Back to Journals » Cancer Management and Research » Volume 13
Prognostic Value of the TP53 Mutation Location in Metastatic Breast Cancer as Detected by Next-Generation Sequencing
Authors Bai H, Yu J, Jia S, Liu X, Liang X, Li H ![]()
Received 23 December 2020
Accepted for publication 19 March 2021
Published 15 April 2021 Volume 2021:13 Pages 3303—3316
DOI https://doi.org/10.2147/CMAR.S298729
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Bilikere Dwarakanath
Han Bai,1 Jianjun Yu,2 Shidong Jia,2 Xiaoran Liu,1 Xu Liang,1 Huiping Li1
1Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education/Beijing), Department of Breast Oncology, Peking University Cancer Hospital and Institute, Beijing, 100142, People’s Republic of China; 2Huidu Shanghai Medical Sciences, Shanghai, 201499, People’s Republic of China
Correspondence: Xu Liang; Huiping Li
Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education/Beijing), Department of Breast Oncology, Peking University Cancer Hospital and Institute, No. 52, Fucheng Road, Haidian District, Beijing, 100142, People’s Republic of China
Tel/Fax +86-10-88196739
Email [email protected]; [email protected]
Purpose: The status of TP53 mutations was measured in cell-free DNA from patients with metastatic breast cancer (MBC) to investigate disease characteristics and the prognostic role of different locations of the TP53 mutation site.
Patients and Methods: Blood samples were taken from a total of 187 patients diagnosed with MBC who were treated at the Department of Breast Oncology, Peking University Cancer Hospital between January 2013 and March 2020. Next-generation sequencing was used to investigate the TP53 mutation spectra of circulating free DNA in these blood samples.
Results: Among the 187 MBC patients, TP53-mutated patients had a significantly shorter median disease-free survival (DFS) and overall survival (OS) compared with TP53 wild-type patients (P=0.001 and P=0.006, respectively). Additionally, in hormone receptor positive/HER2 negative (HR+/HER2-) and triple negative (TNBC) cohorts, TP53-mutated patients had a significantly shorter median DFS than TP53 wild-type patients (P=0.038 and P=0.023, respectively). The 79 patients with TP53 mutations carried 87 somatic TP53 mutations, of which most (77.0%) mapped to the DNA-binding domain (DBD) of the protein encoded by TP53 exons 5– 8. In patients with TP53 mutations, those occurring in the non-DBD had a significantly shorter median DFS and OS than TP53 wild type (P< 0.001 and P=0.001, respectively). Additionally, patients with non-missense mutations in the DBD had a significantly shorter median DFS and OS than TP53 wild-type patients (P=0.001 and P< 0.001, respectively). TP53-mutated patients had a significantly shorter DFS than TP53 wild-type patients in the adjuvant endocrine therapy sensitive group (P=0.008), but differences in the endocrine therapy resistant group were not significant.
Conclusion: TP53-mutated MBC patients had a significantly worse outcome than TP53 wild-type patients especially those in HR+/HER2– and TNBC cohorts. Of TP53-mutated patients, those with non-missense mutations in the DBD had worse breast cancer-related survival. TP53 mutations were also associated with endocrine resistance.
Keywords: advanced breast cancer, TP53 mutation, NGS, adjuvant endocrine therapy, DNA-binding domain
Introduction
As a tumor suppressor and DNA binding transcription factor, TP53 is actively involved in the regulation of the cell cycle, apoptosis, and genomic stability.1,2 TP53 is one of the most frequently mutated genes in human cancers, including breast cancer,3 and numerous studies have reported it as a biomarker for predicting an aggressive and metastatic phenotype in breast cancer.4–7 Most of these studies used first-generation sequencing or automated DNA extraction from formalin-fixed and paraffin-embedded tissue (FFPE). However, real-time (RT)-PCR results and first-generation sequencing could not be used to detect all TP53 mutations to further investigate TP53 status more accurately and reliably.
TP53 is located on chromosome 17p13.1 and contains 11 exons and 10 introns. Most TP53 mutations map to exons 5–8, which encodes the DNA binding domain (DBD), and most are missense mutations.8–10 Hotspot codons 175, 213, 245, 248, 273, and 282 account for at least 2% of all mutations within the DBD.2 Patients with acute myeloid leukemia carrying TP53 mutations in the DBD had a worse prognosis than those with wild-type TP53.11 Furthermore, another clinical trial showed that truncating mutations in the DBD had a significant independent prognostic value in breast cancer, being associated with increased recurrence compared with patients with non-modified p53 proteins.12
Early studies using first-generation sequencing or automated DNA extraction from FFPE found that TP53 mutations were associated with poor prognosis in hormone receptor-positive (HR+) breast cancer patients.13–15 Moreover, in an HR+ cohort, TP53 signaling was enriched in resistant tumors (38% in the aromatase inhibitor-resistant group vs 17% in the sensitive group) such that HR+ tumors with TP53 mutations were mostly aromatase inhibitor-resistant.5 No significant result was obtained from human epidermal growth factor receptor 2 positive (HER2+) or triple-negative breast cancer (TNBC) cohorts.16 Other studies found that TP53 mutations were associated with tumor recurrence and apoptosis, which were more common in HER2-positive and TNBC cohorts.17,18
While the significance of TP53 mutations has been shown by RT-PCR and first-generation sequencing, most clinical laboratories do not use next-generation sequencing (NGS) to determine the p53 mutational status because of high costs and complex interpretation. Therefore, it is difficult to understand the clinical applications of TP53.19 In the present study, we collected peripheral blood samples from Chinese patients with freshly diagnosed metastatic breast cancer (MBC) and examined the whole exons and introns of TP53 by NGS to further investigate the relationship between TP53 mutations, prognosis, and therapy.
Materials and Methods
Patients
From January 2013 to March 2020, patients past first-line treatment and those for whom blood samples were not available were excluded, leaving a total of 194 at the stage of first-line treatment at the Department of Breast Oncology, Peking University Cancer Hospital. Of these, 187 consented with enrollment and had complete clinic-pathological information (Figure 1).
