cell division.178 At the same time, HIF-2 suppresses autophagy. When HIF-2 was removed, there was a notable increase in the autophagy response in the chondrocytes of the growth plate cartilage in mice. This increase was linked to a decrease in the activity of protein kinase B (Akt), mTOR, and the expression of B cell lymphoma-2 (Bcl-2).180
Autophagy and Aging
Autophagy is involved in the development and final differentiation of the growth plate in hypertrophic chondrocytes.181 The temporary rise in the levels of ULK1, Beclin1, and LC3 suggests that autophagy may serve as a protective response to cellular stress in the initial degenerative phase of OA. However, as the pathological changes advance to mild OA, the amount of autophagy proteins in the surface of the joint cartilage diminishes. In the advanced phase of OA, there are defects in autophagy across the entire articular cartilage layer, affecting both the middle and deeper regions, which eventually results in the death of chondrocytes.182,183 Chondrocyte autophagy takes place before cartilage damage happens, and its function gradually declines with age, which aligns with the deterioration of chondrocytes.184 Additionally, a recent study indicates that the progression of OA deteriorates with the aging of fibroblast-like synovial cells, which is associated with reduced autophagy and increased levels of the senescence-associated secretory phenotype (SASP). The restoration of autophagy counteracts the effects of aging and the secretion of the SASP, while also reducing cartilage damage by limiting GATA4 activity.185 The data indicates that autophagy serves as a protective mechanism for joint cartilage and synovial tissue in response to different stress conditions. However, prolonged exposure to the damage caused by excessive stress that exceeds the repair capacity of autophagy can worsen cartilage degeneration.184 OA worsens as people age, and this may be linked to apoptosis that depends on caspases.186 In GFP-LC3 transgenic mice, LC3 and ATG5 are most highly expressed in the chondrocytes found in the superficial and middle layers. Additionally, the removal of mouse ATG5 results in the early aging of these chondrocytes. In older mice, there is a notable decrease in the quantity of autophagosomes within chondrocytes, and the amount of vesicles in the subchondral bone declines significantly. Dysfunctional autophagy worsens OA-like alterations in the synovial tissue.187 In healthy adults, significant autophagy is present in the outer layer of cartilage, while cartilage in older individuals exhibits reduced autophagy levels.182 This suggests that impaired autophagy contributes to premature aging in cartilage, which in turn causes irregular cartilage production, abnormal expression of catabolic genes, and damage to the ECM. In addition, mechanical injury to cartilage can influence the autophagy of cartilage cells. Research has indicated that mechanical injury can suppress autophagy.188 This is associated with a notable reduction in the expression of ULK1, Beclin1, and LC3 in the outer region of the cartilage within 48 hours post-injury, whereas the stimulation of external autophagy can restore cartilage damage resulting from mechanical injury.188 In contrast, suitable mechanical stress can help reduce aging and inflammation in chondrocytes, manage autophagy, improve chondrocyte function, and lessen cartilage deterioration.189,190 In a mouse model of OA, suitable mechanical stress boosted autophagy in cartilage, leading to higher levels of LC3 and lower levels of p62/SQSTM1. This ultimately facilitated the formation of cartilage cells and improved the recovery from cartilage damage.191
Autophagy and Inflammation
The levels of inflammatory factors in the synovial tissue and subchondral bone are closely linked to the degenerative alterations in the structure of cartilage affected by OA.27 Research indicates that these inflammatory factors can speed up the aging process of cartilage cells by hindering autophagy and triggering inflammatory reactions, which in turn worsens OA.192 The inflammatory response triggered by IL-1β significantly increases the phosphorylation of PI3K, Akt, and mTOR proteins, resulting in a reduction of autophagy activity, this results in an obstruction of the G1 phase in chondrocytes and a decrease in the S phase. Additionally, inhibiting the PI3K-Akt-mTOR pathway can enhance autophagy in chondrocytes of OA rats and reduce inflammatory reactions.193 In OA mice, the targeted reduction of PPARγ results in an increased generation of inflammatory markers such as inducible nitric oxide synthase and cyclooxygenase-2. This is linked to the heightened expression of mTOR and a decrease in important autophagy markers, which in turn intensifies the inflammatory response in the cartilage and accelerates the development of OA.194 The mTOR-mediated autophagy pathway is significantly linked to synovitis in OA. Inhibiting mTOR triggers the activation of autophagy, which in turn decreases the formation of synovial osteoclasts and reduces IL-1 expression in joint cartilage, thereby safeguarding the cartilage from erosion.195 The activation of the MAPK/NF-κb pathway is advantageous for reducing autophagy levels in kidney cells during OA.196 Recent studies have revealed that epigenetic changes play a role in the connection between autophagy and inflammation in patients with OA.197 Methyltransferase-like 3 enhances the stability of Bcl-2 mRNA via the M6A/Yth m6A RNA binding protein 1 pathway, this limits the autophagy process driven by Beclin1 in the body, which in turn increases the apoptosis of cartilage cells and the deterioration of subchondral bone in mice with OA.197
Autophagy and Oxidative Stress
Oxidative stress arises when there is an imbalance between the generation of ROS in the mitochondria and the capacity to eliminate them.198 In OA chondrocytes, different factors like mechanical damage and aging trigger the activation of cytokine receptors and Toll-like receptors (TRL), which in turn increases the production of superoxide (O— 2) in the mitochondria and decreases the levels of antioxidant enzymes superoxide dismutase 1 and superoxide dismutase 2, this eventually results in the buildup of ROS and the impairment of mitochondrial function.139,199 The oxidative alteration of both intracellular and extracellular elements, along with the suppression of collagen production and the activation of the MMP and ADAMTS families, contributes to the deterioration and breakdown of cartilage.200 The breakdown products continue to worsen joint deterioration, creating a harmful cycle.201 During the early stages, the autophagy process in OA cartilage is increased, but it diminishes in the later stages. This indicates that autophagy may serve as a protective mechanism against oxidative stress. As a result, some individuals propose that enhancing autophagy could aid in eliminating the excessive buildup of inflammatory substances and ROS.202 The excessive buildup of p62 in OA chondrocytes due to oxidative stress results in disrupted autophagy and heightened chondrocyte apoptosis. On the other hand, treatment with alginate specifically addresses the autophagic damage caused by oxidative stress and promotes autophagy by activating Bcl2 interacting protein 3 (BNIP3).203
Harmful Autophagy in Chondrocytes and Cartilage
The studies mentioned indicate that in models of aging or OA, the autophagy function in joint cartilage or chondrocytes is compromised. Restoring proper autophagy levels may help mitigate the pathological damage resulting from inflammation and oxidative stress in OA, ultimately leading to a reduction in the condition.172,204,205 However, an overactivation of autophagy could negatively impact the regulation of OA. Mitofusin2 is an important molecule that plays a role in controlling autophagy and inflammation. When it is overexpressed, it can greatly suppress the PI3K/AKT/mTOR pathway, leading to an overproduction of autophagy. This disrupts the normal balance of autophagy in chondrocytes, raises the levels of ROS, and triggers inflammatory responses, ultimately resulting in cartilage degeneration.206 Chang172 et al discovered that the autophagy levels in the cartilage of OA patients are considerably greater than those in young injured cartilage or in cartilage from older individuals. The cell clusters in the upper region of the cartilage show a strong expression of Beclin-1 and LC3, and there is evidence of autophagic cell death occurring in the chondrocytes of OA patients. This may be linked to an overactivation of autophagy.172 When chondrocytes are grown in an acidic environment for 48 hours, the acid-sensing ion channel 1a, which is activated by the external acidity, triggers chondrocyte senescence by continuously activating autophagy. This indicates that consistently elevated autophagy levels might contribute negatively to the senescence of OA chondrocytes.154,207 Moreover, oxidative damage to DNA can result in overactivation and improper regulation of autophagy. When healthy cartilage cells experience acute external oxidative stress, the expression of autophagy-related genes rises initially but returns to normal within 6 to 24 hours.208 In contrast, the autophagy levels in OA cartilage cells stay elevated for an additional 24 hours.209 In OA chondrocytes, oxidative stress causes mitochondrial impairment, leading to a higher generation of ROS. This ROS serves as the membrane source for both immature and mature autophagosomes in OA chondrocytes. This results in the creation of a reservoir that preemptively manages autophagy, which in turn reduces autophagy activity to handle further variations in ROS levels. As a result, this group that is susceptible to autophagy is why OA chondrocytes do not quickly decrease their autophagy response when faced with oxidative stress, which may even result in the death of chondrocytes due to autophagy.209,210
In summary, throughout the process of OA development, autophagy responds to several influences including aging, low oxygen levels, mechanical stress, inflammation, and oxidative stress. During this process, autophagy levels may rise as a defensive reaction to different pathological changes in the joint microenvironment.19,204,211 However, if autophagy adaptation fails, it can result in the advancement of degenerative changes in the cartilage. However, even as we emphasize the protective function of autophagy, we must remain mindful of its dual nature. We propose that the bidirectional control of autophagy in relation to the survival or death of chondrocytes may be influenced by the specific type of cellular stress and the length of time the stress is applied.212 Nonetheless, research on the impact of excessive autophagy on cartilage or chondrocyte function remains scarce. More investigation is required in the future to determine the thresholds that differentiate protective autophagy from excessive autophagy in chondrocytes across various pathological conditions. Consequently, we think that autophagy could serve as an important therapeutic target for articular cartilage in different stress scenarios. However, the therapeutic range and dosage of targeted autophagy medications still require additional investigation.
Cuproptosis and Osteoarthritis
Cuproptosis is a unique type of cell death characterized by the buildup of copper in cells. This accumulation causes mitochondrial lipidation proteins to aggregate and destabilizes Fe-S cluster proteins, resulting in a form of cell death that is distinct from that induced by oxidative stress.213,214
Disorders of Copper in Cartilage and Bone
About 66% of the copper within the human body is located in the muscles and skeletal structure. Numerous research have validated the significant function of copper in managing bone metabolism.215,216 Copper has the ability to affect collagen production and bone matrix metabolism, thereby supporting healthy bone growth. Copper can improve bone mass and durability, in addition to encouraging bone growth.215,217,218 Similarly, copper is essential for preserving the structure and balance of cartilage tissue. Under typical circumstances, copper (Cu) maintains a stable presence in cells, with its stability primarily dependent on three copper transporter proteins: SLC31A1 and ATP7A/B. SLC31A1 is involved in the absorption of copper, whereas ATP7A/B is involved in its elimination.219,220 Figure 4. However, disruptions in copper balance, whether due to excessive accumulation or deficiency, can cause copper levels in cells to exceed or drop below the limits maintained by homeostatic processes.221 This could result in harmful effects on bone and cartilage cells. Research has indicated that the level of copper in cartilage is greater than that found in other types of tissues within the knee joint, such as the meniscus and synovium.222 Cu²⁺ can enhance regeneration of cartilage and aid in the rebuilding of the junction between bone and cartilage, which helps reduce damage to cartilage tissue and speeds up the repair process.223 Copper can influence the balance of cartilage by adjusting the function of hypoxia-inducible transcription factors and proteins associated with cartilage. Additionally, copper significantly boosts the chondrogenic differentiation of mesenchymal stem cells derived from bone marrow.224,225 During the process of collagen production, copper functions as a cofactor of lysyl oxidase (LOX), which plays a key role in interlinking collagen in tissues that are abundant in cartilage. Furthermore, copper is involved in the metabolism of bone and cartilage by enhancing immune responses in cartilage and facilitating the development of articular cartilage and underlying bone.225,226 Insufficient copper levels may interfere with the development of bones and cartilage, leading to potential harm to their structural integrity. Copper deficiency could impact the reactions facilitated by different enzymes that require copper functioning as a cofactor. A significant result is the deterioration of LOX, which hinders the linking of peptide chains.227 Consequently, the matrix of bone is weakened, the cartilage becomes more delicate, and the body’s vulnerability to cartilage breaks increases. In the end, the cartilage’s integrity is weakened.228 This damage may be associated with HIF activation that is mediated by copper. An interruption in the HIF-1α signaling pathway can affect the production of energy and the synthesis of biological molecules in cells, potentially resulting in problems related to skeletal development.226,229,230 However, elevated copper levels can hinder the process of rat bone marrow mesenchymal stem cells (MSCs) differentiating into osteoblasts. It is widely thought that this phenomenon happens due to the stabilization of the HIF-1α protein and the reduced expression of a gene known as Runx2, both of which play crucial roles in normal bone development.217 Copper can directly prevent the breakdown of bone carried out by osteoclasts.231 The experiment showed a notable reduction in the number of identified mature osteoclasts when the Cu²⁺ concentration reached 20 µm or more. As the levels of Cu²⁺ increase, there is a notable reduction in the number of osteoclasts.232 Earlier research has indicated a link to mitochondrial processes, copper proteins are capable of effectively removing ROS from both external and internal sources, thus helping to restore mitochondrial function and manage chondrocyte metabolism.233 Nevertheless, differing opinions continue to exist, and a definitive conclusion has not been achieved. A lack of copper can decrease the function of monoamine oxidase and Lox, which can result in the production of reactive ROS by cells. As a result, the redox balance in osteoblasts becomes disrupted, which reduces their survival capacity and hinders their development.234,235 Concurrently, the solubility of collagen rises, the peptide chains break down, and the collagen loses its stability, which ultimately results in reduced bone strength.236,237 In tissue engineering, particularly regarding regeneration of cartilage, the addition of metal ions can improve cell adhesion, improve cell performance, and enhance the potential for regenerating cartilage tissue.238,239 Currently, the research on copper ions is facing some limitations. The research into whether raising copper concentrations in injured cartilage, which has low copper content, can promote the healing of cartilage tissue is a topic that merits additional investigation, especially regarding copper’s role.240 Too much copper may harm cartilage, but the damage can be effectively repaired based on the severity of the cartilage injury. In the initial phases of WD, research indicated the presence of copper deposits in the cartilage and synovial tissue of patients with OA.241,242 Li243 et al also noted that copper is found in cartilage that is significantly damaged, as opposed to cartilage that shows only mild wear. This discovery indicates that an increase in copper levels is associated with the exacerbation of cartilage damage.243 Crucially, adding copper to chondrocyte cultures could result in changes to the cell’s cytoskeleton, such as differences in microfilaments, microtubules, and the arrangement of fibronectin.244 The deactivation of integrin-associated kinase in chondrocytes could lead to a decrease in levels of cyclin D1, which could hinder the proliferation of chondrocytes.245 Nonetheless, the exact ways in which copper accumulation impacts cartilage require further investigation. Conducting thorough research on how cell therapy affects copper levels, both in the body and in connection with relevant scaffolds, alternative materials, and other groundbreaking innovations, is anticipated to provide valuable insights for future research initiatives.
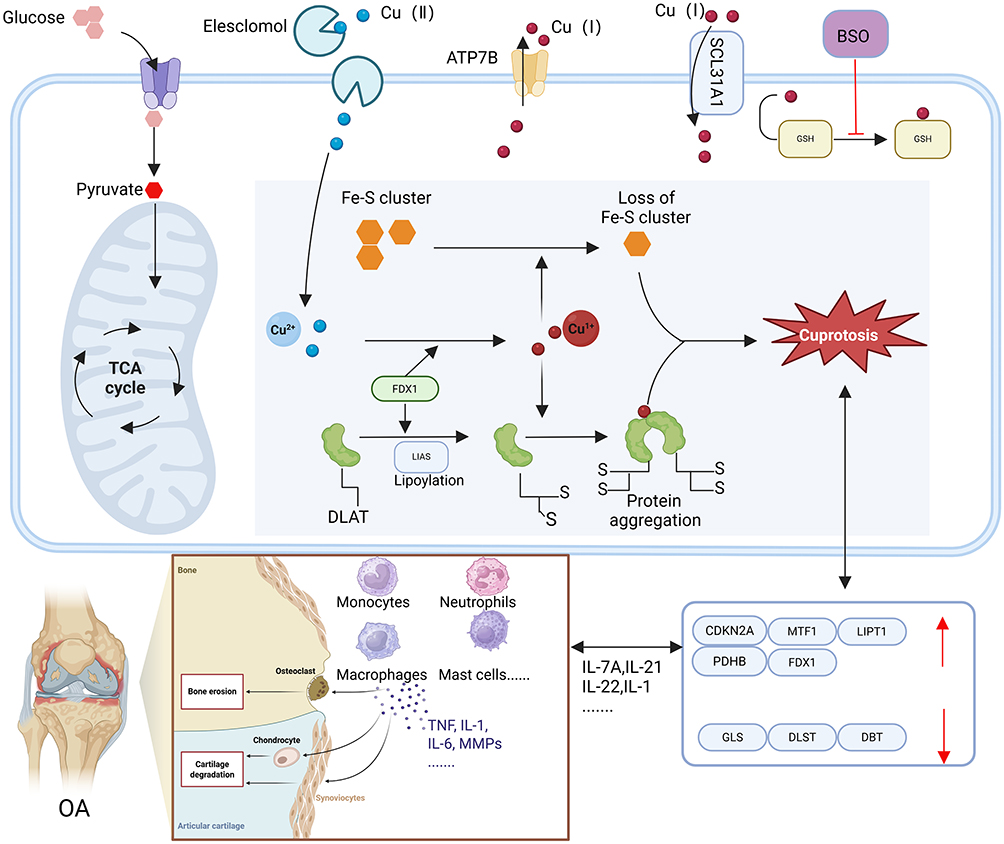 |
Figure 4 The mechanism of cuproptosis and its impact on OA. DLAT: Dihydroceramide S-acetyltransferase, FDX1: Ferroxidase 1, LIAS: Thiosulfate synthase, TCA: Tricarboxylic acid cycle, Fe-S cluster: Iron-sulfur cluster protein. Under ordinary conditions, copper maintains a stable level inside cells, The steady state of Cu predominantly relies on three copper transporters, namely ATP7A/BandSLC31A1. SLC31A1 is accountable for the uptake of copper into cells, whereas ATP7A/B is in charge of copper efflux. The Cu ion carrier ethylchelmonate is capableof transporting Cu2+ into the cell, resulting in the accumulation of Cu within the cell. BSO restrains the chelating activity of the intracellular Cu chelator GSH. In the event of disruption of copper equilibrium, on the one hand, when there is an excessive accumulation of Cu2+ in cells that are dependent on mitochondrial respiration, Cu2+combines with sulfinic acid DLAT, resulting in abnormal oligomerization of DLAT. The augmentation of insoluble DLAT gives rise to cytotoxicity and triggers cell death; On the other hand, FDX1 converts Cu2+ into a more toxic form of Cu+, this will result in the suppression of Fe-S cluster protein synthesis and trigger cell death. Furthermore, CRGs are associated with cellular factors influencing synovial cells, it has an impact on diverse cell types in OA, like neutrophils, monocytes, and macrophages. Created in BioRender. Jiwei, H (2025) https://BioRender.com/n41i521. |
Cuproptosis in Bone and Cartilage Biology
In March 2022, Tsvetkov et al were the first to suggest that copper ions could lead to cell death.246 They found a distinct type of cell death triggered by copper, which is different from the three types of cell death mentioned above. They named the recently identified copper-dependent cell death mechanism “cuprotosis.” The primary way copper causes cell death is through the buildup of copper ions inside the cell.246 Copper is transported into the cell through the Cu²⁺ carrier elesclomol, while the reduction in endogenous Cu chelator GSH in the cell leads to an increase in intracellular Cu²⁺ levels.247 A higher concentration of intracellular Cu²⁺ could influence The association of α-lipoic acid within the tricarboxylic acid cycle, potentially interrupting the cycle and resulting in protein clumping and imbalance.214 Copper ions additionally reduce the levels of iron-sulfur (Fe-S) cluster proteins, leading to stress caused by protein toxicity.214 The process of DLAT oligomerization and the decrease in Fe-S stability ultimately result in cell death246 Figure 4. In the past few years, the importance of copper in the biology of bone and cartilage has been increasingly uncovered. CRGs are the genes associated with cell death triggered by copper ions, including GLS, MTF1, DLAT, CDKN2A, PDHA1, PDHB, as well as DLD. Research has demonstrated that these genes affect the biology of bone and cartilage by regulating the functions of chondrocytes, osteoclasts, and osteoblasts.248 DLAT is abundantly expressed in chondrocytes, resulting in their death and subsequent cartilage damage. At the same time, blocking DLAT could lead to a reduction in chondrocyte death.249 MTF1 is significantly expressed in chondrocytes and FLS, which contributes to the development of osteophytes, subchondral bone sclerosis, cartilage damage, and inflammation of the synovial membrane. On the other hand, the lack of the MTF1 gene can successfully prevent the breakdown of cartilage and subchondral bone hardening in mice.250 GLS can influence the differentiation of kidney cells by modulating histone acetylation.251 The regulation of cell fate and bone formation is influenced by GLS and glutamine metabolism during the differentiation process. Furthermore, the osteogenic and adipogenic differentiation of SSCs can also be affected by GLS and glutamine metabolism.252 The protein of P16 (INK4A), which is produced by the CDKN2A gene, is a common indicator of cellular aging. An overproduction of this protein could hinder the mineralization process in osteoblasts and lead to a rise in the quantity of osteoclasts.253 Earlier research has also outlined the influence of CRGs on the formation of osteoclasts and osteoblasts. Inhibiting PDHA1 could encourage the growth of osteoblasts, whereas activating PDHA1 may influence both macrophages and osteoblasts by reducing the secretion of inflammatory substances.220 PDHB could impact the differentiation of osteoclasts by interacting with kinase 10 associated with nima, or it may also influence the differentiation of osteoblasts by affecting mitochondrial balance.254 DLD influences glycolysis, which in turn impacts the differentiation of osteoclasts.254 In synovitis tissue, the CRGs that are expressed at high levels include PDHA, LIPT1, FDX1, PDHB, and CDKN2A, whereas DBT and DLST are expressed at lower levels. It is believed that certain CRGs may influence the normal physiological functions of the synovium and modulate the activity of synovial cells255,256 Figure 4. At present, the effects of CRGs on the biology of bone and cartilage remains uncertain, and the possible mechanisms behind CRGs are not well understood. However, earlier research has indicated that many genes influence bone formation and resorption, either directly or indirectly. In the forthcoming future, these associated genes have significant potential for progress in bone and cartilage biology. Therefore, additional study is needed to investigate them further.
