")
Back to Journals » Journal of Blood Medicine » Volume 6
Prognostic significance of ASXL1, JAK2V617F mutations and JAK2V617F allele burden in Philadelphia-negative myeloproliferative neoplasms
Authors Yonal-Hindilerden I, Daglar-Aday A, Akadam-Teker B, Yilmaz C, Nalcaci M, Yavuz AS, Sargin D
Received 7 December 2014
Accepted for publication 3 February 2015
Published 1 June 2015 Volume 2015:6 Pages 157—175
DOI https://doi.org/10.2147/JBM.S78826
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Martin H Bluth
Ipek Yonal-Hindilerden, Aynur Daglar-Aday, Basak Akadam-Teker, Ceylan Yilmaz, Meliha Nalcaci, Akif Selim Yavuz, Deniz Sargin
Division of Hematology, Department of Internal Medicine, Istanbul Medical Faculty, Istanbul University, Fatih-Istanbul, Turkey
Background: Despite insights into the genetic basis of Philadelphia-negative myeloproliferative neoplasms (Ph-negative MPNs), a significant proportion of essential thrombocythemia (ET) and primary myelofibrosis (PMF) patients present with no known MPN disease alleles. There were no previous studies investigating the impact of ASXL1 mutations in Ph-negative MPNs in Turkey. In the current study, we investigated the prognostic significance of ASXL1 mutations in Turkish MPN patients. We also aimed to determine the prognostic significance of JAK2V617F allele burden and the relationship of JAK2V617F mutation with ASXL1 mutations in Ph-negative MPNs.
Methods: About 184 patients from a single center diagnosed with Ph-negative MPNs were screened for ASXL1, JAK2V617F mutations, and JAK2V617F allele burden: 107 ET and 77 PMF.
Results: A total of 29 ASXL1 mutations were detected in 24.7% of PMF and 8.4% of ET patients. ASXL1-mutated ET patients showed a trend toward an increase in the incidence of cerebrovascular events and higher total leukocyte counts. ASXL1-mutation in PMF was associated with older age and a higher prevalence of bleeding complications. In univariate analysis, overall survival (OS) was significantly reduced in ASXL1-mutated PMF patients. In multivariate analysis, Dynamic International Prognostic Scoring System-plus high-risk category and ASXL1 mutation status were independently associated with shorter survival in PMF. In PMF, mutational status and allele burden of JAK2V617F showed no difference in terms of OS and leukemia-free survival.
Conclusion: We conclude that ASXL1 mutations are molecular predictors of short OS in PMF.
Keywords: Philadelphia-negative myeloproliferative neoplasms (Ph-negative MPNs), ASXL1, JAK2V617F, JAK2V617F allele burden
Introduction
Chronic myeloproliferative neoplasms (MPNs) are a group of heterogeneous diseases of clonal origin characterized by abnormal proliferation of one or several myeloid lineages.1 The classical Philadelphia (Ph)-negative MPNs mainly comprise polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF). Although these disorders originate from a common multipotent progenitor, they differ in phenotypic properties and clinical courses, with ET the most favorable and PMF the most dismal outcome.2–4 It was not until the last 8 years that the molecular basis for Ph-negative MPNs became known. In 2005, several studies identified JAK2V617F as a major molecular event in a large proportion of patients with Ph-negative MPNs.5–9 JAK2V617F mutation was described in virtually all patients with PV and about half of the patients with ET and PMF. However, the role of the JAK2V617F mutation in the pathogenesis of three phenotypically disparate Ph-negative MPNs remained unclear. In addition, a small number of PV and about half of ET and PMF patients tested negative for the JAK2V617F mutation, which eventually led to a number of gene discovery efforts in Ph-negative MPNs. After the discovery of JAK2V617F mutation, a series of mutations in the thrombopoietin gene MPL and exon 12 mutations of JAK2 were described in 2006 and 2007, respectively.10,11 MPL515 mutations were reported in up to 8%–10% of PMF and 3%–5% of ET patients.12–15 JAK2 exon 12 mutations were identified in 2.5%–3.4% of total PV patients and in about 30% of patients diagnosed as JAK2V617F-negative PV.11,16 In view of the fact that a variable percentage of ET and PMF patients lack both JAK2 and MPL mutations, the molecular basis of these patients still remains largely unclear. In recent years, mutations in epigenetic modifiers, such as mutations of IDH1/2, TET2, EZH2, ASXL1, and a mutation in the gene that encodes the endoplasmic reticulum-associated, calcium-binding protein calreticulin known as CALR, have been identified in Ph-negative MPNs.17–22 Despite insights into the genetic basis of MPNs, a proportion of ET and PMF patients present with no known MPN disease alleles. A series of observations led to the hypothesis that the initiating genetic events directing the development of MPNs have not yet been clarified. ASXL1 mutations have been proposed as prognostic markers for risk stratification in PMF.20,23–26 Thus far, studies on the impact of ASXL1 mutations on survival in PMF have mostly comprised a small number of patients.20,23,24 The study by Vannucchi et al25 investigated the prognostic importance of ASXL1 mutations in a large number of PMF patients. Subsequently, the study by Tefferi et al26 examined the ASXL1 mutation-based molecular prognostication in another large series of PMF patients. To further characterize the role of ASXL1 mutations in Ph-negative MPNs, we analyzed a relatively large cohort of 184 MPN cases diagnosed on the basis of WHO criteria, and correlated the findings with clinical data, laboratory characteristics, overall survival (OS), and leukemia-free survival (LFS). In one recent study, the frequency of ASXL1 mutations in ET patients with CALR mutation was 16% (8/50), and 2.1% (2/97) in ET patients with JAK2V617F mutation.27 Our study investigated the impact of ASXL1 mutations in 107 ET patients. Although most studies published so far have been retrospective in nature, several studies have reported poor prognostic correlation of low JAK2V617F allele burden in PMF.28,29 The other aim of the current study was to determine the prognostic significance of JAK2V617F allele burden and the relationship of JAK2V617F mutation with ASXL1 mutations in Ph-negative MPNs.
Materials and methods
Patients
The study group consisted of a whole cohort of 184 patients with Ph-negative MPNs – 107 ET (58 females, 49 males) and 77 PMF (43 females, 34 males) patients – diagnosed according to the 2008 WHO criteria.30 The study was approved by the local ethics committee of Istanbul University Istanbul Medical Faculty and was performed according to the principles of the Declaration of Helsinki. Informed written consent for study sample collection and permission for use in research were obtained from all participants. Patients included in the study were under follow-up between May 1995 and July 2013. Patient samples were collected at the time of referral (including both newly and previously diagnosed patients) to the outpatient clinic of the Hematology department at Istanbul University Istanbul Medical Faculty between January 2011 and July 2013. Data obtained at study entry included demographics; diagnostic features, such as blood counts, lactate dehydrogenase (LDH) levels, and clinical complications, such as bleeding and thrombosis. Medical history of red blood cell transfusion, phlebotomy, medications, splenectomy, and allogeneic hematopoietic stem cell transplantation (AHSCT) was obtained. Risk factors for cardiovascular diseases – diabetes mellitus, hypertension, cigarette smoking, and dyslipidemia – were reported. LDH levels were measured at the biochemistry laboratory of our department. Spleen with a longitudinal diameter of ≥130 mm up to 160 mm and ≥160 mm on ultrasound was evaluated as mild and massive splenomegaly, respectively. The karyotype of bone marrow or peripheral blood cells was analyzed whenever assessable metaphases could be harvested in PMF patients. Karyotypes were described according to International System for Human Cytogenetic Nomenclature (ISCN) guidelines.31 Risk stratification of PMF patients was done according to Dynamic International Prognostic Scoring System (DIPSS)-plus.32 Unfavorable karyotypes consist of complex karyotype or sole or two abnormalities including +8, −7/7q−, i(17q), inv(3), −5/5q−, 12p−, or 11q23 rearrangement.33 Other cytogenetic abnormalities were defined as favorable karyotype abnormalities. Patients with no cytogenetic abnormalities were grouped to have normal karyotype. OS and LFS were calculated as months from the time of diagnosis to death or last follow-up and the time of diagnosis to leukemic transformation, respectively.
Genomic analysis
Patient samples were sent from the outpatient clinic of our Hematology department to the Molecular Hematology Laboratory of Istanbul University between January 2011 and July 2013. For molecular analysis, all samples were processed at our Molecular Hematology Laboratory. Genomic DNA was extracted from peripheral blood granulocytes using conventional methods. Genomic DNA was isolated using a High Pure polymerase chain reaction (PCR) Template Preparation Kit (Roche Diagnostic, Mannheim, Germany). The concentration of DNA extracted was assessed with a Nano-Drop-2000 Spectrophotometer (Thermo Scientific, Wilmington, DE, USA).
Genotyping for ASXL1 mutation
A 339-bp PCR fragment (covering codons for aa 575–687), containing the mutational region of ASXL1 (exon 12), was amplified from genomic DNA using the following primers: forward: 5′-CCACCCTGGGTGGTTAAAG-3′, reverse: 5′-TCGCTGTAGATCTGACGTAC-3′.34 In the study by Pratcorona et al,34 83% of all known ASXL1 mutations within exon 12 had been identified using this method. PCR amplifications were performed in a total volume of 25 μL PCR mix containing 1.5 μL of deoxyribonucleotide triphosphate, 3 μL solution of PCR buffer, 1.2 μL of MgCl2 concentration, 0.4 μL of each primer, 0.3 μL of Taq polymerase, 50 ng/μL of DNA template, and a sufficient amount of dH2O to complete the volume to 25 μL. Cycling parameters for all amplifications were as follows: initial denaturation step of 95°C for 5 minutes, 35 cycles of an initial melt at 95°C for 1 minute, 35 cycles of annealing at 56°C for 1 minute, 35 cycles of extension at 72°C for 1 minute, and a final extension step of 72°C for 7 minutes. After amplification, products were purified using Invisorb Spin DNA Extraction Kit (Invitek GmbH, Berlin, Germany), and quality and size were assessed on electrophoresis through 2% agarose gel. PCR products were bidirectionally sequenced on ABI 3730xl DNA Analyzers, including PCR primers (Applied Biosystems, Foster City, CA, USA). We analyzed sequencing traces both manually and with the ClustalW2 multiple sequence alignment program. Information of genetic alterations identified were obtained from the Ensembl Genome Browser (http://www.ensembl.org). Known single nucleotide polymorphisms were identified by searching the NCBI dbSNP database (http://www.ncbi.nlm.nih.gov/snp).
