Back to Journals » Vascular Health and Risk Management » Volume 17
Prognostic Significance of Systemic Arterial Stiffness Evaluated by Cardio-Ankle Vascular Index in Patients with Idiopathic Pulmonary Hypertension
Authors Radchenko GD ![]() , Sirenko YM
, Sirenko YM ![]()
Received 2 December 2020
Accepted for publication 27 January 2021
Published 9 March 2021 Volume 2021:17 Pages 77—93
DOI https://doi.org/10.2147/VHRM.S294767
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Konstantinos Tziomalos
Ganna D Radchenko, Yuriy M Sirenko
Department of Symptomatic Hypertension, “National Scientific Center “The M.D. Strazhesko Institute of Cardiology”” of National Academy of Medical Science, Kyiv, Ukraine
Correspondence: Ganna D Radchenko
Department of Symptomatic Hypertension, “National Scientific Center “The M.D. Strazhesko Institute of Cardiology”” of National Academy of Medical Science, 5, Narodnogo Opolchenia Str, Kyiv, 03680, Ukraine
Tel +380 667128405
Email [email protected]
Background: In a previous study, the cardio-ankle vascular index (CAVI) was increased significantly in idiopathic pulmonary arterial hypertension (IPAH) patients compared to the healthy group and did not much differ from one in systemic hypertensives. In this study the relations between survival and CAVI was evaluated in patients with IPAH.
Patients and Methods: We included 89 patients with new-diagnosed IPAH without concomitant diseases. Standard examinations, including right heart catheterization (RHC) and systemic arterial stiffness evaluation, were performed. All patients were divided according to CAVI value: the group with CAVI ≥ 8 (n = 18) and the group with CAVI < 8 (n = 71). The mean follow-up was 33.8 ± 23.7 months. Kaplan–Meier and Cox regression analysis were performed for the evaluation of our cohort survival and the predictors of death.
Results: The group with CAVI≥ 8 was older and more severe compared to the group with CAVI< 8. Patients with CAVI≥ 8 had significantly reduced end-diastolic (73.79± 18.94 vs 87.35± 16.69 mL, P< 0.009) and end-systolic (25.71± 9.56 vs 33.55± 10.33 mL, P< 0.01) volumes of the left ventricle, the higher right ventricle thickness (0.77± 0.12 vs 0.62± 0.20 mm, P < 0.006), and the lower TAPSE (13.38± 2.15 vs 15.98± 4.4 mm, P< 0.018). RHC data did not differ significantly between groups, except the higher level of the right atrial pressure in patients with CAVI≥ 8– 11.38± 7.1 vs 8.76± 4.7 mmHg, P< 0.08. The estimated overall survival rate was 61.2%. The CAVI≥ 8 increased the risk of mortality 2.34 times (CI 1.04– 5.28, P = 0.041). The estimated Kaplan–Meier survival in the patients with CAVI ≥ 8 was only 46.7 ± 7.18% compared to patients with CAVI < 8 - 65.6 ± 4.2%, P = 0.035. At multifactorial regression analysis, the CAVI reduced but saved its relevance as death predictor - OR = 1.13, CI 1.001– 1.871.
Summary: We suggested the CAVI could be a new independent predictor of death in the IPAH population and could be used to better risk stratify this patient population if CAVI is validated as a marker in a larger multicenter trial.
Keywords: idiopathic pulmonary arterial hypertension, systemic arterial stiffness, pulse wave velocity, cardio-ankle vascular index, survival
Introduction
Idiopathic pulmonary arterial hypertension (IPAH) is an orphan progressive disease with poor prognosis, which is characterized by small pulmonary artery remodeling, increased pulmonary vascular resistance (PVR) ≥3 Wood, and mean pulmonary artery pressure (mPAP) >20 mmHg, and by excessive right heart overloading.1 The prevalence of this disease in the general population ranges from 6 to 9 in a million adults.2–5 IPAH has traditionally been considered a disease of young women, but more recently a larger proportion of patients have been diagnosed after age 50.2,5
A lot of studies reported the different predictors of death in pulmonary arterial hypertension (PAH). Usually with poor prognosis there are associations with: age, male gender, connective tissue diseases, portal hypertension, a low functional capacity (III–IV WHO functional class, 6-minute walk test distance <440 m, low diffusing capacity for carbon monoxide, low O2 consumption), cardiac biomarkers, increased right atrial area, low systolic function of the right ventricle and cardiac output, high mean right atrial blood pressure, and PVR.6–11 Recently some risk stratification calculators were proposed for practical work.12–16 At the 6th World Congress on pulmonary hypertension (NICE, 2018) there was confirmed the usefulness of risk stratification, but some limitations were indicated of the existent risk assessment methods such as the retrospective character of analyses and not including some complications in the risk scale.1 Studies searching for new diagnostic and prognostic markers (especially non-invasive ones) still continue.17
Some investigations were devoted to the evaluation of inflammation markers (interleukin-1 receptor family member ST2, IL-32, tumor necrosis factor-α, interferon-γ) in patients with pulmonary hypertension.17–21 As is well known, the pathogenesis of PAH includes the small pulmonary artery remodeling that is displayed by hypertrophy, proliferation, plexiform injury, vasoconstriction, microthrombosis, and endothelial cell dysfunction. The inflammatory cytokines support these changes.22–26 In patients with IPAH and heritable PAH (HPAH) the levels of tumor necrosis factor-α, interferon-γ, and some interleukins were increased compared to the control group and the elevated level of interleukin-6 was a predictor of worse survival.26 The prognostic role of the inflammation was confirmed in other studies,27–29 but it is still unclear if the inflammation is the cause or consequence of pulmonary vascular remodeling. Some investigations reported about the associations between severity of the inflammation and changes in pulmonary arteries30,31 as well as about the positive influence of the anti-inflammatory therapy (by imatinib) on the clinical course in patients with PAH.32–34
Systemic inflammation takes a prominent position in the development of other cardiovascular diseases (atherosclerosis, systemic arterial hypertension, heart failure, etc.) through the direct and non-direct effects on the heart and arteries.35–40 In some studies there were reported about the association of the inflammation markers with increasing systemic arterial stiffness.41,42 In a recent publication, N. Nickel et al proposed that pulmonary hypertension could have systemic manifestations that realize from different mechanisms: abnormal endothelial responses, metabolic dysregulation, inflammation, and BMPR-2 (Bone Morphogenetic Protein Receptor Type 2) mutation.43 The authors indicated the morphological changes in systemic vessels and their abnormal endothelial responses could be part of the clinical presentation and could have a negative influence on the patient's functional capacity. They concluded even though these extrapulmonary manifestations in PAH patients are mild they could have prognostic significance, but this needs to be proved in clinical trials.
In our previous study we hypothesized if the inflammation is important for PAH and systemic hypertension, the activated cytokines and other proinflammatory substances in IPAH could affect not only the pulmonary arteries but the systemic arteries also. We evaluated the systemic artery stiffness parameters in patients with newly diagnosed IPAH and in essential hypertensive patients.44 We found the mean cardio-ankle vascular index (CAVI), which is independent of the blood pressure characteristic of arterial stiffness,45 was increased significantly in IPAH patients compared to the healthy group and did not much differ from one in systemic hypertensives matched to age. Also, the CAVI correlated with IPAH patient functional capacity and right ventricle function. But we did not analyze the association of CAVI with the prognosis.
The aim of this study was to evaluate the relationship between survival and CAVI and to compare it with other traditional predictors of death in patients with IPAH.
Patients and Methods
Patients
We provided a single-center, retrospective cohort study with analysis based on the incident cases of IPAH/HPAH. IPAH/HPAH was diagnosed in conformity with ESC-2015 guidelines5 after right heart catheterization (RHC): precapillary pulmonary hypertension (mPAP ≥25 mmHg, PVR ≥ 240 dynes*s/cm,5 and pulmonary artery wedge pressure (PAWP) ≤15 mmHg at rest) in absence of other etiologies for PAH. The family history of PAH has only a few patients. The genetic analysis was not provided because it has not been available in the Ukraine till now.
In the retrospective analysis, we included the patients with IPAH/HPAH, who corresponded to inclusion/exclusion criteria, and were treated and followed in a single Ukrainian referral center from May 2014 till April 2020. The inclusion criteria were a newly diagnosed IPAH/HPAH, the absence of exclusion criteria, age ≥18 and <60 years, and signing of the informed consent form for personal data analysis. Patients were excluded if they had concomitant diseases or states which could influence systemic arterial stiffness: i.e. diabetes mellitus, moderate-severe chronic kidney disease, ischemic heart disease, peripheral vascular disease, obesity, systemic arterial hypertension, history of stroke or other systemic artery or rheumatic diseases, or age ≥60 years. All patients were followed till the endpoint development or the end of the study. The study size was limited only by the period of inclusion and the inclusion/exclusion criteria.
All patients signed an informed consent form for the personal data process. The study was approved by the local ethics committee of the State Institution “National Scientific Center “The M.D. Strazhesko Institute of Cardiology”” of National Academy of Medical Science (Kyiv, Ukraine) and has been performed according to the Good Clinical Practice guidelines and the Declaration of Helsinki.
Methods
A routine physical examination was conducted on all patients. Systolic (SBP) and diastolic (DBP) arterial blood pressure (BP) was evaluated three times by the Omron M-10 (Omron Healthcare Co., Ltd., Kyoto, Japan) in a sitting position after 10 minutes of rest. The mean of three measurements was included in the database. Heart rate (HR) was assessed after the second BP measurement. Body mass and height were evaluated with SECA 220 (Seca GmbH & Co, Hamburg, Germany) and a body mass index calculation was done. For exclusion of the systemic arterial hypertension, ambulatory blood pressure monitoring was provided by device ABPM-04 (Meditech, Budapest, Hungary).