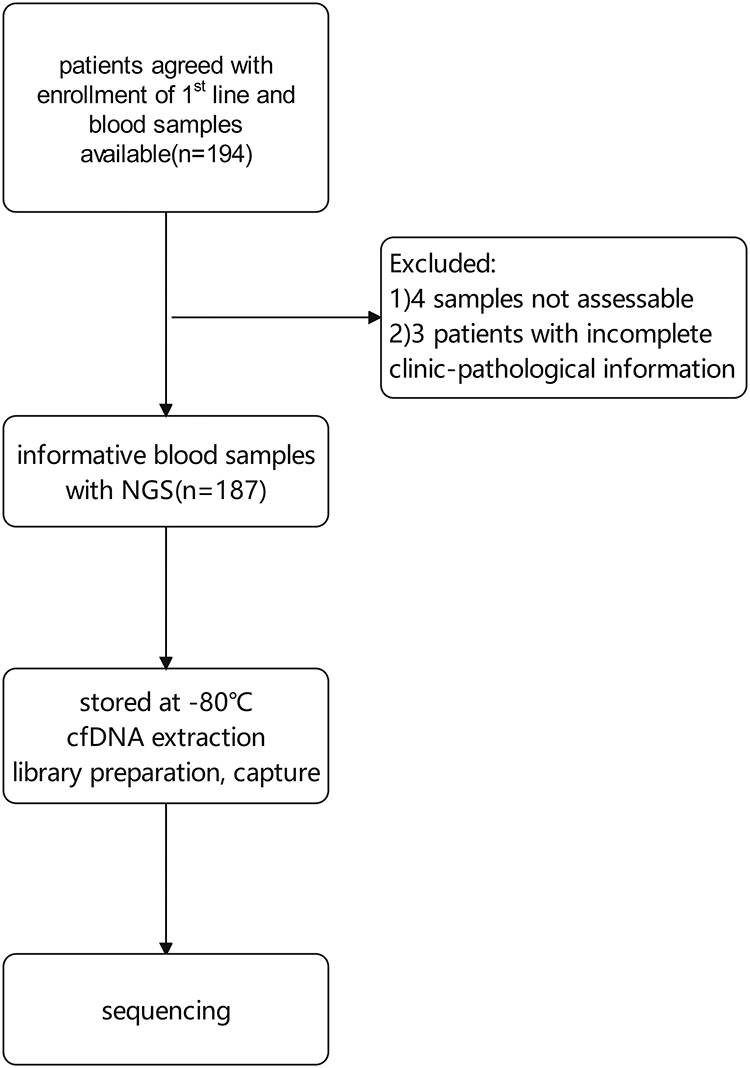 |
Figure 1 Flowchart of patient inclusion. |
We defined estrogen receptor (ER), progesterone receptor (PR), and HER2 status according to recommended guidelines,20,21 which identified three subtypes: the HER2+ cohort, HR+/HER2- cohort, and TNBC cohort.
HR+/HER2- patients who accepted adjuvant endocrine therapy were divided into two groups: endocrine-resistant patients were defined as patients relapsing during adjuvant endocrine therapy, or <12 months after its completion. Endocrine-sensitive patients were defined as patients relapsing ≥12 months after completing adjuvant endocrine therapy in the early breast cancer stage.22
Samples
Peripheral blood samples before first-line therapy were collected in EDTA Vacutainer tubes and centrifuged at 2000 g for 10 min at 4°C. The supernatant was then removed, and each sample of 3 mL plasma was stored at –80°C.
Circulating Free DNA Extraction
Circulating free (cf)DNA was extracted using a QIAamp Circulating Nucleic Acid Kit (QIAamp, Venlo, the Netherlands) from EDTA and citrate anticoagulant plasma. The average volume of plasma used for extraction was 2.6 mL (range, 0.7–3.9 mL). The quantity and quality of the purified cfDNA were checked using a Qubit 3.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) and Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). For samples with severe genomic contamination from peripheral blood cells, size selection was performed to remove large genomic fragments with AMPure XP beads (Beckman Coulter, Brea, CA, USA). Samples with a total yield <5 ng were considered inadequate for NGS and were removed from any further sequencing methods.
Library Preparation, Capture, and Sequencing
cfDNA was end-repaired before the dA-tailing process, and then ligated with proprietary UMI adapters. The library yield was measured after PCR amplification using a Qubit and Bioanalyzer 2100. Samples yielding >700 ng proceeded to the hybridization step. Library capture was conducted using biotin-labeled DNA probes (Thermo Fisher Scientific). In brief, the library was hybridized using PredicineCARE panel (Huidu Shanghai Medical Sciences, Inc.) overnight and captured on Dynabeads M-270 Streptavidin (Thermo Fisher Scientific).23,24 Unbound fragments were washed away, and the enriched fragments were amplified via PCR. For library preparation, the purified product was checked using Bioanalyzer 2100 and loaded into the HiSeq X Ten system (Illumina, San Diego, CA, USA) for NGS with paired-end 150 bp sequencing kits.
Analyses of NGS Data Generated from cfDNA
Consensus binary alignment map (BAM) files were derived by merging paired-end reads that originated from the same molecules (based on mapping location and unique molecular identifiers) as single-strand fragments. Single-strand fragments from the same double-strand DNA molecules were merged to be double-stranded for suppressing sequencing and PCR errors during this process. NGS quality-checking was performed by examining the percentage of targeted regions with >1500x unique consensus coverage. Samples with <80% regions having >1500x unique coverage were deemed to be QC failed and excluded. Candidate variants, consisting of point mutations, small insertions and deletions, were identified using Huidu proprietary bioinformatics pipeline. Candidate variants with low base quality, mapping scores, and other quality metrics were filtered. Candidate variants in repeat regions were also excluded.
A variant identified in cfDNA was considered to be a candidate somatic mutation-based if all of the following pre-defined criteria were present. These criteria were 1) the presence of at least 4 distinct paired reads in the mutation in the plasma; 2) the number of distinct paired reads containing a particular mutation in the plasma is at least 0.1% of the total distinct read pairs (if the nucleotide change and amino acid change are identical to an alteration observed in ≥20 cancer cases reported in the COSMIC database or previously reported as a cancer hotspot [http://www.cancerhotspots.org]) or the number of distinct paired reads containing a particular mutation in the plasma was at least 0.25% of the total distinct read pairs (if the nucleotide change and amino acid change are not a frequent alteration in COSMIC database or reported as a cancer hotspot previously); 3) the variant is not present in public databases of common germline variants, including 1000 genomes, ExAC, gnomAD, and KAVIAR, with population allele frequency >0.5%; 4) the variant is not present in matched PBMC samples (unpublished data, manuscript in preparation).
Candidate somatic mutations were further filtered based on gene annotation to identify those occurring in protein-coding regions. Intronic and silent changes were excluded, and mutations resulting in missense mutations, nonsense mutations, frameshifts, or splice site alterations were retained. Mutations annotated as benign or likely benign in ClinVar database were also filtered.
Evaluation
Clinical outcome was evaluated as disease-free survival (DFS) and overall survival (OS). Disease-free survival (DFS) was defined as the interval between surgery and time of recurrence for relapsed patients so that patients with stage IV were not included. OS was defined as the time from diagnosis to the date of death or last follow-up. According to Response Evaluation Criteria in Solid Tumors version 1.1 guidelines,25 we evaluated the response assessment by a computed tomography scan or magnetic resonance imaging every 6–12 weeks or as the patient’s condition deteriorated.