Quantification of JAK2V617F allele burden
JAK2 MutaScreen Kit (Ipsogen, Luminy Biotech, Marseille, France) was used for the detection of JAK2V617F status and quantitative JAK2V617F allele burdens in genomic DNA using TaqMan allelic discrimination.35,36 The assay is based on the simultaneous use of two specific TaqMan probes and the measurement of the respective fluorescence of the two alleles (FAM for V617F and VIC for wild-type) to differentiate the amplification of each allele. PCR amplifications were done in a total volume of 25 μL PCR mix containing 12.5 μL of TaqMan Universal Master Mix, 2.5 μL of primers and probes mix, 5 μL of nuclease-free PCR grade water, and 50 ng/μL of DNA template. PCR conditions were as follows: initial denaturation step of 95°C for 15 minutes, followed by 50 cycles of amplification consisting of denaturation at 95°C for 15 seconds, annealing at 60°C for 1 minute, and extension 60°C for 20 seconds. Quantification of mutant alleles was performed by using a Rotor-Gene 3000 PCR instrument (Corbett Research, Sydney, Australia). Mutant allele burden was reported as the percentage of total JAK2 represented by JAK2V617F (that is, JAK2V617F + JAK2 wild type). The mutant allele burden was estimated by six-scaled standards of JAK2 V617F mutant allele (2%, 5%, 12.5%, 31%, 50%, and 78%). JAK2V617F mutant allele burden equal to or less than 50% and greater than 50% were named as low and high JAK2V617F allele burden, respectively.
Statistical analyses
SPSS version 16 (Prentice Hall, Upper Saddle River, NJ, USA) was used for association analysis. Continuous variables were summarized as mean (SD). The chi-square statistics were used to compare categorical variables between the groups. Analysis of continuous variables among the groups was performed using the Mann–Whitney U test (two groups) or the Kruskal–Wallis test for multiple comparison. The odds ratios (OR) are accompanied by Cornfield 95% confidence interval (CI). Two-tailed P-values <0.05 were considered significant. Parametric and nonparametric correlation analyses were used to detect associations with ASXL1 mutations. OS curves were constructed by the Kaplan–Meier method for ET and PMF patients. Also, Kaplan–Meier estimation was used to plot LFS curves for PMF patients. Log-rank test was used to calculate the difference of OS and LFS between the groups. Variables attaining a significant level at the univariate analysis were included in a multivariate Cox proportional hazard regression analysis for assessing their independent association with OS. Cumulative risk of death was calculated by the use of OR.
Results
A total of 184 consecutive patients diagnosed as ET and PMF were included. The mean age of the whole cohort at the time of the study was 58.2 years (SD, 14.68), and 54.9% were females.
Comparison of patients with ET and PMF
According to the 2008 WHO criteria, 107 patients were diagnosed as ET and 77 as PMF.30 The mean age of ET (54.2% females) and PMF (55.8% females) was 56.3 years (SD, 14.5) and 60.8 years (SD, 14.5), respectively (P=0.647).
Prevalence of JAK2V617F mutation was significantly higher in PMF than in ET patients (75.3% and 59.8%, respectively; P=0.028). Moreover, the frequency of JAK2V617F-positive patients with mutant allele burden in the upper quartile ranges (allele burden >50%) was higher in PMF compared with ET (23.4% and 4.7%, respectively; P=0.001).
The analysis of the variations detected by conventional PCR using bidirectional sequencing identified 20 mutations of ASXL1 (exon 12) in 19 PMF patients (24.7% of PMF patients) (one patient presented with two distinct mutations) (Table 1). Of these, 14 (18.2%) had nonsense, four (5.2%) missense mutations, while one patient harbored missense along with nonsense mutations. The most frequent variation of ASXL1 mutation in PMF patients was c.1934dupG, which constituted 55% of mutations (11 in 20 mutations). The second most common lesions were c.1900_1922del and c.1954G>A, with the same frequency (3 in 20 mutations, 15%, each). 73.6% (14 in 19) of ASXL1-mutated PMF patients carried JAK2V617F. Fourteen of 77 PMF patients (18.2%) harbored, simultaneously, ASXL1 and JAK2V617F mutations. Among 107 ET patients studied, ASXL1 was mutated in 9 cases (8.4%) (Table 2). Among them, 5 displayed nonsense and 4 missense mutations (4.7% and 3.7%, respectively). The most common variations of ASXL1 mutation in ET were c.1934dupG and c.1954G>A, with the same frequency (3 in 9 mutations, 33.3%, each). About 66.6% (6 in 9) of ASXL1-mutated ET patients carried JAK2V617F mutation. Six of 107 ET patients (5.6%) displayed, simultaneously, ASXL1 and JAK2V617F mutations.
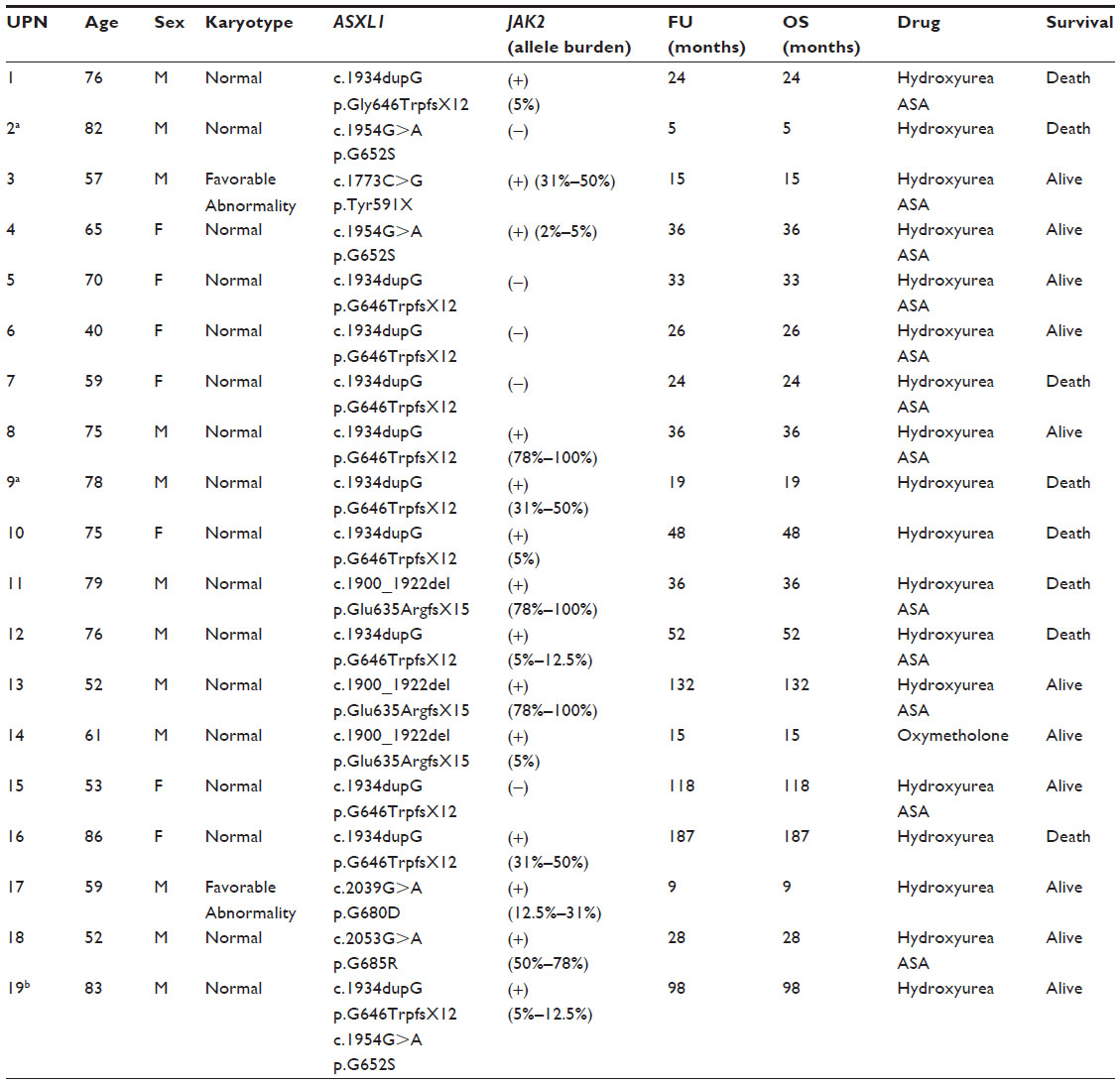 | Table 1 Clinical and molecular characteristics of ASXL1-mutated PMF patients |
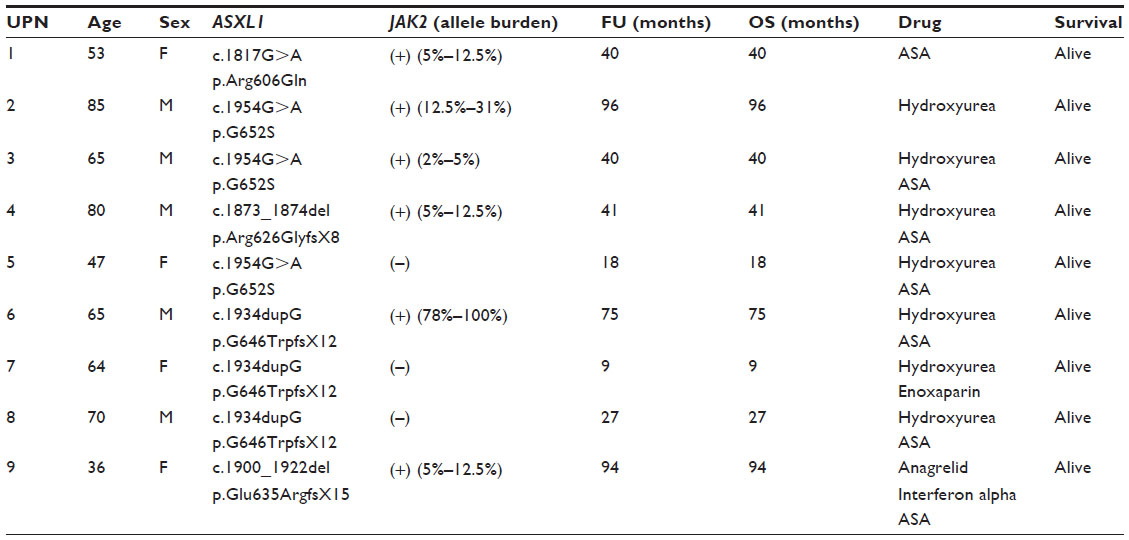 | Table 2 Clinical and molecular characteristics of ASXL1-mutated ET patients |
Clinical and molecular characteristics of ASXL1-mutated PMF and ET patients are outlined in Tables 1 and 2.
The frequency of ASXL1 mutations (exon 12) was significantly higher in PMF patients (24.7%) compared with ET patients (8.4%) (P=0.005). Moreover, the frequency of combined JAK2V617F and ASXL1 mutations was significantly higher in PMF than in ET (18.2% and 5.6%, respectively; P=0.014).