According to the American Thoracic Society guidelines, a 6-minute walking test (6MWT) was provided twice on different days and the best result was used in our analysis.45,46 Additionally, the HR, BP, oxygen saturation (SaO2), and dyspnea score (Borg Dyspnoea Score) were fixed before and just after the tests.
Transthoracic echocardiography (TTE) was conducted using standard measurements (Artida, Toshiba, Tokyo, Japan) by the same specialist in accordance with the American Society of Echocardiography and European Association of Cardiovascular Imaging recommendations.47 We determined the left and right ventricle/atrium sizes (areas and index values), the ejection fraction of the left ventricle, stroke volume, pulmonary valve velocity values, tricuspid annular plane systolic excursion (TAPSE), tricuspid regurgitation velocity, calculated systolic pulmonary BP (SPBP), dimension and collapse of the inferior vena cava, and indices of right/left ventricle eccentricity in diastole and systole. Also, we evaluated the left ventricle diastolic dysfunction: the relationship between the early and late left ventricle filling (E- and A-wave – mitral E/A ratio); time that the rapid flow velocity declines in early diastole (E-wave deceleration time = DT); the velocity for filling of the left ventricle to start after the ventricle relaxes (length of the isovolumetric relaxation time = IVRT), the relationship between the maximum velocity of the E-wave of mitral valve inflow and the maximal velocity of E (E/eʹ ratio).
Using SphygmoCor (AtCor Medical Pty Ltd., Sydney, Australia) we measured pulse wave velocity (PWV) and central BP in standard methods: probes were placed at the right common carotid artery, right femoral artery, and right radial artery; the distance between sensors was measured by a centimeter ribbon. Segment [carotid artery – femoral artery] was used for the evaluation of the elastic artery stiffness (PWVe), and the segment [carotid artery – radial artery] was used for the assessment of the muscular artery stiffness (PWVm).48 The central BP was calculated by the Sphygmocor-PVx device too.
CAVI measurements were conducted by Sphygmomanometer and Sphygmograph VaSera-1500N (Fukuda, Tokyo, Japan) by standard methods.49,50 After obtaining the distance from the level of the aortic valve (brachial level) to the measuring point (the ankle) and the time delay between the closing of the aortic valve to the identified change in arterial pressure wave at the set point CAVI was calculated with help of the device software.51 PWV, SBP, and DBP as well as arterial pulse waveform analysis were used for the automatic CAVI calculation on the left and right sides. It could be attained through the electrocardiogram, cardiac phonogram, and the pressure cuffs on the testing subject at the reference points. In our analysis, we included the highest value among two (from the right or left side) measurements. The normal value of CAVI is supposed <8.0 as per the manufacturer’s instructions. The value ≥9.0 is considered as a sign of increased systemic stiffness.44
All tests, including CAVI analysis, were provided in our center before the RHC was done and before the specific therapy was administered.
In all patients RHC was performed via jugular venous access, with zero reference leveled at the mid-chest in the supine position. Parameters of the pulmonary circulation were measured as follows: the mean right atrial, right ventricular, pulmonary artery, and the wedge pressures, cardiac output (the thermodilution method with Swan-Ganz catheter). The calculations of the stroke volume, the cardiac index (CI), PVR, systemic vascular resistance (SVR), and total vascular resistance (TVR) were done by standard formulas.52 The vasoreactivity test with inhaled iloprost as one available in Ukraine was conducted in all pulmonary hypertensive patients in compliance with ESC-2015 guideline baseline and 4 months later in the case of a positive result.5
The routine biochemical analysis was used for assessing such parameters as glucose, cholesterol, creatinine, bilirubin, electrolytes, and uric acid serum levels by an automatic photometer (Cormay Livia Chemistry Analyzer, Lublin, Poland). The glomerular filtration rate (GFR) was calculated by the EPI formula from serum creatinine concentration. N-terminal pro-B-type natriuretic peptide (NT-proBNP), ferritin and thyroid-stimulating hormone (TSH) levels were determined also.
All examinations were done by specialists who were not involved directly in the study. All patients underwent the same methods of examination. Dividing the groups was done after stopping the follow-up.
Treatment
Baseline all procedures were conducted in patients before specific therapy administration. Sildenafil and iloprost were the main prescribed specific pulmonary hypertension drugs as those have been available in the Ukraine since 2014 and included in the National drug supply program. Since 2019 bosentan and ambrisentan could be added in some patients as a third specific drug or as a second one in the case of iloprost or sildenafil intolerance. Calcium channel blockers were used in the chronic positive vasoreactive patients. Supportive therapy included loop diuretics, spironolactone, iron supplement drugs, warfarin, and digoxin.
The endpoint of the study was death for any reason. The information about death was selected from relatives and based on a copy of the death certificate.
Statistic Methods
All variables are presented as mean and standard deviation (SD) or as a proportion. Nonparametric sensitivity analysis (Mann–Whitney test) results are presented as mean and (minimal – maximal) values. Statistics Package for Social Sciences (SPSS version 13.0, SPSS Inc, Chicago, IL, USA) was used for statistical analyses. The analyzed parameters were compared between two groups of patients: those who had CAVI ≥8 and those who had CAVI <8. The ANOVA one way test was used for normally distributed continuous variables, and the Mann–Whitney U-test was used for abnormally distributed parameters. For differences of categorical variables in groups, the chi-square analysis was used. The Pearson correlation analysis was done for the evaluation of the relationships between the CAVI, the left ventricle volumes, and the parameters of the left ventricular diastolic function.
A Kaplan–Meier method and Long rank Mantel–Cox tests were applied to estimate study population survival (± standard error) and determine the predictive significance of variables. For evaluation of the death predictors, we used the discrete and continuous variables. The choice of the category boundaries was done on the ‘external criteria’ approach. We used the criteria accepted for risk stratification in guidelines. The discrete variable CAVI ≥8 was accepted as a well-known marker of abnormal arterial stiffness. The continuous variables (right atrium area, right atrium pressure, duration of symptoms) were categorized as a hazard ratio per 1 unit increase. Univariate Cox regression analysis was used to examine the relationship between survival and main parameters, those usually accepted as influenced on prognosis (age, functional class, gender, 6 MWT distance, anticoagulant therapy, NT-proBNP, bilirubin, TAPSE, right atrium area and blood pressure, PVR, cardiac output, etc.). The multivariate Cox regression analysis based on the proportional-hazard model was provided to determine the independency of parameters influenced on prognosis in our population with a focus on CAVI. Statistical significance was expressed by a P-value < 0.05.
Results
Baseline Characteristics of the Study Population
During the study period in our center, 387 patients with pulmonary hypertension were treated and only 103 had the newly diagnosed IPAH/HPAH. In the analysis we included 89 patients with IPAH/(HPAH), who corresponded to inclusion/exclusion criteria. Fourteen patients were not included due to not matching eligible criteria. Among them, 10 had diabetes mellitus, 1 a history of stroke, and 6 had systemic arterial hypertension. Most included patients were women (84.3%), at functional class III (64%) with a mean age of 42.7±13.1 years. Only 3 patients had a family history of PAH. The first line-specific sildenafil treatment was administered to 75 (84.3%) patients and iloprost was added for 24 (27%) participants. The initial double combination was prescribed only for 24 (27%) patients. Thirteen (14.6%) patients were chronic calcium channel blocker responders. Endothelin receptor blockers were used only for 2 patients as the third drug, because of its late inclusion in the National drug supply program and more preferable prescribing for patients with PAH associated with congenital heart diseases. The mean follow-up period was 33.7±23.7 (min 6 – max 79) months.
We accepted CAVI value ≥8 as an indicator of abnormal systemic arterial stiffness and the study population was divided into two groups based on its parameter. Eighteen patients with IPAH/HPAH had CAVI ≥8 and 71 patients a CAVI <8. Demographic, clinical, and laboratory characteristics of groups are summarized in Table 1. Patients with increased systemic arterial stiffness were older and had the higher levels of NT-proBNP and hematocrit, the higher administered dose of sildenafil, the more loop diuretic and warfarin usage rate, the higher the international normalization ratio, and the lower glomerular filtration rate. Their functional capacity at the time of admittance to our center was reduced (less 6MWT distance and oxygen saturation) compared to the group with CAVI <8. But patients with normal CAVI had more cases of thyroid disorders.