Statistical Analysis
SPSS software version 20 was used to analyze the TP53 status and categorical patient characteristics. DFS and OS were estimated by the Kaplan–Meier method and comparisons between groups were conducted by the log rank test. P values <0.05 were considered significant. For multivariable analysis, Cox proportional hazards method was used to evaluate clinical outcome. The association between the TP53 status and clinical characteristics was examined using the Chi-square test.
Results
Patient Characteristics
Of 187 patients, 79 carried TP53 mutations and 108 had wild-type TP53. Detailed baseline clinical information of all patients is shown in Table 1. The median age in the TP53 mutated group was 48 years (range: 27–69 years old) versus 46 years of age in the TP53 wild-type group (range: 26–80 years old) (P = 0.702). We also found that 73.4% (58/79) of TP53-mutated patients and 86.1% (93/108) of TP53 wild-type patients were HER2 negative (P=0.030).
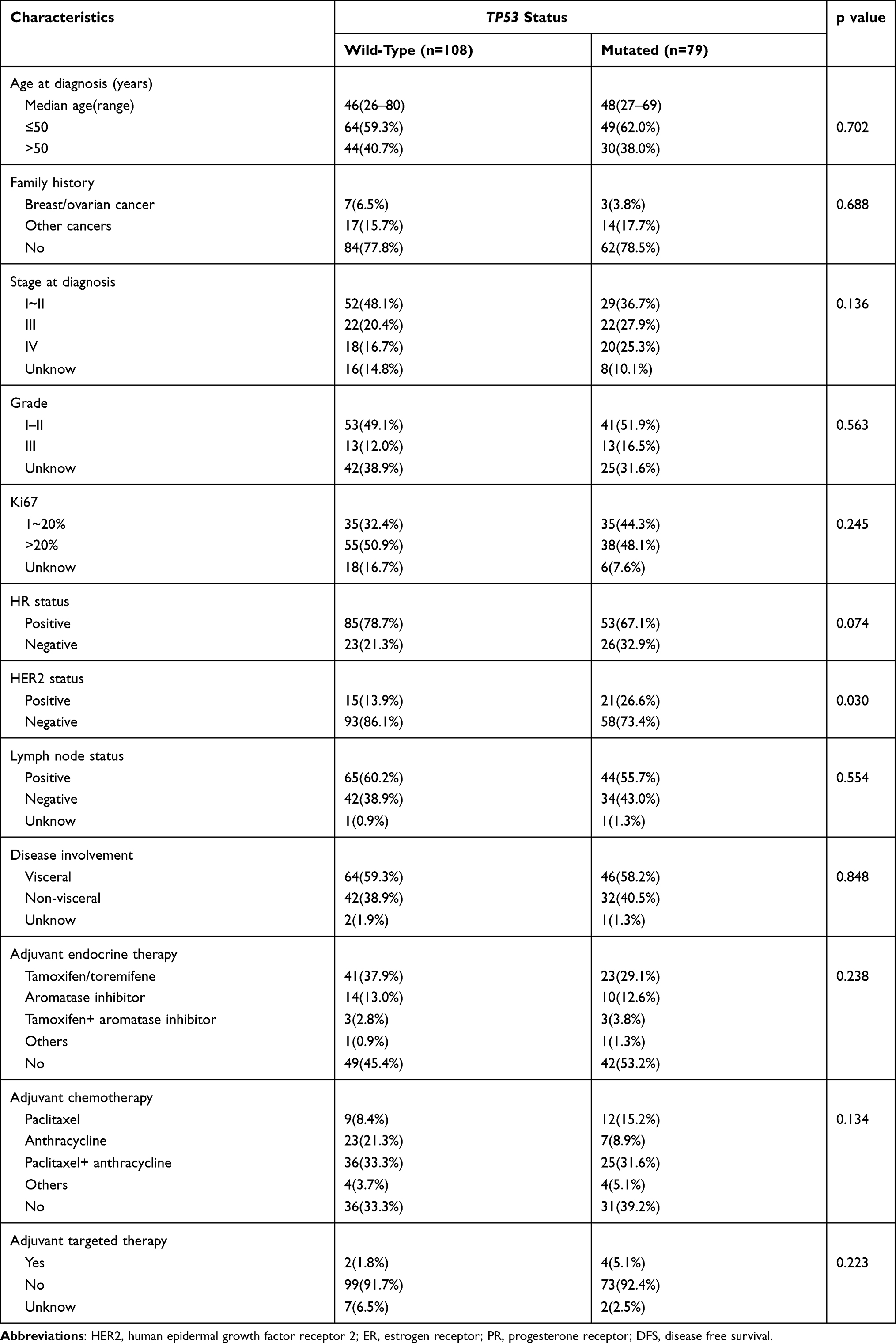 |
Table 1 Baseline Clinical Characteristics of TP53 Wild-Type and -Mutated Metastatic Breast Cancer Patients (n=187) |
In univariate analysis of DFS (Table 2), HER2 status (P=0.024) and HR status (P=0.000) were significant predictors in TP53 wild-type patients and TP53-mutated patients, respectively, and Ki67 status was also a significant predictor for TP53 wild-type patients (P=0.001) and TP53-mutated patients (P=0.022). After multivariable analysis of DFS (Table 2), Ki67 status (P=0.003) and HR status (P=0.000) in TP53 mutated group remained significant predictors and patients with stage III had a higher risk of relapse after surgery than stage I–II (p=0.030) in TP53 wild-type cohort.
Characteristics of TP53-Mutated Patients
A total of 87 somatic TP53 mutations were identified in the 79 TP53-mutated patients. Sixty-seven of these (77.0%) were located in exons 5–8, which span the DBD of the protein (Supplementary Table S1). Codons 175, 220, and 248 within the DBD were the locations of 4.6% of all mutations, respectively, which were all missense mutations (Figure 2). Of the 87 mutations, there were 46 missense mutations (43 was in DBD, 1 was in TD, 1 was in TAD, and 1 was outside the p53 protein domain) and 41 non-missense mutations (18 nonsense mutations, 3 splicing mutations, 16 frameshift mutations, 4 in-frame mutations).
 |
Figure 2 The mutational spectra of TP53 in TP53-mutated patients. ( Notes: According to http://www.cbioportal.org, truncating mutations included nonsense mutations, frameshift mutations and splicing mutations. |






TP53 Status in Different Subtypes
We found that the median DFS of TP53-mutated patients was significantly shorter at 33.0 months (95% confidence interval [CI]=21.4–44.6) than that of TP53 wild-type patients at 51.0 months (95% CI=39.1–60.9) (hazard ratio=1.89, 95% CI=1.31–2.71, P=0.001) (Figure 3A). Similarly, the median OS of TP53-mutated patients was significantly shorter at 67.0 months (95% CI=44.4–89.6) than that of TP53 wild-type patients at 140.0 months (95% CI=119.5–160.5) (hazard ratio=1.99, 95% CI=1.21–3.26, P=0.006) (Figure 3B).