Among the PMF patients, the frequencies of nonsense (including frameshift mutations) and missense mutations in ASXL1 were 18.2% (14 in 77) and 5.2% (4 in 77), respectively. One PMF patient (1.3%) presented with simultaneous nonsense and missense mutations in the ASXL1 gene. In ET patients, the prevalence of nonsense mutations and missense mutations in the ASXL1 gene was 4.7% (5 in 107) and 3.7% (4 in 107), respectively. The frequency of nonsense mutations in the ASXL1 gene was significantly higher in PMF compared with ET patients (18.2% and 4.7%, respectively; P=0.012). As regards the distribution of ASXL1 mutations, the most common mutation in PMF was c.1934dupG (14.3%), while c.1934dupG and c.1954G>A were the most frequent mutations in ET (2.8% each) (P=0.05).
Outcomes of ASXL1 (exon 12) mutations
Twenty-nine ASXL1 alterations were determined in 28 of 184 study samples of Ph-negative MPNs (15.2%). The most prevalent mutation was a frameshift mutation, named c.1934dupG, that comprised 48.2% of ASXL1 mutations (14 in 29) (Figure 1A). The second most frequent mutation was c.1954G>A, a single-base exchange leading to missense mutation (6 in 29, 20.6%) (Figure 1B). c.1900_1922 del, a nonsense alteration consisting of a 23-bp deletion in ASXL1 presumed to truncate the plant homeodomain finger domain (PHD), was detected as the third most prevalent mutation (4 in 29, 13.7%) (Figure 1C).
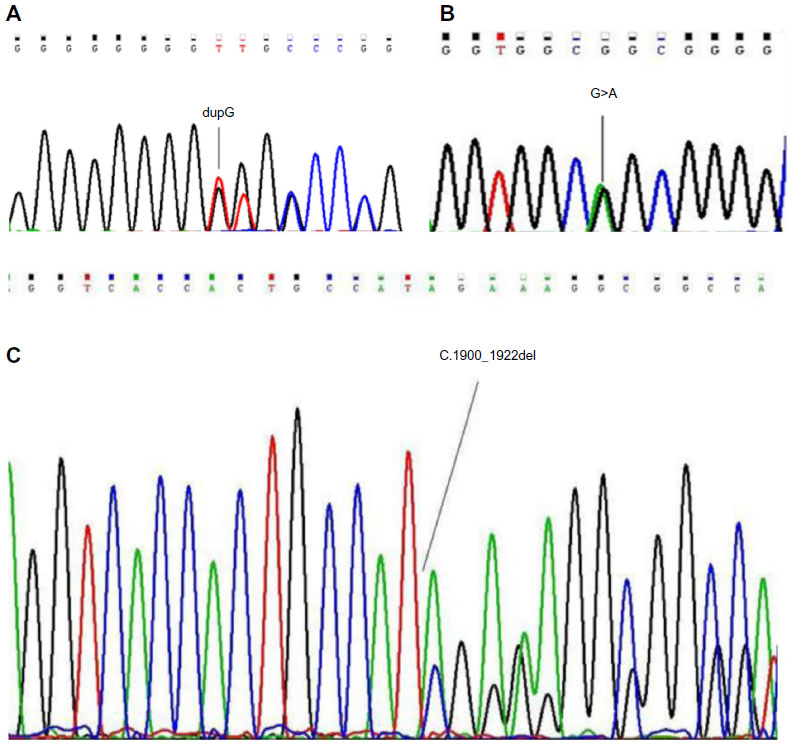 | Figure 1 DNA sequence chromatograms demonstrating mutations at ASXL1 gene. |
Comparison of ET patients according to the ASXL1 mutational status
Clinical and laboratory characteristics of the patients with ET divided by ASXL1 mutational status are summarized in Table 3.
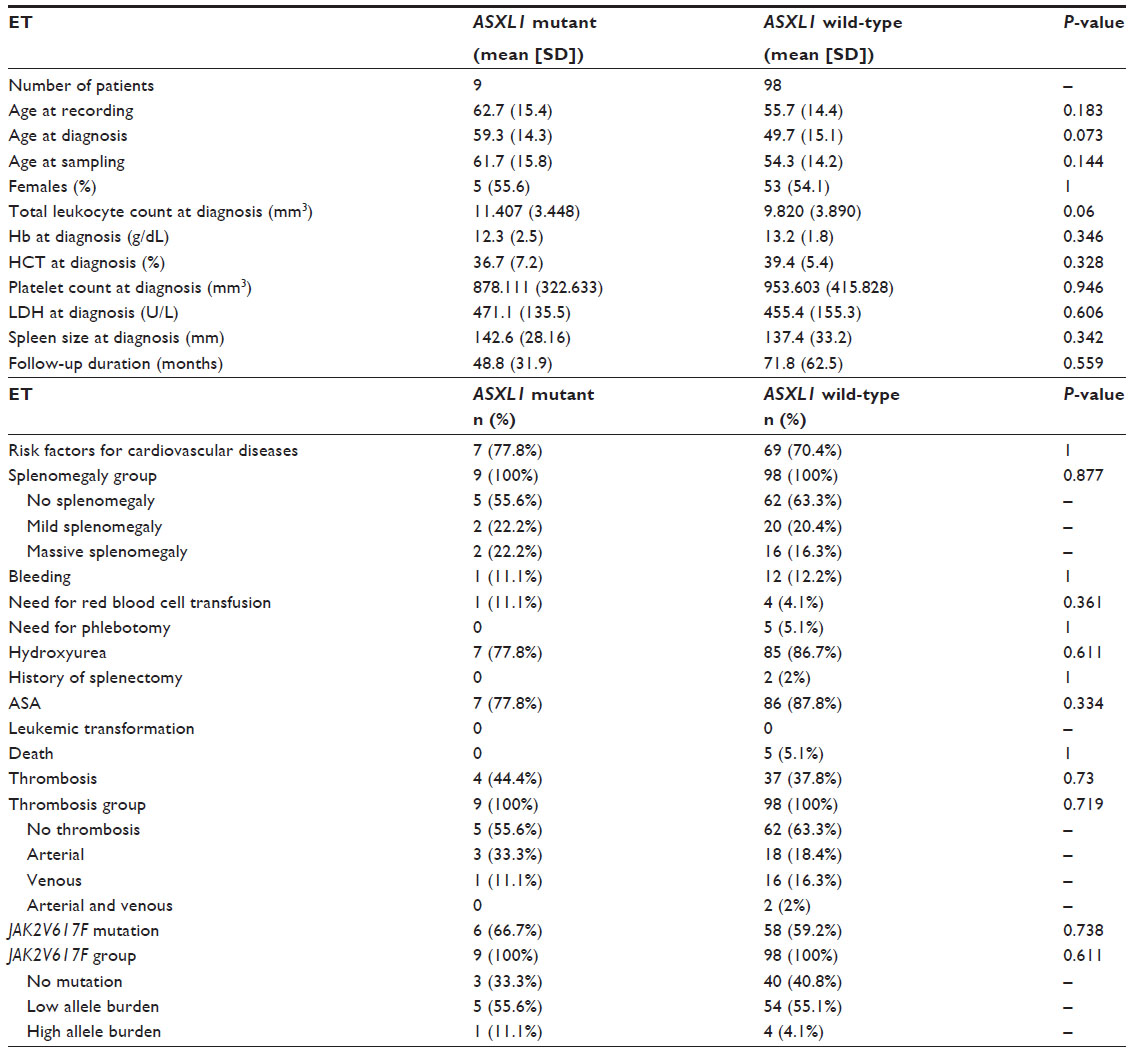 | Table 3 Clinical and laboratory features of patients with ET according to ASXL1 mutational status |
No significant differences were observed in age at recording and sampling, while there was a trend toward older age at diagnosis in ASXL1-mutated ET patients compared with wild-type patients (P=0.183; P=0.144, and P=0.073, respectively). There was no significant difference in sex between ASXL1 mutant and wild-type ET patients. ASXL1-mutated ET patients showed similar levels of hemoglobin (Hb), hematocrit (HCT), and platelet counts with wild-type patients. A trend toward higher total leukocyte counts was observed in ASXL1-mutated ET patients with respect to ASXL1 wild-type ET patients (P=0.06).
There was no significant difference in LDH levels between ET cases with ASXL1 mutation and cases without ASXL1 mutation. Likewise, the mean spleen size and the degree of splenomegaly were not significantly different between ASXL1 mutant and wild-type ET patients. No significant differences were observed in the rate of phlebotomy and the presence of risk factors for cardiovascular diseases between ET patients with and without ASXL1 mutation. The need for red blood cell transfusion was similar between two groups.
There were no significant differences in the rate of bleeding events and the localization of bleeding complications between ASXL1-mutated and wild-type ET patients (P=1 and P=0.994, respectively). The prevalence of major thrombotic events and venous thrombosis did not significantly differ among ASXL1-mutated and wild-type ET patients (P=0.73 and P=1, respectively). In addition, no significant difference was observed in the rate of arterial thrombosis between ASXL1 mutant and wild-type ET patients (33.3% and 20.4%, respectively; P=0.416). As regards the localization of arterial thrombosis among ET patients, 11.1% of ASXL1-mutated ET patients experienced coronary arterial disease, 11.1% cerebrovascular accident, 11.1% cerebrovascular accident concomitant with peripheral arterial disease; while coronary arterial disease developed in 10.2% and cerebrovascular accident in 5.1% of ASXL1 wild-type patients (P=0.059). Consequently, ASXL1-mutated ET patients showed a trend toward an increase in the incidence of cerebrovascular events (P=0.059).
The frequency of JAK2V617F mutation did not differ among ASXL1-mutated and wild-type ET patients (66.7% and 59.2%, respectively; P=0.738). ASXL1 mutant ET patients have similar quantitative JAK2V617F allele burdens compared with their wild-type counterparts (P=0.405). Moreover, the frequency of JAK2V617F-positive patients with mutant allele burden in the upper quartile ranges was not different between ASXL1 mutant and wild-type ET patients (11.1% and 4.1%, respectively; P=0.611). ASXL1 mutation was detected in 6 of 64 (9.4%) JAK2V617F-positive and 3 of 43 (7%) JAK2V617F-negative ET cases (P=0.738). ET patients with JAK2V617F allele burden data were divided into three groups: JAK2V617F mutation-negative (n=43), JAK2V617F-positive with mutant allele burden in the lower quartile (allele burden ≤50%, n=59) and upper quartile ranges (allele burden >50%, n=5). The prevalence of ASXL1 mutation did not differ across all these three groups: 7% (3 of 43) in JAK2V617F wild-type, 8.5% (5 of 59) in patients with low JAK2V617F allele burden, and 20% (1 of 5) in patients with high JAK2V617F allele burden (P=0.611).