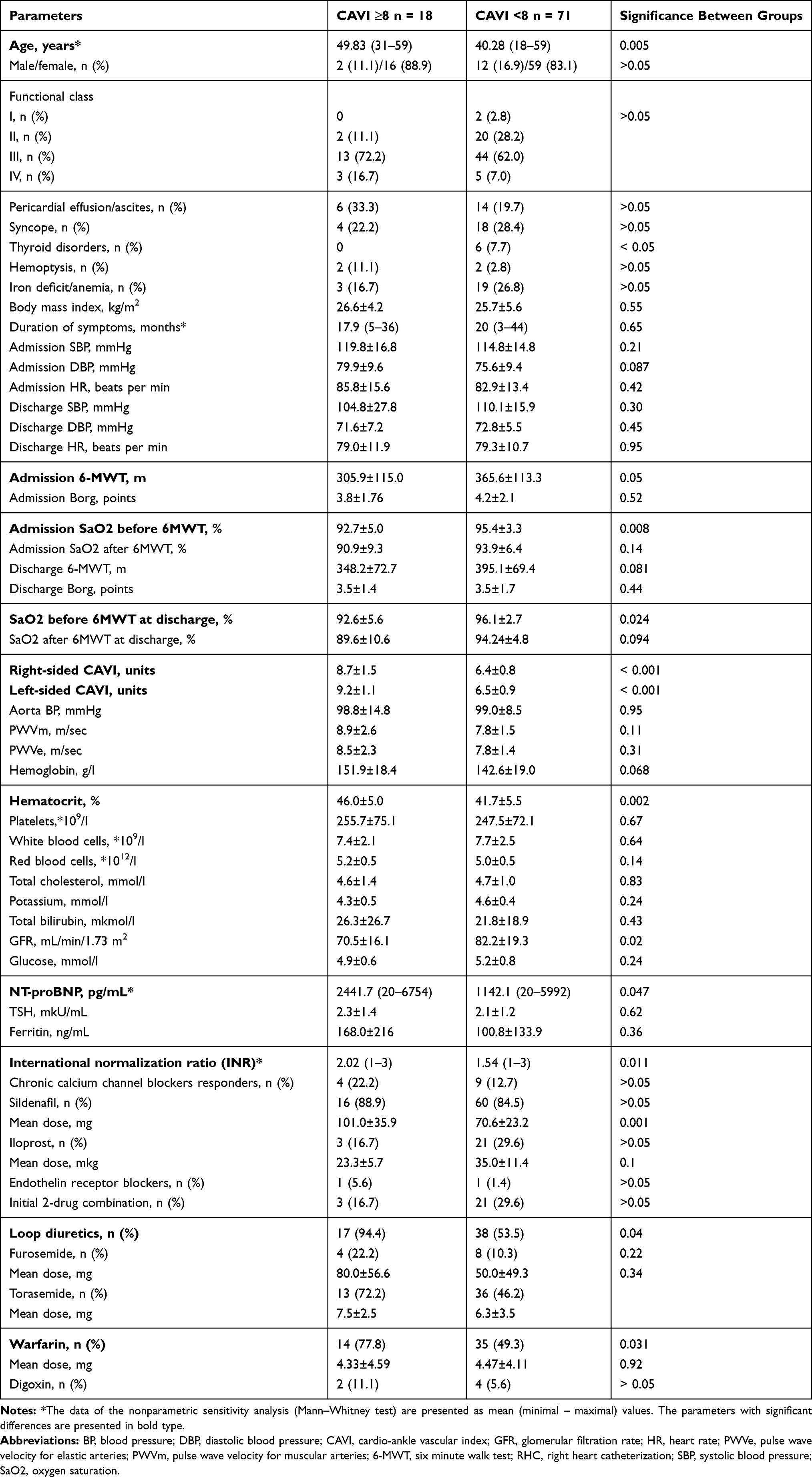 |
Table 1 Characteristics of Patients with Idiopathic Pulmonary Arterial Hypertension in Groups of CAVI ≥8 and <8 (M±SD, n (%)) |
The TTE and RHC parameters of our groups are shown in Table 2. Patients with CAVI ≥8 had a significantly reduced end-diastolic (73.79±18.94 vs 87.35±16.69 mL, P < 0.009) and end-systolic (25.71±9.56 vs 33.55±10.33 mL, P < 0.01) volumes of the left ventricle, the higher right ventricle thickness (0.77±0.12 vs 0.62±0.20 mm, P < 0.006), and the lower TAPSE (13.38±2.15 vs 15.98±4.4 mm, P < 0.018). We noted the significant differences between groups in the parameters which are used for the determination of the left ventricular diastolic dysfunction. Patients with abnormal systemic arterial stiffness had the reduced E/A ratio and the higher deceleration time, which could be the results of the more prominent impairing of the left ventricle diastolic function and could explain the more increased left atrial square in this group than in the group with CAVI <8 – 20.01±13.95 vs 16.49±3.5 cm2, P = 0.055. The Pearson correlation analysis confirmed the significant relationships between CAVI and the end-diastolic (r = −0.28, P = 0.042) and the end-systolic (r = −0.25, P = 0.065) left ventricle volumes; E/A ratio (r = −0.36, P = 0.014) and DT (r = 0.33, P = 0.02).
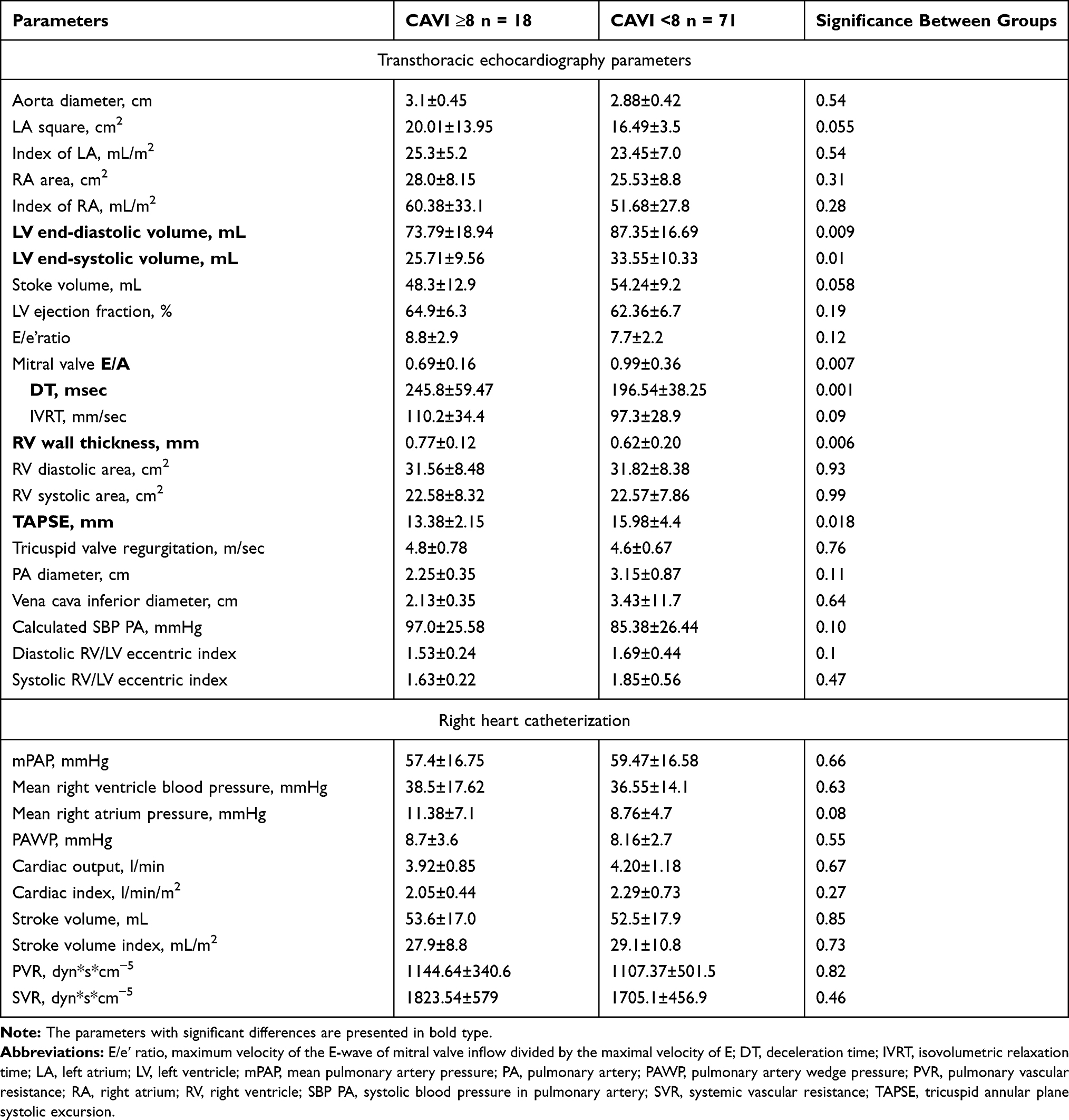 |
Table 2 Echocardiography and Right Heart Catheterization Parameters in Groups of CAVI ≥8 and <8 (M±SD) |
RHC data did not differ significantly between groups, except a tendency to the higher level of the right atrial pressure in patients with CAVI ≥8 – 11.38±7.1 vs 8.76±4.7 mmHg, P < 0.08.
Survival in the Study Population and Predictors of Poor Prognosis
During the follow-up period 24 patients died mainly due to the progression of right heart failure or sudden cardiac death. In one patient the cause of death was bowel cancer with perforation, which was operated on, but the increasing right heart failure complicated the patient’s recovery. Cumulative study population survival is present in Figure 1. According to Kaplan–Meier analysis estimated survival was 61.2±3.8% with 95% CI 53.8–68.6. In non-survived patients, the mean CAVI was significantly higher than in patients who survived – 7.8±1.56 vs 6.8±1.2, P = 0.004. But they did not differ significantly by the PWVe and PWVm – 8.1±1.8 and 8.1±2.39 m/sec vs 8.39±2.25 and 7.99±1.56 m/sec, respectively, NS.
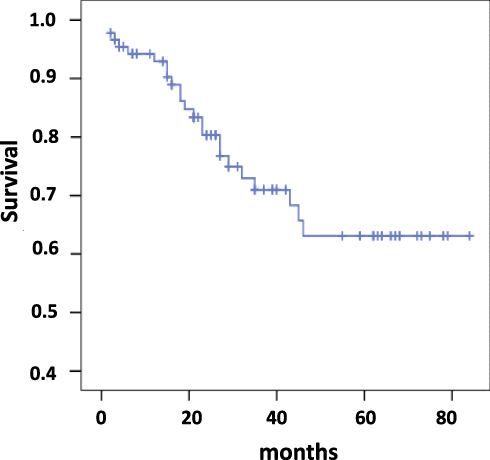 |
Figure 1 Survival since the time of study initiation (n = 89). |
We provided the univariate analysis for the evaluation of the significant survival predictors in our study population. The patient characteristics which correlated with prognosis in reported literature data6,14 were included in this analysis and are present in Table 3.