The Mechanisms of Chondrocyte Apoptosis in Osteoarthritis
Apoptosis is an active cell death process regulated by intrinsic genetic mechanisms under specific physiological or pathological conditions. It is also known as programmed cell death (PCD) or type I cell death.24 Morphologically, it is characterized by cell shrinkage, loss of connections, and separation from surrounding cells, accompanied by cytoplasmic condensation, disappearance of mitochondrial membrane potential, nuclear mass aggregation, nucleolar fragmentation, and degradation of DNA into 180–200 bp fragments. Finally, apoptotic vesicles with intact cytoplasmic structures are formed without triggering an inflammatory response.257,258 Apoptosis can be categorized into exogenous (death receptor pathway) and endogenous (mitochondrial pathway) types.258 In the exogenous pathway, death receptors (such as Fas, TNFR1, TRAILR, members of the TNFRSF family)259 bind to death ligands, causing conformational changes in the cell membrane and activating caspases, ultimately leading to cell apoptosis.257 In osteoarthritis (OA), apoptosis plays a crucial role in the deterioration of articular cartilage. Exogenously, synovial inflammation is common in OA patients, and inflammatory mediators (such as ROS, IL, NO, matrix-degrading enzymes, etc) promote chondrocyte death through specific pathways.135 For instance, the death domain of TNFR-1 binds to the FADD protein, recruits and activates Caspase-3, 7, and 8, forms a death-inducing signaling complex, and triggers chondrocyte apoptosis.260,261 Certain drugs (such as oxflo and sparflo) can also promote chondrocyte apoptosis by activating the TNFR signaling pathway. Endogenously, factors like DNA damage, endoplasmic reticulum stress, hypoxia, high cytoplasmic Ca²⁺ concentration, and severe oxidative stress can trigger the mitochondrial apoptosis pathway.258 This causes the opening of the mitochondrial permeability transition pore, release of cytC, Smac, AIF, etc., and activation of the caspase cascade, resulting in chondrocyte apoptosis. For example, an increase in intracellular Ca²⁺ activates calpain, which cleaves membrane-bound AIF, and the soluble AIF fragments are released outside the mitochondria to induce apoptosis.258 In addition, inflammatory mediators such as ROS and inducible nitric oxide synthase,262 as well as IL-1β (which can damage mitochondrial DNA, interfere with chondrocyte metabolism, enhance TNF-α activity, etc).,263 are closely related to chondrocyte apoptosis in OA. High levels of ROS can also damage cell membranes, nucleic acids, and mitochondria, exacerbating inflammation and cell death.264 Currently, the molecular regulatory mechanisms of signaling pathways related to apoptosis in OA and the correlations among various factors remain incompletely understood. Future research on targeted drugs based on these findings is expected to provide new insights for the early prevention and treatment of OA.263–266
ResultsProgrammed Cell Death Drugs in Clinical Application for Osteoarthritis
Targeted Therapy for Ferroptosis in Osteoarthritis
The elements of traditional Chinese medicine are significant in the regulation of ferroptosis. Icariin (ICA) has the ability to alleviate the decrease in GPX4, SLC7A11, and SLC3A2L levels in synovial cells affected by OA due to LPS, and it also reduces the levels of malondialdehyde (MDA) and iron. Studies indicate that ICA is capable of preventing ferroptosis in synovial cells induced by LPS affected by osteoarthritis by downregulating the iron transport protein TFR1 and enhancing the system Xc/GPX4 regulatory pathway.267 Myristicin (CAD), a compound derived from ginger plants, has been shown to lower levels of iron, ROS, and MDA in chondrocytes that have deteriorated due to IL-1β exposure. Additionally, it enhances the expression of the proteins GPX4 and SLC7A11.This indicates that CAD could greatly reduce ferroptosis in chondrocytes that is triggered by IL-1β.268 The proposed mechanism could activate the GPX4/SLC7A11 signaling pathway, which might help decrease ferroptosis in cartilage cells.268 Biochanin A (BCA) is an active ingredient found in Astragalus. In a study involving iron overload in chondrocytes derived from mice treated with FAC, BCA has the ability to counteract the increased levels of the iron transport protein TFR1 and the decreased levels of FPN, thus helping to manage the labile iron pool (LIP) in chondrocytes.98 It may also boost the expression levels of the Xc-system, GSH, and GPX4 proteins, which would strengthen the protective processes against lipid peroxidation and decrease the buildup of ROS caused by excess iron. Research indicates that BCA might reduce cellular iron levels by blocking TFR1 and enhancing the expression of FPN.98 Additionally, controlling the Xc-system/GPX4 pathway is crucial for regulating the buildup of ROS, which helps prevent lipid peroxidation and slows down the advancement of OA.98 Research indicates that Baicalin, a substance derived from astragalus extract, can lower the levels of MMP13, ADAMTS5, and Col1 that are triggered by IL-1β in chondrocytes impacted by osteoarthritis.269 Additionally, it enhances the expression of Col2 while reducing the amounts of lipid ROS and Fe²⁺. The findings indicate that Baicalein can prevent the breakdown of the ECM caused by IL-1β. Additionally, it encourages ECM production in human chondrocytes impacted by OA by inhibiting ferroptosis, which may help slow down the progression of OA.269 The polyphenolic compound Theaflavin-3,3′-dialate (TF3), found in black tea, has demonstrated the ability to counteract the decrease of FTH1, GPX4, and SLC7A11 caused by erastin in chondrocytes from individuals with OA.270 Furthermore, it lowers the levels of ROS in cells and enhances Fe²⁺ storage through FTH1, thereby aiding in the reduction of ferroptosis associated with OA.270 The findings indicate that focusing on important molecules associated with ferroptosis, including ROS, GPX4/SLC7A11, GSH, FTH1, TFR1, FPN, and Fe²⁺, through the active ingredients of traditional Chinese medicine could provide a hopeful approach for treating OA in the future.(Table 1) Research shows that semaphorin 5A prevents ferroptosis, upregulates GPX4, and modulates the SREBP1/SCD-1 pathway via the PI3K/AKT/mTOR axis in rheumatoid arthritis. Given the shared synovial inflammation and tissue degradation in OA, semaphorin 5A likely plays a similar regulatory role in OA, making it a potential target for both conditions.271 Metformin has demonstrated promise in the treatment of OA by enhancing the expression of Gpx4, which helps safeguard cartilage cells from dying. This process not only relieves OA symptoms but also contributes to some improvement in subchondral bone sclerosis and decreases angiogenesis in mouse models.272 Research studies have demonstrated that sitosterol derived from Achyranthes bidentata can inhibit pro-inflammatory factors in chondrocytes and reduce the breakdown of the ECM.273 Cheng274 and his team discovered that stigmasterol can prevent ferroptosis triggered by IL-1β, thereby improving cell survival. This process works by inhibiting the interaction between sterol regulatory element-binding transcription factor 2 (SREBTF2) and the sterol regulatory element.274 D-Mannose is a natural substance present in various plants and their excretions, which has demonstrated the ability to reduce the vulnerability of chondrocytes to cell death, particularly in relation to ferroptosis induced by HIF-2α. This effect could potentially help relieve symptoms related to OA.274 Iron chelators are compounds that attach to iron, helping to remove it from the body. These medications function by inhibiting the Fenton reaction and providing antioxidant benefits in the body.275 Desferrioxamine (DFO) is a potent iron-binding agent utilized in the treatment of conditions associated with the buildup of excess iron in the body.276,277 The findings indicate that DFO may help to slow the advancement of OA, probably by stimulating the Nrf2 pathway. This stimulation could play a role in lowering Fe²⁺ levels in OA chondrocytes and preventing ferroptosis associated with OA.94 Fer1 acts as a fat-soluble antioxidant and specifically prevents ferroptosis. It aids in eliminating oxygen free radicals generated by iron in lipid hydroperoxides, It has the ability to generate an effect that counteracts ferroptosis, akin to that of GPX4.278 Research has indicated that Fer1 reduced the elevated levels of ROS, malondialdehyde (MDA), and Fe²⁺, while also counteracting the reduced expression of GPX4, GSH, and the GSH/GSSG ratio caused by TBHP in chondrocytes impacted by OA.279 Additionally, it restored the mRNA expression levels of GPX4 and FTH1 that had been lowered. The findings suggest that Fer1, recognized for its function as a blocker of iron-related cell death, is capable of stopping TBHP-triggered ferroptosis in chondrocytes affected by OA.90 It does this by decreasing lipid peroxidation, regulating iron buildup, and preserving the activity of GPX4/GSH.90 In conclusion, focusing on specific molecules related to iron overload and lipid peroxidation could be a significant strategy for preventing and treating OA (Table 2).
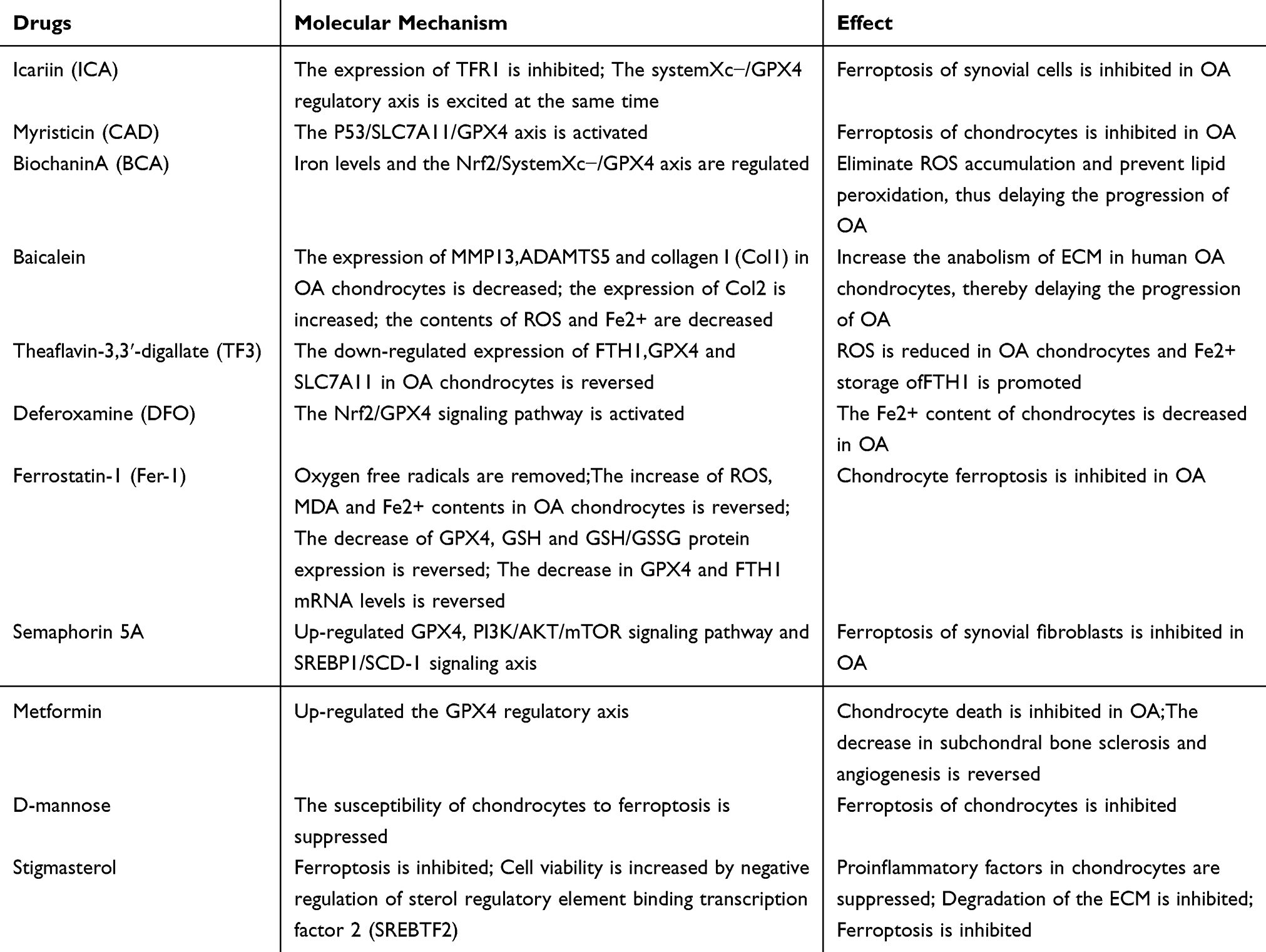 |
Table 1 Potential Drugs That Can Inhibit Ferroptosis in OA |
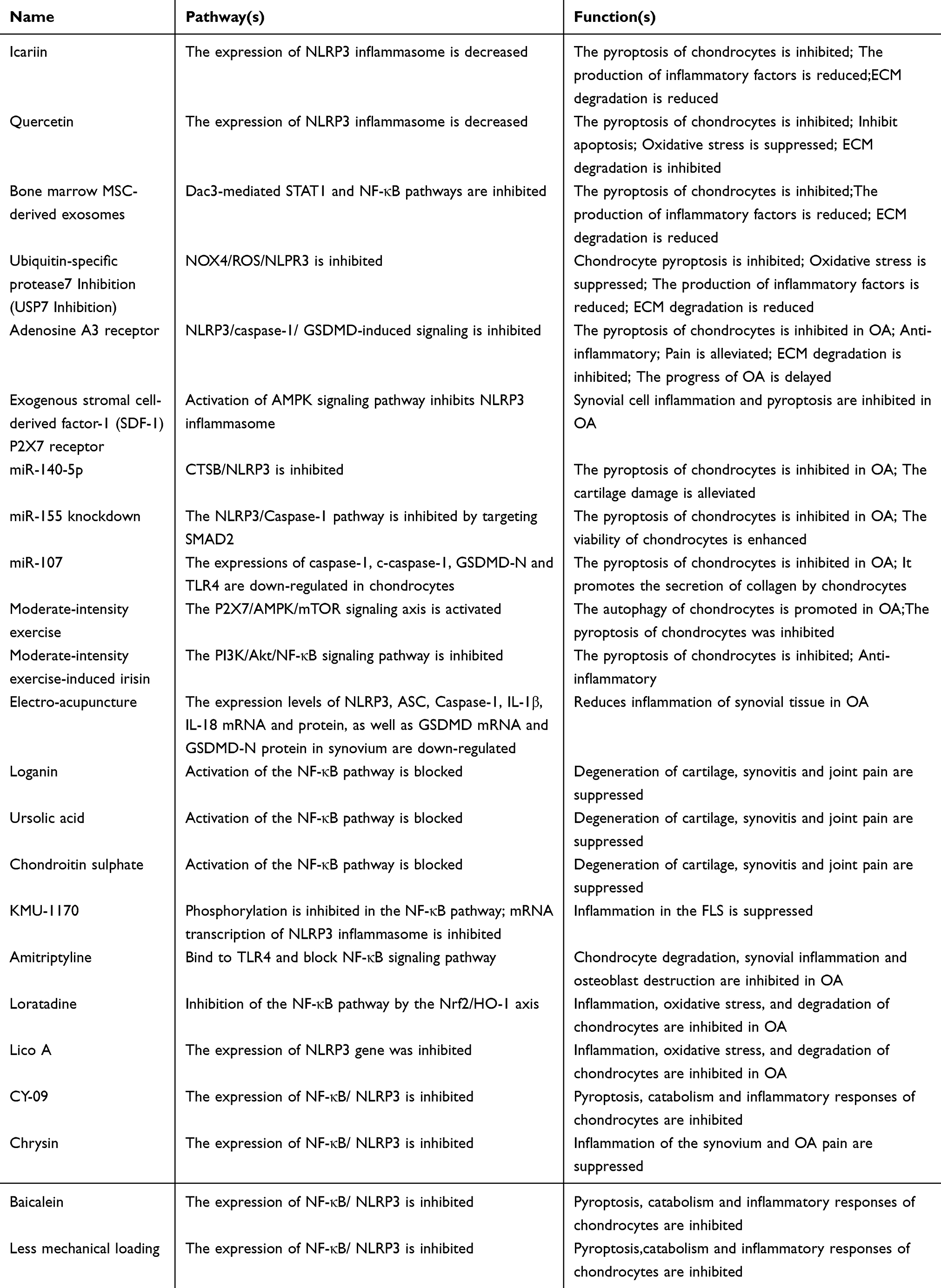 |
Table 2 Strategies to Inhibit Chondrocyte Pyroptosis, Including Phytochemicals, Exosomes, microRNAs, Growth Factors, Biological,Enzymes, and Physical Therapy |
Targeted Therapy for Cell Pyroptosis in Osteoarthritis
Since pyroptosis is strongly linked to inflammation, it is important to take both factors into account when creating a treatment plan for OA.280 Matrine and Morroniside can decrease chondrocyte pyroptosis and the inflammatory response by blocking the activation of NF-κB.281,282 The research indicated that a diet high in unsaturated fatty acids may have anti-inflammatory and anti-pyroptotic properties by blocking the TLR4/NF-κB signaling pathway as well as the NLRP3, caspase-1 or GSDMD pathway, which could help lessen the impact of OA.283 In the OA rat model, metformin, a medication often used to treat diabetes, lowers NLRP3 levels and helps reduce pyroptosis in cartilage cells due to its inhibitory properties.284 Exosomes obtained from mesenchymal stem cells, along with microRNA, phytochemicals, growth factors, and biological enzymes, have been shown to decrease chondrocyte pyroptosis, indicating their promising ability to inhibit this process. Furthermore, blocking pathways associated with pyroptosis can help relieve OA by decreasing pyroptosis124,285–297 (Table 2). Inflammatory bodies play a crucial role in starting and regulating pyroptosis by detecting different DAMPs and PAMPs, which activates pro-inflammatory caspases. Therefore, inhibiting the activation and formation of inflammasomes could safeguard cells from undergoing pyroptosis and the related issues that come with it. At present, scientists are concentrating on the NLRP3 inflammasome and the signaling pathways that control it, as it can react to different stimuli that lead to cellular disruptions.298 As previously stated, the NF-κB pathway is essential for the triggering of the NLRP3 inflammasome. Therefore, blocking the activation or transmission of the NF-κB pathway is the preferred method for targeted inhibition of pyroptosis.299 Creating a regulatory pathway involving NF-κB/NLRP3 inflammation using Marasmius, Mrsolic acid, and chondroitin sulfate to reduce cartilage deterioration and alleviate synovitis and OA pain in rats.300–302 In 2021, Baek303 developed a novel multi-protein kinase inhibitor known as KMU-1170, which is derived from indole-2-one. They subsequently found that it could successfully inhibit nearly all phosphorylation activities in the NF-κB signaling pathway, leading to a decrease during the mRNA transcription process of the NLRP3 inflammasome.303 Additionally, the Franco-Trepat team found that the antidepressant amitriptyline has the ability to bind to TLR4, effectively inhibiting the NF-κB signaling pathway. This action prevents the activation of NLRP3 in OA chondrocytes, synovial cells, and osteoblasts, ultimately impeding the secondary immune response.304 Furthermore, the NLRP3 inflammasome needs a secondary signal for its activation and assembly following the receipt of the primary activation signal. Research has indicated that ROS are essential for the activation and formation of the NLRP3 inflammasome.305 As a result, enhancing the state of oxidative stress within cells has emerged as another effective approach. The antihistamine loratadine’s component licochalcone A (Lico A) and licorice extracts serve as regulatory agents for Nrf2, helping to reduce the NLRP3 inflammasome in response to ROS stimulation. The first mechanism reduces the NF-κB pathway through the Nrf2/HO-1 axis, while the second enables Nrf2 to enter the cell nucleus and directly inhibit the triggering of the NLRP3 gene.306,307 Numerous pharmacological treatments, including the NLRP3 inhibitor CY-09,124 Curcumin,308 and Baicalin (delivered via intra-articular injection),120 along with physical therapy methods like decreasing mechanical stress on the knee joint119 and applying electrical stimulation,309 have demonstrated effectiveness in lowering NLRP3 inflammasome expression. These approaches help prevent chondrocyte pyroptosis, metabolic degradation, and inflammatory reactions, ultimately leading to improved cartilage health. Nonetheless, these research works failed to investigate the molecular mechanisms within animal models, indicating that future research should focus on further exploring and advancing this area. GSDMD experiences pyroptosis as it processes a wide array of both internal and external signals from the inflammasome.310,311 If the GSDMD pore does not form properly, it will impede the function of most previously activated inflammasomes, along with their related cysteine proteases and biochemical signals. By inhibiting the widespread activation of GSDMD, which initiates pyroptosis and subsequently diminishes the inflammatory response, this approach aids in preventing pyroptosis. As a result, inhibiting GSDMD demonstrates greater selectivity than using particular inflammasome inhibitors or treatments aimed at IL-1β and IL-18. Consequently, from a theoretical standpoint, focusing on the inhibition of GSDMD could be the most promising approach for future treatments. Currently, strategies aimed at specifically inhibiting GSDMD can be categorized into two main approaches: the first approach focuses on decreasing the cleavage of GSDMD by inflammatory cysteine proteases, while the second approach targets the prevention of oligomerization and/or membrane insertion of GSDMD-nto.309 Yang312 developed a GSDMD-derived inhibitor called N-Acetyl-phenylalanine-leucine-threonine-aspartic acid-chloromethyl ketone (Ac-FLTD-CMK) by pinpointing the cleavage site at which GSDMD interacts with caspase. They discovered that it substituted for full-length GSDMD and interacted with the previously mentioned caspase, inhibiting cleavage.312 Furthermore, experiments involving covalent modification of human GSDMD cyc191 and mouse GSDMD cyc192 revealed that disulfiram and necrosulfonamide, which are inhibitors of pyroptosis, were unable to trigger the oligomerization of GSDMD-NT, It can successfully prevent the formation of pores both in living organisms and in laboratory settings.313,314 Our research indicates that the chemical compound LDC7559 exhibits a level of effectiveness comparable to other medications aimed at GSDMD, likely because it interacts with GSDMD-NT.315 Additionally, cell biology researchers have explored the potential for cells to mend GSDMD pores in their membranes by forming vesicles that can enclose and later release these pores into the surrounding environment.316 Furthermore, another study investigated the function of GSDMD in post-traumatic OA. Eliminating GSDMD reduces cartilage alterations, synovitis, and subchondral bone hardening.317 In conclusion, GSDMD represents a significant advancement in the treatment and management of conditions associated with pyroptosis. Nevertheless, because there is insufficient detailed research on the pyroptotic molecular mechanisms in cell types beyond immune cells, and given that inhibitors targeting GSDMD do not influence the initial inflammatory processes associated with pyroptosis, further experiments are required to clarify the precise molecular mechanisms and effects of GSDMD-targeted inhibitors across various diseases.