There was no significant difference in the prevalence of hydroxyurea use, acetylsalicylic acid (ASA) use, and the rate of splenectomy between ASXL1 mutation-positive and -negative ET patients. Of the ASXL1 mutant ET patients, one had never received any form of cytoreductive treatment (Table 2). Duration of follow-up was longer in ASXL1 wild-type ET than mutant type but with no statistical significance (mean 71.8 months [SD, 62.5] and 48.8 months [SD, 31.9], respectively; P=0.559). During follow-up, 5 of 98 (5.1%) ASXL1 wild-type ET patients deceased, whereas at the end of the data collection period, all ASXL1 mutant ET patients were alive. Rates of death were similar among ASXL1 wild-type and mutant ET patients (5.1% and 0%, respectively; P=1).
In this population, the presence of ASXL1 mutation did not correlate with HCT level, total leukocyte count, platelet count, LDH level, spleen size, bleeding complications, total thrombotic events, arterial thrombosis, and venous thrombosis (r<0.2). In addition, no correlations were observed between the aforementioned parameters and combined ASXL1 and JAK2V617F mutations (r<0.2).
Comparison of PMF patients according to the ASXL1 mutational status
Clinical and laboratory features of PMF patients depending on ASXL1 mutational status are outlined in Table 4.
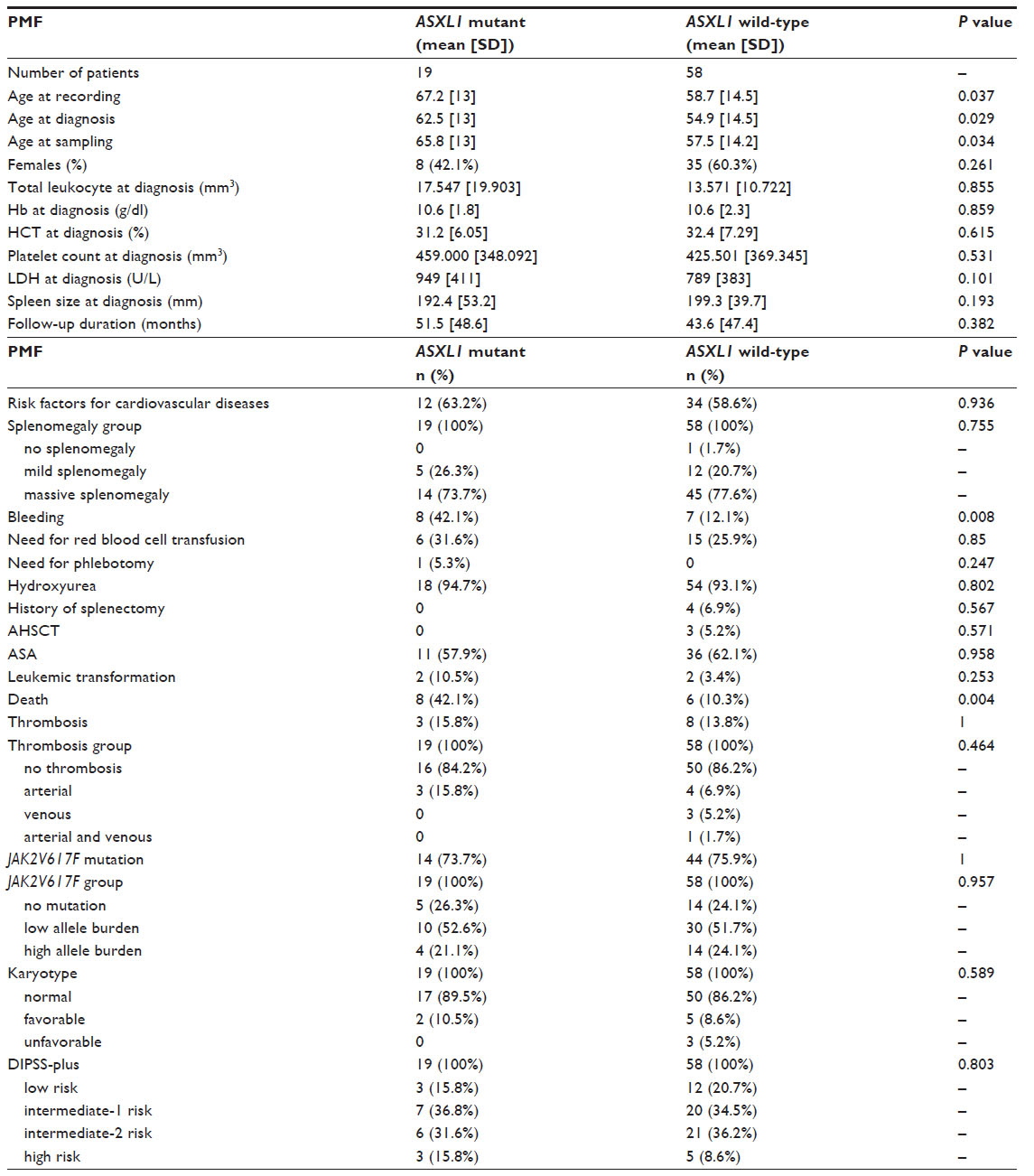 | Table 4 Clinical and laboratory characteristics of ASXL1-mutated and wild-type PMF patients |
The age at recording, diagnosis, and sampling was significantly higher in ASXL1-mutated PMF patients with respect to wild-type counterparts (P=0.037; P=0.029, and P=0.034, respectively). The sex did not differ among ASXL1-mutated and wild-type PMF patients. No significant differences were observed in Hb level, HCT level, total leukocyte count, platelet count, and LDH level between the two groups.
There were no significant differences in mean spleen size, degree of splenomegaly, need for red blood cell transfusion, rate of phlebotomy, and presence of risk factors for cardiovascular diseases in ASXL1-mutated PMF patients as compared with wild-type patients. The prevalence of major thrombotic events, arterial thrombosis, and venous thrombosis did not significantly differ among ASXL1-mutated and wild-type PMF patients (P=1, P=0.437, and P=0.567, respectively).
The prevalence of bleeding complications was significantly higher in ASXL1-mutant PMF patients than in those without ASXL1 mutation (42.1% and 12.1%, respectively; P=0.008). As regards the localization of bleeding events for PMF patients, 21.1% of ASXL1 mutant PMF patients experienced gastrointestinal bleeding and 10.5% intracranial hemorrhage; while gastrointestinal hemorrhage occurred in 1.7% of ASXL1 wild-type PMF patients. No intracranial hemorrhage developed in PMF patients without ASXL1 mutation (P=0.001). Consequently, the incidence of severe bleeding was significantly higher in ASXL1-mutated PMF patients compared with ASXL1 wild-type PMF patients (P=0.001).
The frequency of JAK2V617F mutation did not differ between ASXL1-mutated and wild-type PMF patients (73.7% and 75.9%, respectively; P=1). There was no significant difference in the quantitative JAK2V617F allele burdens between PMF patients with ASXL1 mutation and without the mutation (P=0.838). Moreover, the prevalence of JAK2V617F-positive patients with mutant allele burden in the upper quartile ranges did not differ between ASXL1 mutant and wild-type PMF patients (21.1% and 24.1%, respectively; P=0.957). Fourteen of 58 (24.1%) JAK2V617F-positive and 5 of 19 (26.3%) JAK2V617F-negative PMF patients displayed ASXL1 mutations (exon 12) (P=0.535). As regards the JAK2V617F allele burden, the prevalence of ASXL1 mutation did not differ across all three groups: 5 of 19 (26.3%) in JAK2V617F wild-type, 10 of 40 (25%) in patients with low JAK2V617F allele burden, and 4 of 18 (22.2%) in patients with high JAK2V617F allele burden (P=0.957).
The prevalence of hydroxyurea use, ASA use, rate of AHSCT, and history of splenectomy showed no difference between PMF patients with and without ASXL1 mutation. Similarly, no significant differences were observed in the use of other medical treatments between two groups (P>0.05). One of the ASXL1-mutated PMF patients had received oxymetholone but no cytoreductive treatment (Table 1).
DIPSS-plus risk stratification did not differ between PMF patients with and without ASXL1 mutation (P=0.803). Distribution of karyotype categories was similar between two groups: 89.5% normal, 10.5% favorable, and 0% unfavorable karyotype in ASXL1-mutated PMF patients and 86.2% normal, 8.6% favorable, and 5.2% unfavorable karyotype in ASXL1 wild-type PMF patients (P=0.589).
Duration of follow-up was similar between PMF patients with and without ASXL1 mutation (mean 51.5 months [SD, 48.6] and 43.6 months [SD, 47.4], respectively; P=0.382). ASXL1 mutant PMF patients showed higher rates of death compared with ASXL1 wild-type PMF patients (42.1% and 10.3%, respectively; P=0.004). The rate of leukemic transformation was higher in ASXL1-mutated PMF than in wild-type counterparts, but with no statistical significance (10.5% and 3.4%, respectively; P=0.253).
In PMF patients, the presence of ASXL1 mutation did not correlate with HCT level, total leukocyte count, platelet count, LDH level, mean spleen size, total thrombotic events, arterial thrombosis, and venous thrombosis (r<0.2). In PMF, a mild positive correlation was found between ASXL1 mutation and bleeding complications (r=0.327). Further on, we observed a mild positive correlation between bleeding events and the copresence of ASXL1 and JAK2V617F mutations, whereas no such correlations were found between the other aforementioned parameters and combined ASXL1 and JAK2V617F mutations (r=0.363 and r<0.2, respectively).
Survival curves
Kaplan–Meier survival curves in ET patients
In ET patients, parameters including mutational status of ASXL1, JAK2V617F, sequence variations within ASXL1 exon 12, and allele burden of JAK2V617F mutation were tested in univariate survival analysis for influence on OS.
OS was similar between ASXL1 mutation-positive (n=9) and ASXL1 mutation-negative (n=98) ET cases (P=0.737) (Figure 2A). In ASXL1 mutant ET patients, 5 displayed nonsense and 4 missense mutations. OS did not differ among ET patients with respect to ASXL1 sequence variations (P=0.945) (Figure 2B).