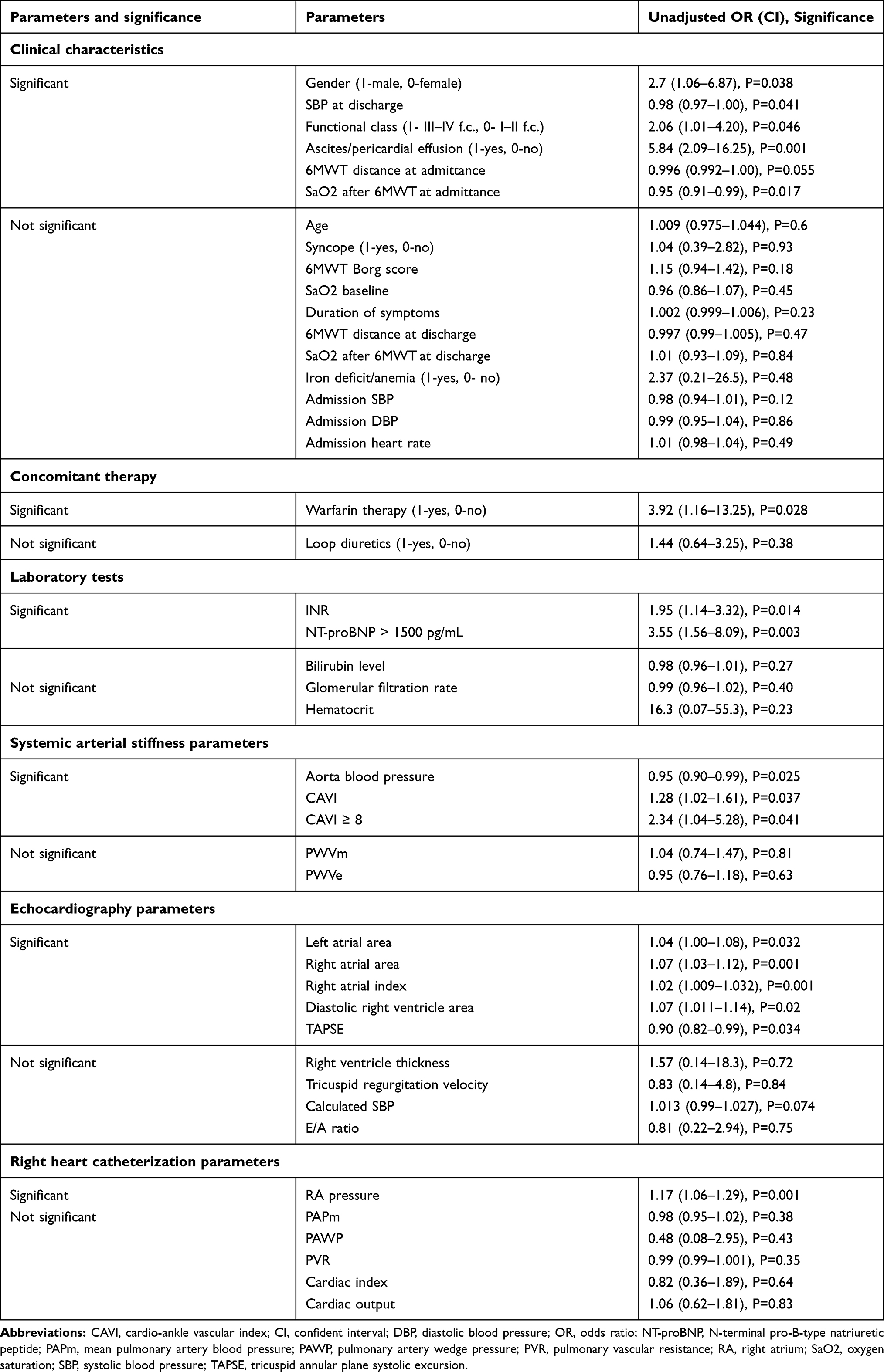 |
Table 3 Cox Proportional Hazards Regression Analysis in General IPAH/HPAH Population |
Male gender, the NT-proBNP level ≥1500 pg/mL, the signs of ascites/pericardial effusion, III–IV functional class WHO, the oxygen saturation after 6MWT, the warfarin therapy, the international normalization ratio, the right atrium area, and the right atrium pressure confirmed the significant influence on death rate in our IPAH/HPAH patients. Also, higher SBP level at the time of admission was linked with the lowest odds ratio. The left atrial area, the right ventricle area in diastole, and TAPSE were associated with a higher rate of death too. The iron deficit/anemia status, age, 6-MWT distance at discharge, thyroid disorders, and other TTE/RHC parameters did not associate significantly with prognosis.
The CAVI ≥8 increased significantly the risk of mortality by2.34 times (CI 1.04–5.28, P = 0.041). The estimated Kaplan–Meier survival (Figure 2) in the patients with CAVI ≥8 was only 46.7±7.18% (CI 32.62–60.76) compared to patients with CAVI <8 – 65.6±4.2% (CI 57.39–73.91), Log rank χ2 = 4.46, P = 0.035.
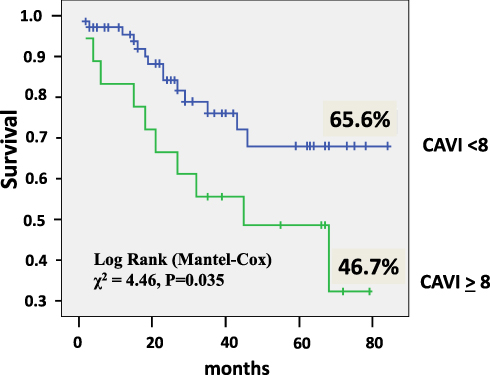 |
Figure 2 Kaplan–Meier survival of idiopathic pulmonary arterial hypertension patients in relation to cardio-ankle vascular index (CAVI). |
Multivariate Cox regression analysis based on the proportional-hazard models was used for the evaluation of the CAVI independency in the prediction of death in IPAH patients (Table 4). The male gender (OR = 2.21, CI 1.1–10.0), the III–IV functional class WHO (OR = 1.33, CI 1.11–1.99), the NT-proBNP level >1500 pg/mL (OR = 2.2, CI 1.063–7.6), the oxygen saturation after 6MWT at time of admittance (OR = 0.87, CI 0.79–0.96), and the higher right atrial pressure (OR = 1.21, CI 1.07–1.36) demonstrated the independent significance in the increasing of the mortality risk in our study population. The CAVI reduced but saved its relevance (OR = 1.13, CI 1.001–1.871). Such parameters as the presence of ascites/pericardial effusion (OR = 5.8, CI 0.88–7.2), the SBP at discharge (OR = 0.99, CI 0.97–1.006), and the international normalization ratio (OR = 2.54, CI 0.95–6.7) were close to being significant.