Targeted Treatment of Mitochondrial Autophagy in Osteoarthritis
A thorough and detailed knowledge of the targets associated with mitochondrial autophagy and targeted therapies is crucial for the treatment of OA. Focusing on mitochondrial autophagy and swiftly removing damaged mitochondria is crucial for maintaining the normal physiological function of chondrocytes.175,318 This strategy can effectively prevent and reduce cartilage degeneration associated with OA. The primary therapeutic agents aimed at modulating mitochondrial autophagy in OA consist of active ingredients from conventional Chinese medicine, dissolvable amino acids, and beneficial bacteria generated through gut microbiota metabolism, the trace element zinc, metformin, and exosomes.83,319,320 The elements of traditional Chinese medicine are significant in the regulation of mitosis. Baicalin, a natural substance extracted from the desiccated root of Scutellaria baicalensis, has demonstrated properties that reduce inflammation, combat tumors, and modulate the immune system.321 Baicalin enhances the level of Bcl-1, Beclin3, LC3-II/LC3-I, p-Drp1, PINK1, and Parkin in chondrocytes of rats stimulated by IL-1β. This action supports cell survival, autophagy, and mitophagy in these chondrocytes, while also elevating MMP levels.322 It has been noted that Baicalin can prevent the induction of apoptosis in rat chondrocytes by IL-1β. The findings indicate that Baicalin could stimulate IL-1β-induced mitophagy via the PINK1/Parkin pathway, which may help reduce damage to chondrocytes.322 Curcumin, a polyphenolic compound derived from turmeric rhizomes, has the potential to greatly improve the gene and protein expression of mitotic markers such as PINK1, Parkin, LC3B, P62, and Beclin1 in chondrocyte degeneration caused by IL-1β.323 It can also suppress the expression of the genes and proteins associated with the markers of inflammation IL-1β and MMP13. This suggests that Curcumin (turmeric extract) may slow down the deterioration of cartilage by stimulating mitochondrial autophagy in chondrocytes.323 These research findings indicate that the mitochondrial autophagy influenced by the active ingredients in traditional Chinese medicine could be a promising area for future OA therapies.(Table 3) Studies have shown that Irisin constitutes a soluble peptide made up of 112 amino acids, playing a role in the regulation of bone remodeling and the maintenance of muscle balance.324 Irisin has the ability to boost the levels of PINK1 and Parkin in chondrocytes of mice stimulated by IL-1β, promote mitochondrial autophagy, decrease excessive mitochondrial fission, enhance mitochondrial fusion, and slow down the advancement of OA.325 This indicates that the recombinant Irisin protein can enhance mitochondrial autophagy triggered by IL-1β in mouse chondrocytes, manage mitochondrial dynamics, and ultimately help prevent the advancement of OA.325 Research indicates that zinc provides a protective benefit to SW1353 human chondrosarcoma cells that have been exposed to methyl iodide acetate (MIA).326 Zinc has the ability to mitigate cartilage damage resulting from MIA treatment by restoring low ATP levels and enhancing mitochondrial autophagy pathways associated with Parkinson’s disease. This suggests that zinc may offer protective benefits against OA.326 Research has shown that metabolites from the gut microbiome, including uric acid (UA), can stimulate the PINK1/Parkin pathway in patients with OA.327 This activation helps to prevent the degeneration of cartilage cells and enhances mitochondrial autophagy, leading to potential therapeutic benefits.327 Additionally, certain research has indicated that metformin can enhance mitochondrial autophagy by increasing levels of SIRT3, PINK1, and Parkin, along with the LC3II/LC3I ratio, in OA degenerative chondrocytes activated by IL-1β in mice. Metformin can reduce the expression of MMP13 and MMP3 while increasing the expression of Col2, which helps to mitigate the ECM degradation caused by IL-1β in OA degenerative cartilage cells in mice.328 However, the SIRT3 inhibitor 3-TYP can counteract these effects, indicating that SIRT3 may serve as an upstream target in the mitophagy pathway regulated by PINK1/Parkin. Metformin can activate the PINK1/Parkinson’s disease-dependent mitophagy pathway through SIRT3, counteracting the metabolic imbalance of synthesis and degradation in osteoarthritic chondrocytes induced by IL-1β.328 Therefore, the mitophagy pathway that Parkinson’s relies on plays a crucial part in hindering the advancement of OA (Table 3). Tang et al caused cell damage in primary rat articular chondrocytes by exposing them to advanced glycation end products (AGEs). They discovered that exosomes from bone marrow stem cells (BMSCs) increased the levels of proteins related to autophagy LC3-II/LC3-I and Beclin-1, and enhanced autophagy in chondrocytes that had been treated with AGEs. The increased levels of Drp1 enhance cell apoptosis by inhibiting the expression of proteins related to autophagy, including Beclin-1 and LC3-I/LC3-II. Exosomes obtained from bone marrow mesenchymal stem cells (BMSCs) can counteract the suppression of autophagy caused by the increased levels of Drp1. As a result, BMSC exosomes influence mitochondrial autophagy through DRP1 to inhibit chondrocyte apoptosis, making it a potential target for OA treatment.329 These outcomes suggest that improving mitophagy in osteoarthritis chondrocytes could be a novel approach for preventing and treating OA, and may also provide a valuable target for future drug screening aimed at OA prevention and treatment (Table 3).
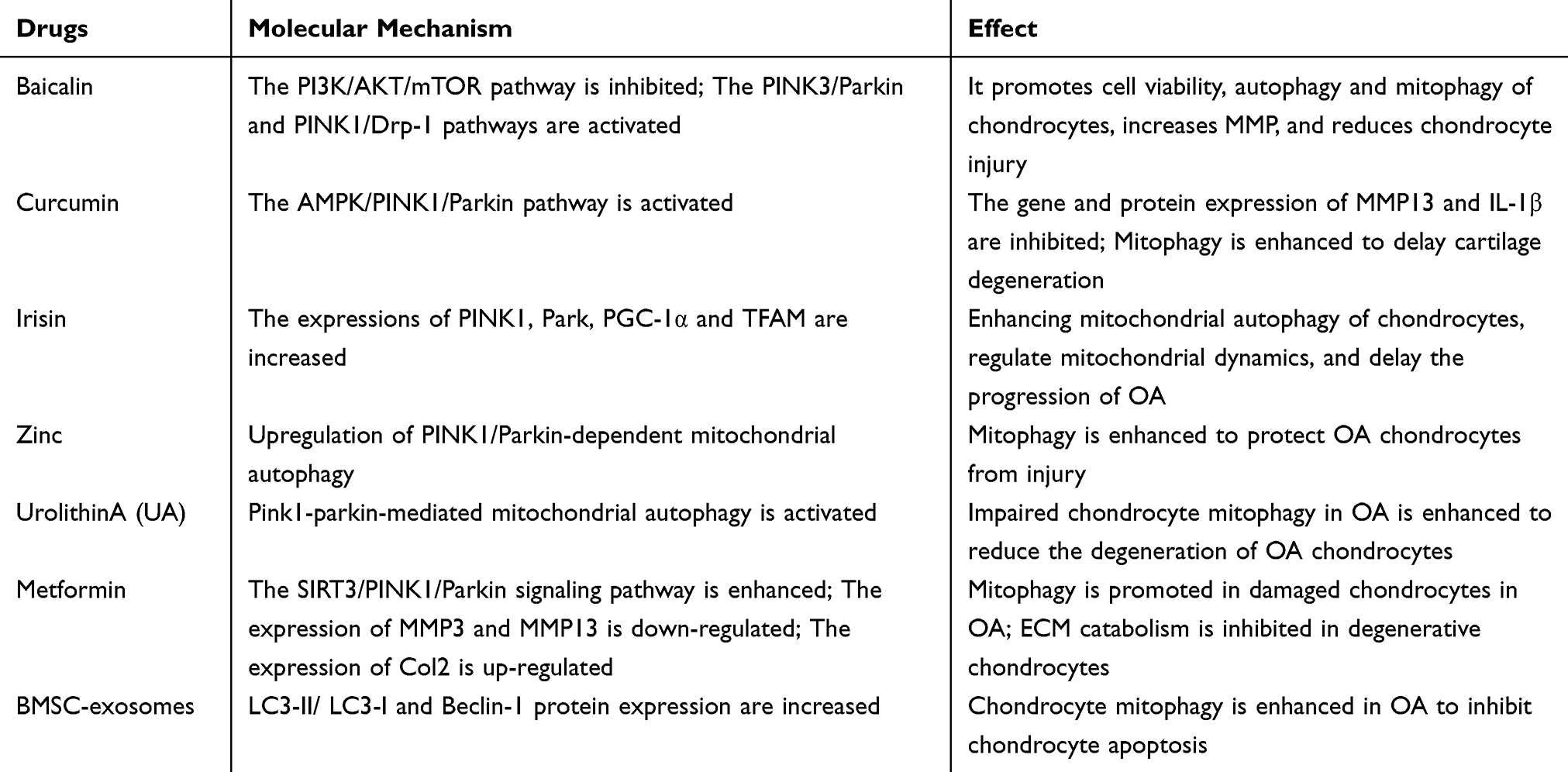 |
Table 3 Potential Drugs That Can Regulate Mitophagy in OA |
Clinical Application of Cuproptosis Targeted Therapy Strategy in Osteoarthritis
We have gained insights into the alterations in copper balance associated with osteoarthritic conditions, and when combined with the results from CRGs, this offers a potential therapeutic target for addressing these diseases. The balance of copper in the body is typically reestablished in two ways: by using copper chelators to decrease the availability of copper and by using copper carriers to transport copper into cells, thereby raising the intracellular copper levels.219 Common agents that chelate copper consist of penicillamine, zinc sulfate (ZS), trientine, and ammonium tetrathiomolybdate (ATTM)330 (Table 4). Copper chelators have the ability to restrain cell growth by attaching to the surplus copper present within the cells. They can also generate a group of ROS through redox reactions to initiate cell apoptosis, specifically target and remove aged chondrocytes, and successfully enhance cartilage formation to address OA.219 This unique benefit differentiates them from other medications and offers significant opportunities for future advancement. There is still significant potential for the creation of new copper chelating agents, and continued research in this area is highly anticipated. Research indicates that copper ion carriers like Escitalopram,331 Disulfiram (DSF),332 and analogs of Copper(II) (atsm333 and gtsm334) can elevate copper concentrations within cells, especially in the mitochondria (Table 4). At present, copper ion carriers are not commonly used in the treatment of bone and joint conditions. However, it has been noted that copper levels have increased in most cases of arthritis. Carriers of copper ions can enhance the copper levels inside cells and provide unique benefits in comparison to other medications. The advancement and study of “New carriers of copper ions” hold significant promise, and there is a strong anticipation for more research in this area. Moreover, stress in the endoplasmic reticulum can disturb the balance of non-erythrocyte cells and activate the unfolded protein response (UPR). Prolonged or permanent endoplasmic reticulum stress may lead to cell death that is initiated by the UPR. As a result, focusing on endoplasmic reticulum stress alone or disrupting the UPR signal to decrease cartilage cell death could be a promising approach for treating OA in the future.335 Nanoparticles are often integrated into the contemporary approaches of cartilage tissue engineering to improve chondrocyte function and hold considerable potential for treating degenerative joint diseases.336 The thermosensitive hydrogel with multi-functional composite properties (HPP@Cu gel) successfully reduces inflammation by neutralizing reactive oxygen and nitrogen species, shifting macrophage polarization from the M1 to M2 phenotype, and suppressing the release of inflammatory mediators. Research using a rat model of OA has demonstrated that these activities can help alleviate cartilage deterioration and decrease the generation of inflammatory substances.337
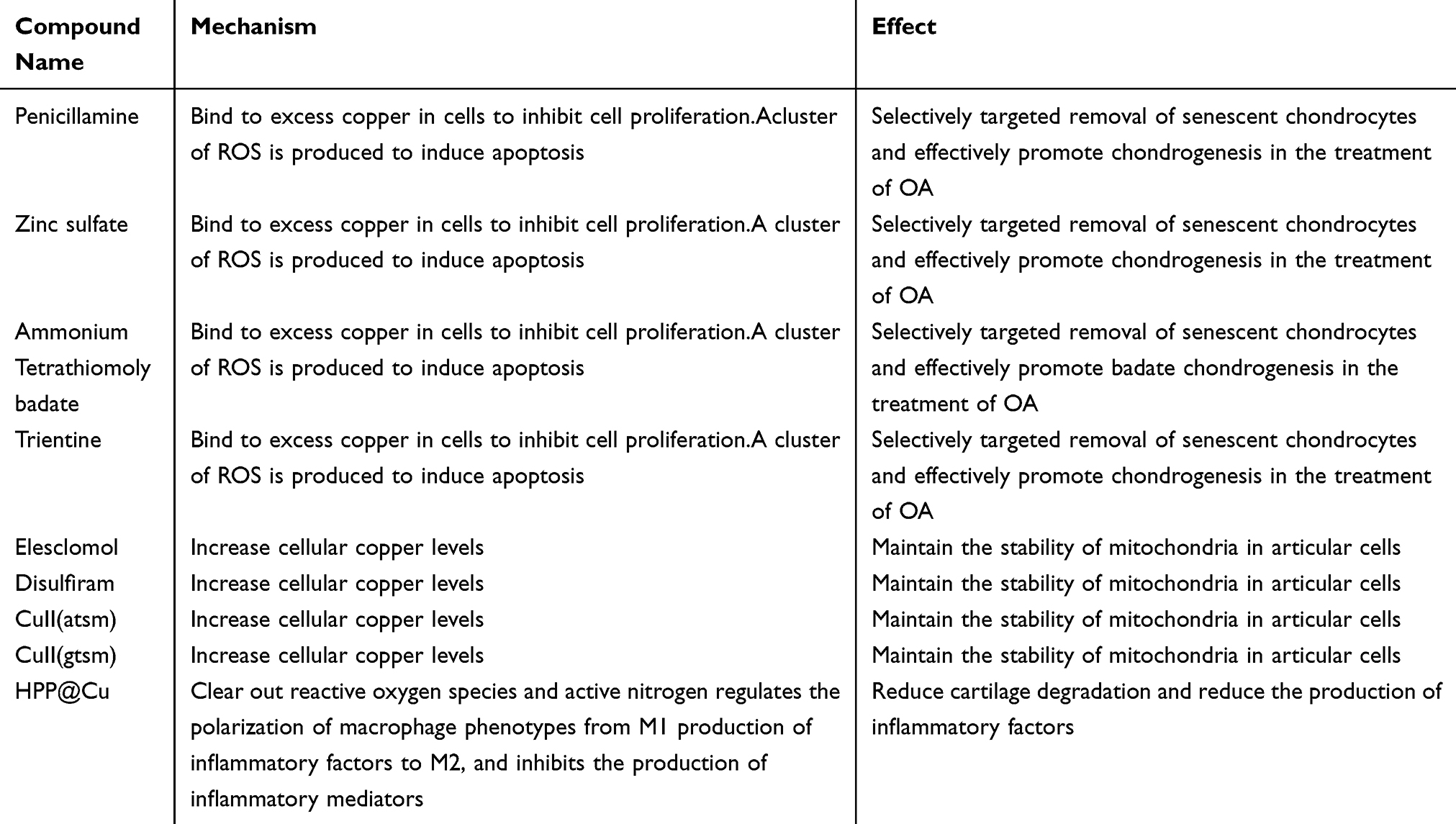 |
Table 4 The Clinical Application of Cu-Modulating Agents |
Strategies for Inhibiting Chondrocyte Apoptosis
Currently, numerous strategies aim to delay or reverse OA, with inhibiting chondrocyte apoptosis being a central research focus. These strategies mainly fall into the following five categories. In the realm of non - coding RNAs, microRNAs (miRNAs), a class of 20–24 nucleotide - long molecules encoded by endogenous genes, regulate downstream pathways by modulating mRNA stability and translation and inhibiting target gene expression.338 For example, miR - 93 suppresses chondrocyte apoptosis by downregulating the TLR4/NF - κB signaling pathway; miR - 107 inhibits the NF - κB pathway by targeting TRAF3, reducing AKT/mTOR activity, promoting autophagy, and ultimately inhibiting apoptosis.339 miR - 27a, miR - 206, and miR - 186 regulate the PI3K/AKT/mTOR and PI3K/AKT pathways to activate autophagy and inhibit apoptosis in OA chondrocytes.340,341 Additionally, miR - 199 - 3p and DNMT3B protect chondrocytes through targeted gene regulation, while miR - 1236 promotes chondrocyte apoptosis.342–345 Long non - coding RNAs (lncRNAs), such as MINCR, LncRNA - p21, and HOTAIR, often regulate chondrocyte apoptosis by suppressing miRNA expression.346–352 Circular RNAs (circRNAs) interact with miRNAs, modulating target gene expression and influencing chondrocyte apoptosis.353,354 Phytochemicals, including Morroniside, Loganin, Angelica polysaccharide, and Genistein, possess antioxidant, anti - inflammatory, autophagy - promoting, and anti - apoptotic properties.355–358 They inhibit chondrocyte apoptosis, reduce synovial inflammation, and balance extracellular matrix metabolism by regulating signaling pathways like NF - κB, ERK1/2, and SIRT1/AMPK.300,359–367 Traditional Chinese medicine formulas, such as Danzi kang knee granules, can promote stem cell proliferation and cartilage differentiation in OA rats and decrease chondrocyte apoptosis rates.368 Exosomes play a significant role in inhibiting chondrocyte apoptosis. Exosomes derived from human bone marrow mesenchymal stem cells (BMSCs - exo) can promote type II collagen synthesis, regulate glutamine metabolism, reduce inflammatory cytokine levels, and decrease chondrocyte apoptosis.369,370 Exosome gels alleviate IL - 1β/LPS - induced chondrocyte apoptosis and degeneration by inhibiting NF - κB and STAT signaling.371 Proteins and cytokines, such as NRG4, LMP - 1, Dlx5, and DRP1, inhibit chondrocyte apoptosis by regulating signaling axes like MAPK/JNK. Combining them with drug delivery systems, such as hydrogels and exosomes, can enhance treatment safety and efficacy.27,372–379 Melatonin can regulate TGF - β, MAPK, or NF - κB signaling pathways, promoting cartilage matrix synthesis, inhibiting chondrocyte apoptosis, and reducing inflammation.380–382 Biomaterials, such as melatonin - loaded hydrogels, show promise in promoting chondrocyte adhesion, proliferation, and matrix synthesis.383,384 Although these strategies for inhibiting chondrocyte apoptosis have made progress, large - scale animal experiments and clinical trials are still needed to further verify their effectiveness and safety.