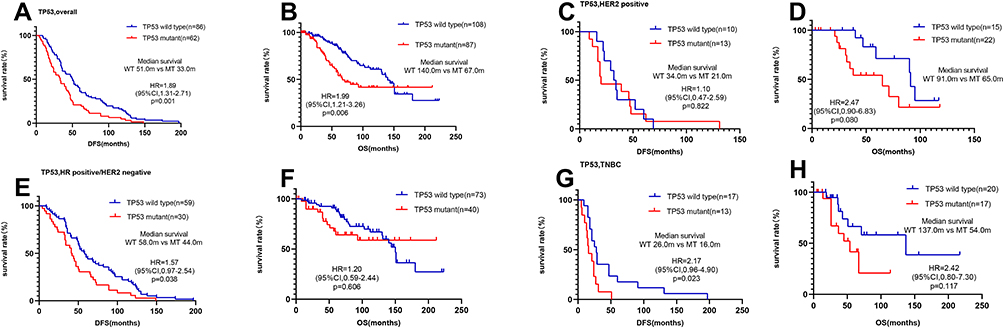 |
Figure 3 Survival analyses by Kaplan–Meier according to TP53 status in MBC patients. (A and B) TP53 wild-type patients had a significantly better clinical outcome than TP53-mutated patients. (C and D) there were no significant differences between TP53 wild-type and -mutated patients in the HER2-positive cohort. (E and F) TP53 wild-type patients had a significantly longer median DFS and OS than TP53-mutated patients in the HR+/HER2– cohort. (G and H) TP53 wild-type patients had a significantly longer median DFS than TP53-mutated patients in the TNBC cohort. |
In the HER2+ cohort (n=36, 21 of whom were TP53-mutated patients), there was no significant difference regarding TP53 status with respect to DFS (34.0 vs 21.0 months, P=0.822) (Figure 3C) or OS (91.0 vs 65.0 months, P=0.080) (Figure 3D).
In the HR+/HER2- cohort (n=113, 40 of whom were TP53-mutated patients), the median DFS of TP53 mutated patients of 44.0 months (95% CI=35.9–52.1) was significantly shorter than the 58.0 months (95% CI=46.2–69.8) of TP53 wild-type patients (hazard ratio=1.57, 95% CI=0.97–2.54, P=0.038) (Figure 3E). No significant difference was observed for OS (P=0.606) (Figure 3F).
In the TNBC cohort (n=38, 18 of whom were TP53-mutated patients), the median DFS of TP53-mutated patients of 16.0 months (95% CI=7.8–24.2) was significantly shorter than the 26.0 months (95% CI=16.6–35.4) of TP53 wild-type patients (hazard ratio=2.17, 95% CI=0.96–4.90, P=0.023) (Figure 3G). There was no significant difference regarding TP53 status with respect to OS (137.0 vs 54.0 months, P=0.117) (Figure 3H).
DBD Missense Mutations Were Associated with Improved Survival
We next classified the 187 patients into three groups by mutation domain: TP53 mutations in the DBD, TP53 mutations in the non-DBD, and TP53 wild-type groups. The median DFS for these patients was 36.6 (95% CI=25.3–42.7), 22 (95% CI=16.1–25.9), and 51 (95% CI=39.1–60.9) months, respectively, while the median OS was 80 (95% CI=46.3–113.7), 51 (95% CI=41.2–60.8), and 140 (95% CI=119.5–160.5) months, respectively.
TP53 wild-type patients had a significantly better clinical outcome than those with TP53 mutations in the DBD with respect to DFS (P=0.008, Figure 4A) and OS (P=0.003, Figure 4B). Similarly, TP53 wild-type patients had a significantly better clinical outcome than those with TP53 mutations in the non-DBD with respect to DFS (P<0.001, Figure 4A) and OS (P=0.001, Figure 4B). There were no significant differences in DFS or OS between patients with TP53 mutations in the DBD compared with those in the non-DBD.
 |
Figure 4 Survival analyses by Kaplan–Meier according to TP53 mutation sites in MBC patients. (A) Patients with a mutation in the non-DNA binding domain had a significantly shorter median DFS than TP53 wild-type patients and those with mutations in the DNA-binding domain. (B) Patients with a mutation in the non-DNA binding domain had shorter median OS than TP53 wild-type patients and those with mutations in the DNA-binding domain. (C) Patients with protein non-stable mutation had shortest median DFS than patients with protein stable mutation and TP53 wild-type patients. (D) Patients with protein non-stable mutation had shortest median OS than patients with protein stable mutation and TP53 wild-type patients. Notes: Protein stable mutations would include non-truncating and non-frame altering mutations outside of the p53 tetramerization domain, and protein non-stable mutations would include all truncating and frame-altering mutations, as well as mutations in the tetramerization domain. |
And then, we divided patients into three groups: TP53 wild-type group; protein stable mutations group (non-truncating and non-frame altering mutations outside of the p53 tetramerization domain); protein non-stable mutations group (all truncating and frame-altering mutations, and mutations in the tetramerization domain).
Patients with protein non-stable mutations had significantly shorter DFS (21.0 months vs 49.0 months, respectively, hazard ratio=2.82, 95% CI=1.63–4.87, P<0.001, Figure 4C) and OS (57.0 months vs 140.0 months, respectively, hazard ratio=4.05, 95% CI=1.95–8.40, P<0.001, Figure 4D) than TP53 wild-type patients. Moreover, the median DFS of protein stable mutations was 43.5 months, longer than protein non-stable mutations (hazard ratio=0.54, 95% CI=0.31–0.93, P=0.025, Figure 4C). There were no significant differences in DFS or OS between patients with protein stable mutations and TP53 wild type.
Furthermore, we wanted to study mutations in DBD so that we classified them into missense (n=43) and non-missense mutations (n=24, including nonsense mutations, splicing mutations, frameshift mutations and in-frame mutations). Patients with non-missense mutations in the DBD had significantly shorter DFS (20.0 months vs 51.0 months, respectively, hazard ratio=3.26, 95% CI=1.58–6.71, P=0.001, Figure 5A) and OS (57.0 months vs 140.0 months, respectively, hazard ratio=10.45, 95% CI=3.79–28.8, P<0.001, Figure 5B) than TP53 wild-type patients. Moreover, the median OS of patients with non-missense mutations in the DBD was significantly shorter than those with missense mutations in the DBD (hazard ratio=2.45, 95% CI=1.05–5.09, P=0.015, Figure 5B). There were no significant differences in DFS or OS between patients with missense mutations in the DBD and wild-type TP53 patients.