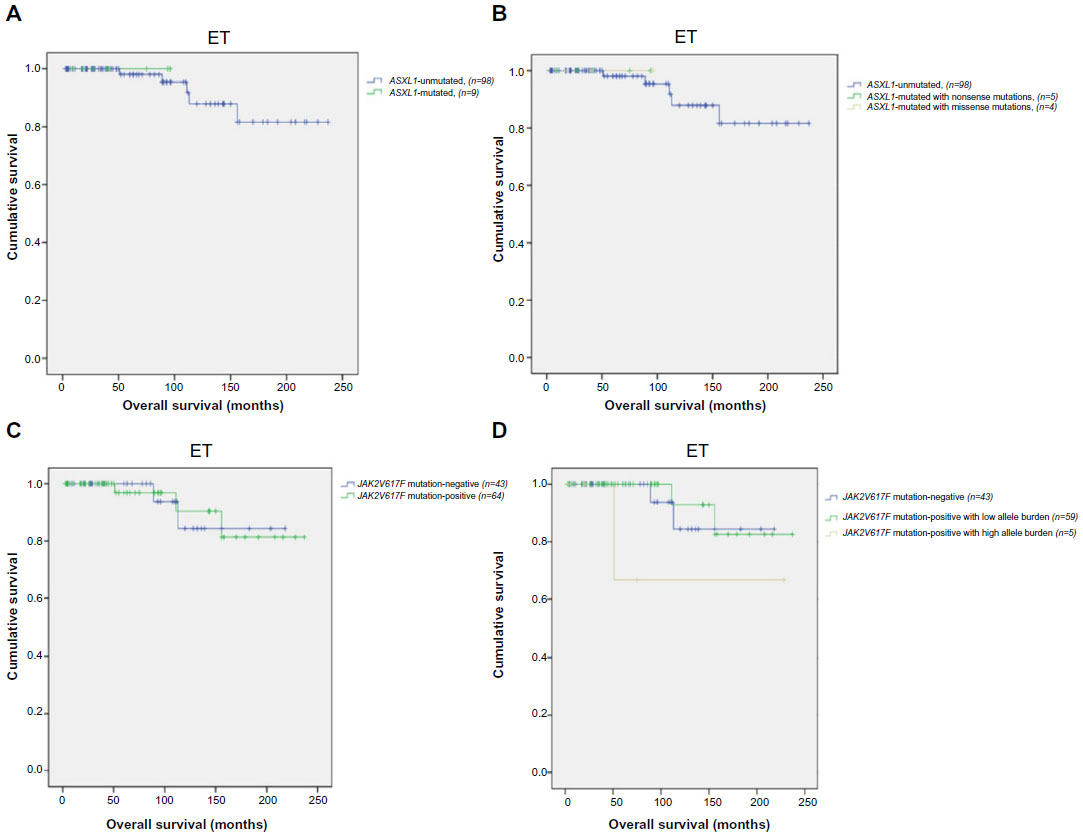 | Figure 2 Survival outcomes in ET patients (n=107). |
The mean OS of ET patients with or without JAK2V617F mutation was 215 months (95% CI: 193–238) and 200 months, respectively (95% CI: 177–223) (P=0.958) (Figure 2C). ET patients with JAK2V617F allele burden data were divided into three groups: JAK2V617F mutation-negative (n=43), V617F-positive with mutant allele burden in the lower quartile (allele burden ≤50%, n=59) and upper quartile ranges (allele burden >50%, n=5). Comparison across all three groups revealed no significant difference in OS (mean 200 months; 95% CI: 177–223, 219 months; 95% CI: 197–241, and 169 months; 95% CI: 74–263, respectively; P=0.249) (Figure 2D).
Kaplan–Meier survival curves in PMF patients
In PMF patients, the following parameters were tested in univariate survival analysis for impact on OS and LFS: DIPSS-plus risk stratification, mutational status of ASXL1, JAK2V617F, sequence variations within ASXL1 exon 12, and allele burden of JAK2V617F mutation. Variables that reached a significant level at the univariate analysis were included in a multivariate analysis by Cox proportional hazard model for assessing their independent association with OS.
Kaplan–Meier plots revealed significantly inferior OS in ASXL1-mutated PMF patients (n=19) as compared with ASXL1 wild-type (n=58) PMF patients (mean 108 months; 95% CI: 62–153 and 202 months; 95% CI: 123–282, respectively; P=0.025) (Figure 3A). In ASXL1-mutated PMF patients, 14 harbored nonsense, 4 missense, and 1 nonsense concomitant with missense mutations. Univariate analysis revealed a trend toward shorter OS in the presence of ASXL1 nonsense sequence variations compared with wild-type and other sequence variations (P=0.09) (Figure 3B).
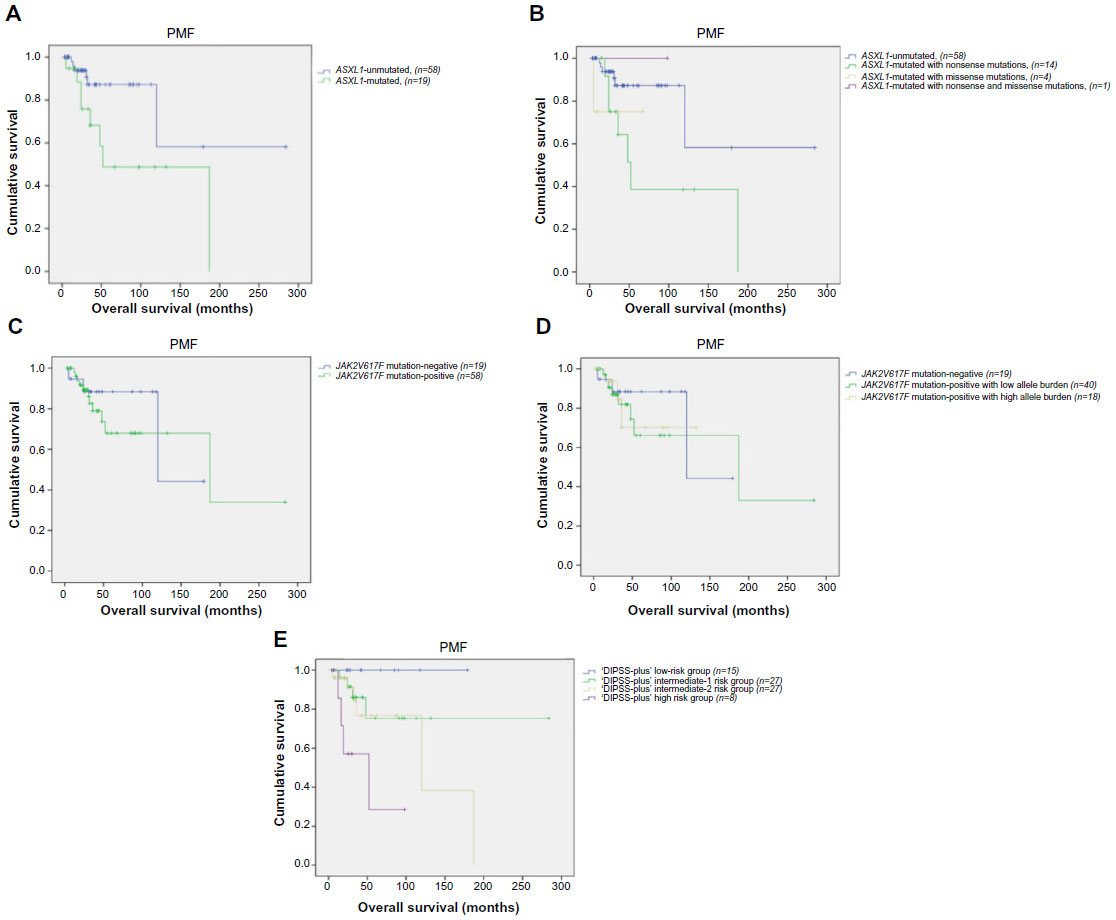 | Figure 3 Survival outcomes in PMF patients (n=77). |
In PMF patients, the presence of JAK2V617F mutation showed no difference in terms of OS (mean 170 months; 95% CI: 112–229 and 133 months; 95% CI: 92–175, respectively; P=0.589) (Figure 3C). With regard to JAK2V617F allele burden data, PMF patients were divided into three groups: JAK2V617F mutation-negative (n=19), V617F-positive with mutant allele burden in the lower quartile (n=40) and upper quartile ranges (n=18). OS was not different between these three groups (mean 133 months; 95% CI: 92–175, 167 months; 95% CI: 104–230, and 101 months; 95% CI: 71–132, respectively; P=0.857) (Figure 3D).
In univariate analysis, DIPSS-plus high-risk patients did not live as long as those with other risk groups (P=0.007) (Figure 3E).
Cox analysis demonstrated that the association between inferior OS and DIPSS-plus risk score was sustained during multivariate analysis, which included ASXL1 mutation, ASXL1 sequence variations, and DIPSS-plus risk stratification as covariates (OR: 3.19; 95% CI:1.09–9.2; P=0.002). In addition, multivariate analysis confirmed the independent prognostic value of ASXL1 mutation (OR: 2.75; 95% CI: 1.37–5.5; P=0.033). Univariate analysis revealed a trend toward shorter OS in the presence of ASXL1 nonsense sequence variations compared with wild-type and other sequence variations (P=0.09), whereas prognostic significance was not sustained for ASXL1 nonsense sequence variations in multivariate analysis (P=0.131). To conclude, multivariate analysis of OS confirmed the independent prognostic relevance of the mutant ASXL1 and DIPSS-plus high-risk group in PMF patients (OR: 2.75; 95% CI: 1.37–5.5; P=0.033 and OR: 3.19; 95% CI: 1.09–9.2; P=0.002, respectively).
The difference of LFS between ASXL1 mutant (n=19) and ASXL1 wild-type PMF patients (n=58) was not significant (mean 166 months; 95% CI: 140–193 and 270 months; 95% CI: 251–289, respectively; P=0.275) (Figure 4A). In addition, LFS was similar when PMF patients were grouped according to the ASXL1 sequence variations (P=0.207) (Figure 4B).