Discussion
Cell death is essential for maintaining the proper functioning of a healthy human body. OA, a complicated long-term condition, is significantly affected by the process of programmed cell death in cells. Different types of programmed cell death have specific functions in OA. In this review, we first explore the connection between PCD and OA. For instance, the key features of iron-induced ferroptosis include lipid peroxidation and excess iron, which can result in abnormal chondrocyte death by elevating levels of ROS and MMP13, while reducing levels of type II collagen. The activation of the NLRP3 inflammasome can trigger pyroptosis in chondrocytes. Autophagy can slow down the development of OA by influencing the function of chondrocytes. A novel type of cell death caused by copper ions, referred to as cuproptosis, may also contribute to OA. An overload of copper can disrupt the TCA cycle, causing protein toxicity and resulting in the death of chondrocytes. It’s important to highlight that nearly all forms of PCD can generate various inflammatory factors, potentially leading to intense inflammatory reactions and worsening OA. Furthermore, we identified several molecules, including NF-κB, Jak, mTOR, and IER1, that are crucial in the regulation of PCD during the progression of OA. NF-κB has a dual function in the apoptosis of cartilage cells, acting to both promote and inhibit the process. mTOR plays a vital role in autophagy, particularly via the PI3K/AKT/mTOR signaling pathway. mTORC1 can suppress autophagy, which helps reduce the symptoms of OA. Inflammatory agents can activate the JAK/STAT signaling pathway, resulting in the death of chondrocytes.
Conclusions
In conclusion, we think it is important to investigate various forms of PCD and the signaling pathways associated with the progression of OA for the purpose of developing targeted therapies for the condition. Substances that block PCD may be used as treatment options for OA. We strongly believe that, despite numerous challenges, thorough experiments and clinical studies can position PCD as a significant target for creating therapeutic drugs aimed at the causes of OA, while also reducing the likelihood of side effects and other complications as much as possible.
Data Sharing Statement
Data are contained within the article.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This publication is funded in part by the National Natural Science Foundation of China (Project Approval number: 82060394), the internal funding from the First Hospital of Lanzhou University (Project number: ldyyyn2022-73), and Lanzhou Talent Innovation and Entrepreneurship Project (Project number: 2020-RC-45).
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Safiri S, Kolahi -A-A, Smith E. et al. Global, regional and national burden of osteoarthritis 1990-2017: a systematic analysis of the Global Burden of Disease Study 2017. Ann Rheum Dis. 2020;79(6):819–828. doi:10.1136/annrheumdis-2019-216515
2. Lorenz H, Richter W. Osteoarthritis: cellular and molecular changes in degenerating cartilage. Prog Histochem Cytochem. 2006;40(3):135–163. doi:10.1016/j.proghi.2006.02.003
3. Sanchez-Lopez E, Coras R, Torres A, et al. Synovial inflammation in osteoarthritis progression. Nat Rev Rheumatol. 2022;18(5):258–275. doi:10.1038/s41584-022-00749-9
4. Battistelli M, Favero M, Burini D, et al. Morphological and ultrastructural analysis of normal, injured and osteoarthritic human knee menisci. Eur J Histochem. 2019;63(1). doi:10.4081/ejh.2019.2998
5. Fontanella CG, Belluzzi E, Pozzuoli A, et al. Exploring Anatomo-Morphometric Characteristics of Infrapatellar, Suprapatellar Fat Pad, and Knee Ligaments in Osteoarthritis Compared to Post-Traumatic Lesions. Biomedicines. 2022;10(6). doi:10.3390/biomedicines10061369
6. He Y, Li Z, Alexander PG, et al. Pathogenesis of Osteoarthritis: risk Factors, Regulatory Pathways in Chondrocytes, and Experimental Models. Biology. 2020;9(8). doi:10.3390/biology9080194
7. Man GS, Mologhianu G. Osteoarthritis pathogenesis - a complex process that involves the entire joint. J Med Life. 2014;7(1):37–41.
8. Kloppenburg M, Namane M, Cicuttini F. Osteoarthritis. Lancet. 2025;405(10472):71–85. doi:10.1016/S0140-6736(24)02322-5
9. Robinson WH, Lepus CM, Wang Q, et al. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2016;12(10):580–592. doi:10.1038/nrrheum.2016.136
10. Zheng L, Han Z, Luo D, et al. IL-6, IL-1β and TNF-α regulation of the chondrocyte phenotype: a possible mechanism of haemophilic cartilage destruction. Hematology. 2023;28(1):2179867. doi:10.1080/16078454.2023.2179867
11. Mehana E-SE, Khafaga AF, El-Blehi SS. The role of matrix metalloproteinases in osteoarthritis pathogenesis: an updated review. Life Sci. 2019;234:116786. doi:10.1016/j.lfs.2019.116786
12. Najm A, Alunno A, Gwinnutt JM, et al. Efficacy of intra-articular corticosteroid injections in knee osteoarthritis: a systematic review and meta-analysis of randomized controlled trials. Joint Bone Spine. 2021;88(4):105198. doi:10.1016/j.jbspin.2021.105198
13. Oray M, Abu Samra K, Ebrahimiadib N, et al. Long-term side effects of glucocorticoids. Expert Opin Drug Saf. 2016;15(4):457–465. doi:10.1517/14740338.2016.1140743
14. Liu S, Pan Y, Li T, et al. The Role of Regulated Programmed Cell Death in Osteoarthritis: from Pathogenesis to Therapy. Int J Mol Sci. 2023;24(6):5364. doi:10.3390/ijms24065364
15. An S, Hu H, Li Y, et al. Pyroptosis Plays a Role in Osteoarthritis. Aging Dis. 2020;11(5):1146–1157. doi:10.14336/AD.2019.1127
16. Franceschini A, Capece M, Chiozzi P, et al. The P2X7 receptor directly interacts with the NLRP3 inflammasome scaffold protein. FASEB J. 2015;29(6):2450–2461. doi:10.1096/fj.14-268714
17. Yan HF, Zou T, Tuo QZ, et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther. 2021;6(1):49. doi:10.1038/s41392-020-00428-9
18. Tong L, Yu H, Huang X, et al. Current understanding of osteoarthritis pathogenesis and relevant new approaches. Bone Res. 2022;10(1):60. doi:10.1038/s41413-022-00226-9
19. Wang J, Zhang Y, Cao J, et al. The role of autophagy in bone metabolism and clinical significance. Autophagy. 2023;19(9):2409–2427. doi:10.1080/15548627.2023.2186112
20. Guo W, Wei B, Sun J, et al. Suppressive oligodeoxynucleotide-induced dendritic cells rein the aggravation of osteoarthritis in mice. Immunopharmacol Immunotoxicol. 2018;40(5):430–436. doi:10.1080/08923973.2018.1523927
21. Farinelli L, Aquili A, Mattioli-Belmonte M, et al. Synovial mast cells from knee and Hip osteoarthritis: histological study and clinical correlations. J Exp Orthop. 2022;9(1):13. doi:10.1186/s40634-022-00446-2
22. Li Y, Gao H, Brunner TM, et al. Menstrual blood-derived mesenchymal stromal cells efficiently ameliorate experimental autoimmune encephalomyelitis by inhibiting T cell activation in mice. Stem Cell Res Ther. 2022;13(1):155. doi:10.1186/s13287-022-02838-8
23. Wang Z, Wang B, Zhang J, et al. Chemokine (C-C Motif) Ligand 2/Chemokine Receptor 2 (CCR2) Axis Blockade to Delay Chondrocyte Hypertrophy as a Therapeutic Strategy for Osteoarthritis. Med Sci Monit. 2021;27:e930053. doi:10.12659/MSM.930053
24. Rathmell JC, Thompson CB. Pathways of apoptosis in lymphocyte development, homeostasis, and disease. Cell. 2002;109:S97–107. doi:10.1016/s0092-8674(02)00704-3
25. Yuan H, Yi N, Li D, et al. PPARgamma regulates osteoarthritis chondrocytes apoptosis through caspase-3 dependent mitochondrial pathway. Sci Rep. 2024;14(1):11237. doi:10.1038/s41598-024-62116-w
26. Berenbaum F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!). Osteoarthritis Cartilage. 2013;21(1):16–21. doi:10.1016/j.joca.2012.11.012
27. Kapoor M, Martel-Pelletier J, Lajeunesse D, et al. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol. 2011;7(1):33–42. doi:10.1038/nrrheum.2010.196
28. Rahmati M, Mobasheri A, Mozafari M. Inflammatory mediators in osteoarthritis: a critical review of the state-of-the-art, current prospects, and future challenges. Bone. 2016;85:81–90. doi:10.1016/j.bone.2016.01.019
29. Hou CH, Tang CH, Chen PC, et al. Thrombospondin 2 Promotes IL-6 Production in Osteoarthritis Synovial Fibroblasts via the PI3K/AKT/NF-kappaB Pathway. J Inflamm Res. 2021;14:5955–5967. doi:10.2147/JIR.S314747
30. Scanzello CR. Chemokines and inflammation in osteoarthritis: insights from patients and animal models. J Orthop Res. 2017;35(4):735–739. doi:10.1002/jor.23471
31. Lee JK, Kim SH, Lewis EC, et al. Differences in signaling pathways by IL-1beta and IL-18. Proc Natl Acad Sci U S A. 2004;101(23):8815–8820. doi:10.1073/pnas.0402800101
32. Tabeian H, Betti BF, Dos Santos Cirqueira C, et al. IL-1beta Damages Fibrocartilage and Upregulates MMP-13 Expression in Fibrochondrocytes in the Condyle of the Temporomandibular Joint. Int J Mol Sci. 2019;20(9). doi:10.3390/ijms20092260
33. Lechner J, Chen M, Hogg RE, et al. Peripheral blood mononuclear cells from neovascular age-related macular degeneration patients produce higher levels of chemokines CCL2 (MCP-1) and CXCL8 (IL-8). J Neuroinflammation. 2017;14(1):42. doi:10.1186/s12974-017-0820-y
34. Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001;11(9):372–377. doi:10.1016/s0962-8924(01)02064-5
35. Bodmer JL, Schneider P, Tschopp J. The molecular architecture of the TNF superfamily. Trends Biochem Sci. 2002;27(1):19–26. doi:10.1016/s0968-0004(01)01995-8
36. Yu H, Li M, Wen X, et al. Elevation of alpha-1,3 fucosylation promotes the binding ability of TNFR1 to TNF-alpha and contributes to osteoarthritic cartilage destruction and apoptosis. Arthritis Res Ther. 2022;24(1):93. doi:10.1186/s13075-022-02776-z
37. Liacini A, Sylvester J, Li WQ, et al. Induction of matrix metalloproteinase-13 gene expression by TNF-alpha is mediated by MAP kinases, AP-1, and NF-kappaB transcription factors in articular chondrocytes. Exp Cell Res. 2003;288(1):208–217. doi:10.1016/s0014-4827(03)00180-0
38. Hwang HS, Kim HA. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. Int J Mol Sci. 2015;16(11):26035–26054. doi:10.3390/ijms161125943
39. Scheller J, Chalaris A, Schmidt-Arras D, et al. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. 2011;1813(5):878–888. doi:10.1016/j.bbamcr.2011.01.034
40. Wu B, Guo X, Yan X, et al. TSG-6 inhibits IL-1beta-induced inflammatory responses and extracellular matrix degradation in nucleus pulposus cells by activating the PI3K/Akt signaling pathway. J Orthop Surg Res. 2022;17(1):572. doi:10.1186/s13018-022-03468-9
41. Deshmane SL, Kremlev S, Amini S, et al. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. 2009;29(6):313–326. doi:10.1089/jir.2008.0027
42. Borzi RM, Mazzetti I, Cattini L, et al. Human chondrocytes express functional chemokine receptors and release matrix-degrading enzymes in response to C-X-C and C-C chemokines. Arthritis Rheum. 2000;43(8):1734–1741. doi:10.1002/1529-0131(200008)43:8<1734::AID-ANR9>3.0.CO;2-B
43. Zhao XY, Yang ZB, Zhang ZJ, et al. CCL3 serves as a potential plasma biomarker in knee degeneration (osteoarthritis). Osteoarthritis Cartilage. 2015;23(8):1405–1411. doi:10.1016/j.joca.2015.04.002
44. Lieberthal J, Sambamurthy N, Scanzello CR. Inflammation in joint injury and post-traumatic osteoarthritis. Osteoarthritis Cartilage. 2015;23(11):1825–1834. doi:10.1016/j.joca.2015.08.015
45. Russo RC, Garcia CC, Teixeira MM, et al. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert Rev Clin Immunol. 2014;10(5):593–619. doi:10.1586/1744666X.2014.894886
46. Merz D, Liu R, Johnson K, et al. IL-8/CXCL8 and growth-related oncogene alpha/CXCL1 induce chondrocyte hypertrophic differentiation. J Immunol. 2003;171(8):4406–4415. doi:10.4049/jimmunol.171.8.4406
47. Galliera E, Locati M, Mantovani A, et al. Chemokines and bone remodeling. Int J Immunopathol Pharmacol. 2008;21(3):485–491. doi:10.1177/039463200802100301
48. Sun F, Zhang Y, Li Q. Therapeutic mechanisms of ibuprofen, prednisone and betamethasone in osteoarthritis. Mol Med Rep. 2017;15(2):981–987. doi:10.3892/mmr.2016.6068
49. Rasheed Z, Akhtar N, Haqqi TM. Advanced glycation end products induce the expression of interleukin-6 and interleukin-8 by receptor for advanced glycation end product-mediated activation of mitogen-activated protein kinases and nuclear factor-kappaB in human osteoarthritis chondrocytes. Rheumatology (Oxford). 2011;50(5):838–851. doi:10.1093/rheumatology/keq380
50. Marquez-Curtis LA, Janowska-Wieczorek A. Enhancing the migration ability of mesenchymal stromal cells by targeting the SDF-1/CXCR4 axis. Biomed Res Int. 2013;2013:561098. doi:10.1155/2013/561098
51. Belluzzi E, Olivotto E, Toso G, et al. Conditioned media from human osteoarthritic synovium induces inflammation in a synoviocyte cell line. Connect Tissue Res. 2019;60(2):136–145. doi:10.1080/03008207.2018.1470167
52. Belluzzi E, Stocco E, Pozzuoli A, et al. Contribution of Infrapatellar Fat Pad and Synovial Membrane to Knee Osteoarthritis Pain. Biomed Res Int. 2019;2019:6390182. doi:10.1155/2019/6390182
53. Guilak F, Nims RJ, Dicks A, et al. Osteoarthritis as a disease of the cartilage pericellular matrix. Matrix Biol. 2018;71-72:40–50. doi:10.1016/j.matbio.2018.05.008
54. Peng Z, Sun H, Bunpetch V, et al. The regulation of cartilage extracellular matrix homeostasis in joint cartilage degeneration and regeneration. Biomaterials. 2021;268:120555. doi:10.1016/j.biomaterials.2020.120555
55. Yang Y, Jiang G, Zhang P, et al. Programmed cell death and its role in inflammation. Mil Med Res. 2015;2:12. doi:10.1186/s40779-015-0039-0
56. Zou Z, Hu W, Kang F, et al. Interplay between lipid dysregulation and ferroptosis in chondrocytes and the targeted therapy effect of metformin on osteoarthritis. J Adv Res. 2025;69:515–529. doi:10.1016/j.jare.2024.04.012
57. Berndt C, Alborzinia H, Amen VS, et al. Ferroptosis in health and disease. Redox Biol. 2024;75:103211. doi:10.1016/j.redox.2024.103211