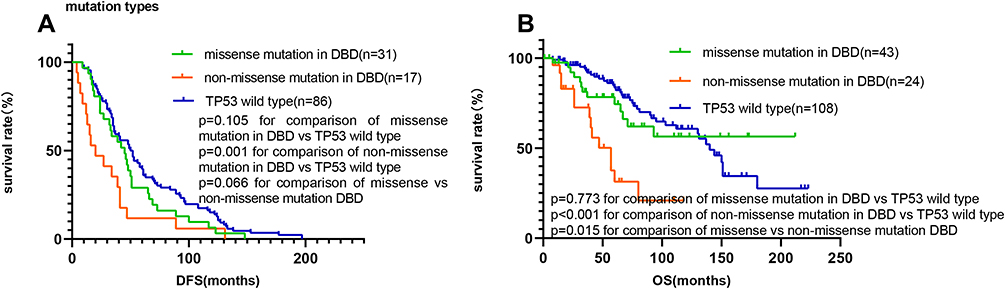 |
Figure 5 Survival analyses by Kaplan–Meier according to TP53 mutation type in the DNA binding domain. (A and B) Patients with non-missense mutations in the DNA binding domain had a significantly shorter median DFS and OS than TP53 wild-type patients and those with missense mutations in the DNA binding domain. |
TP53 Status Was Associated with Adjuvant Endocrine Therapy Response
A total of 96 patients who received adjuvant endocrine therapy were selected to evaluate the relationship between TP53 mutation status and the response to endocrine therapy. As shown in Table 3, we found that 84.7% (50/59) of patients accepted adjuvant chemotherapy in TP53 wild-type group, whereas 78.4% (29/37) of patients accepted adjuvant chemotherapy treatment in TP53 mutant patients. There was no significant difference between TP53 status and adjuvant chemotherapy (P=0.467). As well known, ESR1 mutations are associated with acquired endocrine resistance in breast cancer so that we took ESR1 mutation rate into consideration in Table 3, but there were no significant differences in ESR1 mutation rate (p=0.558) between the two groups.
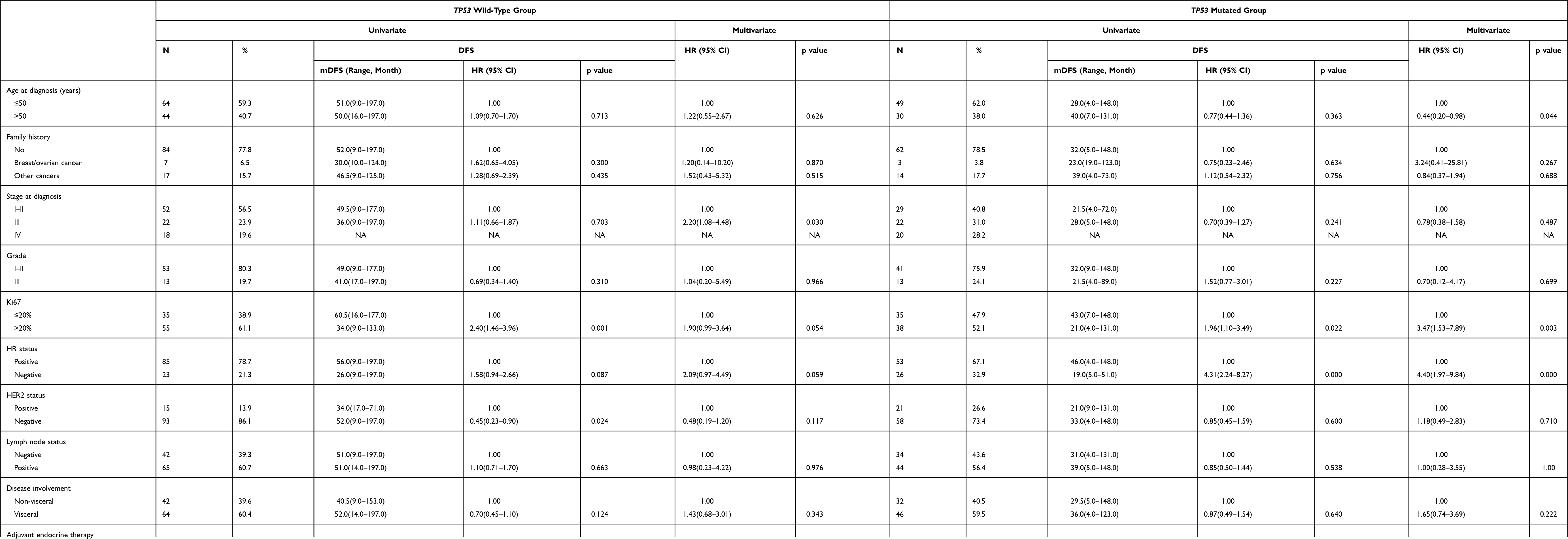 |
Table 2 Univariate and Multivariate Cox Regression Analysis of DFS in TP53 Wild-Type and -Mutated Patients |
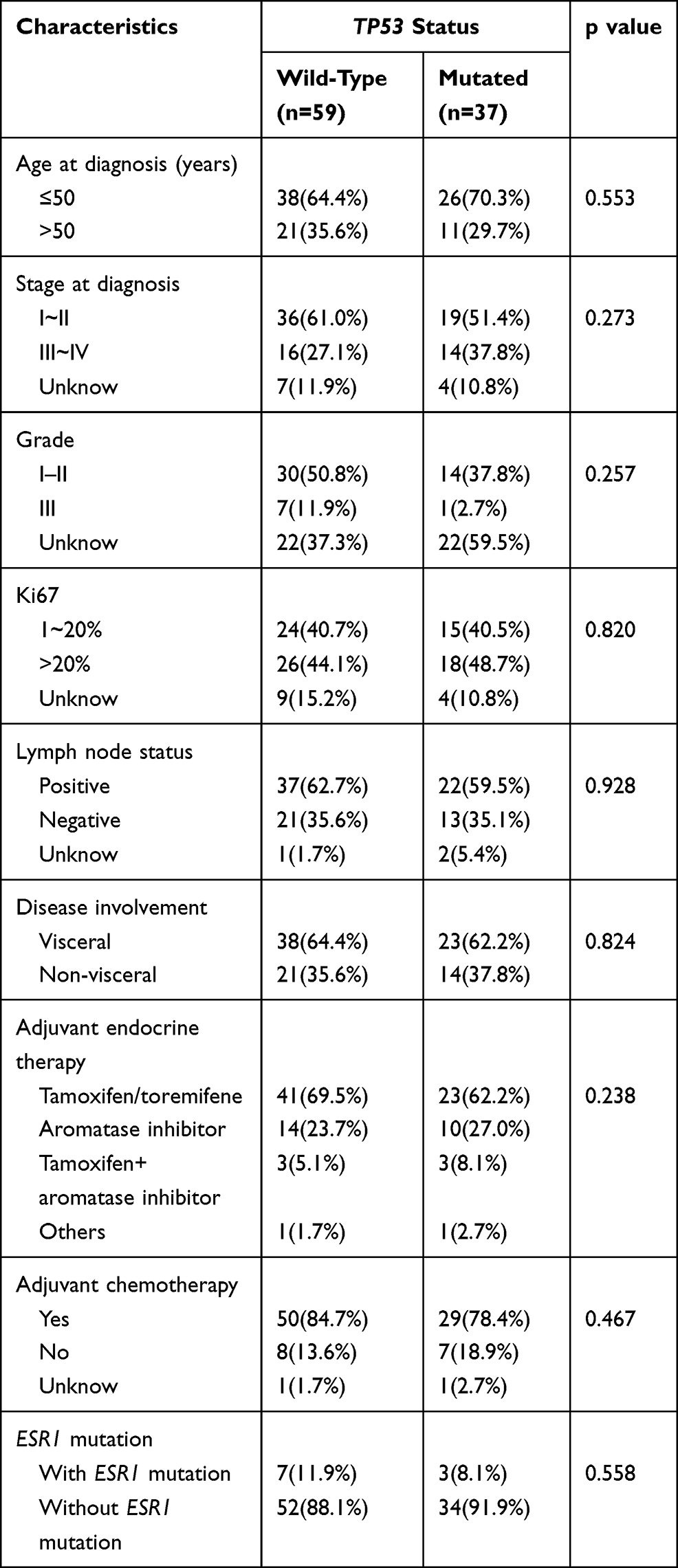 |
Table 3 Clinical Characteristics of Patients Receiving Adjuvant Endocrine Therapy (n=96) |
To further explore the relationship between TP53 status and treatment response, we classified patients into the adjuvant endocrine therapy-resistant group and the adjuvant endocrine therapy sensitive group. Interestingly, we found that in the adjuvant endocrine therapy sensitive group, patients with TP53 mutations had a significantly shorter DFS than TP53 wild-type patients (69.0 months vs 108.0 months, respectively, hazard ratio=3.22, 95% CI=0.70–14.77, P=0.008) (Figure 6B). No significant DFS differences between TP53-mutated and TP53 wild-type patients were seen in the endocrine therapy-resistant group (34.0 months vs 40.0 months, respectively, P=0.903) (Figure 6A).
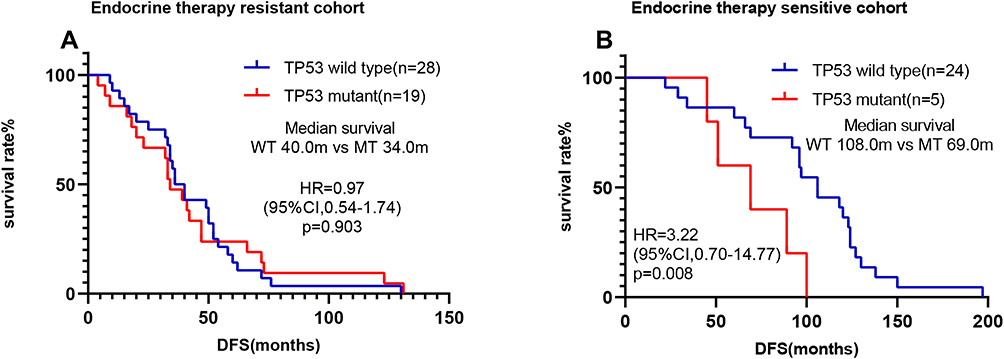 |
Figure 6 Survival analyses by Kaplan–Meier according to TP53 status in MBC receiving adjuvant endocrine therapy. (A) There was no significant difference in TP53 status in the endocrine therapy-resistant cohort. (B) TP53 wild-type patients had a significantly better clinical outcome than TP53-mutated patients in the endocrine therapy sensitive cohort. |
Discussion
In our study, we used NGS to detect TP53 mutations in the cfDNA, which might affect tumor temporal and spatial heterogeneity, of 187 Chinese MBC patients. Our results indicated that TP53 mutations could be used as a prognostic marker for worse outcome in MBC and for the response of adjuvant endocrine therapy.
We established genomic profiles of patients which revealed a TP53 mutation frequency of 42.2%, similar to that seen in the Guangdong Provincial People’s Hospital cohort (45.0%) but higher than in the TCGA breast cancer cohort (30.0%).26 Another recent study on cfDNA molecular profiling in Chinese patients with MBC reported a TP53 mutation rate of 64.1% compared with 52% in Caucasian patients.27,28 These discrepancies could reflect differences between patient ethnicities, such as in the median age of breast cancer patients with TP53 mutations in our study of 48 years compared with 55.2 years in Caucasians.29
The p53 pathway was previously shown to rank top in the basal-like breast cancer subtype, but not in the HER2-enriched type; therefore, TP53 mutations were not associated with poor prognosis in the HER2-enriched group.6 In support of this, our data indicated that the TP53 mutation status was an independent predictive factor of survival especially in HR+/HER2– and TNBC cohorts, but not in the HER2-positive cohort.