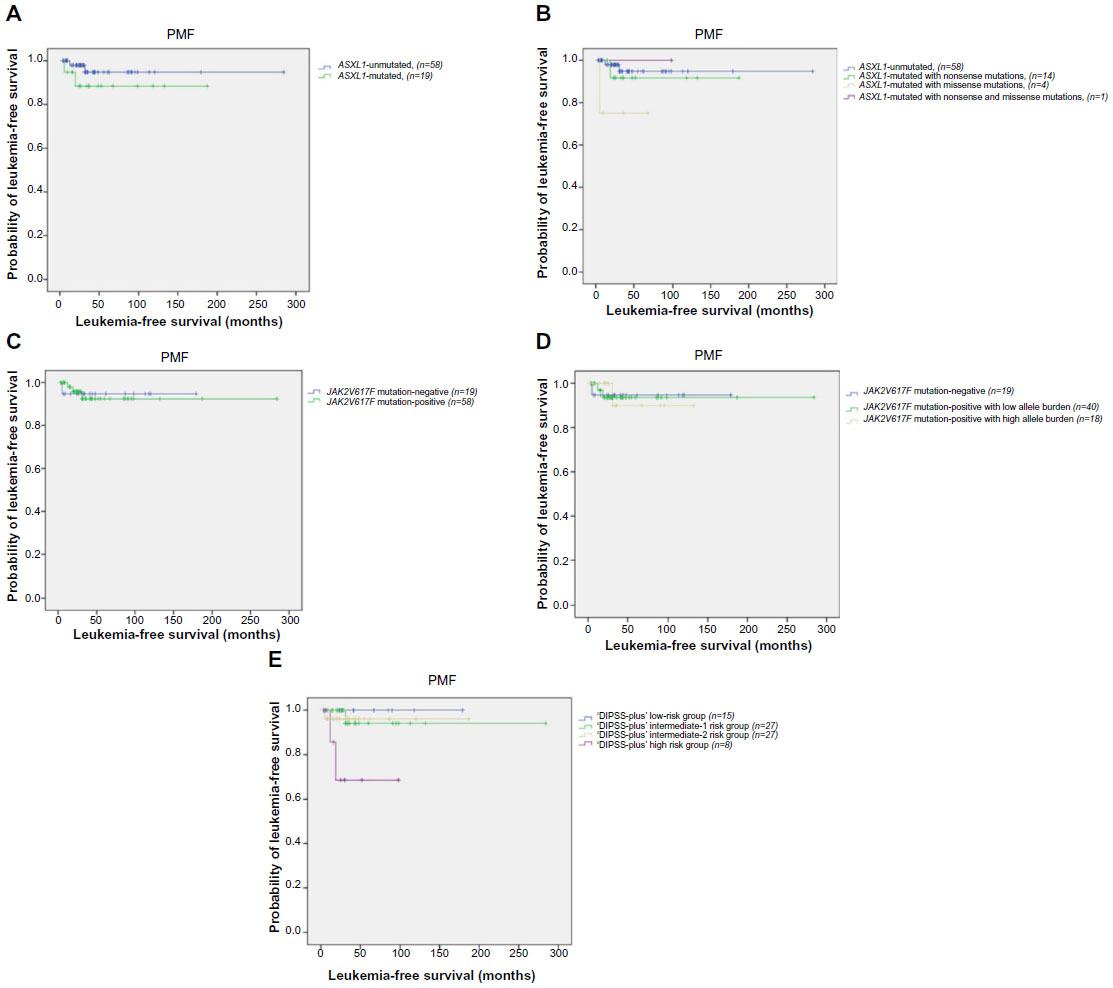 | Figure 4 Leukemia-free survival in PMF patients (n=77). |
LFS was not different in JAK2V617F mutation-positive PMF patients compared with JAK2V617F mutation-negative PMF patients (mean 264 months; 95% CI: 242–286 and 169 months; 95% CI: 152–187, respectively; P=0.934) (Figure 4C). Comparison across JAK2V617F mutation-negative PMF patients (n=19), JAK2V617F-positive patients with low allele burden (n=40), and high allele burden (n=18) showed no significant difference in LFS (mean 169 months; 95% CI: 152–187, 266 months; 95% CI: 243–289, and 121 months; 95% CI: 103–140, respectively; P=0.997) (Figure 4D).
Univariate analysis revealed worse LFS in DIPSS-plus high-risk patients compared with other risk groups (P=0.032) (Figure 4E).
Discussion
During the last 8 years, several additional novel molecular abnormalities (such as IDH1/2, TET2, EZH2, ASXL1, CALR mutations) have been identified in Ph-negative MPNs.5,21,22,37 The precise role in pathogenesis of these genetic alterations is nowadays under investigation, yet none of them seem to be disease specific. We believe that the identification of the new genetic lesions in Ph-negative MPNs increases our understanding of the complex molecular pathogenesis of these disorders and supplies new specific diagnostic, prognostic, and therapeutic approaches for the treatment of these patients.
There are a limited number of studies analyzing the frequency of ASXL1 mutational status in Ph-negative MPNs.18,20,23,25 Mutations in ASXL1 were reported to be present in approximately 10% of MPN patients.20 Studies analyzing ASXL1 mutations separately in Ph-negative MPNs described these mutations in 2%–5% of PV, 5%–10% of ET, 13%–26% of PMF, and 22%–38.5% of post-PV/ET myelofibrosis patients.18,20,23 Abdel-Wahab et al23 identified ASXL1 mutations in 13% of PMF patients (6 in 46) and 23% of post-PV/ET myelofibrosis (5 in 22) among the study samples from Mayo Clinic. In the same study, among the 25 Harvard PMF patients, 3 (12%) harbored ASXL1 mutations.23 In another study, by Brecqueville et al,24 the frequences of ASXL1 mutations were reported as roughly 7% (2 in 30) in PV, 4% (2 in 53) in ET, 20% (6 in 30) in PMF, 50% (2 in 4) in post-PV myelofibrosis, and 10% (1 in 10) in post-ET myelofibrosis. Carbuccia et al20 found ASXL1 mutations in 3 of 10 PMF patients (30%) and in 1 of 35 ET patients (3%). In the study by Stein et al,38 mutations of ASXL1 were identified in 32% (15 in 47) of patients with PMF and 2% (1 in 42) of PV patients. No ET patients (n=41) harbored ASXL1 mutation.38 In another study, the mutational frequencies in PMF and post-PV/ET myelofibrosis were 55% (23 in 42) and 22% (5 in 23), respectively.39 Patient cohorts, which studied ASXL1 mutations in Ph-negative MPNs so far, have been too small to draw any conclusion.18,20,23 The largest study that investigated the impact of ASXL1 mutations in PMF included 279 patients from Mayo Clinic and 483 patients from the European cohort.25 In that study, the frequencies of ASXL1 mutations in PMF patients from Mayo Clinic and the European cohort were 31% and 21.7%, respectively.25 In the study by Tefferi et al,26 the frequencies of ASXL1 mutations in 293 PMF patients from the Florence University and in 277 PMF patients from Mayo Clinic cohorts were 19.4% and 30.6%, respectively.
Differences in the prevalence of ASXL1 mutations among Ph-negative MPNs may be based on several factors: the number of patients included in the study, the identification of polymorphisms instead of true mutations, and the presence of c.1934 dupG (p.G646TrpfsX12), which remains controversial. To our knowledge, there are no previous studies that investigated ASXL1 mutations in Ph-negative MPNs in Turkey. Our study allows the estimation of the incidence of ASXL1 mutations in a relatively large Turkish cohort of Ph-negative MPNs.
We performed the conventional PCR system for the ASXL1 mutation analysis in ET and PMF patients, using primers as described by Pratcorona et al34 In our study, ASXL1 mutations were more frequent in PMF (19 in 77, 24.7%) than in ET patients, which confirmed previous reports (9 in 107, 8.4%).24,38 In summary, the prevalence of ASXL1 mutations among our PMF patients was similar to that in the European cohort in the previous largest study (24.7% and 21.7%, respectively).25 Our study suggests the approach of Pratcorona et al34 as a safe, rapid, cost-effective, and efficient screening technique for ASXL1 mutation detection in Ph-negative MPNs.
The most prevalent variant in the ASXL1 gene is c.1934dupG, resulting in a frameshift (p.G646TrpfsX12), which accounts for more than 50% of the reported ASXL1 mutations.38,40–42 However, the most frequently reported ASXL1 alteration, c.1934dupG (p.G646TrpfsX12), has been suspected not to be a true mutation.43 Abdel-Wahab et al43 detected this alteration in DNA from normal tissues of ASXL1 mutant patients and in more than 25% of samples from healthy individuals. Thus, Abdel-Wahab et al43 reported it as a sequencing artifact rather than a somatic mutation. Based on the results of the aforementioned study, patients with c.1934dupG (p.G646TrpfsX12) were censored from the analysis in another study investigating the incidence of ASXL1 mutations in malignant myeloid disorders by Abdel-Wahab et al.23 Yet in other studies, c.1934dupG (p.G646TrpfsX12) was not detected in the DNA of healthy volunteers and germ-line DNAs.38,42,44 Ricci et al39 analyzed ASXL1 mutations in granulocytes and purified CD3+ T-lymphocytes of patients with myelofibrosis and showed that c.1934dupG (p.G646TrpfsX12) was restricted solely to the myeloid compartment. Pratcorona et al34 screened ASXL1 mutations in 91 samples from healthy individuals, and none of them revealed c.1934dupG (p.G646TrpfsX12). Schnittger et al45 showed that acute myeloid leukemia (AML) patients with c.1934dupG (p.G646TrpfsX12) remained positive and c.1934dupG wild-type samples remained negative upon repeated testing of the samples. As a whole and by taking into account the recent papers, we regarded the c.1934dupG (p.G646TrpfsX12) as a real somatic mutation in our study.34,38,39,42,44,45 In the study by Stein et al,38 the two most common lesions in ASXL1 mutation-positive MPN patients were c.1934dupG (p.G646TrpfsX12) and c.1900_1922del (p.Glu635ArgfsX15) (38% and 8%, respectively). In our total MPN cohort, 29 ASXL1 variations were detected in 184 patients (15.2%). In our study, similar to the study by Stein et al,38 the most prevalent variation was c.1934dupG, which comprised 48.2% (14/29) of ASXL1 mutations. However, the c.1954G>A was the second most common alteration in our MPN cohort, which differed from the study mentioned above (6 in 29, 20.6%). The third most frequent alteration observed in our study was c.1900_1922del (4 in 29, 13.7%). Distribution of ASXL1 mutations among disease subgroups revealed that the most common alterations of the ASXL1 gene in our ET patients were c.1934dupG and c.1954G>A, with the same frequency (3 in 9 mutations, 33.3%, each). The most prevalent ASXL1 variant in our PMF patients was c.1934dupG, which comprised 55% of mutations (11 in 20 mutations). The second most common alterations in PMF were c.1900_1922del and c.1954G>A, with the same frequency (3 in 20 mutations, 15%, each). Stein et al38 found 28 nonsense mutations in the ASXL1 gene in 23 myelofibrosis patients, but none in ET patients. In that study, all of the 3 mutant ET patients had missense mutations, whereas all of the 23 mutant myelofibrosis patients had nonsense mutations.38 Similar to the previously published finding by Stein et al,38 the frequency of nonsense mutations in the ASXL1 gene was significantly higher in our PMF patients than in ET patients (18.2% and 4.7%, respectively).
There are a limited number of studies investigating the association of sex and age with ASXL1 mutational status in Ph-negative MPNs.23–25,38 Two different series reported that ASXL1-mutated myelofibrosis patients were similar to their wild-type counterparts in terms of age and sex distribution.23,38 However, in a series of 127 patients with Ph-negative MPNs, ASXL1 mutation was associated with older age.24 Likewise, in the Mayo cohort of the study by Vannucchi et al,25 ASXL1 mutations clustered with older PMF patients. In our study, there was a trend toward older age at diagnosis in ASXL1-mutated ET patients compared with wild-type patients, whereas no significant difference in sex between ASXL1 mutant and wild-type ET patients was observed. In addition, we found a significant association between ASXL1 mutation and older age in PMF patients, whereas the sex did not differ between ASXL1-mutated and wild-type PMF patients.
