Back to Journals » OncoTargets and Therapy » Volume 12
Prognostic significance and biological function of Lamina-associated polypeptide 2 in non-small-cell lung cancer
Authors Liu C ![]() , Yu H, Shen X, Qiao J
, Yu H, Shen X, Qiao J ![]() , Wu X, Chang J, Zhu X, Wang J
, Wu X, Chang J, Zhu X, Wang J ![]() , Shen X
, Shen X
Received 11 July 2018
Accepted for publication 26 April 2019
Published 16 May 2019 Volume 2019:12 Pages 3817—3827
DOI https://doi.org/10.2147/OTT.S179870
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr XuYu Yang
Chang Liu,1,2,* Hui Yu,1,2,* Xuxia Shen,2,3 Jie Qiao,1,2 Xianghua Wu,1,2 Jianhua Chang,1,2 Xunxia Zhu,4 Jialei Wang,1,2 Xiaoyong Shen4
1Department of Medical Oncology, Fudan University Shanghai Cancer Center, Shanghai 200032, People’s Republic of China; 2Department of Oncology, Shanghai Medical College, Fudan University, Shanghai 200032, People’s Republic of China; 3Department of Pathology, Fudan University Shanghai Cancer Center, Shanghai 200032, People’s Republic of China; 4Department of Thoracic Surgery, The Affiliated Huadong Hospital of Fudan University, Shanghai 200040, People’s Republic of China
*These authors contributed equally to this work
Purpose: Lamina-associated polypeptide 2 (LAP2; encoded by TMPO), is a nuclear protein that may affect chromatin regulation and gene expression through dynamically binding to nuclear lamin. TMPO (LAP2) plays dual roles of either suppressing or promoting proliferation of cells, depending on the status of the cell. It has been reported that TMPO is up-regulated in various cancer types. However, its function in lung cancer has not been studied yet.
Materials and methods: A series of clinical microarray datasets for lung cancer were investigated to demonstrate the expression of TMPO. The transcription of TMPO gene in human lung cancer was analyzed using Oncomine platform (www.oncomine.org) according to the standardized procedures described previously. Four separate datasets (Hou Lung, Okayama Lung, Beer Lung, and Garber Lung) were analyzed.
Results: Here, we show that TMPO is over-expressed in lung cancer tissues, and that a high level of TMPO indicates a poor prognosis in lung cancer patients. Knockdown of TMPO in lung cancer cells inhibits cell proliferation and induces apoptosis. Also, down-regulation of TMPO leads to an impaired metastatic ability of tumor cells. A nude mice tumor model show that knockdown of TMPO suppresses tumor formation in vivo.
Conclusion: Collectively, this study suggests TMPO as an oncogene and a novel prognostic gene in lung cancer.
Keywords: TMPO, LAP2, non-small-cell lung cancer, prognostic factor, oncogene
Introduction
Lung cancer is the leading cause of cancer-related deaths among men and women worldwide.1 In 2012, about 1.8 million new cases of lung cancer were diagnosed worldwide.2 According to their histological features, lung cancer is sub-divided into four major histological subtypes, including small-cell lung cancer, squamous cell carcinoma (SCC), adenocarcinoma (ADC), and large-cell carcinoma (LCC). The latter three types of cancer are collectively referred to as non-small-cell lung cancer (NSCLC), accounting for 80% of lung cancers.3 Despite advances in early diagnosis, target and immune therapies, lung cancer is often diagnosed at an advanced stage and has a poor prognosis, with a 5-year survival rate of 16.8% after cancer diagnosis.4
Targeted therapy shed a light on NSCLC treatment. NSCLC with an epidermal growth factor receptor (EGFR) mutation respond well to tyrosine kinase inhibitor (TKI), including gefitinib and erlotinib. EGFR mutation happens in 30~40% of adenocarcinomas in East Asians and 10~15% of those in Caucasians and African Americans. However, NSCLC patients with EGFR wild type cannot benefit from TKI therapy. Moreover, tumors will progress on EGFR TKI therapy within approximately 8–14 months, often as a result of acquired resistance mutations.5 These findings indicate that NSCLC are far more complicated than just an EGFR mutation, and other gene signaling pathways may be involved in the tumorigenesis and drug resistance, which need further investigation.
Lamina-associated polypeptide 2 (LAP2), also recognized as Thymopoietin (TMPO), is allocated in genome 12q21.2, and can give rise to six nucleus-localized isoforms (α, β, γ, δ, ε, ζ) by alternative splicing. The isoforms can be divided into two groups, one of which has a trans-membrane domain that localizes to the inner nuclear membrane, while the other one has a unique C-terminus and localizes to nucleoplasm and bind to pRb protein.6–9 All the isoforms interact with lamin A/C and chromatin. The nucleoplasmic LAP2α has dual functions of either suppressing or promoting proliferation depending on different cellular status.10,11 LAP2α and LAP2β were found to be over-expressed in malignant hematologic diseases,12 digestive tract carcinomas,13 glioblastoma14 and other cancers.11 However, until now, the prognostic value of TMPO in lung cancer has not been studied yet.