58. Deng L, He S, Guo N, et al. Molecular mechanisms of ferroptosis and relevance to inflammation. Inflamm Res. 2023;72(2):281–299. doi:10.1007/s00011-022-01672-1
59. Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369–379. doi:10.1038/cdd.2015.158
60. Tang D, Kroemer G. Ferroptosis. Curr Biol. 2020;30(21):R1292–R1297. doi:10.1016/j.cub.2020.09.068
61. Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–98. doi:10.1038/nchembio.2239
62. Li J, Cao F, Yin HL, et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11(2):88. doi:10.1038/s41419-020-2298-2
63. Lin X, Ping J, Wen Y, et al. The Mechanism of Ferroptosis and Applications in Tumor Treatment. Front Pharmacol. 2020;11:1061. doi:10.3389/fphar.2020.01061
64. Zou Y, Li H, Graham ET, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16(3):302–309. doi:10.1038/s41589-020-0472-6
65. Bebber CM, Muller F, Prieto Clemente L, et al. Ferroptosis in Cancer Cell Biology. Cancers. 2020;12(1). doi:10.3390/cancers12010164
66. Aschner M, Skalny AV, Martins AC, et al. Ferroptosis as a mechanism of non-ferrous metal toxicity. Arch Toxicol. 2022;96(9):2391–2417. doi:10.1007/s00204-022-03317-y
67. Patane GT, Putaggio S, Tellone E, et al. Ferroptosis: emerging Role in Diseases and Potential Implication of Bioactive Compounds. Int J Mol Sci. 2023;24(24). doi:10.3390/ijms242417279
68. Fujii J, Osaki T, Soma Y, et al. Critical Roles of the Cysteine-Glutathione Axis in the Production of gamma-Glutamyl Peptides in the Nervous System. Int J Mol Sci. 2023;24(9). doi:10.3390/ijms24098044
69. Macchi V, Stocco E, Stecco C, et al. The infrapatellar fat pad and the synovial membrane: an anatomo-functional unit. J Anat. 2018;233(2):146–154. doi:10.1111/joa.12820
70. Zhou S, Maleitzke T, Geissler S, et al. Source and hub of inflammation: the infrapatellar fat pad and its interactions with articular tissues during knee osteoarthritis. J Orthop Res. 2022;40(7):1492–1504. doi:10.1002/jor.25347
71. Jing X, Lin J, Du T, et al. Iron Overload Is Associated With Accelerated Progression of Osteoarthritis: the Role of DMT1 Mediated Iron Homeostasis. Front Cell Dev Biol. 2020;8:594509. doi:10.3389/fcell.2020.594509
72. Kennish L, Attur M, Oh C, et al. Age-dependent ferritin elevations and HFE C282Y mutation as risk factors for symptomatic knee osteoarthritis in males: a longitudinal cohort study. BMC Musculoskelet Disord. 2014;15:8. doi:10.1186/1471-2474-15-8
73. Van Vulpen LF, Roosendaal G, Van Asbeck BS, et al. The detrimental effects of iron on the joint: a comparison between haemochromatosis and haemophilia. J Clin Pathol. 2015;68(8):592–600. doi:10.1136/jclinpath-2015-202967
74. Morquette B, Shi Q, Lavigne P, et al. Production of lipid peroxidation products in osteoarthritic tissues: new evidence linking 4-hydroxynonenal to cartilage degradation. Arthritis Rheum. 2006;54(1):271–281. doi:10.1002/art.21559
75. Shi Q, Vaillancourt F, Cote V, et al. Alterations of metabolic activity in human osteoarthritic osteoblasts by lipid peroxidation end product 4-hydroxynonenal. Arthritis Res Ther. 2006;8(6):R159. doi:10.1186/ar2066
76. Abusarah J, Bentz M, Benabdoune H, et al. An overview of the role of lipid peroxidation-derived 4-hydroxynonenal in osteoarthritis. Inflamm Res. 2017;66(8):637–651. doi:10.1007/s00011-017-1044-4
77. Vaillancourt F, Fahmi H, Shi Q, et al. 4-Hydroxynonenal induces apoptosis in human osteoarthritic chondrocytes: the protective role of glutathione-S-transferase. Arthritis Res Ther. 2008;10(5):R107. doi:10.1186/ar2503
78. Mao X, Fu P, Wang L, et al. Mitochondria: potential Targets for Osteoarthritis. Front Med Lausanne. 2020;7:581402. doi:10.3389/fmed.2020.581402
79. Yao X, Sun K, Yu S, et al. Chondrocyte ferroptosis contribute to the progression of osteoarthritis. J Orthop Translat. 2021;27:33–43. doi:10.1016/j.jot.2020.09.006
80. Camacho A, Simao M, Ea HK, et al. Iron overload in a murine model of hereditary hemochromatosis is associated with accelerated progression of osteoarthritis under mechanical stress. Osteoarthritis Cartilage. 2016;24(3):494–502. doi:10.1016/j.joca.2015.09.007
81. Karim A, Bajbouj K, Shafarin J, et al. Iron Overload Induces Oxidative Stress, Cell Cycle Arrest and Apoptosis in Chondrocytes. Front Cell Dev Biol. 2022;10:821014. doi:10.3389/fcell.2022.821014
82. Rochette L, Dogon G, Rigal E, et al. Lipid Peroxidation and Iron Metabolism: two Corner Stones in the Homeostasis Control of Ferroptosis. Int J Mol Sci. 2022;24(1). doi:10.3390/ijms24010449
83. An F, Zhang J, Gao P, et al. New insight of the pathogenesis in osteoarthritis: the intricate interplay of ferroptosis and autophagy mediated by mitophagy/chaperone-mediated autophagy. Front Cell Dev Biol. 2023;11:1297024. doi:10.3389/fcell.2023.1297024
84. Burton LH, Radakovich LB, Marolf AJ, et al. Systemic iron overload exacerbates osteoarthritis in the strain 13 Guinea pig. Osteoarthritis Cartilage. 2020;28(9):1265–1275. doi:10.1016/j.joca.2020.06.005
85. Jing X, Du T, Li T, et al. The detrimental effect of iron on OA chondrocytes: importance of pro-inflammatory cytokines induced iron influx and oxidative stress. J Cell Mol Med. 2021;25(12):5671–5680. doi:10.1111/jcmm.16581
86. Foret MK, Do Carmo S, Lincoln R, et al. Effect of antioxidant supplements on lipid peroxidation levels in primary cortical neuron cultures. Free Radic Biol Med. 2019;130:471–477. doi:10.1016/j.freeradbiomed.2018.11.019
87. Ai Y, Yan B, Wang X. The oxidoreductases POR and CYB5R1 catalyze lipid peroxidation to execute ferroptosis. Mol Cell Oncol. 2021;8(2):1881393. doi:10.1080/23723556.2021.1881393
88. Gladkova Capital Ie C. Role of Imbalance of Lipid Peroxidation and Articular Cartilage Remodeling in the Pathogenesis of Early Primary and Post-Traumatic Gonarthrosis in Rats. Bull Exp Biol Med. 2022;172(4):415–418. doi:10.1007/s10517-022-05405-6
89. Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radic Biol Med. 2020;152:175–185. doi:10.1016/j.freeradbiomed.2020.02.027
90. Miao Y, Chen Y, Xue F, et al. Contribution of ferroptosis and GPX4’s dual functions to osteoarthritis progression. EBioMedicine. 2022;76:103847. doi:10.1016/j.ebiom.2022.103847
91. Chen H, Zhong Y, Sang W, et al. Protopine protects chondrocytes from undergoing ferroptosis by activating Nrf2 pathway. Biochem Biophys Res Commun. 2024;710:149599. doi:10.1016/j.bbrc.2024.149599
92. Pan YN, Jia C, Yu JP, et al. Fibroblast growth factor 9 reduces TBHP-induced oxidative stress in chondrocytes and diminishes mouse osteoarthritis by activating ERK/Nrf2 signaling pathway. Int Immunopharmacol. 2023;114:109606. doi:10.1016/j.intimp.2022.109606
93. Wang S, Li W, Zhang P, et al. Mechanical overloading induces GPX4-regulated chondrocyte ferroptosis in osteoarthritis via Piezo1 channel facilitated calcium influx. J Adv Res. 2022;41:63–75. doi:10.1016/j.jare.2022.01.004
94. Guo Z, Lin J, Sun K, et al. Deferoxamine Alleviates Osteoarthritis by Inhibiting Chondrocyte Ferroptosis and Activating the Nrf2 Pathway. Front Pharmacol. 2022;13:791376. doi:10.3389/fphar.2022.791376
95. Xiang Z, Zhang P, Jia C, et al. Piezo1 channel exaggerates ferroptosis of nucleus pulposus cells by mediating mechanical stress-induced iron influx. Bone Res. 2024;12(1):20. doi:10.1038/s41413-024-00317-9
96. Kong R, Ji L, Pang Y, et al. Exosomes from osteoarthritic fibroblast-like synoviocytes promote cartilage ferroptosis and damage via delivering microRNA-19b-3p to target SLC7A11 in osteoarthritis. Front Immunol. 2023;14:1181156. doi:10.3389/fimmu.2023.1181156
97. Sheng W, Liao S, Wang D, et al. The role of ferroptosis in osteoarthritis: progress and prospects. Biochem Biophys Res Commun. 2024;733:150683. doi:10.1016/j.bbrc.2024.150683
98. He Q, Yang J, Pan Z, et al. Biochanin A protects against iron overload associated knee osteoarthritis via regulating iron levels and NRF2/System xc-/GPX4 axis. Biomed Pharmacother. 2023;157:113915. doi:10.1016/j.biopha.2022.113915
99. Yu P, Zhang X, Liu N, et al. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. 2021;6(1):128. doi:10.1038/s41392-021-00507-5
100. Vasudevan SO, Behl B, Rathinam VA. Pyroptosis-induced inflammation and tissue damage. Semin Immunol. 2023;69:101781. doi:10.1016/j.smim.2023.101781
101. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci. 2017;42(4):245–254. doi:10.1016/j.tibs.2016.10.004
102. Li D, Wu M. Pattern recognition receptors in health and diseases. Signal Transduct Target Ther. 2021;6(1):291. doi:10.1038/s41392-021-00687-0
103. Van Opdenbosch N, Lamkanfi M. Caspases in Cell Death, Inflammation, and Disease. Immunity. 2019;50(6):1352–1364. doi:10.1016/j.immuni.2019.05.020
104. He W-T, Wan H, Hu L, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25(12):1285–1298. doi:10.1038/cr.2015.139
105. Liu X, Zhang Z, Ruan J, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535(7610):153–158. doi:10.1038/nature18629
106. Bolivar BE, Vogel TP, Bouchier‐Hayes L. Inflammatory caspase regulation: maintaining balance between inflammation and cell death in health and disease. FEBS J. 2019;286(14):2628–2644. doi:10.1111/febs.14926
107. Xu Z, Kombe Kombe AJ, Deng S, et al. NLRP inflammasomes in health and disease. Mol Biomed. 2024;5(1):14. doi:10.1186/s43556-024-00179-x
108. Han X, Lin D, Huang W, et al. Mechanism of NLRP3 inflammasome intervention for synovitis in knee osteoarthritis: a review of TCM intervention. Front Genet. 2023;14:1159167. doi:10.3389/fgene.2023.1159167
109. Sandstrom A, Mitchell PS, Goers L, et al. Functional degradation: a mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science. 2019;364(6435). doi:10.1126/science.aau1330
110. Ghimire L, Paudel S, Jin L, et al. The NLRP6 inflammasome in health and disease. Mucosal Immunol. 2020;13(3):388–398. doi:10.1038/s41385-020-0256-z
111. Mullins B, Chen J. NLRP9 in innate immunity and inflammation. Immunology. 2021;162(3):262–267. doi:10.1111/imm.13290
112. He Y, Hara H, Nunez G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci. 2016;41(12):1012–1021. doi:10.1016/j.tibs.2016.09.002
113. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477–489. doi:10.1038/s41577-019-0165-0
114. Sundaram B, Kanneganti TD. Advances in Understanding Activation and Function of the NLRC4 Inflammasome. Int J Mol Sci. 2021;22(3). doi:10.3390/ijms22031048
115. Lugrin J, Martinon F. The AIM2 inflammasome: sensor of pathogens and cellular perturbations. Immunol Rev. 2018;281(1):99–114. doi:10.1111/imr.12618
116. Yin Y, Li X, Sha X, et al. Early hyperlipidemia promotes endothelial activation via a caspase-1-sirtuin 1 pathway. Arterioscler Thromb Vasc Biol. 2015;35(4):804–816. doi:10.1161/ATVBAHA.115.305282
117. Tall AR, Westerterp M. Inflammasomes, neutrophil extracellular traps, and cholesterol. J Lipid Res. 2019;60(4):721–727. doi:10.1194/jlr.S091280
118. Pazar B, Ea HK, Narayan S, et al. Basic calcium phosphate crystals induce monocyte/macrophage IL-1beta secretion through the NLRP3 inflammasome in vitro. J Immunol. 2011;186(4):2495–2502. doi:10.4049/jimmunol.1001284
119. He Z, Nie P, Lu J, et al. Less mechanical loading attenuates osteoarthritis by reducing cartilage degeneration, subchondral bone remodelling, secondary inflammation, and activation of NLRP3 inflammasome. Bone Joint Res. 2020;9(10):731–741. doi:10.1302/2046-3758.910.BJR-2019-0368.R2
120. Bai H, Yuan R, Zhang Z, et al. Intra-articular Injection of Baicalein Inhibits Cartilage Catabolism and NLRP3 Inflammasome Signaling in a Posttraumatic OA Model. Oxid Med Cell Longev. 2021;2021:6116890. doi:10.1155/2021/6116890
121. Wang M, Sampson ER, Jin H, et al. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res Ther. 2013;15(1):R5. doi:10.1186/ar4133
122. Fosang AJ, Beier F. Emerging Frontiers in cartilage and chondrocyte biology. Best Pract Res Clin Rheumatol. 2011;25(6):751–766. doi:10.1016/j.berh.2011.11.010
123. Zhang L, Ma S, Su H, et al. Isoliquiritigenin Inhibits IL-1beta-Induced Production of Matrix Metalloproteinase in Articular Chondrocytes. Mol Ther Methods Clin Dev. 2018;9:153–159. doi:10.1016/j.omtm.2018.02.006
124. Zhang Y, Lin Z, Chen D, et al. CY-09 attenuates the progression of osteoarthritis via inhibiting NLRP3 inflammasome-mediated pyroptosis. Biochem Biophys Res Commun. 2021;553:119–125. doi:10.1016/j.bbrc.2021.03.055
125. Chen Y, Zeng D, Wei G, et al. Pyroptosis in Osteoarthritis: molecular Mechanisms and Therapeutic Implications. J Inflamm Res. 2024;17:791–803. doi:10.2147/JIR.S445573
126. Sun FF, Hu PF, Xiong Y, et al. Tricetin Protects Rat Chondrocytes against IL-1beta-Induced Inflammation and Apoptosis. Oxid Med Cell Longev. 2019;2019:4695381. doi:10.1155/2019/4695381
127. Thomson A, Hilkens CMU. Synovial Macrophages in Osteoarthritis: the Key to Understanding Pathogenesis? Front Immunol. 2021;12:678757. doi:10.3389/fimmu.2021.678757
128. Eitner A, Muller S, Konig C, et al. Inhibition of Inducible Nitric Oxide Synthase Prevents IL-1beta-Induced Mitochondrial Dysfunction in Human Chondrocytes. Int J Mol Sci. 2021;22(5). doi:10.3390/ijms22052477
129. Guerne PA, Carson DA, Lotz M. IL-6 production by human articular chondrocytes. Modulation of its synthesis by cytokines, growth factors, and hormones in vitro. J Immunol. 1990;144(2):499–505.
130. Aigner T, Mckenna L, Zien A, et al. Gene expression profiling of serum- and interleukin-1 beta-stimulated primary human adult articular chondrocytes--a molecular analysis based on chondrocytes isolated from one donor. Cytokine. 2005;31(3):227–240. doi:10.1016/j.cyto.2005.04.009
131. Pulsatelli L, Dolzani P, Piacentini A, et al. Chemokine production by human chondrocytes. J Rheumatol. 1999;26(9):1992–2001.
132. Wojdasiewicz P, Poniatowski LA, Szukiewicz D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm. 2014;2014:561459. doi:10.1155/2014/561459
133. Olee T, Hashimoto S, Quach J, et al. IL-18 is produced by articular chondrocytes and induces proinflammatory and catabolic responses. J Immunol. 1999;162(2):1096–1100.