Several previous studies that investigated the laboratory characteristics of Ph-negative MPNs divided by ASXL1 mutational status reported conflicting results.24,25,38 Brecqueville et al24 reported that the presence of ASXL1 mutation did not influence total leukocyte count and platelet count, but that the Hb level was lower in ASXL1-mutated MPN patients. In the study by Stein et al,38 no differences in total leukocyte count, platelet count, and Hb level were observed between ASXL1-mutated and wild-type myelofibrosis patients. A recent study of 483 PMF patients from the European cohort revealed that ASXL1 mutations were associated with leukocytosis and anemia.25 In the same study, which included 279 PMF patients from Mayo Clinic, ASXL1 mutations were clustered in patients with leukocytosis, whereas there was no significant association with anemia.25 In line with the study of Stein et al,38 levels of Hb, HCT, total leukocyte, and platelet counts did not differ between our ASXL1 mutant and wild-type PMF patients. In a previous study with a small number of ET patients, a trend toward lower HCT levels was observed in ASXL1-mutated ET patients compared with ASXL1 wild-type counterparts, but with no significant difference in Hb level, total leukocyte, and platelet counts between ET patients with and without ASXL1 mutation.24 In a recent study, ET patients showing the co-expression of ASXL1 and CALR mutations had lower Hb levels, while there was no impact on total leukocyte and platelet counts.27 Our study showed that the presence of ASXL1 mutation had a trend toward higher total leukocyte counts, but did not influence Hb level, HCT level, and platelet counts.
A limited number of studies examined the relationship of ASXL1 mutation to spleen size in Ph-negative MPNs.25,38 In a previous study, spleen size did not differ between PMF patients with and without ASXL1 mutation.38 On the other hand, another study of 483 PMF patients from the European cohort showed a significant association between ASXL1 mutation and splenomegaly.25 Like the study by Stein et al,38 our study did not show any relationship between ASXL1 mutation and spleen size in PMF patients. To our knowledge, this is the first report of an evaluation of the relationship between spleen size and ASXL1 mutation in ET patients. We did not find any difference in spleen size between ET patients with and without ASXL1 mutation.
Only one previous study that included 53 ET and 30 PV patients had examined the relationship between thrombosis and ASXL1 mutation.24 In that study, the rate of thrombosis did not differ between ASXL1-mutated and unmutated MPN patients.24 In our study, no significant differences were observed in the rate of total thrombotic events, arterial thrombosis, and venous thrombosis between ASXL1-mutated and wild-type ET patients. However, as regards the localization of arterial thrombosis among our ET patients, ASXL1-mutated ET patients showed a trend toward an increase in the incidence of cerebrovascular events. We did not find differences in the rate of total thrombotic events, arterial thrombosis, and venous thrombosis between PMF patients with and without ASXL1 mutation.
To the best of our knowledge, no previous study has examined the relationship of ASXL1 mutation with bleeding events and LDH level in MPN. In our study, we observed no significant differences in LDH level between ASXL1-mutated and wild-type PMF patients. Similarly, mean LDH level did not differ between ET patients with and without ASXL1 mutation. In our study, no significant difference in the rate of bleeding events was observed between ASXL1-mutated and wild-type ET patients. However, the prevalence of bleeding complications was significantly higher in our ASXL1-mutated PMF patients than in our wild-type PMF patients (42.1% and 12.1%, respectively). Also, in PMF patients, we found a mild positive correlation between ASXL1 mutation and bleeding complications. As regards the localization of bleeding events among PMF patients, 21.1% of ASXL1 mutant PMF patients experienced gastrointestinal bleeding, and 10.5% experienced intracranial hemorrhage. On the other hand, 1.7% of ASXL1 wild-type PMF patients experienced gastrointestinal hemorrhage, whereas no intracranial hemorrhage developed in PMF patients without ASXL1 mutation. Consequently, the incidence of severe bleeding was significantly higher in ASXL1-mutated PMF patients compared with ASXL1 wild-type PMF patients.
Several studies showed that ASXL1 mutations were found with the same frequency in JAK2V617F mutant and JAK2V617F wild-type MPN patients.24,38 In our study, the incidence of ASXL1 mutation did not differ between JAK2V617F-positive and -negative ET patients (9.4% and 7%, respectively). In addition, there was no difference in the rate of ASXL1 mutation between PMF patients with and without JAK2V617F mutation (24.1% and 26.3%, respectively). In accordance with these findings, the prevalence of ASXL1 mutation did not differ when our ET and PMF patients were divided according to JAK2V617F allele burden separately.
There are a limited number of studies comparing the median values of JAK2V617F allele burden and the frequency of JAK2V617F mutation among myelofibrosis patients with and without ASXL1 mutation.38,39 Ricci et al39 reported that the frequency of JAK2V617F mutation was significantly higher in myelofibrosis patients without ASXL1 mutation than in those with ASXL1 mutation (74% and 48%, respectively). Stein et al38 found no difference in median values of JAK2V617F allele burden between myelofibrosis patients with and without ASXL1 mutation (median 57% and 61%, respectively). Also, in that study, the frequency of JAK2V617F mutation showed no difference between ASXL1-mutated and wild-type myelofibrosis patients (68% and 71%, respectively).38 In line with the aforementioned observation, we found no significant difference in the frequency of JAK2V617F mutation between PMF patients with and without ASXL1 mutation (73.7% and 75.9%, respectively). Moreover, in our study group, there was no significant difference in JAK2V617F mutant allele load between PMF patients with and without ASXL1 mutation. To the best of our knowledge, no previous study has compared JAK2V617F mutant allele load and the frequency of JAK2V617F mutation between ET patients with and without ASXL1 mutation. In our study, quantitative JAK2V617F allele burden and the frequency of JAK2V617F mutation did not differ between ASXL1 mutant and wild-type ET patients.
A study of 483 PMF patients from the European cohort revealed that ASXL1 mutations were not associated with cytogenetic risk groups.25 In the same study, which included 279 PMF patients from Mayo Clinic, ASXL1 mutations were shown to occur more likely in the presence of normal karyotype, but showed no difference in the presence of favorable and unfavorable cytogenetic categories.25 In our study, we found no difference in the distribution of karyotype categories between ASXL1-mutated and wild-type PMF patients. Several prognostic scoring systems have been developed to risk stratify patients with PMF.32,46,47 A limited number of studies have investigated the association of DIPSS-plus risk distribution with ASXL1 mutational status in PMF patients.24,25 In a small series of PMF patients, ASXL1-mutated patients showed a tendency to be included in the DIPSS-plus high-risk group.24 In the Mayo cohort of the study by Vannucchi et al,25 ASXL1 mutations significantly clustered in the PMF patients with DIPSS-plus high-risk category. In the European cohort of the same study, ASXL1 mutations were found to be significantly enriched in the PMF patients with International Prognostic Scoring System (IPSS) high-risk group.25 Risk stratification of our PMF patients was done according to DIPSS-plus.32 In contrast to the aforementioned studies, we found that DIPSS-plus risk distribution between our ASXL1 mutant and wild-type PMF patients was similar.
Several reports mostly including a small number of patients have highlighted the impact of ASXL1 mutations on outcomes of PMF patients.20,23,24 A study of 44 myelofibrosis patients (30 PMF, 10 post-ET myelofibrosis, and 4 post-PV myelofibrosis) showed that patients harboring ASXL1 mutations had a shorter 5-year OS compared with ASXL1 wild-type patients (56% and 87%, respectively).24 Another study that included 46 PMF patients from Mayo Clinic and 25 PMF patients from Harvard Institutes showed worse survival in the presence of ASXL1 mutation in univariate analysis.23 The largest study that investigated the impact of ASXL1 mutations in PMF patients included 279 patients from Mayo Clinic and 483 patients from the European cohort.25 In the European cohort, OS and LFS were independently predicted by ASXL1 mutations. On the other hand, ASXL1-mutated PMF patients from Mayo Clinic showed inferior OS but no difference in LFS.25 In another large study, the presence of ASXL1 mutation was significantly associated with shortened survival in both Mayo and Florence cohorts.26
In our study, ASXL1 mutant PMF patients showed higher rates of death compared with ASXL1 wild-type PMF patients (42.1% and 10.3%, respectively). Moreover, we observed shorter OS in our PMF patients with ASXL1 mutation (n=19) than in our patients without ASXL1 mutation (n=58). Multivariate analysis confirmed the independent prognostic value of ASXL1 mutation in our PMF patients. However, LFS was similar between our ASXL1-mutated and unmutated PMF patients. We also attempted to determine the prognostic impact of ASXL1 mutation variants among PMF patients. In our study, univariate analysis revealed a trend toward shorter OS in the presence of ASXL1 nonsense sequence variations with respect to wild-type and other sequence variations, whereas ASXL1 nonsense sequence variations did not retain its prognostic effect in PMF in multivariate analysis. In addition, LFS was similar when PMF patients were grouped according to the ASXL1 sequence variations. To our knowledge, this is the first report of an evaluation of the impact of ASXL1 mutations on outcome in patients with ET. Our ET patients with and without ASXL1 mutation showed no difference in terms of OS.
In our PMF patients, ASXL1 mutations clustered with normal karyotype, yet portended poor patient survival. This observation suggests that ASXL1 mutations may represent an independent prognostic biomarker in patients with PMF.
In our study, the frequency of combined JAK2V617F and ASXL1 mutations was significantly higher in PMF patients than in ET patients (18.2% and 5.6%, respectively). In PMF patients, no correlations were seen between HCT level, total leukocyte count, platelet count, LDH level, mean spleen size, death, total thrombotic events, arterial thrombosis, venous thrombosis, and combined ASXL1 and JAK2V617F mutations. Also, in ET patients, no correlations were observed between the aforementioned parameters and combined ASXL1 and JAK2V617F mutations. In PMF patients, a mild positive correlation was observed between bleeding events and combined ASXL1 and JAK2V617F mutations, whereas no such correlation was found among ET patients. Further investigations are required to determine the impact of the copresence of JAK2V617F and ASXL1 mutations on disease course and complications in Ph-negative MPNs.
In our study, approximately 7% (3 in 43) of JAK2V617F-negative ET patients showed the ASXL1 mutations. The frequency of ASXL1 mutations in our JAK2V617F-negative PMF patients was 26.3% (5 in 19). From our findings, it may be deduced that analysis of the ASXL1 genes provided an additional 7% increase in ET and a 26.3% increase in PMF patients with a detected molecular marker of clonality.