In the present study, the differential expression and prognostic significance of TMPO in lung cancer were analyzed through data mining from the Oncomine and Proggene databases. A siRNA recognizing the consensus region within N-terminus of TMPO gene were designed to study the effect of downregulated expression of TMPO on the biological phenotype and tumorigenicity of lung cancer cells.
Materials and methods
Data mining and Oncomine platform analysis
A series of clinical microarray datasets for lung cancer were investigated to demonstrate the expression of TMPO. The transcription of TMPO gene in human lung cancer was analyzed using Oncomine platform (
Survival analysis of lung cancer patients using an online PROG gene database
Using the median gene expression value as the bifurcating point, samples were divided into High and Low gene expression groups in PROG gene. Six representative microarray datasets (GSE11969, GSE13213, GSE17710, GSE30219, GSE37745 and GSE31210) were enrolled to calculate the overall survival or relapse-free survival of lung cancer patients. GSE11969 contains expression profiles in 90 patients with non-small cell lung cancers (NSCLC); GSE13213 has 117 patients with lung adenocarcinoma expression profiles; GSE17710 has 56 patients with lung squamous cell carcinoma; GSE30219 has 282 lung tumor samples analyzed; GSE37745 is composed of fresh frozen tissue of 96 NSCLC patients;GSE31210 contains a profile of 226 lung adenocarcinomas. p-values were calculated by log-rank test, and Hazard ratio (HR) value was utilized to evaluate the potential risk of death related to high expression of TMPO. HR value greater than 1.0 means patients with higher expression level of target gene will have greater probability of death.
Cell culture
Human lung cancer cell lines including H1299, A549, H460, PC-9 and PC-9R were obtained from Shanghai Institute of Cell Biology, Chinese Academy of Science. H1299, H460, PC-9 and PC-9R cells were cultured in RPMI-1640 (Hyclone, GE Healthcare Sciences, Marlborough, MA, USA) supplemented with 10% FBS (Hyclone) at 37°C humidified atmosphere containing 5% CO2. A549 cells were cultured in DMEM (Hyclone) supplemented with 10% FBS (Hyclone) at 37°C humidified atmosphere containing 5% CO2.
Lentiviral vector construction and virus infection
The sequence (5′-CTTGTGAAATACGGAGTGAAT-3′) was used as the target sequence to down-regulate the TMPO (NM_001032283.2) gene level. The negative control small interfering RNA (siRNA) was 5ʹ-TTCTCCGAACGTGTCACGT-3ʹ. The stem-loop-stem oligos (shRNAs) were synthesized, annealed and ligated into the BamH I/EcoR I-linearized pFH-GFPvector. The lentiviral-based shRNA-expressing vectors were checked by DNA sequencing. The generated plasmids were named as pFH-GFP-shTMPO and pFH-GFP-shCon.
HEK293T cells (1.0×106) were seeded onto 10-cm dishes and cultured for 24 h to reach 70–80% confluence. Two hours before transfection, the medium was replaced with serum-free DMEM. Three plasmids including 10 µg pFH plasmid, 7.5 µg packaging vector pHelper 1.0 and 5 µg expression plasmid pHelper 2.0 were added to 0.95 ml Opti-MEM and 30 µl of Lipofectamine 2000. The mixture was added to the cells and incubated for 6 hrs before replacing the medium with 10 ml of complete DMEM medium (with 10% PBS). Lentiviral particles were harvested at 48 h after transfection. As the lentivirus carries green fluorescence protein (GFP), the viral titer was determined by end-point dilution assay through counting the numbers of GFP-expressing HEK293T cells under fluorescence microscopy at 72 h after transduction.