Several studies have shown that the DBD is the most frequently mutated TP53 region in breast cancer. In line with this, codons 175, 220, and 248 located within the DBD were the site of many TP53 mutations in our study, of which most were missense mutations. DBD mutations were previously reported to have prognostic value,30,31 while non-missense mutations were associated with a worse outcome in MBC.32 A recent study showed that missense mutation in the DNA-binding domain had dominant-negative effects (DNE).33 There was no difference in survival between patients with dominant-negative p53 mutant tumors and those with TP53 mutations that are predicted to be non-dominant negative.34,35 In our study, TP53 missense mutations in the DBD were associated with improved survival. Further analysis showed that patients with TP53 mutations in the non-DBD had a significantly shorter DFS than those in the TP53 non-mutation cohort.36 In order to investigate the prognostic value of p53 protein further, we divided them into TP53 wild-type group; protein stable mutations group and protein non-stable mutations group. In our study, patients with protein non-stable mutations had significantly shorter DFS and OS than TP53 wild-type patients. Moreover, protein non-stable mutations included all truncating and frame-altering mutations, and mutations in the tetramerization domain so that mutations in TD had a worse clinical outcome. The reasons were that mutations in TD could either abolish or reduce binding of p53 protein to DNA and transcriptional activation, and TP53 mutation in TD domain had dominant-negative effects (DNE) that inactivate TP53 wild type in some cases.37 Other researchers also found mutations in TD domain were associated with cancer-associated development.38 Not all missense mutations cause protein accumulation, while non-missense mutations are true loss-of-function mutations. Thus, missense mutations have generally been associated with higher protein expression compared with non-missense mutations.16
Some clinical trials showed us TP53 might be the potential to be a therapeutic biomarker. Studies on the role of TP53 mutation in breast cancer response to chemotherapy are conflicting.39–42 Data on the association between TP53 mutations and endocrine therapy response were also controversial.43–45 When it came to the association between hormone therapy and chemotherapy, some researchers found that adding hormone therapy to chemotherapy could improve the survival for TP53 wild-type patients not for TP53 mutation patients.46,47 While in our research, 84.7% of patients in TP53 wild-type group and 78.4% patients in TP53 mutant group all accepted adjuvant chemotherapy and endocrine therapy treatment in Table 3, and the distribution of patients with adjuvant chemotherapy was balanced in two groups, which did not exert an influence on the analysis of endocrine therapy and TP53 status. In our study, we also found TP53 mutations were associated with endocrine resistance. TP53-mutated patients had a shorter DFS than TP53 wild-type patients in the adjuvant endocrine therapy sensitive group. Previously, increased expression of estrogen-related receptor (ERR)α was associated with increased levels of p53 in ERα-positive cases. ERα and ERRα share only 33% homology in their ligand-binding domains, resulting in the insensitivity of ERRα to tamoxifen.48 Additionally, TP53 wild-type tumors might be more responsive to endocrine therapy because this disrupts the ERα–p53 interaction and reactivates p53.49
The retrospective nature of our study resulted in a number of limitations. DFS might have influenced the survival analysis, which was retrospectively calculated. Additionally, we lacked matched primary and recurrence samples for analysis. Finally, we did not analyze p53 protein expression to verify our results.