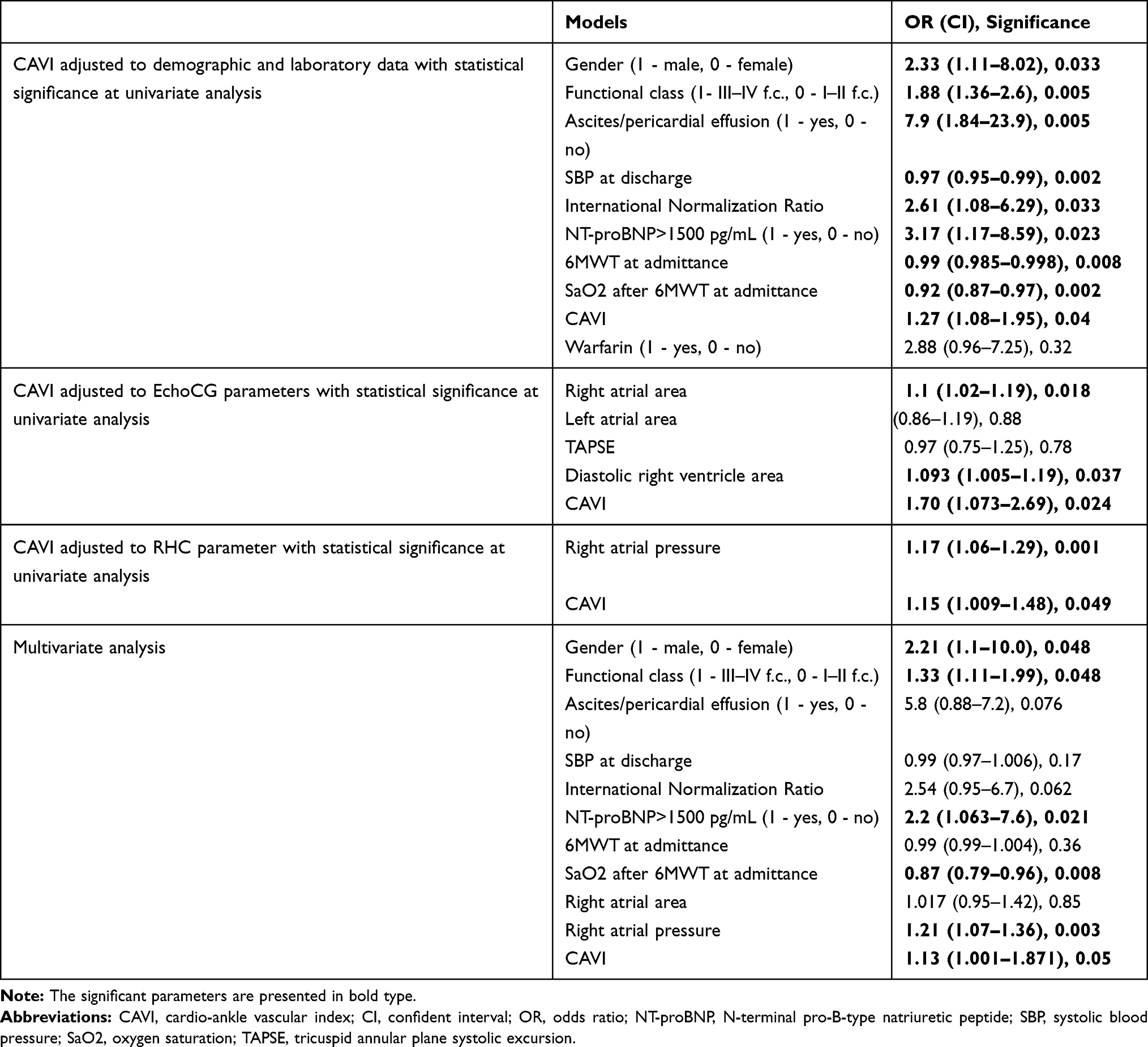 |
Table 4 Cox Proportional Hazards Regression Model Analysis in General IPAH/HPAH Population |
Discussion
The systemic arterial stiffness in patients with pulmonary hypertension was evaluated in some studies. The objectives to provide these studies were excellently summarized in the review of N. Nickel et al.43 The most well-known is that endothelial dysfunction plays a role in the development of PAH and is the target for specific drugs in the treatment of PAH patients.5 But it is hard to imagine the isolated damage to the pulmonary arteries and no changes in the systemic arteries. Indeed, now we have some evidence which indicates the systemic artery changes in pulmonary hypertension: the tendency to increase the coronary heart disease prevalence in PAH patients,53,54 the reduction in brachial artery dilation in 2.7% of patients with IPAH and 6.3% of patients with scleroderma and PAH,55 the lower cerebral blood flow at rest and with exercise in patients with PAH during the measuring of mean flow velocity in the middle cerebral artery,56 the significant albumin excretion (as a marker of the endothelial dysfunction) in 15–23% of patients with PAH without known kidney disease and traditional CV risk factors,57 and the morphological changes in nailfold capillaries and sublingual vessels.58
These systemic artery changes could be mediated by some factors. The first one is genetic. The BMPR2 mutation could be found in 75% of HPAH24 and its presence was associated with proteinuria and increased C-reactive protein level in NP. In Nickel et al's study,57 the authors considered BMPR2 signaling has an impact on vascular homeostasis through the expression of collagen 4, ephrin A1, endothelial nitric oxide, caveolin-1, and others59,60 and therefore, reduced BMPR2 signaling could influence the renal perfusion and protein handling (leads to albuminuria) and cytokine circulation.
The second factor is the systemic inflammation. A study by E. Soon and colleagues found increased levels of tumor necrosis factor-α, interferon-γ and some interleukins in patients with IPAH and HPAH compared to the control group.26 The patients with elevated interleukin-6 had worse survival rates.26 The role of inflammation markers in impairing the aorta stiffness was reported in many publications.61–63 Therefore, if PAH patients have the increased inflammation factors in circulation it could lead to systemic artery damage.
The third factor is metabolic disorders mediated by peripheral circulation worsening, hypoxia, and thyroid disorders. The poor peripheral circulation as a result of the reduced cardiac output and the sedentary life of PAH patients might lead to skeletal muscle dysfunction and insulin resistance.64 Hypoxia and thyroid dysfunction (subclinical hypothyroidism rate at PAH is ranged from 20% to 49.1%)65–67 are associated with lipid disorders, increased oxidative stress, and the circulation of proinflammation factors that correlated with PAH parameters (PVR, right ventricle function). Thus, in PAH patients, the same metabolic disturbances could be found as in patients of high cardiovascular risk and they might be linked with increased systemic artery stiffness.35–40
The systemic arterial stiffness could be evaluated by different methods. A summarized comparison of some methods is presented in Table 5. 45 The current gold standard is PWV assessment based on measurements of the time for the pressure pulse to go between two points. This time depends on arterial stiffness and blood pressure. The CAVI is a more direct measure of the systemic artery stiffness and it has some advantages: blood pressure independence of arterial elasticity, the longer evaluated arterial segment (includes the ascending aorta and ankle), and relative simplicity with low cost. The two indices (the stiffness parameter β and the Bramwell-Hill formula) were combined for the CAVI calculation.45,68,69 The weak correlation with systolic blood pressure and significantly better reproducibility compared to brachial-ankle PWV was demonstrated before.45,70 The PWV could depend on gender and heart rate also, which is not reported for CAVI. A study by Schillaci et al showed that CAVI, but not PWV, was associated with inappropriately high left ventricular masses for a given cardiac workload and with low midwall systolic function.71 It means that CAVI may have a relationship to the left ventricular structure and function that is independent of blood pressure levels and it is more sensitive than PWV.
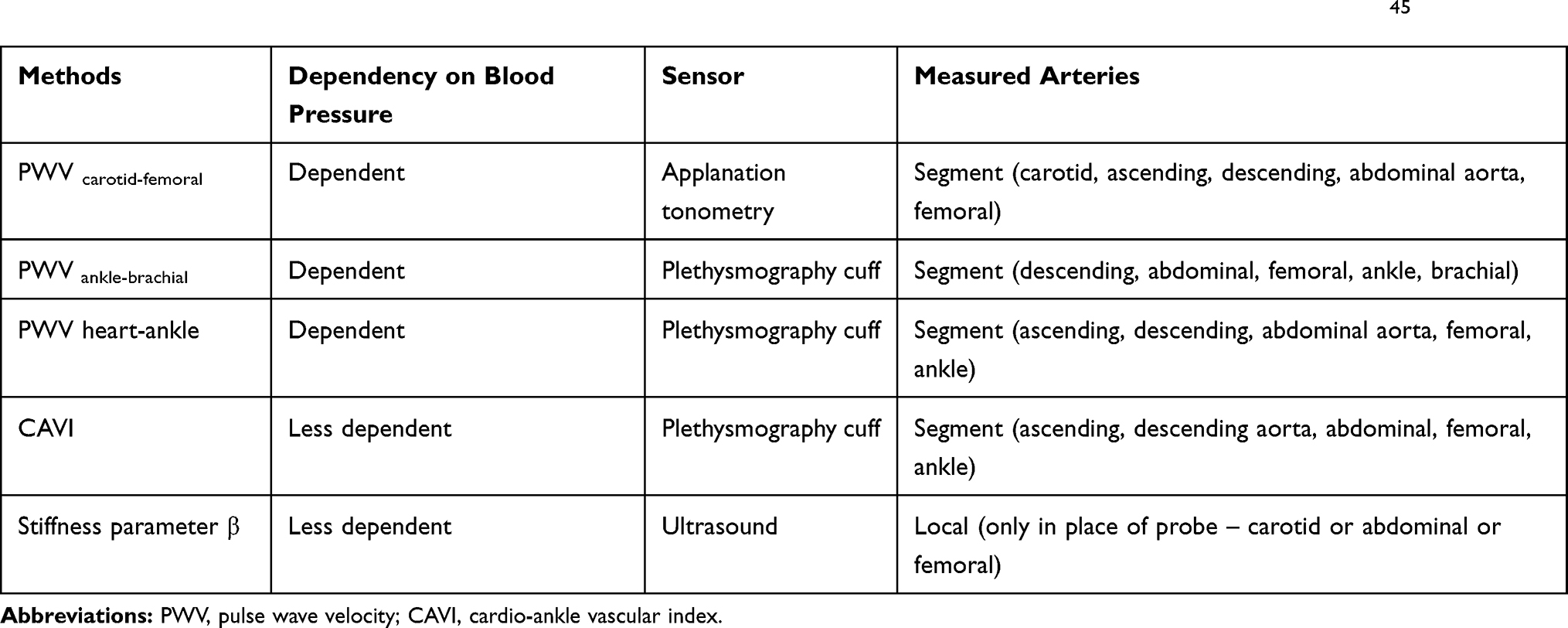 |
Table 5 The Comparison of Different Arterial Stiffness Measurement Methods (Adapted with T. Miyoshi and H. Ito)45 |
The CAVI has a very strong correlation with the stiffness parameter β that was measured by transesophageal EchoCG72 and ECG-gated multidetector CT scan.73 The stiffness parameter β represents the logarithmic change in blood pressure required to increase the arterial diameter. That is why it is relatively independent of blood pressure. But this parameter evaluation is difficult in practice, because it needs synchronous, precise assessment of both pressure and diameter changes in a given artery.68 Furthermore, these measurements are obtained by assessing the only local segment of the artery, while CAVI reflects the artery stiffness from the origin of the aortic valve to the ankle region, and blood pressure is measured at the upper arm.