Western blot analysis
Cells were washed twice with ice-cold PBS and lysed in 2XSDS sample buffer [100 mM Tris-HCl (pH 6.8), 10 mM EDTA, 4% SDS and 10% glycine]. Equal amount of proteins (30 µg) were loaded and separated by electrophoresis (50 V, 3 h) on 10% SDS-PAGE gels. Western blot was performed according to previous report.20 The primary antibodies used were as follows: Anti-TMPO, BAD, Bcl-2, Caspase-3, BCL-xl, PARP, CDK2, CDK4, Cyclin A2, Cyclin B1, Cyclin D1, E-cadherin, N-cadherin, and Vimentin. Horseradish peroxidase-conjugated antibody was used as secondary antibody (1:5,000 dilution, #SC-2054; Santa Cruz Biotechnology, Inc., Dallas, TX, USA). An anti-GAPDH antibody (1:500,000, 10494-1-AP; ProteinTech group, Chicago, IL, USA) was used as the loading control. In addition, 9 proteins including Bad, Bcl-2, cleaved caspase-3, PARP, Cyclin B1, CDK2, Cyclin A2, Cyclin D1 and GAPDH were separated on 10% SDS-PAGE at the same time in different lanes. According to protein molecular weight, SDS-PAGE was cut before transferring electrophoretically onto a PVDF membrane. Bad, CDK2 and cyclin B1 were cut in the same lane at molecular weight 15–25 KD, 25–40 and 40–100 KD. Bcl-2 and cyclin A2 were cut in the same lane at molecular weight 15–35 and 40–70 KD. Cleaved caspase-3 and cyclin D1 were cut at molecular weight 15–25 and 25–40 KD. GAPDH and PARP were cut at molecular weight 15–40 and 55–170 KD.
Flow cytometry analysis
H1299 cells were cultivated in 6-well plates and inoculated with lentiviruses at a MOI of 20. Cells were transferred to 6-cm dishes at a cell density of 2×105 after 3-day incubation. The cells were harvested after 2-day incubation period and then cell apoptosis was determined by Annexin V-APC/7-AAD staining method before cell confluency reached 80% of the dishes. Triple samples were repeated by FAC Scan Flow Cytometer (Becton Dickinson, San Jose, CA, USA) in accordance with the manufacturer’s guideline.
Wound healing assay and transwell invasion assay
H1299 cells stably transduced with Lv-shTMPO were seeded on a 24-well culture plate (Corning, Corning, NY, USA), and the wound healing assay were performed at a cell confluence of 80%. Gently and slowly scratch the monolayer with a new 0.2 ml pipette tip across the center of the well. The resulting gap distance therefore equals to the outer diameter of the end of the tip. After scratching, gently wash the well twice with PBS to remove the detached cells. The cells were grown for an additional 48 hours, then fixed with 4% paraformaldehye for 30 minutes. The photos of cell monolayer were taken under a light microscope. Each experimental group was set in triplicate, and three randomly chosen views were photographed.
Transwell chambers (8-µm pore filter; BD Biosciences, Franklin Lakes, NJ, USA) were coated with Matrigel™ Basement Membrane Matrix (BD Biosciences) and incubated at 37°C for 4 hours to allow solidification. Following 24-hour incubation with Lv-shCon, Lv-shTMPO, 2×105 H1299 cells suspended in serum-free DMEM were added to the upper chamber, and DMEM containing 10% FBS was added to the lower chamber as a chemoattractant. Non-invading cells were removed 48 h ours later, using cotton swabs (Sigma-Aldrich), and invasive cells located on the lower surface of the chamber were then stained with 0.1% crystal violet (Sigma-Aldrich) dissolved in 20% methanol (Sigma-Aldrich). Cell invasiveness was determined by counting the number of penetrating cells in ten random high-power fields under a phase-contrast microscope (A19.2703; Nikon Corporation, Tokyo, Japan) at ×200 magnification.
Xenograft assay of tumors in animals
Six-week-old female Balb/C nude mice were randomly divided into Lv-shCon control group and Lv-shTMPO knockdown group (n=10 in each group). H1299/Lv-shCon or H1299/Lv-shTMPO at 2×106/100 µL cell suspension in PBS was injected s.c. into the dorsal flanks of each mouse using a 25-gauge needle. Tumor volume was measured twice a week post-injection. Tumor weight was measured at the end of experiment after mice were sacrificed.
Statistical analysis
Statistical analysis was carried by graphpad prism software. Group difference was compared by using Student’s t-test or log-rank test, and p<0.05 was considered as statistically significant.
Results
TMPO is overexpressed in non-small cell lung cancer compared to normal tissue
Data mining on mRNA expression profiles between normal and malignant lung tissue sets indicated that TMPO could be one of the candidate biomarkers. In this study, four independent datasets were enrolled to identify the differential TMPO mRNA expression in lung cancer. TMPO was over-expressed in lung squamous carcinoma, adenocarcinoma and large cell carcinoma comparing to normal tissues (p<0.05) in Hou Lung, and in lung adenocarcinoma Okayama Lung, Beer Lung and Garber Lung datasets (Figure 1). These data suggested that TMPO might play a critical role in the transformation of lung cancer, and could be a biomarker for lung cancer.