Current prognostication in PMF is based on the IPSS, DIPSS, and DIPSS-plus.32,46,48 The particular study provides practical information on prognostic classification and decision making regarding treatment in PMF patients. Our ASXL1-mutated PMF patients did not show an increase in adverse karyotypic features. Despite the aforementioned findings, in our relatively large cohort of PMF patients, ASXL1 mutations were associated with shortened OS. Thus, it seems that ASXL1 mutations represent independent prognostic biomarkers in PMF. To conclude, our observations demonstrate additional genetic events in the pathogenesis and prognosis of PMF patients, including mutations in ASXL1. Moreover, these results are consistent with the previous study that the mutation screening for ASXL1 might be added to future clinical trials and prospective observational studies.25
The particular study describes mutations and mutational combinations that have clinical and prognostic significance in Ph-negative MPNs. Our findings support previous observations that mutations in epigenetic regulators might be prognostically more detrimental than those activating JAK-STAT signaling in PMF. Consequently, our results support that detection of ASXL1 mutations may enable a more accurate assessment of the risk stratification and lead to more accurate therapeutic decisions and monitoring of the impact of novel drugs.
Funding
The study was approved by the local ethics committee of Istanbul University Istanbul Medical Faculty (file number: 2012/1571-1245) and supported by the Istanbul University Scientific Research Foundation (project number: 30427).
Acknowledgment
We would like to thank the Molecular Hematology Laboratory in Istanbul University, Istanbul Medical Faculty for assistance with sample processing.
Author contributions
IYH designed the research, supplied samples, analyzed the data and drafted the article. ADA, BAT, and CY performed the laboratory work, helped in acquisition of data and assisted in drafting of the article. MN and ASY made important contributions to the design and concept of the manuscript and revised the article critically for important intellectual content. DS designed the research and revised the article. All authors have reviewed and approved the final version of the manuscript. All authors agree to be accountable for all aspects of the work in ensuring that questions related to accuracy of integrity of any part of the work are appropriately investigated and resolved.
Disclosure
The authors report no conflicts of interest in this work.
References
Anastasi J. The myeloproliferative and overlap, myeloproliferative/myelodysplastic neoplasms. In: Hsi ED, editor. Hematopathology. 2nd ed. St Louis, MO: Elsevier; 2012:479. | |
Adamson JW, Fialkow PJ, Murphy S, Prchal JF, Steinmann L. Polycythemia vera: stem-cell and probable clonal origin of the disease. N Engl J Med. 2007;295(17):913–916. | |
Fialkow PJ, Faguet GB, Jacobson RJ, Vaidya K, Murphy S. Evidence that essential thrombocythemia is a clonal disorder with origin in a multipotent stem cell. Blood. 1981;58(5):916–919. | |
Jacobson RJ, Salo A, Fialkow PJ. Agnogenic myeloid metaplasia: a clonal proliferation of hematopoietic stem cells with secondary myelofibrosis. Blood. 1978;51(2):189–194. | |
James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. | |
Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790. | |
Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. | |
Tefferi A, Gilliland DG. The JAK2V617F tyrosine kinase mutation in myeloproliferative disorders: status report and immediate implications for disease classification and diagnosis. Mayo Clin Proc. 2005;80(7):947–958. | |
Baxter EJ, Scott LM, Campbell PJ, et al. Cancer Genome Project. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. | |
Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3(7):e270. | |
Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356(5):459–468. | |
Ruan GR, Jiang B, Li LD, et al. MPL W515L/K mutations in 343 Chinese adults with JAK2V617F mutation-negative chronic myeloproliferative disorders detected by a newly developed RQ-PCR based on TaqMan MGB probes. Hematol Oncol. 2010;28(1):33–39. | |
Ma W, Zhang X, Wang X, et al. MPL mutation profile in JAK-2 mutation-negative patients with myeloproliferative disorders. Diagn Mol Pathol. 2011;20(1):34–39. doi: 10.1097/PDM.0b013e3181ecd261. | |
Schnittger S, Bacher U, Haferlach C, et al. Characterization of 35 new cases with four different MPL W515 mutations and essential thrombocytosis or primary myelofibrosis. Haematologica. 2009;94(1):141–144. doi: 10.3324/haematol.13224. | |
Lieu CH, Shen YJ, Lai WC, Tsai WH, Hsu HC. Prevalence of MPL W515L/K mutations in Taiwanese patients with Philadelphia-negative chronic myeloproliferative neoplasms. J Chin Med Assoc. 2010;73(10):530–532. doi: 10.1016/S1726-4901(10)70115–5. | |
Pietra D, Li S, Brisci A, et al. Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood. 2008;111(3):1686–1689. | |
Delhommeau F, Dupont S, Della Valle V, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–2301. doi: 10.1056/NEJMoa0810069. | |
Abdel-Wahab O, Manshouri T, Patel J, et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res. 2010;70(2):447–452. doi: 10.1158/0008-5472.CAN-09-3783. | |
Ernst T, Chase AJ, Score J, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010; 42(8):722–726. doi: 10.1038/ng.621. | |
Carbuccia N, Murati A, Trouplin V, et al. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia. 2009;23(11):2183–2186. doi: 10.1038/leu.2009.141. | |
Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013; 369(25):2379–2390. | |
Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–2405. | |
Abdel-Wahab O, Pardanani A, Patel J, et al. Concomitant analysis of EZH2 and ASXL1 mutations in myelofibrosis, chronic myelomonocytic leukemia and blast-phase myeloproliferative neoplasms. Leukemia. 2011;25(7):1200–1202. doi: 10.1038/leu.2011.58. | |
Brecqueville M, Rey J, Bertucci F, et al. Mutation analysis of ASXL1, CBL, DNMT3A, IDH1, IDH2, JAK2, MPL, NF1, SF3B1, SUZ12, and TET2 in myeloproliferative neoplasms. Genes Chromosomes Cancer. 2012;51(8):743–755. doi: 10.1002/gcc.21960. | |
Vannucchi AM, Lasho TL, Guglielmelli P, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27(9):1861–1869. doi: 10.1038/leu.2013.119. | |
Tefferi A, Guglielmelli P, Lasho TL, et al. CALR and ASXL1 mutations-based molecular prognostication in primary myelofibrosis: an international study of 570 patients. Leukemia. 2014;28(7):1494–1500. doi: 10.1038/leu.2014.57. | |
Shen H, Chao H, Ding Z, et al. CALR and ASXL1 mutation analysis in 190 patients with essential thrombocythemia. Leuk Lymphoma. Epub August 13, 2014. | |
Tefferi A, Lasho TL, Huang J, et al. Low JAK2V617F allele burden in primary myelofibrosis, compared to either a higher allele burden or unmutated status, is associated with inferior overall and leukemia-free survival. Leukemia. 2008;22(4):756–761. doi: 10.1038/sj.leu.2405097. | |
Guglielmelli P, Barosi G, Specchia G, et al. Identification of patients with poorer survival in primary myelofibrosis based on the burden of JAK2V617F mutated allele. Blood. 2009;114(8):1477–1483. doi: 10.1182/blood-2009-04-216044. | |
Tefferi A, Vardiman JW. Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia. 2008;22(1):14–22. | |
Shaffer LG, Slovak ML, Campbell LJ, editors. An International System for Human Cytogenetic Nomenclature: Recommendations of the International Standing Committee. Basel, NY: S. Karger; 2009. | |
Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):392–397. doi: 10.1200/JCO.2010.32.2446. | |
Tefferi A. Primary myelofibrosis: 2013 update on diagnosis, risk-stratification, and management. Am J Hematol. 2013;88(2):141–150. doi: 10.1002/ajh.23384. | |
Pratcorona M, Abbas S, Sanders MA, et al. Acquired mutations in ASXL1 in acute myeloid leukemia: prevalence and prognostic value. Haematologica. 2012;97(3):388–392. doi: 10.3324/haematol.2011.051532. | |
Chae H, Lee JH, Lim J, et al. Usefulness of real-time semi-quantitative PCR, JAK2 MutaScreen kit for JAK2 V617F screening. Korean J Lab Med. 2009;29(3):243–248. doi: 10.3343/kjlm.2009.29.3.243. | |
Cankovic M, Whiteley L, Hawley RC, Zarbo RJ, Chitale D. Clinical performance of JAK2 V617F mutation detection assays in a molecular diagnostics laboratory: evaluation of screening and quantitation methods. Am J Clin Pathol. 2009;132(5):713–721. doi: 10.1309/AJCPFHUQZ9AGUEKA. | |
Abdel-Wahab O. Genetics of the myeloproliferative neoplasms. Curr Opin Hematol. 2011;18:117–123. doi: 10.1097/MOH.0b013e328343998e. | |
Stein BL, Williams DM, O’Keefe C, et al. Disruption of the ASXL1 gene is frequent in primary, post-essential thrombocytosis and post-polycythemia vera myelofibrosis, but not essential thrombocytosis or polycythemia vera: analysis of molecular genetics and clinical phenotypes. Haematologica. 2011;96(10):1462–1469. doi: 10.3324/haematol.2011.045591. | |
Ricci C, Spinelli O, Salmoiraghi S, Finazzi G, Carobbio A, Rambaldi A. ASXL1 mutations in primary and secondary myelofibrosis. Br J Haematol. 2012;156(3):404–407. doi: 10.1111/j.1365-2141.2011.08865.x. | |
Gelsi-Boyer V, Brecqueville M, Devillier R, Murati A, Mozziconacci MJ, Birnbaum D. Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J Hematol Oncol. 2012;5:12. doi: 10.1186/1756-8722-5-12. | |
Boultwood J, Perry J, Pellagatti A, et al. Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia. 2010;24(5):1062–1065. doi: 10.1038/leu.2010.20. | |
Chou WC, Huang HH, Hou HA, et al. Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood. 2010;116(20):4086–4094. doi: 10.1182/blood-2010-05-283291. | |
Abdel-Wahab O, Kilpivaara O, Patel J, Busque L, Levine RL. The most commonly reported variant in ASXL1 (c.1934dupG;p.Gly646TrpfsX12) is not a somatic alteration. Leukemia. 2010;24(9):1656–1657. doi: 10.1038/leu.2010.144. | |
Thol F, Friesen I, Damm F, et al. Prognostic significance of ASXL1 mutations in patients with myelodysplastic syndromes. J Clin Oncol. 2011;29(18):2499–2506. doi: 10.1200/JCO.2010.33.4938. | |
Schnittger S, Eder C, Jeromin S, et al. ASXL1 exon 12 mutations are frequent in AML with intermediate risk karyotype and are independently associated with an adverse outcome. Leukemia. 2013;27(1):82–91. doi: 10.1038/leu.2012.262. | |
Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895–2901. doi: 10.1182/blood-2008-07-170449. | |
Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood. 2010;115(9):1703–1708. doi: 10.1182/blood-2009-09-245837. | |
Elena C, Passamonti F, Rumi E, et al. Red blood cell transfusion-dependency implies a poor survival in primary myelofibrosis irrespective of IPSS and DIPSS. Haematologica. 2011;96(1):167–170. doi: 10.3324/haematol.2010.031831. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.