134. Futani H, Okayama A, Matsui K, et al. Relation between interleukin-18 and PGE2 in synovial fluid of osteoarthritis: a potential therapeutic target of cartilage degradation. J Immunother. 2002;25(1):S61–64. doi:10.1097/00002371-200203001-00009
135. Bao J, Chen Z, Xu L, et al. Rapamycin protects chondrocytes against IL-18-induced apoptosis and ameliorates rat osteoarthritis. Aging. 2020;12(6):5152–5167. doi:10.18632/aging.102937
136. Dai SM, Shan ZZ, Nishioka K, et al. Implication of interleukin 18 in production of matrix metalloproteinases in articular chondrocytes in arthritis: direct effect on chondrocytes may not be pivotal. Ann Rheum Dis. 2005;64(5):735–742. doi:10.1136/ard.2004.026088
137. Mathiessen A, Conaghan PG. Synovitis in osteoarthritis: current understanding with therapeutic implications. Arthritis Res Ther. 2017;19(1):18. doi:10.1186/s13075-017-1229-9
138. Scanzello CR, Goldring SR. The role of synovitis in osteoarthritis pathogenesis. Bone. 2012;51(2):249–257. doi:10.1016/j.bone.2012.02.012
139. Liu-Bryan R, Terkeltaub R. Emerging regulators of the inflammatory process in osteoarthritis. Nat Rev Rheumatol. 2015;11(1):35–44. doi:10.1038/nrrheum.2014.162
140. Ni Z, Kuang L, Chen H, et al. The exosome-like vesicles from osteoarthritic chondrocyte enhanced mature IL-1beta production of macrophages and aggravated synovitis in osteoarthritis. Cell Death Dis. 2019;10(7):522. doi:10.1038/s41419-019-1739-2
141. Zhang L, Xing R, Huang Z, et al. Inhibition of Synovial Macrophage Pyroptosis Alleviates Synovitis and Fibrosis in Knee Osteoarthritis. Mediators Inflamm. 2019;2019:2165918. doi:10.1155/2019/2165918
142. Xiao Y, Ding L, Yin S, et al. Relationship between the pyroptosis of fibroblast‑like synoviocytes and HMGB1 secretion in knee osteoarthritis. Mol Med Rep. 2021;23(2). doi:10.3892/mmr.2020.11736
143. Garcia-Arnandis I, Guillen MI, Gomar F, et al. High mobility group box 1 potentiates the pro-inflammatory effects of interleukin-1beta in osteoarthritic synoviocytes. Arthritis Res Ther. 2010;12(4):R165. doi:10.1186/ar3124
144. Kim HM, Kim YM. HMGB1: LPS Delivery Vehicle for Caspase-11-Mediated Pyroptosis. Immunity. 2018;49(4):582–584. doi:10.1016/j.immuni.2018.09.021
145. Zhang L, Zhang L, Huang Z, et al. Increased HIF-1alpha in Knee Osteoarthritis Aggravate Synovial Fibrosis via Fibroblast-Like Synoviocyte Pyroptosis. Oxid Med Cell Longev. 2019;2019:6326517. doi:10.1155/2019/6326517
146. Shi J, Zhao W, Ying H, et al. Estradiol inhibits NLRP3 inflammasome in fibroblast-like synoviocytes activated by lipopolysaccharide and adenosine triphosphate. Int J Rheum Dis. 2018;21(11):2002–2010. doi:10.1111/1756-185X.13198
147. Goldring SR, Goldring MB. The role of cytokines in cartilage matrix degeneration in osteoarthritis. Clin Orthop Relat Res. 2004;427:S27–36. doi:10.1097/01.blo.0000144854.66565.8f
148. Zhu X, Lee CW, Xu H, et al. Phenotypic alteration of macrophages during osteoarthritis: a systematic review. Arthritis Res Ther. 2021;23(1):110. doi:10.1186/s13075-021-02457-3
149. Zhang W, Zhang L, Yang S, et al. Electroacupuncture ameliorates knee osteoarthritis in rats via inhibiting NLRP3 inflammasome and reducing pyroptosis. Mol Pain. 2023;19:17448069221147792. doi:10.1177/17448069221147792
150. Mukherjee S, Nazemi M, Jonkers I, et al. Use of Computational Modeling to Study Joint Degeneration: a Review. Front Bioeng Biotechnol. 2020;8:93. doi:10.3389/fbioe.2020.00093
151. Hirvasniemi J, Thevenot J, Kokkonen HT, et al. Correlation of Subchondral Bone Density and Structure from Plain Radiographs with Micro Computed Tomography Ex Vivo. Ann Biomed Eng. 2016;44(5):1698–1709. doi:10.1007/s10439-015-1452-y
152. Yan J, Feng G, Yang Y, et al. Autophagy attenuates osteoarthritis in mice by inhibiting chondrocyte pyroptosis and improving subchondral bone remodeling. Biomol Biomed. 2023;23(1):77–88. doi:10.17305/bjbms.2022.7677
153. Yu L, Chen Y, Tooze SA. Autophagy pathway: cellular and molecular mechanisms. Autophagy. 2018;14(2):207–215. doi:10.1080/15548627.2017.1378838
154. Yang Q, Xu J, Gu J, et al. Extracellular Vesicles in Cancer Drug Resistance: roles, Mechanisms, and Implications. Adv Sci (Weinh). 2022;9(34):e2201609. doi:10.1002/advs.202201609
155. Kim KH, Lee MS. Autophagy--a key player in cellular and body metabolism. Nat Rev Endocrinol. 2014;10(6):322–337. doi:10.1038/nrendo.2014.35
156. Li H, Li Z, Pi Y, et al. MicroRNA-375 exacerbates knee osteoarthritis through repressing chondrocyte autophagy by targeting ATG2B. Aging. 2020;12(8):7248–7261. doi:10.18632/aging.103073
157. Li J, Chen H, Cai L, et al. SDF-1alpha Promotes Chondrocyte Autophagy through CXCR4/mTOR Signaling Axis. Int J Mol Sci. 2023;24(2). doi:10.3390/ijms24021710
158. Li X, He S, Ma B. Autophagy and autophagy-related proteins in cancer. Mol Cancer. 2020;19(1):12. doi:10.1186/s12943-020-1138-4
159. Li Y, Li G, Guo X, et al. Non-coding RNA in bladder cancer. Cancer Lett. 2020;485:38–44. doi:10.1016/j.canlet.2020.04.023
160. Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20(7):1981–1991. doi:10.1091/mbc.e08-12-1248
161. Di Bartolomeo S, Corazzari M, Nazio F, et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol. 2010;191(1):155–168. doi:10.1083/jcb.201002100
162. Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15(7):741–750. doi:10.1038/ncb2757
163. Burman C, Ktistakis NT. Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett. 2010;584(7):1302–1312. doi:10.1016/j.febslet.2010.01.011
164. Polson HE, De Lartigue J, Rigden DJ, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 2010;6(4):506–522. doi:10.4161/auto.6.4.11863
165. Coutts AS, La Thangue NB. Regulation of actin nucleation and autophagosome formation. Cell Mol Life Sci. 2016;73(17):3249–3263. doi:10.1007/s00018-016-2224-z
166. Mizushima N, Kuma A, Kobayashi Y, et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003;116(Pt 9):1679–1688. doi:10.1242/jcs.00381
167. Huang X, Yao J, Liu L, et al. Atg8-PE protein-based in vitro biochemical approaches to autophagy studies. Autophagy. 2022;18(9):2020–2035. doi:10.1080/15548627.2022.2025572
168. Kimura S, Noda T, Yoshimori T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct Funct. 2008;33(1):109–122. doi:10.1247/csf.08005
169. Noda T, Fujita N, Yoshimori T. The late stages of autophagy: how does the end begin? Cell Death Differ. 2009;16(7):984–990. doi:10.1038/cdd.2009.54
170. Stanley RE, Ragusa MJ, Hurley JH. The beginning of the end: how scaffolds nucleate autophagosome biogenesis. Trends Cell Biol. 2014;24(1):73–81. doi:10.1016/j.tcb.2013.07.008
171. Almonte-Becerril M, Navarro-Garcia F, Gonzalez-Robles A, et al. Cell death of chondrocytes is a combination between apoptosis and autophagy during the pathogenesis of Osteoarthritis within an experimental model. Apoptosis. 2010;15(5):631–638. doi:10.1007/s10495-010-0458-z
172. Chang J, Wang W, Zhang H, et al. The dual role of autophagy in chondrocyte responses in the pathogenesis of articular cartilage degeneration in osteoarthritis. Int J Mol Med. 2013;32(6):1311–1318. doi:10.3892/ijmm.2013.1520
173. Zhang FJ, Luo W, Lei GH. Role of HIF-1alpha and HIF-2alpha in osteoarthritis. Joint Bone Spine. 2015;82(3):144–147. doi:10.1016/j.jbspin.2014.10.003
174. Zhang XA, Kong H. Mechanism of HIFs in osteoarthritis. Front Immunol. 2023;14:1168799. doi:10.3389/fimmu.2023.1168799
175. Lopez De Figueroa P, Lotz MK, Blanco FJ, et al. Autophagy activation and protection from mitochondrial dysfunction in human chondrocytes. Arthritis Rheumatol. 2015;67(4):966–976. doi:10.1002/art.39025
176. Zhu C, Zhang L, Ding X, et al. Non-coding RNAs as regulators of autophagy in chondrocytes: mechanisms and implications for osteoarthritis. Ageing Res Rev. 2024;99:102404. doi:10.1016/j.arr.2024.102404
177. Lu J, Peng Y, Zou J, et al. Hypoxia Inducible Factor-1alpha Is a Regulator of Autophagy in Osteoarthritic Chondrocytes. Cartilage. 2021;13(2_suppl):1030S–1040S. doi:10.1177/19476035211035434
178. Bohensky J, Leshinsky S, Srinivas V, et al. Chondrocyte autophagy is stimulated by HIF-1 dependent AMPK activation and mTOR suppression. Pediatr Nephrol. 2010;25(4):633–642. doi:10.1007/s00467-009-1310-y
179. Woo YM, Shin Y, Lee EJ, et al. Inhibition of Aerobic Glycolysis Represses Akt/mTOR/HIF-1alpha Axis and Restores Tamoxifen Sensitivity in Antiestrogen-Resistant Breast Cancer Cells. PLoS One. 2015;10(7):e0132285. doi:10.1371/journal.pone.0132285
180. Bohensky J, Terkhorn SP, Freeman TA, et al. Regulation of autophagy in human and murine cartilage: hypoxia-inducible factor 2 suppresses chondrocyte autophagy. Arthritis Rheum. 2009;60(5):1406–1415. doi:10.1002/art.24444
181. Shapiro IM, Layfield R, Lotz M, et al. Boning up on autophagy: the role of autophagy in skeletal biology. Autophagy. 2014;10(1):7–19. doi:10.4161/auto.26679
182. Carames B, Taniguchi N, Otsuki S, et al. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010;62(3):791–801. doi:10.1002/art.27305
183. Sasaki H, Takayama K, Matsushita T, et al. Autophagy modulates osteoarthritis-related gene expression in human chondrocytes. Arthritis Rheum. 2012;64(6):1920–1928. doi:10.1002/art.34323
184. Carames B, Olmer M, Kiosses WB, et al. The relationship of autophagy defects to cartilage damage during joint aging in a mouse model. Arthritis Rheumatol. 2015;67(6):1568–1576. doi:10.1002/art.39073
185. Chen X, Gong W, Shao X, et al. METTL3-mediated m(6)A modification of ATG7 regulates autophagy-GATA4 axis to promote cellular senescence and osteoarthritis progression. Ann Rheum Dis. 2022;81(1):87–99. doi:10.1136/annrheumdis-2021-221091
186. Bouderlique T, Vuppalapati KK, Newton PT, et al. Targeted deletion of Atg5 in chondrocytes promotes age-related osteoarthritis. Ann Rheum Dis. 2016;75(3):627–631. doi:10.1136/annrheumdis-2015-207742
187. Pallauf K, Rimbach G. Autophagy, polyphenols and healthy ageing. Ageing Res Rev. 2013;12(1):237–252. doi:10.1016/j.arr.2012.03.008
188. Carames B, Taniguchi N, Seino D, et al. Mechanical injury suppresses autophagy regulators and pharmacologic activation of autophagy results in chondroprotection. Arthritis Rheum. 2012;64(4):1182–1192. doi:10.1002/art.33444
189. Zhang H, Zheng W, Li D, et al. miR-146a-5p Promotes Chondrocyte Apoptosis and Inhibits Autophagy of Osteoarthritis by Targeting NUMB. Cartilage. 2021;13(2_suppl):1467S–1477S. doi:10.1177/19476035211023550
190. Zhang J, Hao X, Chi R, et al. Moderate mechanical stress suppresses the IL-1beta-induced chondrocyte apoptosis by regulating mitochondrial dynamics. J Cell Physiol. 2021;236(11):7504–7515. doi:10.1002/jcp.30386
191. Zheng W, Li X, Liu D, et al. Mechanical loading mitigates osteoarthritis symptoms by regulating endoplasmic reticulum stress and autophagy. FASEB J. 2019;33(3):4077–4088. doi:10.1096/fj.201801851R
192. Salminen A, Kaarniranta K, Kauppinen A. Inflammaging: disturbed interplay between autophagy and inflammasomes. Aging. 2012;4(3):166–175. doi:10.18632/aging.100444
193. Xue JF, Shi ZM, Zou J, et al. Inhibition of PI3K/AKT/mTOR signaling pathway promotes autophagy of articular chondrocytes and attenuates inflammatory response in rats with osteoarthritis. Biomed Pharmacother. 2017;89:1252–1261. doi:10.1016/j.biopha.2017.01.130
194. Vasheghani F, Zhang Y, Li YH, et al. PPARgamma deficiency results in severe, accelerated osteoarthritis associated with aberrant mTOR signalling in the articular cartilage. Ann Rheum Dis. 2015;74(3):569–578. doi:10.1136/annrheumdis-2014-205743
195. Cejka D, Hayer S, Niederreiter B, et al. Mammalian target of rapamycin signaling is crucial for joint destruction in experimental arthritis and is activated in osteoclasts from patients with rheumatoid arthritis. Arthritis Rheum. 2010;62(8):2294–2302. doi:10.1002/art.27504
196. Yao M, Zhang C, Ni L, et al. Cepharanthine Ameliorates Chondrocytic Inflammation and Osteoarthritis via Regulating the MAPK/NF-kappaB-Autophagy Pathway. Front Pharmacol. 2022;13:854239. doi:10.3389/fphar.2022.854239
197. He Y, Wang W, Xu X, et al. Mettl3 inhibits the apoptosis and autophagy of chondrocytes in inflammation through mediating Bcl2 stability via Ythdf1-mediated m(6)A modification. Bone. 2022;154:116182. doi:10.1016/j.bone.2021.116182
198. Luo J, Mills K, Le Cessie S, et al. Ageing, age-related diseases and oxidative stress: what to do next? Ageing Res Rev. 2020;57:100982. doi:10.1016/j.arr.2019.100982
199. Morel F, Rousset F, Vu Chuong Nguyen M, et al. NADPH oxidase Nox4, a putative therapeutic target in osteoarthritis. Bull Acad Natl Med. 2015;199(4–5):673–686.
200. Alcaraz MJ, Megias J, Garcia-Arnandis I, et al. New molecular targets for the treatment of osteoarthritis. Biochem Pharmacol. 2010;80(1):13–21. doi:10.1016/j.bcp.2010.02.017
201. Mazzetti I, Grigolo B, Pulsatelli L, et al. Differential roles of nitric oxide and oxygen radicals in chondrocytes affected by osteoarthritis and rheumatoid arthritis. Clin Sci (Lond). 2001;101(6):593–599.
202. D’adamo S, Cetrullo S, Guidotti S, et al. Spermidine rescues the deregulated autophagic response to oxidative stress of osteoarthritic chondrocytes. Free Radic Biol Med. 2020;153:159–172. doi:10.1016/j.freeradbiomed.2020.03.029
203. Tang Q, Zheng G, Feng Z, et al. Trehalose ameliorates oxidative stress-mediated mitochondrial dysfunction and ER stress via selective autophagy stimulation and autophagic flux restoration in osteoarthritis development. Cell Death Dis. 2017;8(10):e3081. doi:10.1038/cddis.2017.453
204. Vinatier C, Dominguez E, Guicheux J, et al. Role of the Inflammation-Autophagy-Senescence Integrative Network in Osteoarthritis. Front Physiol. 2018;9:706. doi:10.3389/fphys.2018.00706
205. Kong P, Ahmad RE, Zulkifli A, et al. The role of autophagy in mitigating osteoarthritis progression via regulation of chondrocyte apoptosis: a review. Joint Bone Spine. 2024;91(3):105642. doi:10.1016/j.jbspin.2023.105642
206. Deng X, Xu H, Pan C, et al. Moderate mechanical strain and exercise reduce inflammation and excessive autophagy in osteoarthritis by downregulating mitofusin 2. Life Sci. 2023;332:122020. doi:10.1016/j.lfs.2023.122020
207. Yang Y, Ding J, Chen Y, et al. Blockade of ASIC1a inhibits acid-induced rat articular chondrocyte senescence through regulation of autophagy. Hum Cell. 2022;35(2):665–677. doi:10.1007/s13577-022-00676-7
208. Alvarez-Garcia O, Matsuzaki T, Olmer M, et al. Regulated in Development and DNA Damage Response 1 Deficiency Impairs Autophagy and Mitochondrial Biogenesis in Articular Cartilage and Increases the Severity of Experimental Osteoarthritis. Arthritis Rheumatol. 2017;69(7):1418–1428. doi:10.1002/art.40104
209. Goutas A, Syrrou C, Papathanasiou I, et al. The autophagic response to oxidative stress in osteoarthritic chondrocytes is deregulated. Free Radic Biol Med. 2018;126:122–132. doi:10.1016/j.freeradbiomed.2018.08.003
210. Wikstrom JD, Twig G, Shirihai OS. What can mitochondrial heterogeneity tell us about mitochondrial dynamics and autophagy? Int J Biochem Cell Biol. 2009;41(10):1914–1927. doi:10.1016/j.biocel.2009.06.006
211. Lee DY, Bahar ME, Kim CW, et al. Autophagy in Osteoarthritis: a Double-Edged Sword in Cartilage Aging and Mechanical Stress Response: a Systematic Review. J Clin Med. 2024;13(10). doi:10.3390/jcm13103005
212. Lotz MK, Carames B. Autophagy and cartilage homeostasis mechanisms in joint health, aging and OA. Nat Rev Rheumatol. 2011;7(10):579–587. doi:10.1038/nrrheum.2011.109
213. Tang D, Chen X, Kroemer G. Cuproptosis: a copper-triggered modality of mitochondrial cell death. Cell Res. 2022;32(5):417–418. doi:10.1038/s41422-022-00653-7
214. Wang Y, Zhang L, Zhou F. Cuproptosis: a new form of programmed cell death. Cell Mol Immunol. 2022;19(8):867–868. doi:10.1038/s41423-022-00866-1
215. Rondanelli M, Faliva MA, Infantino V, et al. Copper as Dietary Supplement for Bone Metabolism: a Review. Nutrients. 2021;13(7):2246. doi:10.3390/nu13072246
216. Ciosek Z, Kot K, Rotter I. Iron, Zinc, Copper, Cadmium, Mercury, and Bone Tissue. Int J Environ Res Public Health. 2023;20(3):2197. doi:10.3390/ijerph20032197
217. Li S, Wang M, Chen X, et al. Inhibition of osteogenic differentiation of mesenchymal stem cells by copper supplementation. Cell Prolif. 2014;47(1):81–90. doi:10.1111/cpr.12083
218. Medeiros DM. Copper, iron, and selenium dietary deficiencies negatively impact skeletal integrity: a review. Exp Biol Med. 2016;241(12):1316–1322. doi:10.1177/1535370216648805
219. Yu Q, Xiao Y, Guan M, et al. Copper metabolism in osteoarthritis and its relation to oxidative stress and ferroptosis in chondrocytes. Front Mol Biosci. 2024;11:1472492. doi:10.3389/fmolb.2024.1472492
220. Xiang Z, Mei H, Wang H, et al. Cuproptosis and its potential role in musculoskeletal disease. Front Cell Dev Biol. 2025;13:1570131. doi:10.3389/fcell.2025.1570131
221. Lutsenko S, Roy S, Tsvetkov P. Mammalian copper homeostasis: physiological roles and molecular mechanisms. Physiol Rev. 2025;105(1):441–491. doi:10.1152/physrev.00011.2024