 | Figure 1 TMPO is upregulated in non-small cell lung cancers. The expression of TMPO was assessed in non-small cell lung cancers using Oncomine database. (A) In Hou Lung dataset, TMPO is overexpressed in large cell lung carcinoma, lung adenocarcinoma, and squamous cell lung carcinoma, than in normal tissue. (B–D) TMPO is also significantly upregulated in Okayama Lung (B), Beer Lung (C), and Garber Lung (D) datasets. Analyzed sample numbers are shown under each category. The p-value was calculated using two-tailed and unpaired Student’s t-test, and shown above each graph. |
High expression level of TMPO had poorer prognosis in lung cancer patients
To further study the importance of TMPO in lung cancer, we analyzed the data from PROGgene database, a gene expression-based survival analysis web application for multiple cancers. PROGgene have compiled publicly available data from repositories such as Gene Expression Omnibus (GEO), EBI Array Express and recently developed “The Cancer Genome Atlas” (TCGA). With a total of 64 datasets from 18 cancer types, PROGgene is considered the most comprehensive prognostic biomarker identification tool to date.21 Using median gene expression value as bifurcating point, samples were divided into High and Low gene expression groups in PROGgene.
The data indicated that patients with high TMPO expression were more likely to have shorter overall survival and relapse free survival than those with low TMPO expression. Among 17 included overall survival datasets, 13 shows longer median survival or relapse free survival in low TMPO group, six of which were statistically significant. Figure 2 shows representative data downloaded from PROGgene that patients with high TMPO expression have shorter overall survival than those with low TMPO expression in GSE11969, GSE13213, GSE17710, GSE30219 and GSE37745 datasets, and that high TMPO expression predicts a poor relapse-free survival in GSE17710 and GSE31210 datasets. Although patients in GSE31210 dataset had 100% overall survival (data not shown), their relapse free survival were significantly different (Figure 2G). In GSE37745 dataset, the patients with high TMPO level had shorter overall survival, although their relapse free survival were not significant. A close inspection of Oncomine and PROGgene databases showed that TMPO is especially overexpressed and predicts poor prognosis in lung adenocarcinoma.
 | Figure 2 TMPO expression is prognostically relevant in lung cancer patients. (A–E) High expression of TMPO correlates with poor overall survival rates in GSE11969, GSE13213, GSE17710, GSE30219, and GSE37745 datasets from PROG–gene database. (F–H) High expression of TMPO correlates with poor relapse survival in GSE17710 and GSE31210 datasets although not in GSE37745 dataset from PROGgene database. Survival curves were compared using log-rank test. |
TMPO knockdown suppresses proliferation of H1299 lung adenocarcinoma cell line
To identify the role of TMPO in human lung cancer, we studied the endogenous expression level in five model types of lung cancer cells H1299, A549, H460, PC-9 and PC-9R (Figure 3). Then, H1299 a lung adenocarcinoma cell line was chosen for further study. H1299 cells were transducted with shCon and shTMPO by lentiviral vector. The transfection efficiency monitored by an independent green fluorescence protein (GFP) expression is around 90% (Figure 4A). Western blot analysis indicated an efficient knockdown of TMPO in H1299 cells (Figure 4B).
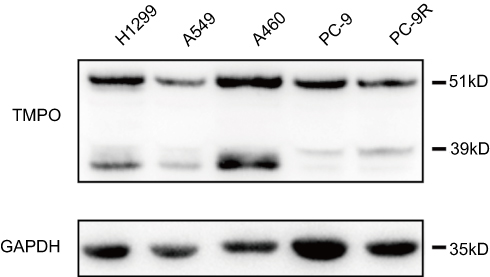 | Figure 3 Expression of TMPO in lung cancer cells. Lung cancer cells were subjected to Western blot analysis for protein expression of TMPO. |
 | Figure 4 Knockdown of TMPO inhibited cell growth and induced cell cycle arrest and apoptosis. (A) H1299 cells were transducted with control vector or shTMPO through lentiviral vector. GFP expression indicated sufficient infection rate of more than 90%. (B) H1299 cells were subjected to Western blotting analysis for TMPO expression. (C) Knockdown of TMPO induced expression alteration of cell cycle and apoptotic regulators. (D) Knockdown of TMPO suppresses proliferation of H1299 lung cancer cells (**p<0.0001). (E) Knockdown of TMPO induces apoptosis in H1299 lung cancer cells (**p<0.0001). (F) Knockdown of TMPO results in G2/M arrest (**p<0.0001). |
MTT assay showed that knockdown of TMPO expression inhibited H1299 proliferation (Figure 4D), probably through induction of apoptosis (Figure 4E, p=0.0004 and <0.0001) and cell cycle arrest at G2/M (Figure 4F). Then, we further studied the molecular profile of cell cycle and apoptotic markers. As shown in Figure 4C, knockdown of TMPO expression resulted in up-regulation of BAD, down-regulation of Bcl-2, and induced a significant cleavage of PARP and caspase-3, indicating an apoptotic activation in H1299 cells. Also, although BCL-xl, CDK2, CDK4, and CyclinA2 were not significantly different between the two groups, TMPO knockdown cause a slight elevation of cyclin D1 and reduction of cyclin B1, which explains why G2/M arrest and lower G1 population were found in TMPO knocked down cells.