Conclusion
In conclusion, TP53 wild-type MBC patients showed better survival than TP53-mutated patients in HR+/HER2– and TNBC cohorts. Missense mutations in the DBD of p53 appeared to be an independent prognostic marker for short DFS, while TP53 mutations were associated with endocrine resistance. This indicates that alternative therapies for HR-positive patients with TP53 mutations should be considered. Large-scale prospective studies are needed to verify our findings.
Data Sharing Statement
We can provide the original data in this manuscript upon request.
Research Ethics and Consent
The written informed consent of this research had been provided by the patients, and this study was approved by the Medical Ethics Committee of Peking University Cancer Hospital & Institute (Approval No.2016KT47) according to the Declaration of Helsinki.
Acknowledgments
We thank Sarah Williams, PhD, from Liwen Bianji, Edanz Editing China, for editing the English text of a draft of this manuscript.
Funding
There were no funding sources for this work.
Disclosure
Jianjun Yu and Shidong Jia are employees and stockholders of Huidu Shanghai Medical Sciences, Ltd. The authors declare that they have no other competing interests.
References
1. Kastenhuber ER, Lowe SW. Putting p53 in context. Cell. 2017;170(6):1062–1078. doi:10.1016/j.cell.2017.08.028
2. Hainaut P, Pfeifer GP. Somatic TP53 mutations in the era of genome sequencing. Cold Spring Harb Perspect Med. 2016;6(11):a026179. doi:10.1101/cshperspect.a026179
3. Stephens PJ, Tarpey PS, Davies H, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486(7403):400–404. doi:10.1038/nature11017
4. Basho RK, de Melo Gagliato D, Ueno NT, et al. Clinical outcomes based on multigene profiling in metastatic breast cancer patients. Oncotarget. 2016;7(47):76362–76373. doi:10.18632/oncotarget.12987
5. Ellis MJ, Ding L, Shen D, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486(7403):353–360. doi:10.1038/nature11143
6. Ren J, Wang B, Li J. Integrating proteomic and phosphoproteomic data for pathway analysis in breast cancer. BMC Syst Biol. 2018;12(Suppl S8):130. doi:10.1186/s12918-018-0646-y
7. Koçak A, Heselmeyer-Haddad K, Lischka A, et al. High levels of chromosomal copy number alterations and TP53 mutations correlate with poor outcome in younger breast cancer patients. Am J Pathol. 2020;190(8):1643–1656. doi:10.1016/j.ajpath.2020.04.015
8. Wang D, Kon N, Lasso G, et al. Acetylation-regulated interaction between p53 and SET reveals a widespread regulatory mode. Nature. 2016;538(7623):118–122. doi:10.1038/nature19759
9. Sullivan KD, Galbraith MD, Andrysik Z, et al. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018;25(1):133–143. doi:10.1038/cdd.2017.174
10. Marchenko ND, Hanel W, Li D, et al. Stress-mediated nuclear stabilization of p53 is regulated by ubiquitination and importin-alpha3 binding. Cell Death Differ. 2010;17(2):255–267. doi:10.1038/cdd.2009.173
11. Parry TE. Mutagenic mechanisms in leukemia and cancer: a new concept cytosine lack could be as mutagenic as cytosine deamination. Leuk Res. 2006;30(9):1079–1083. doi:10.1016/j.leukres.2005.12.019
12. Fernndez-Cuesta L, Oakman C, Falagan-Lotsch P, et al. Prognostic and predictive value of TP53mutations in node-positive breast cancer patients treated with anthracycline- or anthracycline/taxane-based adjuvant therapy: results from the BIG 02–98 Phase III trial. Breast Cancer Res. 2012;14(3):R70. doi:10.1186/bcr3179
13. Meric-Bernstam F, Zheng X, Shariati M, et al. Survival outcomes by TP53 mutation status in metastatic breast cancer. JCO Precis Oncol. 2018;2:1–5. doi:10.1200/PO.17.00245
14. Lopez G, Costanza J, Colleoni M, et al. Molecular insights into the classification of luminal breast cancers: the genomic heterogeneity of progesterone-negative tumors. Int J Mol Sci. 2019;20(3):510. doi:10.3390/ijms20030510
15. Silwal-Pandit L, Vollan HKM, Chin S-F, et al. TP53 mutation spectrum in breast cancer is subtype specific and has distinct prognostic relevance. Clin Cancer Res. 2014;20(13):3569–3580. doi:10.1158/1078-0432.CCR-13-2943
16. Darb-Esfahani S, Denkert C, Stenzinger A, et al. Role of TP53 mutations in triple negative and HER2-positive breast cancer treated with neoadjuvant anthracycline/taxane-based chemotherapy. Oncotarget. 2016;7(42):67686–67698. doi:10.18632/oncotarget.11891
17. Luo Y, Huang W, Zhang H, et al. Prognostic significance of CD117 expression and TP53 missense mutations in triple-negative breast cancer. Oncol Lett. 2018;15(5):6161–6170. doi:10.3892/ol.2018.8104
18. Pileczki V, Pop L, Braicu C, et al. Double gene siRNA knockdown of mutant p53 and TNF induces apoptosis in triple-negative breast cancer cells. Onco Targets Ther. 2016;9:6921–6933. doi:10.2147/OTT.S110719
19. Li J-P, Zhang X-M, Zhang Z, et al. Association of p53 expression with poor prognosis in patients with triple-negative breast invasive ductal carcinoma. Medicine. 2019;98(18):e15449. doi:10.1097/MD.0000000000015449
20. Wolff AC, Hammond MEH, Hicks DG, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. Arch Pathol Lab Med. 2014;138(2):241–256. doi:10.5858/arpa.2013-0953-SA
21. Hammond ME, Hayes DF, Dowsett M, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010;28(16):2784–2795. doi:10.1200/JCO.2009.25.6529