The method of flow-mediated vasodilation in the brachial artery was used in some studies for the evaluation of endothelium dysfunction in PAH.55,74 The abnormal brachial artery dilation was found in 2.7% of patients with IPAH and 6.3% of patients with scleroderma and the severity of the impairment significantly correlated with clinical and hemodynamic parameters of PAH patients.74 But it needs to be noted that the flow-medicated vasodilation method reflects the endothelium dysfunction but not the arterial stiffness. In contrast, in the American Heart Association recommendations, the CAVI obtained a rating “Class I, level of evidence B” for evaluation of the systemic arterial stiffness.75
Thus, the vascular changes in the patients with PAH are not limited to the lungs. We have a lot of evidence of the vascular dysfunction in different organs, not only in the pulmonary arteries and the right heart. These systemic manifestations could be realized through metabolic disorders, inflammation activation, and genetic injury which lead to systemic vascular dysfunction. That is why it is important to find these abnormalities, to evaluate their significance in the risk stratification, to treat them, and improve the PAH patient's survival. The CAVI is a very simple and operator independent parameter with good reproducibility and sensitivity for the systemic arterial stiffness evaluation that was not used in PAH patients before.
Recently we published the results of our study on the comparison of arterial stiffness parameters in patients with IPAH, systemic hypertension, and healthy people who were adjusted to age.44 We established that mean CAVI was the highest in the group with systemic hypertension, but did not differ significantly from mean CAVI in the group newly diagnosed with IPAH. The healthy persons had significantly reduced CAVI, PWVm and PWVe values than patients with IPAH, despite the comparable office and aorta blood pressure. Thus, we confirmed the hypothesis about increasing the systemic artery stiffness in patients with pulmonary hypertension and we were the first who used CAVI for the systemic arterial stiffness evaluation in IPAH patients.
The results of our study about abnormalities in the systemic arteries of PAH patients were in compliance with the other reports. Thus, Chamorro et al detected the impairment of the endothelial function and increased arterial stiffness which were assessed as the reduced flow-mediated dilation of the brachial artery and higher PWVe in patients with PAH in comparison with adjusted health subjects - 10.6±3.9 vs 7.5±6.3 (p < 0.05) and 8.4±2.5 vs 7.3 ± 1.6 m/sec (p < 0.05), respectively.76 Also, in patients with chronic thromboembolic pulmonary hypertension Sznajder et al found significantly higher PWVe than in the control group which was matched to age, gender, and concomitant diseases.77
In a study by Peled et al, it was shown that endothelial dysfunction was present in IPAH and in PAH associated with scleroderma, but the peripheral arterial stiffness was normal in PAH and not to be correlated with PAH per se, and only a tendency to be higher was found in patients with scleroderma, unrelated to the presence of PAH.74 The differences between our and N. Peled’s study results could be explained by the different arterial stiffness method evaluations. We evaluated the aorta segment stiffness, but they - the peripheral arterial stiffness (by finger plethysmography). The peripheral circulation could be poor in PAH patients and methods for the arterial stiffness evaluation should be less sensitive to its changes.
In both indicated studies,76,77 it was suggested that increased systemic arterial stiffness could be associated with poor prognosis in pulmonary hypertension, but only in the present study did we test this suggestion, and confirmed that increased CAVI is associated with a higher rate of death, independent to other traditional risk factors – OR = 1.13, CI 1.001–1.871. In non-survived patients the mean CAVI was significantly higher than in patients who survived – 7.8±1.56 vs 6.8±1.2, P = 0.004, while PWVe and PWVm did not differ significantly and were not associated with the prognosis. Taking into consideration the previous study's73 results about more prominent and strong relations of CAVI with the left ventricle structure compared to PWV we could conclude that CAVI is more sensitive for the evaluation of arterial stiffness changes in PAH patients and should be a preferable marker for survival risk stratification. However, the CAVI had a significant correlation with PWV in our study and in other reports.44,71 Also, we should take into consideration our PAH patients had low systolic blood pressure, which was associated with poor prognosis, and PWV might underestimate arterial stiffness at such low systemic pressure, whereas CAVI is independent from blood pressure. This might be the reason why only CAVI predicted the survival of IPAH.
A very interesting question for discussion is how systemic arterial stiffness could be associated with prognosis. One point of view is the hypothesis of the reverse causality. Patients with a more severe PAH course could have a more enhanced endothelial dysfunction, inflammation activity, and metabolic disorders that lead to an increase of the systemic artery damage, and the poor prognosis is mediated not by this artery damage but by the processes that were the cause of it. From the other side, the arterial stiffness links with the left ventricle structure and function. It was confirmed in many studies.78,79 The diastolic dysfunction could be diagnosed before the left ventricle hypertrophy development in hypertensives and correlated with arterial stiffness.80 The left ventricle function is very important for PAH patients. Usually, the left ventricle has suffered because of the reduced preload and the compression by the enlarged right ventricle. The enhancing of the systemic arterial stiffness could lead to the increasing of the left ventricle afterload and heart structure changes. In our study we found significant correlations between CAVI and the diastolic function parameters in PAH patients that give us the reason to suggest the direct impact of the increased systemic artery stiffness on the left ventricle and, in this way, on prognosis.
In our previous study, we found that the CAVI correlated with age. In this study the group with CAVI ≥8 was older than the group with CAVI <8 as could be expected. For many PAH populations the age was associated with survival,6–11 but not in our study. At the same time the CAVI was linked with death in our population. The Hoeper et al analysis indicated that in recent decades the mean age of PAH patients has increased.81 If patients enrolled between 1981 and 1985 in the US National Institutes of Health (NIH) registry were at mean age 36 years at the time of diagnosis,82 now in the Western countries the mean age has increased - to 65 years in Germany83 and even to 69 years in the Swedish register.84 The older PAH patients are at higher mortality risk because of their age itself and the higher rate of concomitant diseases. In our study we included only patients with newly diagnosed IPAH at age <60 and without concomitant illness which could impact on the arterial stiffness parameters. That is why their prognosis was associated not with age but with PAH parameters and the state of systemic arteries.
Besides CAVI the other traditional death risk factors confirmed their significance: male gender, ascites/pericardial effusion, right atrial blood pressure, functional class IV WHO, NT-proBNP >1500 pg/mL. The higher international normalization ratio was associated with a higher death rate too at unadjusted analysis. In previous reports the anticoagulation treatment demonstrated ambiguous results for survival: the positive in IPAH patients of the COMPERA trial,85 no effect in REVEAL IPAH population86, and the negative influence in patients with connective tissue diseases in both indicated above analyses. We did not find the correlation between the 6-minute walk distance and survival, in spite of many other reports about the predictive role of this parameter. But in the study by Wensel et al, 6MWT results were not significant markers of survival, while the values of the cardiopulmonary test were.87 It could be explained by the influence of different factors on the 6-minute walk distance, including concomitant diseases, age, PAH severity, patient psychological status, and medical staff's skills in carrying out the test. The cardiopulmonary test could be more objective, but more complicated and expensive.
Thus, for our newly-diagnosed IPAH population, the CAVI was an independent predictor of survival as well as some other conventional and commonly accepted parameters. In the future, it may be necessary to provide larger multicentral trials for the validating of this parameter as a novel predictor of death and evaluation of its changes on treatment and their connections with prognosis.
Conclusion
This study provided the results on long-term outcomes in the IPAH/family PAH patients who were treated in the Ukrainian referral center. It demonstrated the overall survival rate was 61.2%. The systemic arterial stiffness was significantly higher in non-survived patients. In the group of patients with abnormal CAVI (≥8), the survival rate was significantly lower compared to the group with CAVI <8 – 46.7±7.18% vs 65.6±4.2% (P = 0.035). Our analysis suggested that abnormal CAVI, but not PWV, could be a new, independent predictor of death in the IPAH population and be used for better risk stratification in this patient population. Also, the CAVI might be the target for treatment. However, these suggestions need to be confirmed in multicenter studies.
Limitations of the Study
The present study is a single-center one with relatively small patient cohort. But in the Ukraine the only department is the referral pulmonary hypertension center that could provide RHC. The criteria of inclusion/exclusion in our study were very restricted – newly-diagnosed IPAH/HPAH, age <60 years, and no concomitant diseases which could impact on the systemic arterial stiffness. In spite of this, we found 89 patients with such an orphan disease like IPAH/HPAH without other abnormalities and could follow them for at least 6 months. Our statistical methods were acceptable for providing survival analysis. The treatment of our patients was limited by only some drugs which are available in the Ukraine – sildenafil and inhaled iloprost. Intravenous prostanoids were not available in the Ukraine till now, which might have influenced the survival results. Despite the national drug supply program existing in the Ukraine, and patients being able to take the specific therapy for free, there are some obstacles in the supply process that lead to irregular treatment. The role of the irregularity was not evaluated in our study, but it could influence results. Also, we did not analyze the effect of PAH treatment on the CAVI, which could be the topic for future studies, as well as the role of this parameter change during the follow-up. The positive parameter dynamic might also be connected with patient prognosis as it was proved for the increasing of functional class, the 6 MWT distance, or the decreasing of the level of NT-proBNP. We did not assess the inflammation markers in our population and their correlations with CAVI that could suggest the directions for future studies.
Acknowledgments
Special thanks to Evgeniy Titov for providing echocardiography and Irina Zhyvilo for providing arterial stiffness measurements.
Disclosure
The authors report no conflicts of interest in this work and no special funds.