TMPO depletion suppresses cell migration as an invasion ability
We further investigated the impact of TMPO on cell mobility. Figure 5A and B showed the wound healing result. Comparing with the control group, the migration ability of TMPO-silencing cells was inhibited (24 hours, p=2.78e−005 and 48 hours, p=2.03e−005, respectively). Matrigel transwell assay indicated that TMPO knockdown suppressed the invasion ability of H1299 cells (Figure 5C and D). To better understand the role of TMPO in lung cancer cells, we studied the expression of EMT markers. As shown in Figure 5E, vimentin was obviously down-regulated, whereas E-cadherin and N-cadherin were slightly up-regulated in shTMPO group compared with the control. These data demonstrated that TMPO might function through the EMT pathway to regulate cell mobility.
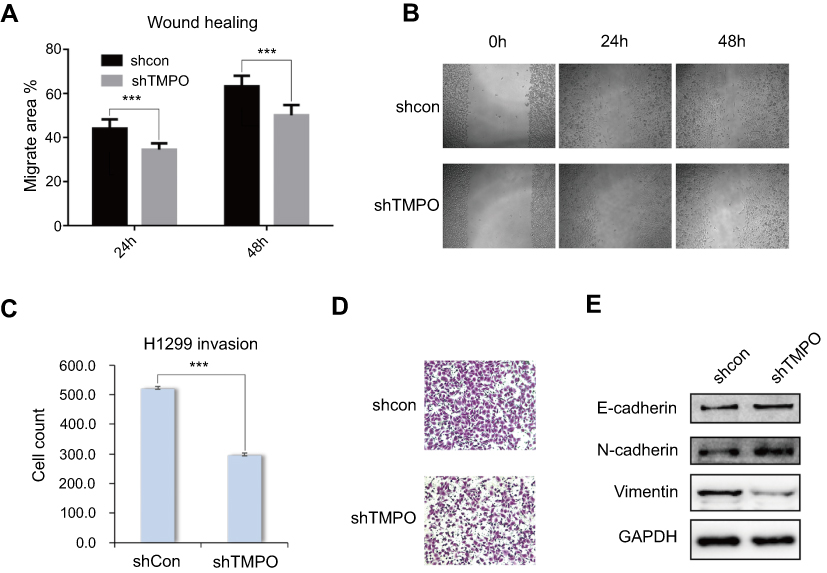 | Figure 5 Knockdown of TMPO expression inhibits the migration and invasion ability of H1299 cells. (A and B) Downregulation of TMPO inhibits the migration ability of H1299 cells by wound healing assay (***p<0.0001). (C and D) Downregulation of TMPO suppresses the invasion ability of H1299 cells by transwell assay (***p<0.0001). (E) Western blot analysis of EMT marker upon TMPO knockdown. |
Silencing TMPO inhibited the tumorigenesis ability of lung cancer cells in vivo
To support the theory we have got in vitro, we performed in vivo study to evaluate the impacts of TMPO expression alteration. In a xenograft nude mice model, H1299/shCon and H1299/shTMPO were injected into nude mice and then the tumor volume was measured twice a week. As shown in Figure 6, the tumor growth ability was significantly inhibited by TMPO knockdown (shCon vs shTMPO) (p=0.0463), accompanied by a reduced final tumor weight (p=0.0441) (5.056±2.014 g vs 0.2200±0.2200 g).
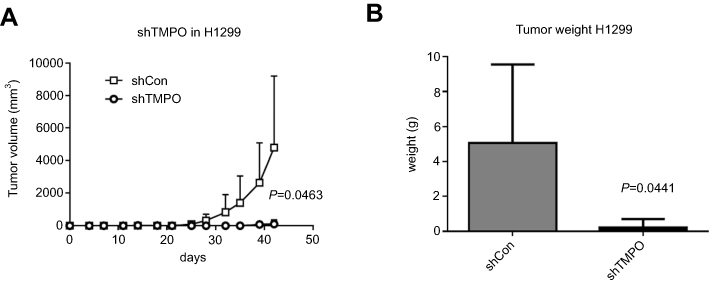 | Figure 6 TMPO knockdown inhibits tumor growth in vivo. (A) Tumor growth curve shows that TMPO knockdown suppresses tumor growth in a xenograft nude mice model (p=0.0463). (B) Final tumor weights were measured after mice were sacrificed. Down-regulated TMPO suppressed tumor growth (p=0.0441). |
Discussion
TMPO/Lap2 is one of LEM domain proteins which are recognized as inner nuclear membrane proteins.22,23 LEM domain proteins play critical roles in DNA replication and regulation of DNA expression. LAP2β, one of LAP2 isoforms, regulates DNA replication in G1 phase of cell cycle by interacting with HA95 protein,24 which was consistent with Kim’s report.13 Here we presented the data demonstrating over-expression of TMPO in lung cancer cells comparing with normal tissues, and down-regulated expression of TMPO inhibited cancer cell growth and invasion ability. This is the first report that expression level of TMPO has significant impacts on lung cancer prognosis.