22. Brandão M, Maurer C, Ziegelmann PK, et al. Endocrine therapy-based treatments in hormone receptor-positive/HER2-negative advanced breast cancer: systematic review and network meta-analysis. ESMO Open. 2020;5(4):e000842. doi:10.1136/esmoopen-2020-000842
23. Newman AM, Lovejoy AF, Klass DM, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34(5):547–555. doi:10.1038/nbt.3520
24. Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–219. doi:10.1038/nbt.2514
25. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–247. doi:10.1016/j.ejca.2008.10.026
26. Zhang G, Wang Y, Chen B, et al. Characterization of frequently mutated cancer genes in Chinese breast tumors: a comparison of Chinese and TCGA cohorts. Ann Transl Med. 2019;7(8):179. doi:10.21037/atm.2019.04.23
27. Rossi G, Mu Z, Rademaker AW, et al. Cell-free DNA and circulating tumor cells: comprehensive liquid biopsy analysis in advanced breast cancer. Clin Cancer Res. 2018;24(3):560–568. doi:10.1158/1078-0432.CCR-17-2092
28. Tao Z, Li T, Feng Z, et al. Characterizations of cancer gene mutations in Chinese metastatic breast cancer patients. Front Oncol. 2020;10:1023. doi:10.3389/fonc.2020.01023
29. Warner ET, Tamimi RM, Hughes ME, et al. Racial and ethnic differences in breast cancer survival: mediating effect of tumor characteristics and sociodemographic and treatment factors. J Clin Oncol. 2015;33(20):2254–2261. doi:10.1200/JCO.2014.57.1349
30. Olivier M, Langer A, Carrieri P, et al. The clinical value of somatic TP53 gene mutations in 1794 patients with breast cancer. Clin Cancer Res. 2006;12(4):1157–1167. doi:10.1158/1078-0432.CCR-05-1029
31. Liu Y, Xu F, Wang Y, et al. Mutations in exon 8 of TP53 are associated with shorter survival in patients with advanced lung cancer. Oncol Lett. 2019;18(3):3159–3169. doi:10.3892/ol.2019.10625
32. Petitjean A, Achatz MIW, Borresen-Dale AL, et al. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007;26(15):2157–2165. doi:10.1038/sj.onc.1210302
33. Boettcher S, Miller PG, Sharma R, et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science. 2019;365(6453):599–604. doi:10.1126/science.aax3649
34. Shahbandi A, Jackson JG. Analysis across multiple tumor types provides no evidence that mutant p53 exerts dominant negative activity. NPJ Precis Oncol. 2019;3(1):1. doi:10.1038/s41698-018-0074-x
35. Giacomelli AO, Yang X, Lintner RE, et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet. 2018;50(10):1381–1387. doi:10.1038/s41588-018-0204-y
36. Terada K, Yamaguchi H, Ueki T, et al. Full-length mutation search of the TP53 gene in acute myeloid leukemia has increased significance as a prognostic factor. Ann Hematol. 2018;97(1):51–61. doi:10.1007/s00277-017-3143-2
37. Gencel-Augusto J, Lozano G. p53 tetramerization: at the center of the dominant-negative effect of mutant p53. Genes Dev. 2020;34(17–18):1128–1146. doi:10.1101/gad.340976.120
38. Muscolini M, Montagni E, Caristi S, et al. Characterization of a new cancer-associated mutant of p53 with a missense mutation (K351N) in the tetramerization domain. Cell Cycle. 2009;8(20):3396–3405. doi:10.4161/cc.8.20.9910
39. Jackson JG, Pant V, Li Q, et al. p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell. 2012;21(6):793–806. doi:10.1016/j.ccr.2012.04.027
40. Bertheau P, Turpin E, Rickman DS, et al. Exquisite sensitivity of TP53 mutant and basal breast cancers to a dose-dense epirubicin−cyclophosphamide regimen. PLoS Med. 2007;4(3):e90. doi:10.1371/journal.pmed.0040090
41. Bertheau P, Espié M, Turpin E, et al. TP53 status and response to chemotherapy in breast cancer. Pathobiology. 2008;75(2):132–139. doi:10.1159/000123851
42. Bonnefoi H, Piccart M, Bogaerts J, et al. TP53 status for prediction of sensitivity to taxane versus non-taxane neoadjuvant chemotherapy in breast cancer (EORTC 10994/BIG 1-00): a randomised Phase 3 trial. Lancet Oncol. 2011;12(6):527–539. doi:10.1016/S1470-2045(11)70094-8
43. Kai K, Nishimura R, Arima N, et al. p53 expression status is a significant molecular marker in predicting the time to endocrine therapy failure in recurrent breast cancer: a cohort study. Int J Clin Oncol. 2006;11(6):426–433. doi:10.1007/s10147-006-0601-6
44. Yamashita H, Toyama T, Nishio M, et al. p53 protein accumulation predicts resistance to endocrine therapy and decreased post-relapse survival in metastatic breast cancer. Breast Cancer Res. 2006;8(4):R48. doi:10.1186/bcr1536
45. Bailey ST, Shin H, Westerling T, et al. Estrogen receptor prevents p53-dependent apoptosis in breast cancer. Proc Natl Acad Sci U S A. 2012;109(44):18060–18065. doi:10.1073/pnas.1018858109
46. Ungerleider NA, Rao SG, Shahbandi A, et al. Breast cancer survival predicted by TP53 mutation status differs markedly depending on treatment. Breast Cancer Res. 2018;20(1):115. doi:10.1186/s13058-018-1044-5
47. Shahbandi A, Nguyen HD, Jackson JG. TP53 mutations and outcomes in breast cancer: reading beyond the headlines. Trends Cancer. 2020;6(2):98–110. doi:10.1016/j.trecan.2020.01.007
48. Thewes V, Simon R, Schroeter P, et al. Reprogramming of the ERRα and ERα target gene landscape triggers tamoxifen resistance in breast cancer. Cancer Res. 2015;75(4):720–731. doi:10.1158/0008-5472.CAN-14-0652
49. Konduri SD, Medisetty R, Liu W, et al. Mechanisms of estrogen receptor antagonism toward p53 and its implications in breast cancer therapeutic response and stem cell regulation. Proc Natl Acad Sci U S A. 2010;107(34):15081–15086. doi:10.1073/pnas.1009575107
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.