References
1. Simonneau G, Montani D, Celermajer D, et al. Number 4 in the series “Proceedings of the 6th World Symposium on Pulmonary Hypertension” Edited by N. Galiè, V.V. McLaughlin, L.J. Rubin and G. Simonneau. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53:1801913. doi:10.1183/13993003.01913-2018
2. Humbert M, Sitbon O, Chaouat A, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med. 2006;173(9):1023–1030. doi:10.1164/rccm.200510-1668OC
3. Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122(2):156–163. doi:10.1161/CIRCULATIONAHA.109.911818
4. Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351:1655–1665. doi:10.1056/NEJMra035488
5. Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016;37(1):67–119. doi:10.1093/eurheartj/ehv317
6. D’Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115:343–349. doi:10.7326/0003-4819-115-5-343
7. Boucly A, Weatherald J, Savale L, et al. Risk assessment, prognosis and guideline implementation in pulmonary arterial hypertension. Eur Respir J. 2017;50:1700889. doi:10.1183/13993003.00889-2017
8. Humbert M, Sitbon O, Yaici A, et al. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J. 2010;36:549–555. doi:10.1183/09031936.00057010
9. Thenappan T, Shah SJ, Rich S, Tian L, Archer S, Gomberg-Maitland M. Survival in pulmonary arterial hypertension: a reappraisal of the NIH risk stratification equation. Eur Respir J. 2010;35:1079–1087. doi:10.1183/09031936.00072709
10. Thenappan T, Glassner C, Gomberg-Maitland M. Validation of the pulmonary hypertension connection equation for survival prediction in pulmonary arterial hypertension. Chest. 2012;141:642–650. doi:10.1378/chest.11-0969
11. Weatherald J, Boucly A, Chemla D, et al. Prognostic value of follow-up hemodynamic variables after initial management in pulmonary arterial hypertension. Circulation. 2018;137(7):693–704. doi:10.1161/CIRCULATIONAHA.117.029254
12. Kylhammar D, Kjellström B, Hjalmarsson C, et al. A comprehensive risk stratification at early follow-up determines prognosis in pulmonary arterial hypertension. Eur Heart J. 2018;39:4175–4181. doi:10.1093/eurheartj/ehx257
13. Hoeper MM, Kramer T, Pan Z, et al. Mortality in pulmonary arterial hypertension: prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur Respir J. 2017;50:1700740. doi:10.1183/13993003.00740-2017
14. Benza RL, Miller DP, Gomberg-Maitland M, et al. Predicting survival in pulmonary arterial hypertension: insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management (REVEAL). Circulation. 2010;122:164–172. doi:10.1161/CIRCULATIONAHA.109.898122
15. Benza RL, Gomberg-Maitland M, Miller DP, et al. The REVEAL registry risk score calculator in patients newly diagnosed with pulmonary arterial hypertension. Chest. 2012;141:354–362. doi:10.1378/chest.11-0676
16. Benza RL, Miller DP, Foreman AJ, et al. Prognostic implications of serial risk score assessments in patients with pulmonary arterial hypertension: a registry to evaluate early and long-term pulmonary arterial hypertension disease management (REVEAL) analysis. J Heart Lung Transplant. 2015;34:356–361. doi:10.1016/j.healun.2014.09.016
17. Anwar A, Ruffenach G, Mahajan A, Eghbali M, Umar S. Novel biomarkers for pulmonary arterial hypertension. Respir Res. 2016;17:88. doi:10.1186/s12931-016-0396-6
18. Benedetto P, Guggino G, Manzi G, et al. Interleukin-32 in systemic sclerosis, a potential new biomarker for pulmonary arterial hypertension. Arthritis Res Ther. 2020;22:127. doi:10.1186/s13075-020-02218-8
19. Luk K, Ip C, Gong M, et al.; and International Health Informatics Study (IHIS) Network. A meta-analysis of soluble suppression of tumorigenicity 2 (sST2) and clinical outcomes in pulmonary hypertension. J Geriatr Cardiol. 2017;14(12):766–771. doi:10.11909/j.issn.1671-5411.2017.12.007.
20. Nold-Petry CA, Nold MF, Zepp JA, Kim S-H, Voelkel NF, Dinarello CA. IL-32-dependent effects of IL-1 on endothelial cell functions. Proc Natl Acad Sci U S A. 2009;106(10):3883–3888. doi:10.1073/pnas.0813334106
21. Nold-Petry CA, Rudloff I, Baumer Y, et al. IL-32 promotes angiogenesis. J Immunol. 2014;192:589–602. doi:10.4049/jimmunol.1202802
22. Versari D, Daghini E, Virdis A, Ghiadoni L, Taddei S. Endothelium-dependent contractions and endothelial dysfunction in human hypertension. Br J Pharmacol. 2009;157:527–536. doi:10.1111/j.1476-5381.2009.00240
23. Thenappan T, Ormiston M, Ryan J, Archer S. Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ. 2018;360:5492. doi:10.1136/bmj.j5492
24. Soubrier F, Chung WK, Machado R, et al. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D13–21. doi:10.1016/j.jacc.2013.10.035
25. Perros F, Dorfmuller P, Humbert M. Current insights on the pathogenesis of pulmonary arterial hypertension. Semin Respir Crit Care Med. 2005;26(4):355–364. doi:10.1055/s-2005-916149
26. Soon E, Holmes AM, Treacy CM, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. 2010;122:920–927. doi:10.1161/CIRCULATIONAHA.109.933762
27. Voelkel NF, Gomez-Arroyo J, Abbate A, Bogaard H, Nicolls M. Pathobiology of pulmonary arterial hypertension and right ventricular failure. Eur Respir J. 2012;40:1555–1565. doi:10.1183/09031936.00046612
28. Cracowski JL, Chabot F, Labarère J, et al. Proinflammatory cytokine levels are linked to death in pulmonary arterial hypertension. Eur Respir J. 2014;43:915–917. doi:10.1183/09031936.00151313
29. Heresi GA, Aytekin M, Hammel JP, Wang S, Chatterjee S, Dweik R. Plasma interleukin-6 adds prognostic information in pulmonary arterial hypertension. Eur Respir J. 2014;43:912–914. doi:10.1183/09031936.00164713
30. Perros F, Dorfmuller P, Montani D, et al. Pulmonary lymphoid neogenesis in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;185:311–321. doi:10.1164/rccm.201105-0927OC
31. Stacher E, Graham BB, Hunt JM, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respiratory Critical Care Med. 2012;186:261–272. doi:10.1164/rccm.201201-0164OC
32. Ghofrani HA, Seeger W, Grimminger F. Imatinib for the treatment of pulmonary arterial hypertension. N Engl J Med. 2005;353(13):1412–1413. doi:10.1056/NEJMc051946
33. Speich R, Treder U, Domenighetti G, Huber LC, Ulrich S. Weaning from intravenous prostanoids and normalization of hemodynamics by long-term imatinib therapy in severe idiopathic pulmonary arterial hypertension. Int J Clin Pharm. 2014;36(2):256–260. doi:10.1007/s11096-013-9881-x
34. Hoeper MM, Barst RJ, Bourge RC, et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation. 2013;127(10):1128–1138. doi:10.1161/CIRCULATIONAHA.112.000765
35. Rodríguez‐Iturbe B, Pons H, Quiroz Y, Lanaspa MA, Johnson RJ. Autoimmunity in the pathogenesis of hypertension. Nat Rev Nephrol. 2014;10:56–62. doi:10.1038/nrneph.2013.248
36. Caillon A, Schiffrin EL. Role of inflammation and immunity in hypertension: recent epidemiological, laboratory, and clinical evidence. Curr Hypertens Rep. 2016;18:21. doi:10.1007/s11906-016-0628-7
37. McMaster WG, Kirabo A, Madhur MS, Harrison DG. Inflammation, immunity, and hypertensive end‐organ damage. Circ Res. 2015;116:1022–1033. doi:10.1161/CIRCRESAHA.116.303697
38. Chen TH, Gona P, Sutherland PA, et al. Long‐term C‐reactive protein variability and prediction of metabolic risk. Am J Med. 2009;122:53–61. doi:10.1016/j.amjmed.2008.08.023
39. Tomiyama H, Shiina K, Matsumoto‐Nakano C, et al. The contribution of inflammation to the development of hypertension mediated by increased arterial stiffness. J Am Heart Assoc. 2017;6:e005729. doi:10.1161/JAHA.117.005729;.
40. Mozos I, Malainer C, Horbańczuk J, et al. Inflammatory Markers for Arterial Stiffness in Cardiovascular Diseases. Front Immunol. 2017;8:1058. doi:10.3389/fimmu.2017.01058
41. Mahmud A, Feely J. Arterial stiffness is related to systemic inflammation in essential hypertension. Hypertension. 2005;46:1118–1122. doi:10.1161/01.HYP.0000185463.27209.b0
42. Arnold N, Gori T, Schnabel R, et al. Relation between arterial stiffness and markers of inflammation and hemostasis data from the population-based gutenberg health study. Sci Rep. 2017;7:6346. doi:10.1038/s41598-017-06175-2
43. Nickel N, Yuan K, Dorfmuller P, et al. Beyond the Lungs: systemic Manifestations of Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 2020;201(2):148–157. doi:10.1164/rccm.201903-0656CI
44. Radchenko G, Zyvylo I, Titov E, Sirenko Y. Systemic arterial stiffness in new diagnosed idiopathic pulmonary arterial hypertension patients. Vasc Health Risk Manag. 2020;16:29–39. doi:10.2147/VHRM.S230041
45. Miyoshi T, Ito H. Assessment of arterial stiffness using the cardio-ankle vascular index. Pulse. 2016;4(1):11–23. doi:10.1159/000445214
46. Guyatt G, Sullivan M, Thompson P, et al. The 6-minute walk: a new measure of exercise capacity in subjects with chronic heart failure. Can Med Assoc J. 1985;132:919–923.