Metastasis of cancer cells significantly affects the prognosis of cancer disease; and 5-year survival rate is remarkably lower than stabilized tumors.25 The mobility of cancer cells is the key factor of metastasis.26 As a consequence, inhibition of migration is effective in carcinoma therapy. And many migration inhibitors are under investigation for lung cancers.27 In this study, we found that TMPO knockdown could arrest cell cycles at S phase, and with further exploration we noted that Cyclin A2 and CDK2/CDK4 were up-regulated. Cyclin A2 and CDK2/CDK4 were known to promote cell cycle to S phase in mammals.28,29 Cyclin D1 binds with CDK4 and this assembled complex phosphorylated retinoblastoma protein Rb.30 Over-expression of Cyclin A2, CDK2 and Cyclin D1/CDK4 complex will accumulate cells to S phase. On the contrary, Cyclin B1 was observed to be down-regulated after TMPO was depleted. Cyclin B1 is a regulatory protein involved in mitosis, and it binds with CDK1 to promote early mitosis.31 Therefore, down regulation of Cyclin B1 resulted in S phase accumulation. Furthermore, it’s been reported that down-regulated Cyclin B1 was associated with tumor regression,32 which was mutually verified in our present study by lung cancer cell growth rate study, in vitro and in vivo.
Early and late cell apoptosis was observed in our study. To further illuminate the mechanism how expression alteration of TMPO affects cell apoptosis, we detected some molecules. B-cell lymphoma proteins (including Bcl-xl and Bcl-2) play a critical role in regulating cell cycles and act as anti-apoptotic proteins, while Bad proteins act as pro-apoptotic role.33 In some cases as reported, down-regulation of Bcl proteins could induce cell apoptosis in cancer cells;34 and Bad was transcriptionally coordinated with p53 to form a Bad/p53 complex and induced apoptosis.35 In our study, the cooperative decreased Bcl-xl/Bcl-2 level and increased Bad proteins suggested that depletion of TMPO could be a tool to induce cell apoptosis. Meanwhile a significant cleavage of PARP and caspase-3 was detected in our study, indicating that TMPO depletion mediated inhibition of PARP and induces apoptosis in death receptor signaling.
In this study we carried out xerograph tumor studies in mice and noticed that TMPO depletion could inhibit tumor cell growth significantly. Meanwhile, in the transwell study we confirmed that the deficiency of TMPO protein could restrain cancer cell invasive capability. In the screening of potential cellular signal pathway alterations, we have detected that Vimentin was decreased and E-cadherin proteins were up-regulated in TMPO depletion lung cancer cells. Vimentin is a type III intermediate filament (IF) protein that is expressed in mesenchymal cells. Some investigators have reported that Vimentin was highly expressed in invasive tumor cells36–38 and was responsible for the cancer cell invasive capacities.39 Down-regulated vimentin could significantly inhibit the cancer invasion in vitro studies.40,41 The data in the present study suggest that TMPO depletion could down-regulate vimentin expression and inhibit the cancer cell metastatic ability. E-cadherin is a transmembrane glycoprotein that mediates calcium-dependent, homotypic cell-cell adhesion. It’s been reported that decreased expression of E-cadherin was associated with increased invasiveness of breast cancer and numerous studies have demonstrated the importance of E-cadherin in maintaining the normal phenotype of epithelial cells.42–44 In this present study E-cadherin was restored by TMPO depletion and the restoration inhibited cancer cell invasiveness. To summarize, our finding of inhibited cancer cell growth and invasion with the progress of vimentin down-regulation was consistent with these reports.
Conclusion
In conclusion, in this present study we have provided evidence that TMPO was over-expressed in lung cancer cells and played critical roles in motility of cancer cells. Although the detailed mechanism needs to be illustrated in future studies, our presented data suggests TMPO could be a novel prognostic factor and therapeutic target for lung cancer patients.
Compliance with ethical standards
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. Approval of animal research was obtained from School of Basic Medical Sciences, Fudan University. All experiments were performed following relevant named institutional and national guidelines and regulations.