222. Roczniak W, Brodziak-Dopierala B, Cipora E, et al. Factors that Affect the Content of Cadmium, Nickel, Copper and Zinc in Tissues of the Knee Joint. Biol Trace Elem Res. 2017;178(2):201–209. doi:10.1007/s12011-016-0927-5
223. Lin R, Deng C, Li X, et al. Copper-incorporated bioactive glass-ceramics inducing anti-inflammatory phenotype and regeneration of cartilage/bone interface. Theranostics. 2019;9(21):6300–6313. doi:10.7150/thno.36120
224. Lu Y, Chen J, Li L, et al. Hierarchical functional nanoparticles boost osteoarthritis therapy by utilizing joint-resident mesenchymal stem cells. J Nanobiotechnology. 2022;20(1):89. doi:10.1186/s12951-022-01297-w
225. Xu C, Chen J, Li L, et al. Promotion of chondrogenic differentiation of mesenchymal stem cells by copper: implications for new cartilage repair biomaterials. Mater Sci Eng C Mater Biol Appl. 2018;93:106–114. doi:10.1016/j.msec.2018.07.074
226. Alshenibr W, Tashkandi MM, Alsaqer SF, et al. Anabolic role of lysyl oxidase like-2 in cartilage of knee and temporomandibular joints with osteoarthritis. Arthritis Res Ther. 2017;19(1):179. doi:10.1186/s13075-017-1388-8
227. Rucker RB, Kosonen T, Clegg MS, et al. Copper, lysyl oxidase, and extracellular matrix protein cross-linking. Am J Clin Nutr. 1998;67(5 Suppl):996S–1002S. doi:10.1093/ajcn/67.5.996S
228. Ciaffaglione V, Rizzarelli E. Carnosine, Zinc and Copper: a Menage a Trois in Bone and Cartilage Protection. Int J Mol Sci. 2023;24(22). doi:10.3390/ijms242216209
229. Burkhead JL, Gogolin Reynolds KA, Abdel-Ghany SE, et al. Copper homeostasis. New Phytol. 2009;182(4):799–816. doi:10.1111/j.1469-8137.2009.02846.x
230. Liu Y, Zhu J, Xu L, et al. Copper regulation of immune response and potential implications for treating orthopedic disorders. Front Mol Biosci. 2022;9:1065265. doi:10.3389/fmolb.2022.1065265
231. Zofkova I, Nemcikova P, Matucha P. Trace elements and bone health. Clin Chem Lab Med. 2013;51(8):1555–1561. doi:10.1515/cclm-2012-0868
232. Bernhardt A, Bacova J, Gbureck U, et al. Influence of Cu(2+) on Osteoclast Formation and Activity In Vitro. Int J Mol Sci. 2021;22(5). doi:10.3390/ijms22052451
233. Cai J, Liu LF, Qin Z, et al. Natural Morin-Based Metal Organic Framework Nanoenzymes Modulate Articular Cavity Microenvironment to Alleviate Osteoarthritis. Research. 2023;6:0068. doi:10.34133/research.0068
234. Nojiri H, Saita Y, Morikawa D, et al. Cytoplasmic superoxide causes bone fragility owing to low-turnover osteoporosis and impaired collagen cross-linking. J Bone Miner Res. 2011;26(11):2682–2694. doi:10.1002/jbmr.489
235. Morikawa D, Nojiri H, Saita Y, et al. Cytoplasmic reactive oxygen species and SOD1 regulate bone mass during mechanical unloading. J Bone Miner Res. 2013;28(11):2368–2380. doi:10.1002/jbmr.1981
236. Sharma-Bhandari A, Park SH, Kim JY, et al. Lysyl oxidase modulates the osteoblast differentiation of primary mouse calvaria cells. Int J Mol Med. 2015;36(6):1664–1670. doi:10.3892/ijmm.2015.2384
237. Vallet SD, Ricard-Blum S. Lysyl oxidases: from enzyme activity to extracellular matrix cross-links. Essays Biochem. 2019;63(3):349–364. doi:10.1042/EBC20180050
238. Ghanbari M, Salavati-Niasari M, Mohandes F. Injectable hydrogels based on oxidized alginate-gelatin reinforced by carbon nitride quantum dots for tissue engineering. Int J Pharm. 2021;602:120660. doi:10.1016/j.ijpharm.2021.120660
239. Ghanbari M, Salavati-Niasari M, Mohandes F. Thermosensitive alginate-gelatin-nitrogen-doped carbon dots scaffolds as potential injectable hydrogels for cartilage tissue engineering applications. RSC Adv. 2021;11(30):18423–18431. doi:10.1039/d1ra01496j
240. Wang Y, Zhang W, Yao Q. Copper-based biomaterials for bone and cartilage tissue engineering. J Orthop Translat. 2021;29:60–71. doi:10.1016/j.jot.2021.03.003
241. Li G, Cheng T, Yu X. The Impact of Trace Elements on Osteoarthritis. Front Med Lausanne. 2021;8:771297. doi:10.3389/fmed.2021.771297
242. Dziezyc K, Litwin T, Czlonkowska A. Other organ involvement and clinical aspects of Wilson disease. Handb Clin Neurol. 2017;142:157–169. doi:10.1016/B978-0-444-63625-6.00013-6
243. Li Y, Chen H, Mou P, et al. Relationship between trace elements in synovial fluid and cartilage and severity of knee osteoarthritis. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi. 2023;37(5):584–588. doi:10.7507/1002-1892.202302008
244. Yuan X, Wang J, Zhu X, et al. Effect of copper on levels of collagen and alkaline phosphatase activity from chondrocytes in newborn piglets in vitro. Biol Trace Elem Res. 2011;144(1–3):597–605. doi:10.1007/s12011-011-9151-5
245. Terpstra L, Prud’homme J, Arabian A, et al. Reduced chondrocyte proliferation and chondrodysplasia in mice lacking the integrin-linked kinase in chondrocytes. J Cell Biol. 2003;162(1):139–148. doi:10.1083/jcb.200302066
246. Tsvetkov P, Coy S, Petrova B, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375(6586):1254–1261. doi:10.1126/science.abf0529
247. Kahlson MA, Dixon SJ. Copper-induced cell death. Science. 2022;375(6586):1231–1232. doi:10.1126/science.abo3959
248. Han J, Luo J, Wang C, et al. Roles and mechanisms of copper homeostasis and cuproptosis in osteoarticular diseases. Biomed Pharmacother. 2024;174:116570. doi:10.1016/j.biopha.2024.116570
249. Hu H, Dou X, Hu X, et al. Identification of a novel cuproptosis-related gene signature for rheumatoid arthritis-A prospective study. J Gene Med. 2023;25(11):e3535. doi:10.1002/jgm.3535
250. Kim JH, Jeon J, Shin M, et al. Regulation of the catabolic cascade in osteoarthritis by the zinc-ZIP8-MTF1 axis. Cell. 2014;156(4):730–743. doi:10.1016/j.cell.2014.01.007
251. Zhang H, Puviindran V, Nadesan P, et al. Distinct Roles of Glutamine Metabolism in Benign and Malignant Cartilage Tumors With IDH Mutations. J Bone Miner Res. 2022;37(5):983–996. doi:10.1002/jbmr.4532
252. Yu Y, Newman H, Shen L, et al. Glutamine Metabolism Regulates Proliferation and Lineage Allocation in Skeletal Stem Cells. Cell Metab. 2019;29(4):966–978e964. doi:10.1016/j.cmet.2019.01.016
253. Farr JN, Xu M, Weivoda MM, et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat Med. 2017;23(9):1072–1079. doi:10.1038/nm.4385
254. Li D, Gao Z, Li Q, et al. Cuproptosis-a potential target for the treatment of osteoporosis. Front Endocrinol. 2023;14:1135181. doi:10.3389/fendo.2023.1135181
255. Wang W, Chen Z, Hua Y. Bioinformatics Prediction and Experimental Validation Identify a Novel Cuproptosis-Related Gene Signature in Human Synovial Inflammation during Osteoarthritis Progression. Biomolecules. 2023;13(1). doi:10.3390/biom13010127
256. Chang B, Hu Z, Chen L, et al. Development and validation of cuproptosis-related genes in synovitis during osteoarthritis progress. Front Immunol. 2023;14:1090596. doi:10.3389/fimmu.2023.1090596
257. Marino G, Kroemer G. Mechanisms of apoptotic phosphatidylserine exposure. Cell Res. 2013;23(11):1247–1248. doi:10.1038/cr.2013.115
258. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. doi:10.1080/01926230701320337
259. Lavrik I, Golks A, Krammer PH. Death receptor signaling. J Cell Sci. 2005;118(Pt 2):265–267. doi:10.1242/jcs.01610
260. Green DR. The Death Receptor Pathway of Apoptosis. Cold Spring Harb Perspect Biol. 2022;14(2). doi:10.1101/cshperspect.a041053
261. Lossi L. The concept of intrinsic versus extrinsic apoptosis. Biochem J. 2022;479(3):357–384. doi:10.1042/BCJ20210854
262. Borrelli Jr J. Chondrocyte apoptosis and posttraumatic arthrosis. J Orthop Trauma. 2006;20(10):726–731. doi:10.1097/01.bot.0000249882.77629.5c
263. Shao J, Ding Z, Peng J, et al. MiR-146a-5p promotes IL-1beta-induced chondrocyte apoptosis through the TRAF6-mediated NF-kB pathway. Inflamm Res. 2020;69(6):619–630. doi:10.1007/s00011-020-01346-w
264. Liu Z, Wang T, Sun X, et al. Autophagy and apoptosis: regulatory factors of chondrocyte phenotype transition in osteoarthritis. Hum Cell. 2023;36(4):1326–1335. doi:10.1007/s13577-023-00926-2
265. Yue S, Su X, Teng J, et al. Cryptotanshinone interferes with chondrocyte apoptosis in osteoarthritis by inhibiting the expression of miR‑574‑5p. Mol Med Rep. 2021;23(6). doi:10.3892/mmr.2021.12063
266. Liang Q, Wang XP, Chen TS. Resveratrol protects rabbit articular chondrocyte against sodium nitroprusside-induced apoptosis via scavenging ROS. Apoptosis. 2014;19(9):1354–1363. doi:10.1007/s10495-014-1012-1
267. Luo H, Zhang R. Icariin enhances cell survival in lipopolysaccharide-induced synoviocytes by suppressing ferroptosis via the Xc-/GPX4 axis. Exp Ther Med. 2021;21(1):72. doi:10.3892/etm.2020.9504
268. Gong Z, Wang Y, Li L, et al. Cardamonin alleviates chondrocytes inflammation and cartilage degradation of osteoarthritis by inhibiting ferroptosis via p53 pathway. Food Chem Toxicol. 2023;174:113644. doi:10.1016/j.fct.2023.113644
269. Wan Y, Shen K, Yu H, et al. Baicalein limits osteoarthritis development by inhibiting chondrocyte ferroptosis. Free Radic Biol Med. 2023;196:108–120. doi:10.1016/j.freeradbiomed.2023.01.006
270. Xu C, Ni S, Xu N, et al. Theaflavin-3,3’-Digallate Inhibits Erastin-Induced Chondrocytes Ferroptosis via the Nrf2/GPX4 Signaling Pathway in Osteoarthritis. Oxid Med Cell Longev. 2022;2022:3531995. doi:10.1155/2022/3531995
271. Cheng Q, Chen M, Liu M, et al. Semaphorin 5A suppresses ferroptosis through activation of PI3K-AKT-mTOR signaling in rheumatoid arthritis. Cell Death Dis. 2022;13(7):608. doi:10.1038/s41419-022-05065-4
272. Yan J, Feng G, Ma L, et al. Metformin alleviates osteoarthritis in mice by inhibiting chondrocyte ferroptosis and improving subchondral osteosclerosis and angiogenesis. J Orthop Surg Res. 2022;17(1):333. doi:10.1186/s13018-022-03225-y
273. Fu C, Qiu Z, Huang Y, et al. Achyranthes bidentata polysaccharides alleviate endoplasmic reticulum stress in osteoarthritis via lncRNA NEAT1/miR-377-3p pathway. Biomed Pharmacother. 2022;154:113551. doi:10.1016/j.biopha.2022.113551
274. Mo Z, Xu P, Li H. Stigmasterol alleviates interleukin-1beta-induced chondrocyte injury by down-regulatingsterol regulatory element binding transcription factor 2 to regulateferroptosis. Bioengineered. 2021;12(2):9332–9340. doi:10.1080/21655979.2021.2000742
275. Adjimani JP, Asare P. Antioxidant and free radical scavenging activity of iron chelators. Toxicol Rep. 2015;2:721–728. doi:10.1016/j.toxrep.2015.04.005
276. Munoz M, Garcia-Erce JA, Remacha AF. Disorders of iron metabolism. Part II: iron deficiency and iron overload. J Clin Pathol. 2011;64(4):287–296. doi:10.1136/jcp.2010.086991
277. Yao X, Zhang Y, Hao J, et al. Deferoxamine promotes recovery of traumatic spinal cord injury by inhibiting ferroptosis. Neural Regen Res. 2019;14(3):532–541. doi:10.4103/1673-5374.245480
278. Miotto G, Rossetto M, Di Paolo ML, et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020;28:101328. doi:10.1016/j.redox.2019.101328
279. Wang X, Liu Z, Peng P, et al. Astaxanthin attenuates osteoarthritis progression via inhibiting ferroptosis and regulating mitochondrial function in chondrocytes. Chem Biol Interact. 2022;366:110148. doi:10.1016/j.cbi.2022.110148
280. Hu S, Wang S, He J, et al. Tetramethylpyrazine alleviates endoplasmic reticulum stress‑activated apoptosis and related inflammation in chondrocytes. Mol Med Rep. 2022;25(1). doi:10.3892/mmr.2021.12528
281. Tian F, Wang J, Zhang Z, et al. LncRNA SNHG7/miR-34a-5p/SYVN1 axis plays a vital role in proliferation, apoptosis and autophagy in osteoarthritis. Biol Res. 2020;53(1):9. doi:10.1186/s40659-020-00275-6
282. Zhang Y, Wang F, Chen G, et al. LncRNA MALAT1 promotes osteoarthritis by modulating miR-150-5p/AKT3 axis. Cell Biosci. 2019;9:54. doi:10.1186/s13578-019-0302-2
283. Jin X, Dong X, Sun Y, et al. Dietary Fatty Acid Regulation of the NLRP3 Inflammasome via the TLR4/NF-kappaB Signaling Pathway Affects Chondrocyte Pyroptosis. Oxid Med Cell Longev. 2022;2022:3711371. doi:10.1155/2022/3711371
284. Yan J, Ding D, Feng G, et al. Metformin reduces chondrocyte pyroptosis in an osteoarthritis mouse model by inhibiting NLRP3 inflammasome activation. Exp Ther Med. 2022;23(3):222. doi:10.3892/etm.2022.11146
285. Zu Y, Mu Y, Li Q, et al. Icariin alleviates osteoarthritis by inhibiting NLRP3-mediated pyroptosis. J Orthop Surg Res. 2019;14(1):307. doi:10.1186/s13018-019-1307-6
286. Wang Q, Ying L, Wei B, et al. Effects of quercetin on apoptosis and extracellular matrix degradation of chondrocytes induced by oxidative stress-mediated pyroptosis. J Biochem Mol Toxicol. 2022;36(2):e22951. doi:10.1002/jbt.22951
287. Xu H, Xu B. BMSC-Derived Exosomes Ameliorate Osteoarthritis by Inhibiting Pyroptosis of Cartilage via Delivering miR-326 Targeting HDAC3 and STAT1//NF-kappaB p65 to Chondrocytes. Mediators Inflamm. 2021;2021:9972805. doi:10.1155/2021/9972805
288. Liu G, Liu Q, Yan B, et al. USP7 Inhibition Alleviates H(2)O(2)-Induced Injury in Chondrocytes via Inhibiting NOX4/NLRP3 Pathway. Front Pharmacol. 2020;11:617270. doi:10.3389/fphar.2020.617270
289. Bai H, Zhang Z, Liu L, et al. Activation of adenosine A3 receptor attenuates progression of osteoarthritis through inhibiting the NLRP3/caspase-1/GSDMD induced signalling. J Cell Mol Med. 2022;26(15):4230–4243. doi:10.1111/jcmm.17438
290. Wang S, Mobasheri A, Zhang Y, et al. Exogenous stromal cell-derived factor-1 (SDF-1) suppresses the NLRP3 inflammasome and inhibits pyroptosis in synoviocytes from osteoarthritic joints via activation of the AMPK signaling pathway. Inflammopharmacology. 2021;29(3):695–704. doi:10.1007/s10787-021-00814-x
291. Li Z, Huang Z, Zhang H, et al. P2X7 Receptor Induces Pyroptotic Inflammation and Cartilage Degradation in Osteoarthritis via NF-kappaB/NLRP3 Crosstalk. Oxid Med Cell Longev. 2021;2021:8868361. doi:10.1155/2021/8868361
292. Zhang L, Qiu J, Shi J, et al. MicroRNA-140-5p represses chondrocyte pyroptosis and relieves cartilage injury in osteoarthritis by inhibiting cathepsin B/Nod-like receptor protein 3. Bioengineered. 2021;12(2):9949–9964. doi:10.1080/21655979.2021.1985342
293. Li G, Xiu L, Li X, et al. miR-155 inhibits chondrocyte pyroptosis in knee osteoarthritis by targeting SMAD2 and inhibiting the NLRP3/Caspase-1 pathway. J Orthop Surg Res. 2022;17(1):48. doi:10.1186/s13018-021-02886-5
294. Qian J, Fu P, Li S, et al. miR-107 affects cartilage matrix degradation in the pathogenesis of knee osteoarthritis by regulating caspase-1. J Orthop Surg Res. 2021;16(1):40. doi:10.1186/s13018-020-02121-7
295. Li Z, Huang Z, Zhang H, et al. Moderate-intensity exercise alleviates pyroptosis by promoting autophagy in osteoarthritis via the P2X7/AMPK/mTOR axis. Cell Death Discov. 2021;7(1):346. doi:10.1038/s41420-021-00746-z
296. Jia S, Yang Y, Bai Y, et al. Mechanical Stimulation Protects Against Chondrocyte Pyroptosis Through Irisin-Induced Suppression of PI3K/Akt/NF-kappaB Signal Pathway in Osteoarthritis. Front Cell Dev Biol. 2022;10:797855. doi:10.3389/fcell.2022.797855
297. Yu YN, Tang CL, Guo X, et al. Effect of electroacupuncture on pyroptosis of synovial tissues in rats with knee osteoarthritis. Zhen Ci Yan Jiu. 2022;47(6):471–478. doi:10.13702/j.1000-0607.20211269
298. Fang Y, Lou C, Lv J, et al. Sipeimine ameliorates osteoarthritis progression by suppression of NLRP3 inflammasome-mediated pyroptosis through inhibition of PI3K/AKT/NF-kappaB pathway: an in vitro and in vivo study. J Orthop Translat. 2024;46:1–17. doi:10.1016/j.jot.2024.04.004
299. Wang YH, Gao X, Tang YR, et al. The Role of NF-kappaB/NLRP3 Inflammasome Signaling Pathway in Attenuating Pyroptosis by Melatonin Upon Spinal Nerve Ligation Models. Neurochem Res. 2022;47(2):335–346. doi:10.1007/s11064-021-03450-7
300. Hu J, Zhou J, Wu J, et al. Loganin ameliorates cartilage degeneration and osteoarthritis development in an osteoarthritis mouse model through inhibition of NF-kappaB activity and pyroptosis in chondrocytes. J Ethnopharmacol. 2020;247:112261. doi:10.1016/j.jep.2019.112261
301. Wang C, Gao Y, Zhang Z, et al. Ursolic acid protects chondrocytes, exhibits anti-inflammatory properties via regulation of the NF-kappaB/NLRP3 inflammasome pathway and ameliorates osteoarthritis. Biomed Pharmacother. 2020;130:110568. doi:10.1016/j.biopha.2020.110568
302. Stabler TV, Huang Z, Montell E, et al. Chondroitin sulphate inhibits NF-kappaB activity induced by interaction of pathogenic and damage associated molecules. Osteoarthritis Cartilage. 2017;25(1):166–174. doi:10.1016/j.joca.2016.08.012
303. Baek HS, Hong VS, Kim SH, et al. KMU-1170, a Novel Multi-Protein Kinase Inhibitor, Suppresses Inflammatory Signal Transduction in THP-1 Cells and Human Osteoarthritic Fibroblast-Like Synoviocytes by Suppressing Activation of NF-kappaB and NLRP3 Inflammasome Signaling Pathway. Int J Mol Sci. 2021;22(3). doi:10.3390/ijms22031194
304. Franco-Trepat E, Alonso-Perez A, Guillan-Fresco M, et al. Amitriptyline blocks innate immune responses mediated by toll-like receptor 4 and IL-1 receptor: preclinical and clinical evidence in osteoarthritis and gout. Br J Pharmacol. 2022;179(2):270–286. doi:10.1111/bph.15707
305. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21(7):677–687. doi:10.1038/nm.3893
306. Gao F, Zhang S. Loratadine Alleviates Advanced Glycation End Product-Induced Activation of NLRP3 Inflammasome in Human Chondrocytes. Drug Des Devel Ther. 2020;14:2899–2908. doi:10.2147/DDDT.S243512
307. Yan Z, Qi W, Zhan J, et al. Activating Nrf2 signalling alleviates osteoarthritis development by inhibiting inflammasome activation. J Cell Mol Med. 2020;24(22):13046–13057. doi:10.1111/jcmm.15905
308. Liao T, Ding L, Wu P, et al. Chrysin Attenuates the NLRP3 Inflammasome Cascade to Reduce Synovitis and Pain in KOA Rats. Drug Des Devel Ther. 2020;14:3015–3027. doi:10.2147/DDDT.S261216
309. Wang Z, Chen M, Wang B, et al. Electroacupuncture Alleviates Osteoarthritis by Suppressing NLRP3 Inflammasome Activation in Guinea Pigs. Evid Based Complement Alternat Med. 2020;2020:5476064. doi:10.1155/2020/5476064
310. Yuan B, Zhou XM, You ZQ, et al. Inhibition of AIM2 inflammasome activation alleviates GSDMD-induced pyroptosis in early brain injury after subarachnoid haemorrhage. Cell Death Dis. 2020;11(1):76. doi:10.1038/s41419-020-2248-z
311. Gao L, Dong X, Gong W, et al. Acinar cell NLRP3 inflammasome and gasdermin D (GSDMD) activation mediates pyroptosis and systemic inflammation in acute pancreatitis. Br J Pharmacol. 2021;178(17):3533–3552. doi:10.1111/bph.15499
312. Yang J, Liu Z, Wang C, et al. Mechanism of gasdermin D recognition by inflammatory caspases and their inhibition by a gasdermin D-derived peptide inhibitor. Proc Natl Acad Sci U S A. 2018;115(26):6792–6797. doi:10.1073/pnas.1800562115
313. Rathkey JK, Zhao J, Liu Z, et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol. 2018;3(26). doi:10.1126/sciimmunol.aat2738
314. Hu JJ, Liu X, Xia S, et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol. 2020;21(7):736–745. doi:10.1038/s41590-020-0669-6
315. Sollberger G, Choidas A, Burn GL, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. 2018;3(26). doi:10.1126/sciimmunol.aar6689
316. Ruhl S, Shkarina K, Demarco B, et al. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science. 2018;362(6417):956–960. doi:10.1126/science.aar7607
317. Yang T, Sun K, Wang C, et al. Gasdermin D deficiency attenuates arthritis induced by traumatic injury but not autoantibody-assembled immune complexes. Arthritis Res Ther. 2021;23(1):286. doi:10.1186/s13075-021-02668-8
318. Valenti MT, Dalle Carbonare L, Zipeto D, et al. Control of the Autophagy Pathway in Osteoarthritis: key Regulators, Therapeutic Targets and Therapeutic Strategies. Int J Mol Sci. 2021;22(5). doi:10.3390/ijms22052700
319. Cheung KCP, Jiao M, Xingxuan C, et al. Extracellular vesicles derived from host and gut microbiota as promising nanocarriers for targeted therapy in osteoporosis and osteoarthritis. Front Pharmacol. 2022;13:1051134. doi:10.3389/fphar.2022.1051134
320. Cheung C, Tu S, Feng Y, et al. Mitochondrial quality control dysfunction in osteoarthritis: mechanisms, therapeutic strategies & future prospects. Arch Gerontol Geriatr. 2024;125:105522. doi:10.1016/j.archger.2024.105522
321. Jiang M, Li Z, Zhu G. Immunological regulatory effect of flavonoid baicalin on innate immune toll-like receptors. Pharmacol Res. 2020;158:104890. doi:10.1016/j.phrs.2020.104890
322. He J, He J. Baicalin mitigated IL-1beta-Induced osteoarthritis chondrocytes damage through activating mitophagy. Chem Biol Drug Des. 2023;101(6):1322–1334. doi:10.1111/cbdd.14215
323. Jin Z, Chang B, Wei Y, et al. Curcumin exerts chondroprotective effects against osteoarthritis by promoting AMPK/PINK1/Parkin-mediated mitophagy. Biomed Pharmacother. 2022;151:113092. doi:10.1016/j.biopha.2022.113092
324. Zhang J, Valverde P, Zhu X, et al. Exercise-induced irisin in bone and systemic irisin administration reveal new regulatory mechanisms of bone metabolism. Bone Res. 2017;5:16056. doi:10.1038/boneres.2016.56
325. Wang FS, Kuo CW, Ko JY, et al. Irisin Mitigates Oxidative Stress, Chondrocyte Dysfunction and Osteoarthritis Development through Regulating Mitochondrial Integrity and Autophagy. Antioxidants. 2020;9(9). doi:10.3390/antiox9090810
326. Huang LW, Huang TC, Hu YC, et al. Zinc protects chondrocytes from monosodium iodoacetate-induced damage by enhancing ATP and mitophagy. Biochem Biophys Res Commun. 2020;521(1):50–56. doi:10.1016/j.bbrc.2019.10.066
327. D’amico D, Olmer M, Fouassier AM, et al. Urolithin A improves mitochondrial health, reduces cartilage degeneration, and alleviates pain in osteoarthritis. Aging Cell. 2022;21(8):e13662. doi:10.1111/acel.13662
328. Wang C, Yang Y, Zhang Y, et al. Protective effects of metformin against osteoarthritis through upregulation of SIRT3-mediated PINK1/Parkin-dependent mitophagy in primary chondrocytes. Biosci Trends. 2019;12(6):605–612. doi:10.5582/bst.2018.01263
329. Tang S, Tang T, Gao G, et al. Bone marrow mesenchymal stem cell-derived exosomes inhibit chondrocyte apoptosis and the expression of MMPs by regulating Drp1-mediated mitophagy. Acta Histochem. 2021;123(8):151796. doi:10.1016/j.acthis.2021.151796
330. Baldari S, Di Rocco G, Toietta G. Current Biomedical Use of Copper Chelation Therapy. Int J Mol Sci. 2020;21(3). doi:10.3390/ijms21031069
331. Nagai M, Vo NH, Shin Ogawa L, et al. The oncology drug elesclomol selectively transports copper to the mitochondria to induce oxidative stress in cancer cells. Free Radic Biol Med. 2012;52(10):2142–2150. doi:10.1016/j.freeradbiomed.2012.03.017
332. Zhu Y, Lei C, Jiang Q, et al. DSF/Cu induces antitumor effect against diffuse large B-cell lymphoma through suppressing NF-kappaB/BCL6 pathways. Cancer Cell Int. 2022;22(1):236. doi:10.1186/s12935-022-02661-4
333. Soon CPW, Donnelly PS, Turner BJ, et al. Diacetylbis(N(4)-methylthiosemicarbazonato) copper(II) (CuII(atsm)) protects against peroxynitrite-induced nitrosative damage and prolongs survival in amyotrophic lateral sclerosis mouse model. J Biol Chem. 2011;286(51):44035–44044. doi:10.1074/jbc.M111.274407
334. Cater MA, Pearson HB, Wolyniec K, et al. Increasing intracellular bioavailable copper selectively targets prostate cancer cells. ACS Chem Biol. 2013;8(7):1621–1631. doi:10.1021/cb400198p
335. Rellmann Y, Eidhof E, Dreier R. Review: ER stress-induced cell death in osteoarthritic cartilage. Cell Signal. 2021;78:109880. doi:10.1016/j.cellsig.2020.109880
336. Sang Y, Zhang J, Liu C, et al. Ameliorating Osteoarthritis in Mice Using Silver Nanoparticles. J Vis Exp. 2023;196. doi:10.3791/65111
337. Zhu C, Han S, Zeng X, et al. Multifunctional thermo-sensitive hydrogel for modulating the microenvironment in Osteoarthritis by polarizing macrophages and scavenging RONS. J Nanobiotechnology. 2022;20(1):221. doi:10.1186/s12951-022-01422-9
338. Lu TX, Rothenberg ME. MicroRNA. J Allergy Clin Immunol. 2018;141(4):1202–1207. doi:10.1016/j.jaci.2017.08.034
339. Ding Y, Wang L, Zhao Q, et al. MicroRNA‑93 inhibits chondrocyte apoptosis and inflammation in osteoarthritis by targeting the TLR4/NF‑kappaB signaling pathway. Int J Mol Med. 2019;43(2):779–790. doi:10.3892/ijmm.2018.4033
340. Cai C, Min S, Yan B, et al. MiR-27a promotes the autophagy and apoptosis of IL-1beta treated-articular chondrocytes in osteoarthritis through PI3K/AKT/mTOR signaling. Aging. 2019;11(16):6371–6384. doi:10.18632/aging.102194
341. Yu Q, Zhao B, He Q, et al. microRNA-206 is required for osteoarthritis development through its effect on apoptosis and autophagy of articular chondrocytes via modulating the phosphoinositide 3-kinase/protein kinase B-mTOR pathway by targeting insulin-like growth factor-1. J Cell Biochem. 2019;120(4):5287–5303. doi:10.1002/jcb.27803
342. Nakano K, Boyle DL, Firestein GS. Regulation of DNA methylation in rheumatoid arthritis synoviocytes. J Immunol. 2013;190(3):1297–1303. doi:10.4049/jimmunol.1202572
343. Gu W, Shi Z, Song G, et al. MicroRNA-199-3p up-regulation enhances chondrocyte proliferation and inhibits apoptosis in knee osteoarthritis via DNMT3A repression. Inflamm Res. 2021;70(2):171–182. doi:10.1007/s00011-020-01430-1
344. Dou P, He Y, Yu B, et al. Downregulation of microRNA-29b by DNMT3B decelerates chondrocyte apoptosis and the progression of osteoarthritis via PTHLH/CDK4/RUNX2 axis. Aging. 2020;13(5):7676–7690. doi:10.18632/aging.103778
345. Wang WT, Huang ZP, Sui S, et al. microRNA-1236 promotes chondrocyte apoptosis in osteoarthritis via direct suppression of PIK3R3. Life Sci. 2020;253:117694. doi:10.1016/j.lfs.2020.117694
346. Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17(1):47–62. doi:10.1038/nrg.2015.10
347. Bridges MC, Daulagala AC, Kourtidis A. LNCcation: lncRNA localization and function. J Cell Biol. 2021;220(2). doi:10.1083/jcb.202009045
348. Kong H, Sun ML, Zhang XA, et al. Crosstalk Among circRNA/lncRNA, miRNA, and mRNA in Osteoarthritis. Front Cell Dev Biol. 2021;9:774370. doi:10.3389/fcell.2021.774370
349. Li D, Wang X, Yi T, et al. LncRNA MINCR attenuates osteoarthritis progression via sponging miR-146a-5p to promote BMPR2 expression. Cell Cycle. 2022;21(22):2417–2432. doi:10.1080/15384101.2022.2099191
350. Tang L, Ding J, Zhou G, et al. LncRNA‑p21 promotes chondrocyte apoptosis in osteoarthritis by acting as a sponge for miR‑451. Mol Med Rep. 2018;18(6):5295–5301. doi:10.3892/mmr.2018.9506
351. Wang B, Sun Y, Liu N, et al. LncRNA HOTAIR modulates chondrocyte apoptosis and inflammation in osteoarthritis via regulating miR-1277-5p/SGTB axis. Wound Repair Regen. 2021;29(3):495–504. doi:10.1111/wrr.12908
352. Jiang M, Liu J, Luo T, et al. LncRNA PACER is down-regulated in osteoarthritis and regulates chondrocyte apoptosis and lncRNA HOTAIR expression. Biosci Rep. 2019;39(6). doi:10.1042/BSR20190404
353. Chen LL, Yang L. Regulation of circRNA biogenesis. RNA Biol. 2015;12(4):381–388. doi:10.1080/15476286.2015.1020271
354. Jiang S, Liu Y, Xu B, et al. Noncoding RNAs: new regulatory code in chondrocyte apoptosis and autophagy. Wiley Interdiscip Rev RNA. 2020;11(4):e1584. doi:10.1002/wrna.1584
355. Chen CY, Kao CL, Liu CM. The Cancer Prevention, Anti-Inflammatory and Anti-Oxidation of Bioactive Phytochemicals Targeting the TLR4 Signaling Pathway. Int J Mol Sci. 2018;19(9). doi:10.3390/ijms19092729
356. Zhu F, Du B, Xu B. Anti-inflammatory effects of phytochemicals from fruits, vegetables, and food legumes: a review. Crit Rev Food Sci Nutr. 2018;58(8):1260–1270. doi:10.1080/10408398.2016.1251390
357. Wu X, Liu Z, Yu XY, et al. Autophagy and cardiac diseases: therapeutic potential of natural products. Med Res Rev. 2021;41(1):314–341. doi:10.1002/med.21733
358. Othman MS, Khaled AM, Al-Bagawi AH, et al. Hepatorenal protective efficacy of flavonoids from Ocimum basilicum extract in diabetic albino rats: a focus on hypoglycemic, antioxidant, anti-inflammatory and anti-apoptotic activities. Biomed Pharmacother. 2021;144:112287. doi:10.1016/j.biopha.2021.112287
359. Yu H, Yao S, Zhou C, et al. Morroniside attenuates apoptosis and pyroptosis of chondrocytes and ameliorates osteoarthritic development by inhibiting NF-kappaB signaling. J Ethnopharmacol. 2021;266:113447. doi:10.1016/j.jep.2020.113447
360. Xu C, Ni S, Zhuang C, et al. Polysaccharide from Angelica sinensis attenuates SNP-induced apoptosis in osteoarthritis chondrocytes by inducing autophagy via the ERK1/2 pathway. Arthritis Res Ther. 2021;23(1):47. doi:10.1186/s13075-020-02409-3
361. Zou Y, Liu Q, Guo P, et al. Anti‑chondrocyte apoptosis effect of genistein in treating inflammation‑induced osteoarthritis. Mol Med Rep. 2020;22(3):2032–2042. doi:10.3892/mmr.2020.11254
362. Liu FC, Wang CC, Lu JW, et al. Chondroprotective Effects of Genistein against Osteoarthritis Induced Joint Inflammation. Nutrients. 2019;11(5). doi:10.3390/nu11051180
363. Wang Q, Zhuang D, Feng W, et al. Fraxetin inhibits interleukin-1beta-induced apoptosis, inflammation, and matrix degradation in chondrocytes and protects rat cartilage in vivo. Saudi Pharm J. 2020;28(12):1499–1506. doi:10.1016/j.jsps.2020.09.016
364. Li W, Wang Y, Tang Y, et al. Quercetin Alleviates Osteoarthritis Progression in Rats by Suppressing Inflammation and Apoptosis via Inhibition of IRAK1/NLRP3 Signaling. J Inflamm Res. 2021;14:3393–3403. doi:10.2147/JIR.S311924
365. Feng K, Chen Z, Pengcheng L, et al. Quercetin attenuates oxidative stress-induced apoptosis via SIRT1/AMPK-mediated inhibition of ER stress in rat chondrocytes and prevents the progression of osteoarthritis in a rat model. J Cell Physiol. 2019;234(10):18192–18205. doi:10.1002/jcp.28452
366. Tang Y, Li Y, Xin D, et al. Icariin alleviates osteoarthritis by regulating autophagy of chondrocytes by mediating PI3K/AKT/mTOR signaling. Bioengineered. 2021;12(1):2984–2999. doi:10.1080/21655979.2021.1943602
367. Mi B, Wang J, Liu Y, et al. Icariin Activates Autophagy via Down-Regulation of the NF-kappaB Signaling-Mediated Apoptosis in Chondrocytes. Front Pharmacol. 2018;9:605. doi:10.3389/fphar.2018.00605
368. Xiaoyu Y, Hui X, Xiaolong L, et al. The mechanism of Danzikang Knee Granule in regulating the chondrogenic differentiation of mesenchymal stem cells based on TGF-beta signaling pathway in cartilage repair in knee osteoarthritis. Cell Mol Biol. 2022;67(5):164–173. doi:10.14715/cmb/2021.67.5.23
369. Jin Y, Xu M, Zhu H, et al. Therapeutic effects of bone marrow mesenchymal stem cells-derived exosomes on osteoarthritis. J Cell Mol Med. 2021;25(19):9281–9294. doi:10.1111/jcmm.16860
370. Jiang K, Jiang T, Chen Y, et al. Mesenchymal Stem Cell-Derived Exosomes Modulate Chondrocyte Glutamine Metabolism to Alleviate Osteoarthritis Progression. Mediators Inflamm. 2021;2021:2979124. doi:10.1155/2021/2979124
371. Zhang Y, Wang X, Chen J, et al. Exosomes derived from platelet-rich plasma administration in site mediate cartilage protection in subtalar osteoarthritis. J Nanobiotechnology. 2022;20(1):56. doi:10.1186/s12951-022-01245-8
372. Loeser RF, Collins JA, Diekman BO. Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2016;12(7):412–420. doi:10.1038/nrrheum.2016.65
373. Wang GX, Zhao XY, Meng ZX, et al. The brown fat-enriched secreted factor Nrg4 preserves metabolic homeostasis through attenuation of hepatic lipogenesis. Nat Med. 2014;20(12):1436–1443. doi:10.1038/nm.3713
374. Ma Y, Gao M, Liu D. Preventing High Fat Diet-induced Obesity and Improving Insulin Sensitivity through Neuregulin 4 Gene Transfer. Sci Rep. 2016;6:26242. doi:10.1038/srep26242
375. Shi L, Xu X, Meng B, et al. Neuregulin 4 Attenuates Osteoarthritis Progression by Inhibiting Inflammation and Apoptosis of Chondrocytes in Mice. Calcif Tissue Int. 2022;110(1):131–142. doi:10.1007/s00223-021-00897-2
376. Lu Y, Zhang C, Jiang S, et al. Anti-Dlx5 Retards the Progression of Osteoarthritis through Inhibiting Chondrocyte Hypertrophy and Apoptosis. Evid Based Complement Alternat Med. 2022;2022:5019920. doi:10.1155/2022/5019920
377. Ansari MY, Novak K, Haqqi TM. ERK1/2-mediated activation of DRP1 regulates mitochondrial dynamics and apoptosis in chondrocytes. Osteoarthritis Cartilage. 2022;30(2):315–328. doi:10.1016/j.joca.2021.11.003
378. Xu C, Zhai Z, Ying H, et al. Curcumin primed ADMSCs derived small extracellular vesicle exert enhanced protective effects on osteoarthritis by inhibiting oxidative stress and chondrocyte apoptosis. J Nanobiotechnology. 2022;20(1):123. doi:10.1186/s12951-022-01339-3
379. Wang J, Zhang L, Zhu J, et al. Hyaluronic Acid Modified Curcumin-Loaded Chitosan Nanoparticles Inhibit Chondrocyte Apoptosis to Attenuate Osteoarthritis via Upregulation of Activator Protein 1 and RUNX Family Transcription Factor 2. J Biomed Nanotechnol. 2022;18(1):144–157. doi:10.1166/jbn.2022.3193
380. Zhang Z, Lin J, Tian N, et al. Melatonin protects vertebral endplate chondrocytes against apoptosis and calcification via the Sirt1-autophagy pathway. J Cell Mol Med. 2019;23(1):177–193. doi:10.1111/jcmm.13903
381. Feng TY, Li Q, Ren F, et al. Melatonin Protects Goat Spermatogonial Stem Cells against Oxidative Damage during Cryopreservation by Improving Antioxidant Capacity and Inhibiting Mitochondrial Apoptosis Pathway. Oxid Med Cell Longev. 2020;2020:5954635. doi:10.1155/2020/5954635
382. Liu X, Gong Y, Xiong K, et al. Melatonin mediates protective effects on inflammatory response induced by interleukin-1 beta in human mesenchymal stem cells. J Pineal Res. 2013;55(1):14–25. doi:10.1111/jpi.12045
383. Kouhi M, Varshosaz J, Hashemibeni B, et al. Injectable gellan gum/lignocellulose nanofibrils hydrogels enriched with melatonin loaded forsterite nanoparticles for cartilage tissue engineering: fabrication, characterization and cell culture studies. Mater Sci Eng C Mater Biol Appl. 2020;115:111114. doi:10.1016/j.msec.2020.111114
384. Atoufi Z, Kamrava SK, Davachi SM, et al. Injectable PNIPAM/Hyaluronic acid hydrogels containing multipurpose modified particles for cartilage tissue engineering: synthesis, characterization, drug release and cell culture study. Int J Biol Macromol. 2019;139:1168–1181. doi:10.1016/j.ijbiomac.2019.08.101
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.