47. ATS Statement. Guidelines for the six minute walk test. Am J Respir Crit Care Med. 2002;166:111–117. doi:10.1164/ajrccm.166.1.at1102
48. Lang RM, Badano LP, Mor-Avi V, et al. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the american society of echocardiography and the european association of cardiovascular imaging. J Am Society Echocardiography. 2015;28(1):1–39. doi:10.1016/j.echo.2014.10.003
49. 2018 ESC/ESH Guidelines for the management of arterial hypertension. The Task Force for the management of arterial hypertension of the European Society of Cardiology (ESC) and the European Society of Hypertension. Eur Heart J. 2018;00:1–98. doi:10.1093/eurheartj/ehy339
50. Fukuda-Denshi Company L. Tokyo, Japan. Available from: https://fukuda.com/products_category/vascular-screening/and.https://fukuda.com/wp-content/uploads/2020/07/VS-2000_Catalog_8Prev2.pdf.
51. Cheuk-Kwan S. Cardio-ankle vascular index (CAVI) as an indicator of arterial stiffness Integrated. Blood Pressure Control. 2013;6:27–38. doi:10.2147/IBPC.S34423
52. Yambe T, Yoshizawa M, Saijo Y, et al. Brachio-ankle pulse wave velocity and cardio-ankle vascular index (CAVI). Biomed Pharmacother. 2004;58(Suppl 1):S95–S98. doi:10.1016/S0753-3322(04)80015-5
53. Shimony A, Eisenberg MJ, Rudski LG, et al. Prevalence and impact of coronary artery disease in patients with pulmonary arterial hypertension. Am J Cardiol. 2011;108:460–464. doi:10.1016/j.amjcard.2011.03.066
54. Anand V, Roy SS, Archer SL, et al. Trends and outcomes of pulmonary arterial hypertension-related hospitalizations in the United States: analysis of the nationwide inpatient sample database from 2001 through 2012. JAMA Cardiol. 2016;1:1021–1029. doi:10.1001/jamacardio.2016.3591
55. Hughes R, Tong J, Oates C, Lordan J, Corris PA. Evidence for systemic endothelial dysfunction in patients and first-order relatives with pulmonary arterial hypertension. Chest. 2005;128(6, Suppl):617S. doi:10.1378/chest.128.6_suppl.617S
56. Malenfant S, Brassard P, Paquette M, et al. Compromised cerebrovascular regulation and cerebral oxygenation in pulmonary arterial hypertension. J Am Heart Assoc. 2017;6:e006126. doi:10.1161/JAHA.117.006126
57. Nickel NP, de Jesus Perez VA, Zamanian RT, et al. Low-grade albuminuria in pulmonary arterial hypertension. Pulm Circ. 2019;9(2):1–9. doi:10.1177/2045894018824564
58. Riccieri V, Vasile M, Iannace N, et al. Systemic sclerosis patients with and without pulmonary arterial hypertension: a nailfold capillaroscopy study. Rheumatology. 2013;52:1525–1528. doi:10.1093/rheumatology/ket168
59. Rhodes CJ, Im H, Cao A, et al. RNA sequencing analysis detection of a novel pathway of endothelial dysfunction in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015;192:356–366. doi:10.1164/rccm.201408-1528OC
60. Austin ED, Ma L, LeDuc C, et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet. 2012;5:336–343. doi:10.1161/CIRCGENETICS.111.961888
61. Desjardins M, Sidibé A, Fortier C, et al. Association of interleukin-6 with aortic stiffness in end-stage renal disease. JASH. 2018;12(1):5–13. doi:10.1016/j.jash.2017.09.013
62. Du B, Ouyang A, Eng J, Fleenor B. Aortic perivascular adipose-derived interleukin-6 contributes to arterial stiffness in low-density lipoprotein receptor deficient mice. Am J Physiol Heart Circ Physiol. 2015;308(11):H1382–H1390. doi:10.1152/ajpheart.00712.2014
63. Reiss A, Siegart N, De Leon J. Interleukin-6 in atherosclerosis: atherogenic or atheroprotective? Clin Lipidol. 2017;12(1):14–23. doi:10.1080/17584299.2017.1319787
64. Zamanian RT, Hansmann G, Snook S, et al. Insulin resistance in pulmonary arterial hypertension. Eur Respir J. 2008;33(2):318–324. doi:10.1183/09031936.00000508
65. Vakilian F, Attaran D, Shegofte M, Lari S, Ghare S. Assessment of thyroid function in idiopathic pulmonary hypertension. Res Cardiovasc Med. 2016;5(2):e29361. doi:10.5812/cardiovascmed.29361
66. Li JH, Safford RE, Aduen JF, Heckman MG, Crook JE, Burger CD. Pulmonary hypertension and thyroid disease. Chest. 2007;132(3):793–797. doi:10.1378/chest.07-0366
67. Udovcic M, Pena RH, Patham B, Tabatabai L, Kansara A. Hypothyroidism and the heart. Methodist DeBakey Cardiovasc J. 2017;13:55–59. doi:10.14797/mdcj-13-2-55
68. Hayashi K, Handa H, Nagasawa S, Okumura A, Moritake K. Stiffness and elastic behavior of human intracranial and extracranial arteries. J Biomech. 1980;13:175–184. doi:10.1016/0021-9290(80)90191-8
69. Bramwell JC, Hill AV. The velocity of the pulse wave in man. Proc R Soc B Biol Sci. 1922;93:298–306. doi:10.1098/rspb.1922.0022
70. Kubozono T, Miyata M, Ueyama K, et al. Clinical significance and reproducibility of new arterial distensibility index. Circ J. 2007;71:89–94. doi:10.1253/circj.71.89
71. Schillaci G, Battista F, Settimi L, Anastasio F, Pucci G. Cardio-ankle vascular index and subclinical heart disease. Hypertens Res. 2015;38:68–73. doi:10.1038/hr.2014.138
72. Takaki A, Ogawa H, Wakeyama T, et al. Cardio-ankle vascular index is a new noninvasive parameter of arterial stiffness. Circ J. 2007;71:1710–1714. doi:10.1253/circj.71.1710
73. Horinaka S, Yagi H, Ishimura K, et al. Cardio-ankle vascular index (CAVI) correlates with aortic stiffness in the thoracic aorta using ECG-gated multi-detector row computed tomography. Atherosclerosis. 2014;235:239–245.
74. Peled N, Shitrit D, Fox BD, et al. Peripheral arterial stiffness and endothelial dysfunction in idiopathic and scleroderma associated pulmonary arterial hypertension. J Rheumatol. 2009;36:970–975. doi:10.3899/jrheum.081088
75. Townsend RR, Wilkinson IB, Schiffrin EL, et al. Recommendations for improving and standardizing vascular research on arterial stiffness: a scientific statement from the American Heart Association. Hypertension. 2015;66:698–722.
76. Chamorro N, Del Pozo R, García-Lucio J, et al. Peripheral arterial stiffness and endothelial dysfunction in pulmonary arterial hypertension. Eur Respir J. 2015;46:PA2452. doi:10.1183/13993003.congress-2015.PA2452
77. Sznajder M, Dzikowska-Diduch O, Kurnicka K, et al. Increased systemic arterial stiffness in patients with chronic thromboembolic pulmonary hypertension. Cardiol J. 2018;7:e009459. doi:10.5603/CJ.a2018.0109
78. Hu Y, Li L, Shen L, Gao H. The relationship between arterial wall stiffness and left ventricular dysfunction. Neth Heart J. 2013;21(5):222–227. doi:10.1007/s12471-012-0353-z
79. Schillaci G, Mannarino MR, Pucci G, et al. Age-specific relationship of aortic pulse wave velocity with left ventricular geometry and function in hypertension. Hypertension. 2007;49(2):317–321. doi:10.1161/01.HYP.0000255790.98391.9b
80. Kim HL, Lim WH, Seo JB, et al. Association between arterial stiffness and left ventricular diastolic function in relation to gender and age. Medicine. 2017;96(1):e5783. doi:10.1097/MD.0000000000005783
81. Hoeper M, Boucly A, Sitbon O. Age, risk and outcomes in idiopathic pulmonary arterial hypertension. Eur Respir J. 2018;51:1800629. doi:10.1183/13993003.00629-2018
82. Rich S, Dantzker DR, Ayres SM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med. 1987;107:216–223. doi:10.7326/0003-4819-107-2-216
83. Hoeper MM, Huscher D, Pittrow D. Incidence and prevalence of pulmonary arterial hypertension in Germany. Int J Cardiol. 2016;203:612–613. doi:10.1016/j.ijcard.2015.11.001
84. Radegran G, Kjellstrom B, Ekmehag B, et al. Characteristics and survival of adult Swedish PAH and CTEPH patients 2000–2014. Scand Cardiovasc J. 2016;50:243–250. doi:10.1080/14017431.2016.1185532
85. Olsson KM, Delcroix M, Ghofrani HA, et al. Anticoagulation and survival in pulmonary arterial hypertension: results from the comparative, prospective registry of newly initiated therapies for pulmonary hypertension (COMPERA). Circulation. 2014;129(1):57–65. doi:10.1161/CIRCULATIONAHA.113.004526
86. Preston IR, Roberts KE, Miller DP, et al. Effect of Warfarin Treatment on Survival of Patients With Pulmonary Arterial Hypertension (PAH) in the registry to evaluate early and long-term PAH disease management (REVEAL). Circulation. 2015;132(25):2403–2411. doi:10.1161/CIRCULATIONAHA.115.018435
87. Wensel R, Francis DP, Meyer FJ, et al. Incremental prognostic value of cardiopulmonary exercise testing and resting hemodynamics in pulmonary arterial hypertension. Int J Cardiol. 2013;167(4):1193–1198. doi:10.1016/j.ijcard.2012.03.135
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.