Acknowledgments
This work is supported by the National Natural Science Foundation of China for Youth (No. 81401892) and the National Natural Science Foundation of China (No. 81572246).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sateia HF, Choi Y, Stewart RW, Peairs KS. Screening for lung cancer. Semin Oncol. 2017;44(1):74–82. doi:10.1053/j.seminoncol.2017.02.003
2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi:10.3322/caac.21262
3.
4. Ridge CA, McErlean AM, Ginsberg MS. Epidemiology of lung cancer. Semin Intervent Radiol. 2013;30(2):93–98. doi:10.1055/s-0033-1342949
5. Langer CJ. Epidermal growth factor receptor inhibition in mutation-positive non-small-cell lung cancer: is afatinib better or simply newer? J Clin Oncol. 2013;31(27):3303–3306. doi:10.1200/JCO.2013.49.8782
6. Dechat T, Vlcek S, Foisner R. Review: lamina-associated polypeptide 2 isoforms and related proteins in cell cycle-dependent nuclear structure dynamics. J Struct Biol. 2000;129(2–3):335–345. doi:10.1006/jsbi.2000.4212
7. Berger R, Theodor L, Shoham J, et al. The characterization and localization of the mouse thymopoietin/lamina-associated polypeptide 2 gene and its alternatively spliced products. Genome Res. 1996;6(5):361–370.
8. Furukawa K, Pante N, Aebi U, Gerace L. Cloning of a cDNA for lamina-associated polypeptide 2 (LAP2) and identification of regions that specify targeting to the nuclear envelope. EMBO J. 1995;14(8):1626–1636.
9. Harris CA, Andryuk PJ, Cline S, et al. Three distinct human thymopoietins are derived from alternatively spliced mRNAs. Proc Natl Acad Sci U S A. 1994;91(14):6283–6287.
10. Dual role for LAP2alpha in progeria cell proliferation. J Cell Sci. 2018;131(3):e0302. doi:10.1242/jcs.216507. Available from: http://jcs.biologists.org/content/131/3/e0302. Accessed May 10, 2019.
11. Brachner A, Foisner R. Lamina-associated polypeptide (LAP)2alpha and other LEM proteins in cancer biology. Adv Exp Med Biol. 2014;773:143–163. doi:10.1007/978-1-4899-8032-8_7
12. Somech R, Gal-Yam EN, Shaklai S, et al. Enhanced expression of the nuclear envelope LAP2 transcriptional repressors in normal and malignant activated lymphocytes. Ann Hematol. 2007;86(6):393–401. doi:10.1007/s00277-007-0275-9
13. Kim HJ, Hwang SH, Han ME, et al. LAP2 is widely overexpressed in diverse digestive tract cancers and regulates motility of cancer cells. PLoS One. 2012;7(6):e39482. doi:10.1371/journal.pone.0039482
14. Zhang L, Wang G, Chen S, et al. Depletion of thymopoietin inhibits proliferation and induces cell cycle arrest/apoptosis in glioblastoma cells. World J Surg Oncol. 2016;14(1):267. doi:10.1186/s12957-016-1018-y
15. Rhodes DR, Kalyana-Sundaram S, Mahavisno V, et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9(2):166–180.
16. Hou J, Aerts J, den Hamer B, et al. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS One. 2010;5(4):e10312. doi:10.1371/journal.pone.0010312
17. Okayama H, Kohno T, Ishii Y, et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012;72(1):100–111. doi:10.1158/0008-5472.CAN-11-1403
18. Beer DG, Kardia SL, Huang CC, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8(8):816–824. doi:10.1038/nm733
19. Garber ME, Troyanskaya OG, Schluens K, et al. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci U S A. 2001;98(24):13784–13789. doi:10.1073/pnas.241500798
20. Zhang J, Liang Q, Lei Y, et al. SOX4 induces epithelial-mesenchymal transition and contributes to breast cancer progression. Cancer Res. 2012;72(17):4597–4608. doi:10.1158/0008-5472.CAN-12-1045
21. Goswami CP, Nakshatri H. PROGgene: gene expression based survival analysis web application for multiple cancers. J Clin Bioinf. 2013;3(1):22. doi:10.1186/2043-9113-3-22
22. Lin F, Blake DL, Callebaut I, et al. MAN1, an inner nuclear membrane protein that shares the LEM domain with lamina-associated polypeptide 2 and emerin. J Biol Chem. 2000;275(7):4840–4847.
23. Wagner N, Krohne G. LEM-domain proteins: new insights into lamin-interacting proteins. Int Rev Cytol. 2007;261:1–46. doi:10.1016/S0074-7696(07)61001-8
24. Martins S, Eikvar S, Furukawa K, Collas P. HA95 and LAP2 beta mediate a novel chromatin-nuclear envelope interaction implicated in initiation of DNA replication. J Cell Biol. 2003;160(2):177–188. doi:10.1083/jcb.200210026
25. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60(5):277–300. doi:10.3322/caac.20073
26. Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9(4):274–284. doi:10.1038/nrc2622
27. Palmer TD, Ashby WJ, Lewis JD, Zijlstra A. Targeting tumor cell motility to prevent metastasis. Adv Drug Deliv Rev. 2011;63(8):568–581. doi:10.1016/j.addr.2011.04.008
28. Yin H, Subrata S, Yang W, Yang G. [Overexpression of cyclin A leads to S-phase arrested apoptosis]. Hua Xi Yi Ke Da Xue Xue Bao. 2001;32(2):188–190.
29. Hosooka T, Ogawa W. A novel role for the cell cycle regulatory complex cyclin D1-CDK4 in gluconeogenesis. J Diabetes Investig. 2016;7(1):27–28. doi:10.1111/jdi.12369
30. Feliers D, Frank MA, Riley DJ. Activation of cyclin D1-Cdk4 and Cdk4-directed phosphorylation of RB protein in diabetic mesangial hypertrophy. Diabetes. 2002;51(11):3290–3299.
31. Porter LA, Donoghue DJ. Cyclin B1 and CDK1: nuclear localization and upstream regulators. Prog Cell Cycle Res. 2003;5:335–347.
32. Yuan J, Yan R, Kramer A, et al. Cyclin B1 depletion inhibits proliferation and induces apoptosis in human tumor cells. Oncogene. 2004;23(34):5843–5852. doi:10.1038/sj.onc.1207757
33. Lan T, Zhao H, Xiang B, Wang J, Liu Y. Suture compression induced midpalatal suture chondrocyte apoptosis with increased caspase-3, caspase-9, Bad, Bak, Bax and bid expression. Biochem Biophys Res Commun. 2017;489(2):179–186. doi:10.1016/j.bbrc.2017.05.120
34. Wu S, Zhu H, Gu J, et al. Induction of apoptosis and down-regulation of Bcl-XL in cancer cells by a novel small molecule, 2[[3-(2,3-dichlorophenoxy)propyl]amino]ethanol. Cancer Res. 2004;64(3):1110–1113.
35. Jiang P, Du W, Heese K, Wu M. The Bad guy cooperates with good cop p53: Bad is transcriptionally up-regulated by p53 and forms a Bad/p53 complex at the mitochondria to induce apoptosis. Mol Cell Biol. 2006;26(23):9071–9082. doi:10.1128/MCB.01025-06
36. Liu X, Wu Y, Zehner ZE, Jackson-Cook C, Ware JL. Proteomic analysis of the tumorigenic human prostate cell line M12 after microcell-mediated transfer of chromosome 19 demonstrates reduction of vimentin. Electrophoresis. 2003;24(19–20):3445–3453. doi:10.1002/elps.200305574
37. Singh S, Sadacharan S, Su S, Belldegrun A, Persad S, Singh G. Overexpression of vimentin: role in the invasive phenotype in an androgen-independent model of prostate cancer. Cancer Res. 2003;63(9):2306–2311.
38. Vasko V, Espinosa AV, Scouten W, et al. Gene expression and functional evidence of epithelial-to-mesenchymal transition in papillary thyroid carcinoma invasion. Proc Natl Acad Sci U S A. 2007;104(8):2803–2808. doi:10.1073/pnas.0610733104
39. Wei J, Xu G, Wu M, et al. Overexpression of vimentin contributes to prostate cancer invasion and metastasis via src regulation. Anticancer Res. 2008;28(1A):327–334.
40. Fite K, Gomez-Cambronero J. Down-regulation of microRNAs (MiRs) 203, 887, 3619 and 182 prevents vimentin-triggered, phospholipase D (PLD)-mediated cancer cell invasion. J Biol Chem. 2016;291(2):719–730. doi:10.1074/jbc.M115.686006
41. Wu KJ, Zeng J, Zhu GD, et al. Silibinin inhibits prostate cancer invasion, motility and migration by suppressing vimentin and MMP-2 expression. Acta Pharmacol Sin. 2009;30(8):1162–1168. doi:10.1038/aps.2009.94
42. Behrens J, Mareel MM, Van Roy FM, Birchmeier W. Dissecting tumor cell invasion: epithelial cells acquire invasive properties after the loss of uvomorulin-mediated cell-cell adhesion. J Cell Biol. 1989;108(6):2435–2447.
43. Chen WC, Obrink B. Cell-cell contacts mediated by E-cadherin (uvomorulin) restrict invasive behavior of L-cells. J Cell Biol. 1991;114(2):319–327.
44. Frixen UH, Behrens J, Sachs M, et al. E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J Cell Biol. 1991;113(1):173–185.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.